

Abstract
Whether chromosomes maintain their nuclear positions during interphase and from one cell cycle to the next has been controversially discussed. To address this question, we performed long-term live-cell studies using a HeLa cell line with GFP-tagged chromatin. Positional changes of the intensity gravity centers of fluorescently labeled chromosome territories (CTs) on the order of several μm were observed in early G1, suggesting a role of CT mobility in establishing interphase nuclear architecture. Thereafter, the positions were highly constrained within a range of ∼1 μm until the end of G2. To analyze possible changes of chromosome arrangements from one cell cycle to the next, nuclei were photobleached in G2 maintaining a contiguous zone of unbleached chromatin at one nuclear pole. This zone was stably preserved until the onset of prophase, whereas the contiguity of unbleached chromosome segments was lost to a variable extent, when the metaphase plate was formed. Accordingly, chromatin patterns observed in daughter nuclei differed significantly from the mother cell nucleus. We conclude that CT arrangements were stably maintained from mid G1 to late G2/early prophase, whereas major changes of CT neighborhoods occurred from one cell cycle to the next. The variability of CT neighborhoods during clonal growth was further confirmed by chromosome painting experiments.
Keywords: nuclear architecture; chromosome territories; chromatin dynamics; cell cycle; photobleaching
Introduction
Chromosome order during cell cycle and cell differentiation has become a focus of research to analyze the nuclear architecture and its functional implications (for reviews see Lamond and Earnshaw, 1998; Cremer et al., 2000; Leitch, 2000; Cremer and Cremer, 2001; Parada and Misteli, 2002). Early attempts to study higher order interphase chromosome arrangements were undertaken in the 19th century (Rabl, 1885). In 1909, based on studies of blastomere nuclei in the nematode Parascaris equorum, Theodor Boveri developed the following hypothesis (Fig. 1 ; Boveri, 1909): (1) chromosomes occupy distinct chromosome territories (CTs)* in the cell nucleus; (2) CT order is stably maintained during interphase; (3) changes of chromosome neighborhoods occur during mitosis, in particular during prometaphase, when chromosomes become attached to the spindle and move toward the metaphase plate. Accordingly, chromosome order in the metaphase plate, and hence, CT order during interphase can profoundly vary from one cell cycle to the next; and (4) chromosomal neighborhoods established in the metaphase plate are maintained during anaphase and telophase in the two separated sets of chromatids. This results in a rather symmetrical CT arrangement in the two daughter nuclei.
Figure 1.

Chromosome arrangements in blastomere nuclei of P. equorum (2 n = 2) drawn by Theodor Boveri. (A) The two nuclei above and below each represent a pair of daughter nuclei from blastomeres studied at prophase of the two-cell stage. Chromosome ends are fixed within evaginations of the nuclear envelope. Note that chromosome arrangements and the positions of the evaginations are similar in each pair, whereas different pairs show striking differences. (B) Interphase blastomere cells from an embryo drawn at the four-cell stage. Chromosome arrangements within the nucleus are invisible, except for nuclear evaginations that indicate telomere positions. Each pair of daughter nuclei shows symmetrical positions of the evaginations, whereas a comparison of the two pairs reveals gross differences.
During the last decade, numerous studies provided conclusive evidence for Boveri's first assumption in various animal and plant species. The chromatin masses of individual interphase chromosomes are arranged within distinct territories (for reviews see Cremer et al., 1993; Leitch, 2000; Cremer and Cremer, 2001). With regard to Boveri's second assumption, the evidence is less clear. A number of reports describe a high degree of stability of large-scale chromatin arrangements in mammalian cell nuclei (Shelby et al., 1996; Abney et al., 1997; Zink and Cremer, 1998; Bornfleth et al., 1999; Chubb et al., 2002; Lucas and Cervantes, 2002), but considerable movements of chromosomal subregions, such as centromeres, were observed during the cell cycle or terminal differentiation (Martou and De Boni, 2000; Cremer et al., 2003). Extensive, rapid movements of chromatin were reported for nuclei of budding yeast and Drosophila (for review see Gasser, 2002; Marshall, 2002). Boveri's third assumption was challenged by Nagele et al. (1995), who noted a precise spatial positioning of chromosomes in prometaphase chromosome rosettes from human fibroblasts and HeLa cells, and suggested permanent associations among adjacent chromosomes due to hypothetical centromere interconnections. Such connections could also provide a mechanism for ordered arrangements of CTs during interphase (Koss, 1998; Nagele et al., 1999). However, other groups reported highly variable neighborhoods of both mitotic chromosomes and CTs (Lesko et al., 1995; Allison and Nestor, 1999; Cremer et al., 2001; Habermann et al., 2001). Several studies provided evidence for a nonrandom radial position of specific CTs in the nuclear interior or periphery (Skalnikova et al., 2000; Sun et al., 2000; Boyle et al., 2001; Cremer et al., 2001). Notably, such a radial order is fully compatible with a highly variable neighborhood of chromosomes (Cornforth et al., 2002). For Boveri's fourth assumption, anecdotal evidence has been reported, but a quantitative, high resolution analysis has not yet been provided. Early studies indicated rather symmetrical locations of nucleoli as well as chromocenters in daughter nuclei from a variety of plant species (Heitz, 1932). FISH experiments suggested a considerable degree of symmetry in the arrangement of centromeres and whole CTs in daughter nuclei (Sun and Yokota, 1999; Habermann et al., 2001).
Considering the conflicting evidence for and against Boveri's assumptions, we decided to test his hypothesis in HeLa cells using state of the art live-cell and 3-D FISH approaches. We chose this cancer cell line for two reasons. First, long-term fluorescence observations of living cells at high resolution carry a risk of phototoxic damage that can result in a stop of the cell cycle and trigger apoptosis. This possibility is reduced in cancer cells, such as HeLa, in which cell cycle checkpoints and apoptotic mechanisms are less effective than in diploid cells. Second, the HeLa cell line used in the present studies expresses GFP-tagged histone H2B (Kanda et al., 1998). This allowed us to perform chromatin bleaching experiments; Criss-cross stripes of photobleached chromatin were produced in nuclei at different stages of interphase, and were followed until fluorescence recovered. In case of large-scale CT movements during the recovery period, we expected the destruction of the stripe pattern, whereas its maintenance would suggest a large-scale stability of CT positions. To analyze possible changes of chromosome arrangements during mitosis, we partially bleached nuclear chromatin in mother cell nuclei during G2 and followed unbleached chromatin from mother nuclei to their daughter nuclei. In case chromosome positions were globally inherited through mitosis, we expected the faithful restoration of the nuclear topology of GFP-labeled chromatin.
Fluorescence recovery of bleached chromatin within a time window of a few hours (Kimura and Cook, 2001) does not allow the detection of very slow, long-range interphase movements of CTs. For a study of long-range movements, we performed fluorescence labeling of a small number of CTs in nuclei of living cells (Schermelleh et al., 2001) and studied their arrangements at all cell cycle stages. 3-D FISH experiments with chromosome paint probes in HeLa cell clones at the two- and four-cell stage allowed us to assess a potential symmetry in daughter cell nuclei, as well as major differences of chromosome arrangements already recognizable at the four-cell stage. The results of these experiments provide evidence that Boveri's hypothesis is valid for HeLa cells.
Results
CT order is stably maintained during interphase
To detect or exclude large-scale CT movements, we produced criss-cross stripes of photobleached chromatin in HeLa cell nuclei with GFP-tagged histone H2B at different stages of interphase (Fig. 2) . In the case of large-scale movements, the stripe pattern should become destroyed. In spite of translational and rotational nuclear movements, the stripe pattern was maintained until it became invisible after 1–2 h due to the replacement of bleached H2B-GFP by unbleached H2B-GFP (Fig. 2 C). This experiment provided evidence for the stability of large-scale CT arrangements during this period.
Figure 2.

Stability of large-scale CT arrangements during interphase of HeLa cells studied by nuclear stripe photobleaching experiments. Cross-stripe (rows A and B) or mesh-like (row C) geometrical patterns bleached into HeLa cell nuclei with GFP-tagged H2B at different stages of the cell cycle. The patterns were maintained until recovery of GFP-fluorescence (1–2 h). (A) Nucleus bleached in G1 (2 h and 20 min after prophase of the mother nucleus). (B) S-phase nucleus. The nucleus was labeled with Cy5-dUTP before bleaching by scratch-replication labeling of the cell, and shows a labeling pattern typical for early S-phase (inset). Stripe photobleaching was performed 2 h later, ensuring that the cell was in middle S-phase at that time. (C) Late S/G2 nucleus. The cell entered mitosis 3 h after stripe bleaching. Note that the mesh-like pattern was stably maintained (arrows), whereas the nucleus rotates. All images present best focus light-optical sections. Bars, 10 μm.
To study the question of whether major changes of CT positions in interphase nuclei may occur during more extended time periods, we made use of a scratch-replication labeling protocol with Cy3-dUTP (Schermelleh et al., 2001). The fluorescent nucleotides, which enter the cell during S-phase, are incorporated into newly synthesized DNA for roughly 1 h. Accordingly, each CT was represented only by a fraction of chromatin foci, called ∼1-Mb chromatin domains (Cremer and Cremer, 2001), that were replicated during the labeling period. After labeling, cells were grown for 5–8 additional cell cycles. During the second and subsequent post-labeling mitoses, Cy3-labeled and unlabeled chromatids were segregated, resulting in nuclei with a steadily decreasing number of labeled CTs. Clusters of domains were assigned to represent a single CT, and the 3-D coordinates of the common intensity gravity center were calculated to represent its nuclear location. It is possible that a larger signal cluster was occasionally assigned to one CT, although it represented several neighboring CTs. Small signal clusters, which consisted of few ∼1-Mb chromatin domains, possibly resulted from sister chromatid exchanges, and therefore represented subchromosomal fragments. In spite of these limitations, the calculated 3-D coordinates served our purpose, namely to distinguish whether individual CTs in HeLa cell nuclei can move to remote nuclear locations during interphase. The 3-D intensity gravity center of the GFP-tagged nuclear chromatin served as the 3-D center of the nucleus (CN). 3-D CT-CN distances, as well as 3-D CT-CT distances were measured at different time points of the total observation period.
24 cells exhibiting nuclei with 1–4 labeled CTs (Table I) were chosen for the analysis of CT movements by time-lapse confocal microscopy with observation periods covering major parts up to a complete HeLa cell cycle of 18–20 h. Evaluation periods in Table I represent the part of the total observation period used for a quantitative analysis. Light-optical nuclear image stacks were sampled at high resolution with intervals between 6 and 30 min. The simultaneous recording of H2B-GFP–tagged chromatin allowed us to monitor translational and rotational nuclear movements as well as changes in nuclear morphology, and to measure the increase of the nuclear volume during interphase. In 12 cells, the observation of a mitotic event preceded an evaluation period that covered G1 to mid S-phase. In 8 cells, a mitotic event resulting in two inconspicuous daughter nuclei was observed after the end of the evaluation period. In these cases, the end of telophase or the beginning of prophase was used as an indicator to estimate the interphase stages, where CT movements were analyzed (Table I), taking into account the approximate length of each cell cycle stage determined for HeLa cells (see Materials and methods). In two cases (Table I, nucleus 10 and 11), the total observation time encompassed two mitotic events. Two cells (Table I, nucleus 15 and 16) did not undergo mitosis during an observation period of 10 h, but showed a nuclear volume increase typical for S-phase.
Table I. Nuclei included in the evaluation of CT-CT and CT-CN distances.
| Nucleus | Approximate cell cycle phases covered |
Start of evaluation after telophase (+) before prophase (−) |
Evaluation period | Volume increase during evaluation |
|---|---|---|---|---|
| h:min | h:min | % | ||
| Fixed | – | 6:00 | +1 | |
| 1d1 | Telophase–early G1 | +0:00 | 2:00 | +116 |
| 2d1 | Telophase–early G1 | +0:00 | 2:15 | +118 |
| 3d1 | Telophase–early G1 | +0:00 | 2:15 | +82 |
| 4 | Telophase–early G1 | +0:00 | 2:36 | +78 |
| 5 | Early G1 | +0:12 | 2:00 | +46 |
| 6d1 | Early G1 | +0:15 | 2:45 | +45 |
| 7 | Early G1 | +0:10 | 2:20 | +42 |
| 8 | Early G1 | +0:15 | 1:30 | +37 |
| 1d2 | Early G1 | +0:15 | 1:45 | +29 |
| 9 | Early G1 | +0:12 | 1:36 | +22 |
| 10 | Early G1 | +0:10 | 2:20 | +25 |
| 11 | Early G1 | +0:10 | 2:40 | +20 |
| 9 | Mid G1 | +1:48 | 2:12 | +7 |
| 2d1 | Mid G1–late G1 | +2:15 | 3:00 | +4 |
| 6d1 | Mid G1–late G1 | +3:00 | 3:00 | +4 |
| 12 | Mid G1–early S | +2:30 | 5:00 | +23 |
| 4 | Mid G1–early S | +2:36 | 5:24 | +22 |
| 5 | Mid G1–early S | +2:12 | 6:00 | +20 |
| 13 | Mid G1–early S | −11:30 | 6:00 | +16 |
| 8 | Mid G1–mid S | +1:45 | 10:00 | +26 |
| 10 | Mid G1–late S/G2 | +2:30/−17:20 | 12:20 | +45 |
| 11 | Late G1–late G2 | +2:50/−17:00 | 16:30 | +63 |
| 14 | Mid G1–late G2 | −15:15 | 15:00 | +55 |
| 3m | Mid G1–late G2 | −13:30 | 13:00 | +48 |
| 15 | LateG1/early S–G2 | ND | 10:00 | +55 |
| 16 | LateG1/early S–G2 | ND | 10:00 | +49 |
| 1m | Late S–late G2 | −7:45 | 7:30 | +36 |
| 2m | Mid S–late G2 | −5:00 | 4:45 | +27 |
| 6m | Late S–late G2 | −4:00 | 3:45 | +19 |
| 17 | Late S–late G2 | −4:00 | 3:45 | +18 |
| 18 | Late S–late G2 | −3:30 | 3:20 | +12 |
| 19 | Early–late G2 | −3:06 | 3:00 | +1 |
Each nucleus is named by an index number (column 1). Nuclei are arranged according to the cell cycle periods covered by the evaluation periods (column 2). Nuclei that were evaluated both during early G1 and later cell cycle stages are listed twice. In five of the nine nuclei recorded during early G1, the telophase stage could not be included in the evaluation because this stage was not completely recorded in z-direction. In four cases, where mother and daughter cell nuclei were evaluated, the mother nucleus is indicated by m, daughter(s) by d1 (and d2). The start of the evaluation period is given in hours after telophase (+) or before prophase (−; column 3). Column 4 provides the length of evaluation period. Column 5 shows the increase of the nuclear volume between the first and the last evaluated time point. A control nucleus was imaged for a period of 6 h after fixation with 4% formaldehyde.
For a typical example, Fig. 3 (A–D) shows the results obtained from nucleus 3m (Table I), which was evaluated from late G1 to late G2 over a time period of 13 h. Optical image stacks from the Cy3 and GFP channels were simultaneously recorded every 15 min. 13.5 h after the start of observation, the cell went into mitosis, yielding two inconspicuous daughter cells. During the evaluation period, the nuclear volume increased from 1,200 μm3 to 1,780 μm3 (Fig. 3, A and B). From a total of 52 3-D image stacks which were recorded during this time, 27 time-image stacks with intervals of 30 min were used for a quantitative analysis of CT-CN and CT-CT distances. The results of these measurements are shown in Fig. 3, C and D. The differences between the maximum and minimum distances, Δ(Max-Min), were taken as a measure of motility. Absolute distances fluctuated and increased slightly over time for distantly located CTs (Fig. 3 C). This increase was likely a passive effect caused by the increase in nuclear volumes because it was no longer noticeable after correction for the nuclear diameter (Fig. 3 D; see Materials and methods). Accordingly, the corrected Δ(Max-Min) values were smaller than the absolute values (compare Fig. 3 C with Fig. 3 D). Fig. 3 D demonstrates that the relative positions of CTs were stably maintained during the entire evaluation period.
Figure 3.

Stability of large-scale CT arrangements studied in nuclei with Cy3-labeled CTs. (A–D) Confocal time-lapse series of a HeLa cell (Table I, nucleus 3m) with replication-labeled CTs and its daughters recorded for a total observation period of 18 h. CT movements in the mother cell nucleus were analyzed from late G1 to late G2 (13-h evaluation period). Time point 0:00 indicates the start of evaluation. (A) Superimposed maximum intensity projections of confocal serial sections from H2B-GFP (green) and Cy3-dUTP (red) together with a transmission image at selected time points (arrowheads in A point to nuclei reconstructed in B). The start of evaluation was assigned to late G1 as the cell entered prophase 13.5 h later. (B) Top and side view of 3-D reconstructions for time points 0:00 (h:min), 4:30, 8:30, and 12:30. Nuclei (outlined in gray) are displayed after correction for rotational movements, and nuclear volumes are indicated. Four signal clusters representing different CTs are shown in different colors for easier discrimination. (C and D) Absolute and corrected distances between the fluorescence gravity center of each CT and the CN (CT-CN), as well as distances between all pairs of CTs (CT-CT) were evaluated at intervals of 30 min. Δ(max-min) indicates the difference between the maximum and minimum value of the respective distances within the complete evaluation period. (E–H) Confocal time-lapse series from another HeLa cell (Table I, nucleus 4) recorded with a time interval of 6 min. The 8-h evaluation period started at telophase (time point 0:00) and ended likely at early S-phase (8:00). The mother cell performed mitosis at time point −0:30. (E) Maximum intensity projections of confocal serial sections as described in A for selected time points (arrowheads in E point to nuclei reconstructed in F). (F) Top and side view of 3-D reconstructions from the evaluated nucleus containing 3 CTs. (G and H) Absolute and corrected distances as described above for C and D. CT-CN and CT-CT distances were evaluated at intervals of 12 min (early G1) and 30 min (mid G1–early S). The transition from early G1 to mid G1 is defined as the time point when the nuclear volume increase reaches a first plateau (see Materials and methods). Note that the higher fluctuation of distances in early G1 as compared with other cell cycle stages. Arrowheads in A and E indicate nuclei shown as 3-D reconstructions in B and F. Bars, 10 μm.
Fig. 3 (E–H) shows nucleus 4 (Table I), which exemplifies the analysis of CT movements covering a period of 8 h from telophase into S-phase. From a total of 80 3-D image stacks recorded at intervals of 6 min, 25 image stacks with intervals of 12 min during early G1 and of 30 min from mid G1 to early S-phase were taken for a quantitative evaluation. Fig. 3 (G and H) provides absolute and corrected CT-CN and CT-CT distance measurements. Δ(Max-Min) values measured during early G1 were higher than the values measured from mid G1 to early S. The Δ(Max-Min) values determined for nucleus 4 during mid G1 to early S were similar to the values noted for nucleus 3m from mid G1 to late G2 (Fig. 3, C and D). Accordingly, more extensive CT movements can occur during early G1.
Table II summarizes the results from 3-D distance analyses of 24 cells and confirms the findings described above for the two example nuclei. Movements of labeled CTs were locally constrained during interphase. However, Δ(Max-Min) values, which express the variability of the measured CT-CN and CT-CT distances, were significantly larger during early G1 (ranging from 0.47 to 4.44 μm for corrected CT-CT distances; 7/19 (37%) of the values were >2 μm), compared with subsequent interphase stages (0.25–2.11 μm for corrected CT-CT distances; only 1/45 (2%) of these values were >2 μm). Notably, the mobility of individual CTs during early G1 varied largely (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200211103/DC1). From mid G1 to late G2, absolute distances increased slightly over time, reflecting the increase in nuclear volume. During early G1, a significantly larger variability was noticed for both absolute and volume-corrected distances. Furthermore, in spite of the increase in nuclear volume, we observed occasional CT-CT and CT-CN distances that became even smaller during early G1 (Fig. 3, G and H). These findings indicate that the more pronounced CT movements during early G1 do not simply reflect the rapid increase of nuclear volume after telophase, but rather the movements of CTs to their final nuclear locations, which are then maintained within a range of 1 μm (maximum ∼2 μm) from mid G1 to late G2. This corresponds to a radius of confinement of 0.5–1 μm.
Table II. Summary of CT-CN/CT-CT distance measurements.
| # nuclei | Δ(Max-Min) CT-CN
|
Δ(Max-Min) CT-CN
|
||||
|---|---|---|---|---|---|---|
| Absolute | Corrected | Absolute | Corrected | |||
| μm | μm | μm | μm | |||
| Fixed | 1 | Median (range) N(Δ >2 μm)/N total |
0.09 (0.07–0.10) 0/3 |
0.11 (0.07–0.14) 0/3 |
0.15 (0.09–0.22) 0/3 |
0.16 (0.15–0.20) 0/3 |
| Telophase–early G1 | 12 | Median (range) N(Δ >2 μm)/N total |
1.34 (0.30–2.46) 6/25 (24%) |
1.26 (0.42–2.75) 3/25 (12%) |
1.76 (0.48–4.10) 8/19 (42%) |
1.53 (0.47–4.44) 7/19 (37%) |
| Mid G1–late G2 | 20 | Median (range) N(Δ >2 μm)/N total |
1.06 (0.32–1.76) 0/48 (0%) |
0.90 (0.24–1.53) 0/48 (0%) |
1.26 (0.40–2.59) 4/45 (9%) |
1.04 (0.25–2.11) 1/45 (2%) |
| P (median) P (Δ >2 μm) |
0.047 0.001 |
0.001 0.037 |
0.135 0.004 |
0.01 0.001 |
||
For each nucleus, distances between intensity gravity centers of CT pairs (CT-CT) as well as distances between CTs and center of nucleus (CT-CN) were measured. Δ(Max-Min) values are provided as a measure of CT motilities and represent the difference between the maximum and minimum CT-CN or CT-CT distances measured for each CT or CT pair during the indicated cell cycle phases. In addition to Δ(Max-Min) values calculated for absolute distances, the respective values for distances corrected for changes in total nuclear volume are presented. Displayed are the median values of Δ(Max-Min) from all CTs and the smallest and largest Δ(Max-Min) value (range) for a fixed control nucleus, for 12 nuclei in telophase–early G1, and for 20 nuclei in mid G1–late G2. The number of Δ(Max-Min) values exceeding 2 μm, N(Δ >2 μm), and the total number of measured Δ(Max-Min) values (N total) are shown. P-values indicate the degree of significance of the differences in median and N(Δ >2 μm) between nuclei in telophase–early G1 and nuclei in mid G1–late G2. The Δ(Max-Min) values obtained for a fixed control nucleus are within one magnitude of order smaller and indicate a minor contribution of mechanical instability in the microscope system. An additional table with all Δ(Max-Min) values is presented as online supplemental material.
In a further experiment, a two-color scratch-replication labeling approach was performed. During the first S-phase, mother nuclei were labeled with Cy3-dUTP. During the next S-phase, daughter nuclei were labeled with Cy5-dUTP. Segregation of labeled and unlabeled chromatids during a post-labeling growth of cells for 4 d resulted in nuclei with a subset of CTs stained in different colors (Fig. 4 ; Video 2). During observation periods of several hours neighboring, differently colored CTs moved repeatedly closer and further apart, confirming locally constrained CT movements in the order of 1 μm. We observed only very little color mixing, indicating the separation of the bulk DNA from different CTs. However, the sensitivity of this approach was not sufficient to exclude intermingling of a fraction of chromatin loops.
Figure 4.

Stability of CT neighborhood during interphase of living HeLa cells. HeLa cells were replication-labeled during S-phase of two consecutive cell cycles (first cycle, Cy3-dUTP, false color red; second cycle, Cy5-dUTP, false color green). Frames of maximum intensity projections from light optical serial sections are displayed for the indicated time points. Insets show GFP signals of confocal nuclear midsections in gray. Images are corrected for translational and rotational nuclear movements. Observation started 4.5 d after the second labeling event. After 3h 50 min, the cell entered prophase. Note that chromosome condensation is a locally confined process with little changes of chromosome arrangements (compare C with D). After completion of mitosis, daughter nuclei were followed for another 3.5 h. Locally constrained movements, but no major rearrangements of differently colored chromatin domains were detected (see Video 2). Bar, 5 μm.
Changes of chromosome neighborhoods occur during mitosis
To find out whether higher order chromatin arrangements change from one cell cycle to the next, we synchronized HeLa cells in early S-phase. 5–6 h after release from the block, when most of the cells were in late S-phase or had entered G2 (Fig. 5 , column I), we bleached the GFP-tagged chromatin, leaving a single, contiguous zone of unbleached chromatin at one nuclear pole (Fig. 5, column II). The cells were then followed by time-lapse confocal microscopy through mitosis into the next G1 phase. 34 cells yielded daughter nuclei with sufficient contrast of unbleached chromatin patches, reflecting the local decondensation of unbleached chromosome segments. 10 cases were excluded, because one or both daughter cells showed nuclei with morphological abnormalities. The remaining 24 cases were analyzed in detail.
Figure 5.

Large-scale CT arrangements in HeLa cell nuclei change from one cell cycle to the next. A–C show examples of live-cell confocal image series from three HeLa cells (for movie sequences, see Video 3). After bleaching of GFP-labeled chromatin, except for a contiguous unbleached region at one nuclear pole, cells were followed through the remaining part of interphase and through mitosis until the formation of daughter nuclei. All images are maximum intensity projections of confocal image stacks. The degree of the redistribution of unbleached chromatin patches in G1 daughter cells (column VI, arrowheads) can vary from “clustered” (A) to “partially clustered” (B) to “scattered” (C). Column I, GFP pattern of HeLa nuclei before bleaching; column II, the same nuclei after partial bleaching; column III, last frame recorded before the formation of the metaphase plate; column IV, metaphase plate. Arrowheads in B and C point to unbleached chromosomal fragments that are remote from the bulk of unbleached chromatin. Columns V and VI, daughter nuclei in anaphase (column V, C), telophase, or very early G1 (column V, A and B) and in early G1 (column VI). Bars, 10 μm.
Until the onset of prophase, the area of unbleached nuclear chromatin retained its location and shape at the nuclear pole. At prophase, several unbleached chromosomal segments became visible within this zone (Fig. 5 C, column III). Mitotic rosettes were typically arranged perpendicular to the surface on which the cells grew. In 9 rosettes, unbleached chromosome segments clustered within a single area (Fig. 5 A, column IV), whereas in 13 rosettes, some or most of these segments were observed in distant locations (Fig. 5, B and C, column IV). Two rosettes could not be evaluated, as the light-optical serial sections did not cover the unbleached segments.
In the 48 daughter cell nuclei, the degree of clustering of unbleached chromatin was scored on maximum intensity projections. 20 nuclei showed a nuclear subregion with a single cluster of unbleached chromatin patches (Fig. 5 A, column V and VI). These cases reflect the best restoration of CT order between the mother nucleus and its daughter nuclei. Nevertheless, the restoration was far from complete because patches of unbleached chromatin were separated by zones of bleached chromatin, suggesting that bleached chromosome segments had moved between unbleached chromosome segments. In 15 daughter cells, most unbleached chromatin patches were clustered, but some patches were located in a remote nuclear area (Fig. 5 B, columns V and VI). In 13 nuclei, patches of unbleached chromatin were distributed over the major part of the nucleus (Fig. 5 C, column VI). Controls excluded the possibility that fluorescent patches simply reflected areas of high chromatin density rather than unbleached chromosome segments. DNA staining of fixed daughter cells with propidium iodide (PI) showed no spatial correlation of intense PI signals with sites of strong GFP fluorescence. In nuclei, which were bleached completely and followed through mitosis, fluorescent patches were not detected (unpublished data). These experiments demonstrate that significant changes of CT order occur during mitosis.
In addition to the visual analysis, we developed a procedure to quantify the degree of nuclear clustering of unbleached chromatin. Pixel fractions with the 10 and 2% highest intensity values, respectively, were thresholded for a representation of the unbleached chromatin (Fig. 6 A). Distances between all possible pairs of pixels representing unbleached chromatin were determined and normalized to the size of the nucleus (relative distance d, see Materials and methods). Distance values were grouped into intervals of increasing relative distances. As a measure of the frequencies of pairs of pixels belonging to each interval, the radial autocorrelation function (RAC) was established (see Materials and methods and Fig. S1). Fig. 6 (B and C) shows the average RAC calculated for both pixel fractions for the 24 mother nuclei and 48 daughter nuclei. Compared with the curve found for mother nuclei, the curve for daughter nuclei shows a decrease of smaller relative distances combined with an increase of larger relative distance values. For a nonparametrical, statistical test (see Materials and methods), median values were calculated from the mean relative distances <d> obtained for each of the 24 mother nuclei immediately after bleaching and before the onset of prophase, and for the 48 daughter nuclei. The comparison of the median values obtained for mother nuclei immediately after bleaching and before the onset of prophase showed a slight increase (4–7%). This increase was significant for the 2% highest intensity pixel fraction (P = 0.03) and suggests minor chromatin movements (Table III). In contrast, the comparison of the median values from mother and daughter nuclei showed a marked (70–95%) and highly significant (P < 0.001) increase. Because daughter nuclei with scattered, unbleached chromatin patches might have contributed decisively to this significance level, we retested the 20 daughter nuclei showing a single cluster of unbleached chromatin patches. Still, a marked (45–60%) and highly significant difference (P < 0.001) was obtained. Our RAC analysis fully confirmed the conclusion derived from the visual inspection; the positions of unbleached chromosome segments studied in daughter nuclei differed significantly from the positions in the mother cell nuclei.
Figure 6.

RAC of the distribution of unbleached chromatin in mother and daughter cell nuclei. (A) Projections of the mid-nuclear optical sections from a mother nucleus immediately after partial bleaching (I) and shortly before the onset of prophase (II), as well as from its two daughters in early G1 (III and IV; compare with Figure 5 C). Nuclear area shown in dark gray. Pixels of the 2% highest intensity fraction shown in black color belong exclusively to the unbleached zone of the mother nucleus, but do not completely cover it. Pixels of the 10% highest intensity fraction shown in white color fully cover the unbleached zone, but include a few pixels from chromatin in the bleached nuclear area (B and C). Average RAC curves calculated from the 10 and 2% highest intensity fraction for 24 mother cell nuclei immediately after bleaching and before prophase, as well as for the 48 daughter cell nuclei. For comparison, the RAC curve for mother nuclei immediately before bleaching is shown, taking into account all nuclear pixels. SDs are within the same order for mother and daughter cell nuclei, and are only shown for the latter.
Table III. Mean relative distances of high intensity chromatin fractions in partially bleached nuclei.
| Median value of <d> (range of <d>)
|
||
|---|---|---|
| 10% highest intensity pixel fraction | 2% highest intensity pixel fraction | |
| 24 mother nuclei after bleaching | 0.179 (0.161–0.209) | 0.116 (0.082–0.184) |
| 24 mother nuclei before prophase | 0.187 (0.161–0.219) | 0.124 (0.087–0.207) |
| 48 daughter nuclei (total) | 0.321 (0.183–0.612) | 0.243 (0.088–0.596) |
| 20 daughter nuclei with a single cluster of unbleached chromatin patches |
0.274 (0.183–0.376) | 0.196 (0.088–0.346) |
Median values and ranges of the mean relative distances for the 10 and 2% high intensity pixel fractions from 24 mother and 48 daughter nuclei. 20 daughter nuclei exhibiting a single cluster of unbleached chromatin patches.
In conclusion, the experiments described in this section support Boveri's hypothesis that changes of chromosome neighborhoods occur when cells pass through mitosis. In the majority of cases (18 of 24), both daughter nuclei showed the same pattern of unbleached patches, suggesting that anaphase–telophase chromosome movements retain a certain level of symmetry. However, in six cases, scattering of unbleached chromatin patches was much more pronounced in one daughter nucleus than in the other (unpublished data). We suggest that different movements of unbleached sister chromatid segments during anaphase–telophase or in early G1 nuclei contributed to this apparent loss of symmetry.
Changes of CT arrangements during clonal growth
To study the variability of arrangements of specific CTs in two-cell clones from independent mitotic events, as well as between the two pairs of daughter nuclei in four-cell clones, we performed two-color 3-D FISH experiments on HeLa cell clones fixed at these stages. Painting probes for chromosomes #7 and #10 were chosen because these chromosome types were present as three free copies and not involved in translocations (unpublished data). Confocal serial sections were obtained from 12 two-cell clones and 10 four-cell clones, and projections were visually compared for translational and mirrorlike similarities of CT arrangements (for examples see Fig. 7 and 8) . Additionally, 3-D reconstructions from daughter nuclei were made, which could be freely rotated allowing the comparison of their CT arrangements from any angle (Fig. 8 B; Videos 4–7). In nuclei of two-cell clones, the CT arrangements showed an obvious (albeit far from perfect) symmetry in most clones, whereas different mitotic cells yielded daughter nuclei with largely different CT arrangements (Fig. 7). In four-cell clones, we could identify pairs of nuclei that showed a notable symmetry of their CT arrangements, whereas strong differences were noted between the two pairs (Fig. 8). In agreement with Boveri's findings (Fig. 1 B), CT arrangements in four-cell clones already differed largely.
Figure 7.

CT #7 and #10 arrangements in nuclei of two-cell clones. Projections of confocal image stacks obtained after painting of chromosome #10 (visualized in red) and #7 (visualized in green). DNA counterstain, blue. A, B, and D represent daughter nuclei with an obvious (although not perfect) symmetry of the relative CT arrangements. F provides an example for a large deviation from a symmetrical arrangement. To facilitate assessment of symmetry, some of the nuclei were rotated and/or mirrored. Bars, 10 μm.
Figure 8.

CT #7 and #10 arrangements in a four-cell clone. (A) Projection of a confocal image stack through nuclei n1 to n4 after painting of chromosome #10 (visualized in red) and #7 (visualized in green). DNA counterstain, blue. (B) 3-D reconstructions of n1 to n4 by volume rendering. Part of the nuclear border is indicated by reconstruction of the counterstain periphery (outside, blue; inside, silver-gray). The top row shows the bottom view, the bottom row the corresponding side view. To show the symmetry CT arrangements in nuclei n1 and n2 more clearly, the x-axis of n2 was inverted. Bar, 10 μm. For interactive videos of nuclear reconstructions, see supplemental information.
Discussion
The present work demonstrates that the location of CTs is stably maintained in HeLa cell nuclei from mid G1 to late G2, but subject to major changes from one cell cycle to the next. Our findings confirm Theodor Boveri's early hypothesis for HeLa cells. In addition, they show that CT movements during early G1 play a role in the final placement of CTs in interphase nuclei.
Chromatin dynamics in interphase nuclei
In the present work, we demonstrate the stability of large-scale interphase chromatin arrangements in HeLa cells using several live-cell approaches. First, bleached cross-stripe patterns produced in histone H2B-GFP–tagged chromatin at different interphase stages were maintained during fluorescence recovery (1–2 h). Zones of unbleached chromatin produced at nuclear poles of late S or G2 nuclei retained their location and shape until the onset of mitosis. These results are consistent with results from Abney et al. (1997), who demonstrated the local persistence of bleached spots in dihydroethidium-stained chromatin of mammalian cells over at least 1 h. Second, our long-term in vivo study of Cy3- or Cy5-labeled CTs showed that CTs were confined within a radius of ∼0.5–1 μm from mid G1 to late G2, whereas more extended positional changes (occasionally exceeding 4 μm) were observed during the first 2–3 h after mitosis. Csink and Henikoff (1998) described heterochromatic associations involving large-scale reorganization of chromosomes during G1 in Drosophila larval nuclei. In a study of painted CTs in fixed human diploid fibroblast nuclei, Bridger et al. (2000) found that the final locations of CT #18 were established during the first 2–4 h of G1. Evidence for an increased mobility during early G1 compared with later interphase stages was presented for GFP-tagged centromeres in living HeLa cells and of other chromosomal sites in CHO cells (Shelby et al., 1996; Tumbar and Belmont, 2001). Together, these results support the idea of a time window of increased chromatin mobility during early G1 until structural components, essential for the stability of CT arrangements, are fully established. More extensive movements during early G1 may also reflect a necessity of CTs to home in to their final nuclear locations and to establish a “polarized” orientation (Sadoni et al., 1999). In all normal and malignant mammalian cell types studied so far, gene-poor chromatin, which replicates in mid S, is preferentially arranged in the nuclear periphery and around the nucleoli, whereas gene-dense, transcriptionally active early replicating chromatin is located in a zone that expands between these two transcriptionally silent compartments (Dimitrova and Berezney, 2002).
A comparison of published studies of chromatin movements in nuclei of living cells should be performed with caution. Long-term measurements of constrained movements of whole CTs performed in the present work with sampling intervals between 6 and 30 min for total observation periods of many hours cannot be directly compared with movements of small chromatin sites analyzed in other cell systems using intervals of minutes or seconds and for much shorter evaluation periods. For lac-operator repeats integrated into different chromosomal sites of human HT-1080 cells, Chubb et al. (2002) found that the maximum range of movements depended on the subnuclear localization with 0.9 μm for a site located in the nuclear periphery and 1.5 μm for a nucleoplasmic site. Diffusion coefficients were in the range of 10−4 μm2/s or lower. In HeLa cell nuclei, initial evaluations of diffusion coefficients for ∼1-Mb chromatin domains and entire CTs recorded with sampling intervals of 20 min hint to values in the order of 10−5 μm2/s (Bornfleth et al., 1999; Edelmann et al., 2001). A higher mobility with diffusion coefficients in the order of 10−3 to 10−2 μm2/s was reported for fluorescence tagged chromosomal sites in Drosophila and yeast (for reviews see Gasser, 2002; Marshall, 2002). In Drosophila early embryos, confinement radii ≤ 0.9 μm were reported for a heterochromatic site (Marshall et al., 1997). In Drosophila spermatocyte nuclei, Vazquez et al. (2001) described long-range motions of integrated lac-operator sites confined to a radius of 3 μm in mid G2 and of ∼0.3 μm in late G2 nuclei. In yeast, the movements of centromeric and telomeric sites were confined to radii ≤ 0.3 μm independent of the cell cycle. In contrast, chromosomal regions close to transcribed genes were found to be less constrained in G1 with a radius of 0.7 μm, showing particularly fast movements of up to 0.5 μm within 10 s. In S-phase, these movements became constrained to a radius ≤ 0.3 μm through a replication-dependent mechanism (Heun et al., 2001). In our HeLa cell study, we did not see changes in the confinement of CTs during S- and G2-phase.
It was shown that motility of a measured target depends on its chromosomal localization (Heun et al., 2001) and its nuclear position (Chubb et al., 2002). The latter paper reported a lower chromatin mobility near the nucleoli and the nuclear periphery, indicating a role of these structures in maintaining the 3-D arrangements of chromatin. Although the lamina-coated nuclear envelope is a proven site for chromatin attachments (Paddy et al., 1990), it is not clear whether the tethering of chromatin to a matrix of branched matrix core filaments plays a major role in constraining chromatin movements (Ma et al., 1999; Cremer et al., 2000). As an alternative, we consider the possibility that the stability of large-scale chromatin arrangements is maintained without the help of an internal nuclear matrix. Movements of a given CT could become constrained mainly by its neighboring CTs.
To interpret higher order chromatin dynamics observed in human, Drosophila, and yeast cells, differences in size of their chromosomes (human, 50–250 Mb; Drosophila, 4.3–70 Mb; yeast, 0.2–1.5 Mb) and genome structure have to be considered. For example, the fraction of transcriptionally active chromatin in yeast amounts to ∼90%, compared with ∼10% in humans. This may correspond with differences of chromatin compaction and rigidity/flexibility of higher order chromatin structures. A “free” yeast chromosome of 1 Mb may behave differently compared with a human ∼1-Mb chromatin domain, which is part of a much larger CT.
Studies of normal cell types are indicated to see whether the large-scale stability of CT order seen in HeLa cell nuclei is a general feature of mammalian interphase nuclei. Such a stability does not exclude more expanded, rapid, and possibly directed movements of subchromosomal regions and genes. Functional implications of changes in higher order chromatin architecture are strongly suggested by the finding that transcriptional activation/gene silencing is correlated with the repositioning of genes and gene clusters (for review see Baxter et al., 2002).
Changes of CT order from one cell cycle to the next
Similarities of CT arrangements in daughter nuclei as well as changes of chromosome arrangements during mitosis were analyzed both by tracking of unbleached chromatin regions through mitosis and by chromosome painting of two-cell and four-cell HeLa cell clones. Coherent unbleached chromatin zones produced in mother nuclei during late S/G2 retained their position until early prophase. In prophase, the unbleached chromatin locally condensed into several unbleached chromosome segments. In daughter nuclei, these segments were recognized as locally decondensed chromatin patches. Our observations that locally constrained chromatin movements are sufficient for the transition of interphase into mitotic chromosomes and vice versa are consistent with findings reported by other groups (Manders et al., 1999; Lemke et al., 2002). Daughter nuclei with distantly located patches of unbleached chromatin demonstrate major changes in the arrangement of chromosome from one cell cycle to the next. Although metaphase plate arrangements of unbleached chromosomes may depend on the orientation of the bleaching axis (parallel or perpendicular to the spindle axis), such differences cannot account for the observation of metaphase plates showing most of the unbleached chromosomes clustered, whereas some had relocated to remote sites. In agreement with Boveri, our observation indicates that changes of chromosome neighborhoods occur during prometaphase. Our data demonstrate that mitosis is of major importance to explain the variability of CT arrangements in interphase nuclei and are in conflict with the findings of Nagele et al. (1995)(1999) that chromosomes in mitotic rosettes have a highly ordered, reproducible arrangement.
As a result of perfectly symmetrical chromatid movements during anaphase/telophase, one would expect daughter nuclei with a translational or mirrorlike symmetry of their CT arrangements. In both chromatin bleaching and 3-D FISH experiments, pairs of daughter nuclei often showed a notable symmetry of the labeled higher order chromatin structures, but this symmetry was never perfect and we noted pairs, where we could not detect an obvious translational or mirrorlike symmetry. Failure to achieve a symmetrical CT distribution may result from an imperfect symmetry of chromatid movements during anaphase. Our finding that CT movements were significantly more pronounced in nuclei studied in early G1 compared with nuclei followed from mid G1 to late G2 provides an additional possibility for movements, which disturb this symmetry.
A comparison of randomly selected pairs of daughter nuclei showed clearly different arrangements of painted CTs. 3-D nuclear reconstructions from four-cell clones demonstrated major differences already in the granddaughters of a cloned cell. We conclude that the variability of CT arrangements in HeLa cell cultures (unpublished data) is caused by drastic changes of CT neighborhoods from one cell cycle to the next, and does not depend on the accumulation of small positional changes during many cell generations. Experiments with other cell lines, including nontransformed diploid cells, are indicated to explore whether the rapid perturbation of chromosome order seen in the clonal progeny of HeLa cells is a special feature or a finding typical for many cell types. Studies of CT arrangements in cell clones from a variety of normal and malignant cell types will answer the question of whether the number of cell cycles required to perturb chromosome order to an extent that reflects the variation observed in an entire cell culture depends on the cell type.
The variability of CT order in HeLa cell nuclei prompts the question of whether CTs may be arranged in an entirely random fashion in this cell line, but this is clearly not the case. In agreement with the nonrandom radial distribution pattern of the gene-dense CTs #19 and gene-poor CTs #18, which was demonstrated in diploid nuclei of lymphocytes and lymphoblastoid cells from man and other primates (Croft et al., 1999; Cremer et al., 2001; Tanabe et al., 2002), we found a more interior location of gene-dense chromosome 19 material in HeLa cell nuclei compared with chromosome 18 material, despite of rearrangements involving these chromosomes (unpublished data).
Materials and methods
Cell culture
HeLa cells expressing histone H2B-GFP (provided by K. Sullivan, Scripps Research Institute, La Jolla, CA; Kanda et al., 1998) were cultured in a humidified incubator with 5% CO2 at 37°C in RPMI 1640 medium with 25 mM Hepes (Biochrom) and 10% FCS.
Scratch-replication labeling
Cells were grown on a 15 × 15-mm2 coverslip to subconfluency. After transferring the coverslip to a new culture dish and draining off excess medium, 10 μl of medium with 50 μM Cy3-dUTP (Amersham Biosciences) was placed onto the cells. A series of scratches was made into the cell layer with a hypodermic needle, and medium was added after 1 min. This procedure led to transient damage of cell membranes in cells along the scratch path, and allowed the uptake of labeled dUTP and incorporation into replicating DNA (Schermelleh et al., 2001). Labeled nuclei showed replication patterns known from 1-h pulse labeling experiments with BrdU and consistent with results from experiments using different in vivo replication labeling methods (Zink et al., 1998; Manders et al., 1999). Cells were further subcultivated for several days to allow segregation of labeled chromatids during subsequent cell cycles. The day before laser scanning observation, cells were harvested by trypsinization and seeded on round coverslips fitting to the live-cell chamber (see below).
For a two-color replication labeling in two consecutive S-phases, the cells were synchronized at G1/S transition by 12 h incubation with aphidicolin (0.15 μg/ml medium). The cells were released from the block by three times washing with CO2-adapted medium, further incubated for 1 h, and scratch-labeled with Cy3-dUTP as described above. After a 1-d incubation, the cells were harvested by trypsinization and seeded 1:1 on a new coverslip to ensure an even distribution of the cells. Cells were blocked by aphidicolin for another 12 h and subsequently scratch-labeled as described above with 100 μM Cy5-dUTP (Amersham Biosciences). Live-cell observation was performed 4 d later.
Long-term observations of cells on the microscope stage
To reduce photodamage, the medium was supplemented with 0.1–0.5 mM of the radical scavenger Trolox™ (Sigma-Aldrich) at least 12 h before the start of live-cell observations. Cells on round coverslips were transferred to a live-cell chamber (FCS2, Bioptechs), mounted on the microscope stage, and kept at 37°C. The objective was operated with an objective heater as part of the FCS2 system. The chamber was perfused with fresh medium using a flexible-tube pump in an airtight assembly to prevent the loss of CO2 and to keep the pH stable at 7.4. A 20-ml syringe connected to the pump and the chamber by a perfusion tube served as a reservoir for CO2-adapted medium. For medium exchange, the pump was automatically run for 10–20 s with a speed of ∼5 μl/s after each imaging time point. With this setup, cells on the microscope stage could be observed over several days from low density until confluence.
Confocal laser scanning of living cells and photobleaching
Live-cell imaging was performed with a microscope (LSM 410; Carl Zeiss MicroImaging, Inc.) using a 63×/1.4 plan-apochromat objective. GFP fluorescence was excited with the Ar+ laser (15 mW, 488 nm) at 30% power, additionally attenuated to 0.1–0.3%, and detected with a band pass filter at 502–542 nm. Cy3 was excited with a 543-nm HeNe-laser (0.5 mW) attenuated to 3% and detected with an LP 570 long pass filter, and Cy5 with a 633-nm HeNe-laser (5 mW) attenuated to 3% and detected with a 665-nm long pass filter. The pinhole was opened to 2–4.5 Airy units, sacrificing some optical sectioning capability for efficient light collection. In scratch-replication labeling experiments, we collected 26–30 serial sections at each imaging time point (256 × 256 pixels or 512 × 512; pixel size, 120–200 nm; z-step, 0.4–0.5 μm; time-interval, 6–30 min; 2× averaging). In photobleaching experiments, 15–25 serial sections were collected (512 × 512 or 256 × 256 pixels; pixel size, 100–180 nm; z-step, 0.8–1 μm; time-interval, 15–60 min; 2× averaging). These imaging parameters were found to be an optimal compromise between a low incidence of apoptosis and the resolution quality of images.
To record time series of 3-D image stacks automatically, we developed a macro for the LSM 410 control software. Within a field of view selected at a low zoom, this macro recorded time series of 3-D image stacks from multiple regions of interest at high zoom. For compensation of z-axis drift, the interface between the coverslip and the cell culture medium was imaged in reflection contrast before each image stack was recorded. The reflected light intensity had a sharp maximum at a distinct z-position, which was used as reference position.
Determination of cell cycle stages
The average cell cycle length of HeLa H2B-GFP cells under live-cell observation conditions was 18–20 h, as determined by the interval between two mitotic events (n = 24). Length of S-phase was defined by BrdU pulse labeling, resulting in 35% BrdU-positive nuclei. As nearly 100% of nuclei were in a proliferative status, as shown by a positive immunostaining of Ki67 protein (a marker for cell proliferation), the length of S-phase was calculated to be ∼7 h. The duration of G1 was estimated by the measurement of nuclear volumes (see below). On average, nuclei showed an ∼2.5-fold increase of the nuclear volume during telophase and early G1. Between 1 h, 45 min and 3 h after telophase, the volumes reached a plateau phase. The period from telophase until the beginning of this plateau phase was defined as early G1 (Table I). The volumes then remained constant for ∼4–5 h (mid-late G1) followed by another 1.5–1.6-fold volume increase during S-phase and G2.
3-D FISH and imaging of HeLa cell clones
HeLa H2B-GFP cells were seeded in low density on gridded coverslips (Bellco) to identify individual cells and cell clones. To ensure the clonal origin of the two- and four-cell clusters, transmission images of defined areas from coverslips were taken 1 h after cell seeding. Single cells with a distance of at least 100 μm to the next cell were identified and further monitored for clonal growth. Because HeLa cells show very low motility, it was easy to keep track of growing clones. Cells were fixed in 4% PFA/1×PBS 28 or 48 h after seeding, when most of the cells had formed two- and four-cell clones, respectively. Permeabilization steps, 3-D FISH, and probe detection were performed according to protocols described in detail elsewhere (Cremer et al., 2001; Solovei et al., 2002). 3-D FISH was performed with painting probes specific for chromosomes #7 (labeled with digoxigenin, detected with Cy3-conjugated antibodies) and #10 (labeled with biotin, detected with avidin-Alexa 488). Nuclei were counterstained with TO-PRO®-3 (Molecular Probes, Inc.) and embedded with antifade solution. Confocal image stacks for all three color channels were recorded with a confocal microscope (LSM 410, Carl Zeiss MicroImaging, Inc.; or TCS-SP, Leica; pixel size, 50–80 nm; z-step, 0.25 μm).
Image processing and 3-D reconstruction
Data were routinely gauss- or median-filtered to reduce noise. Projections were made with the LSM 410 software (Carl Zeiss MicroImaging, Inc.). Images were processed with Adobe Photoshop® 5.5. 3-D reconstructions of confocal image stacks were performed using Amira™ 2.3 (TGS).
Quantitative evaluation of nuclear volumes and CT movements
Intensity gravity centers were determined for CTs, as well as for the entire nucleus counterstained with H2B-GFP. Distances between these gravity centers (CT-CT and CT-CN) as well as nuclear volumes were measured with the plug-in Sync Measure 3D written for ImageJ (http://rsb.info.nih.gov/ij/). The threshold for CT segmentation was set visually. Isolated chromatin foci with low pixel intensities (below 1,000 in 8-bit images) were not considered. To take into account intensity variations due to different degrees of chromatin condensation during the telophase–G1 transition, the threshold for the nuclear volume measurements was determined as follows: all image stacks were normalized to gray levels between 0 and 255. For each data set, one interphase nucleus was chosen, and a profile plot along a central line through a nuclear midsection was made. The mean gray level between the first local maximum within the nucleus and the background was taken as threshold for volume measurement. The measured distances (d) were corrected for the increase in nuclear volume as follows: a volume during the first plateau (mid G1) was set as reference volume (V 0), and ratios V t/V0 were calculated for all volumes V t within a series of measurements. The corrected distances (d c) were calculated as:
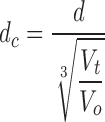 |
Median values of the maximum deviations (Δ(Max-Min)) were compared with the Mann-Whitney U-test (Barlow, 1989). Differences in the proportion of nuclei with Δ(Max-Min) >2 μm were scored with Fisher's exact test (Bland, 2000) using SPSS® software.
RAC analysis
As a measure of pixel distribution, an autocorrelation approach was used (Jähne et al., 1999). In image stacks of partially bleached cell nuclei, the total nuclear area was segmented at low threshold. Thereafter, the threshold for the segmentation of a fraction of high intensity nuclear pixels (2 or 10%) was determined, and was subtracted from all intensity values. The relative distance between all possible pairs of pixels (i and j) was calculated as:
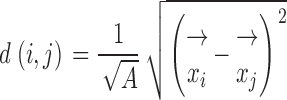 |
with x → i, x→ jbeing the positions of pixels i and j. Α represents the nuclear area. We defined the RAC of an image as:
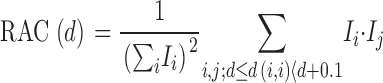 |
with d as the starting value of a relative distance interval; d = 0, 0.1, 0.2,… Ιi, Ιjrepresent fluorescence signal of pixels i and j (minus the threshold intensity).
The RAC is an intensity-weighted measure that reflects how many pairs of pixels fall into each relative distance interval. It was calculated with a plug-in for ImageJ on average intensity projections of 2–4 mid-nuclear optical sections. Other sections were not considered because the upper and lower nuclear borders could not be demarcated clearly due to the lack of a counterstain for the total chromatin.
The mean relative distance <d> was calculated from the RAC as:
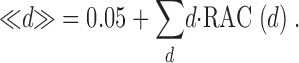 |
For a more intuitive explanation of the RAC, see Fig. S1. The median values of the mean relative distances were compared with the nonparametric Mann-Whitney U-test (Barlow, 1989).
Online supplemental material
Video 1 shows a 4-D time series of individual CTs in HeLa H2B-GFP cells labeled with Cy3-dUTP. Video 2 corresponds to Fig. 4, and shows a time series of a HeLa nucleus with H2B-GFP and CTs labeled with Cy3-dUTP or Cy5-dUTP 4.5 d after the second labeling event. Video 3 corresponds to Fig. 5, and shows maximum intensity projections of H2B-GFP–tagged chromatin from three HeLa cells after partial photobleaching of the whole mother nucleus, except for an unbleached region at one pole. Videos 4–7 show interactive 3-D renderings of the two pairs of daughter cell nuclei from a four-cell clone displayed in Fig. 8. The supplemental table provides additional information to Table I and Table II. For each evaluated cell, the number of evaluated time points and the number of labeled CTs, as well as Δ(Max-Min) values from all CT-CN and CT-CT distances are listed. Fig. S1 provides a schematic explanation of how different distributions of high intensity nuclear signals are represented by the radial autocorrelation procedure. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200211103/DC1.
Supplemental Material
Acknowledgments
The HeLa H2B-GFP cell line was provided by K. Sullivan.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Zi 560/2-2 and Cr59/20-1) and the Wilhelm Sander Stiftung (2001.079.1) to T. Cremer, from the European Community (FIGH-CT1999-00011) to C. Cremer, and by Grants-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan to S. Tashiro.
J. Walter and L. Schermelleh contributed equally to this paper.
The online version of this article includes supplemental material.
S. Tashiro's present address is Department of Biochemistry, Hiroshima University School of Medicine, Hiroshima 734-8551, Japan.
Footnotes
Abbreviations used in this paper: CN, center of the nucleus; CT, chromosome territory; RAC, radial autocorrelation function.
References
- Abney, J.R., B. Cutler, M.L. Fillbach, D. Axelrod, and B.A. Scalettar. 1997. Chromatin dynamics in interphase nuclei and its implications for nuclear structure. J. Cell Biol. 137:1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison, D.C., and A.L. Nestor. 1999. Evidence for a relatively random array of human chromosomes on the mitotic ring. J. Cell Biol. 145:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow, R. 1989. Statistics. Wiley, Chichester, UK. 204 pp.
- Baxter, J., M. Merkenschlager, and A.G. Fisher. 2002. Nuclear organisation and gene expression. Curr. Opin. Cell Biol. 14:372–376. [DOI] [PubMed] [Google Scholar]
- Bland, M. 2000. An Introduction to Medical Statistics. Oxford University Press, Oxford, UK. 424 pp.
- Bornfleth, H., P. Edelmann, D. Zink, T. Cremer, and C. Cremer. 1999. Quantitative motion analysis of subchromosomal foci in living cells using four-dimensional microscopy. Biophys. J. 77:2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri, T. 1909. Die blastomerenkerne von ascaris megalocephala und die theorie der chromosomenindividualität. Archiv für Zellforschung. 3:181–268. [Google Scholar]
- Boyle, S., S. Gilchrist, J.M. Bridger, N.L. Mahy, J.A. Ellis, and W.A. Bickmore. 2001. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum. Mol. Genet. 10:211–219. [DOI] [PubMed] [Google Scholar]
- Bridger, J.M., S. Boyle, I.R. Kill, and W.A. Bickmore. 2000. Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr. Biol. 10:149–152. [DOI] [PubMed] [Google Scholar]
- Chubb, J.R., S. Boyle, P. Perry, and W.A. Bickmore. 2002. Chromatin motion is constrained by association with nuclear compartments in human cells. Curr. Biol. 12:439–445. [DOI] [PubMed] [Google Scholar]
- Cornforth, M.N., K.M. Greulich-Bode, B.D. Loucas, J. Arsuaga, M. Vazquez, R.K. Sachs, M. Bruckner, M. Molls, P. Hahnfeldt, L. Hlatky, and D.J. Brenner. 2002. Chromosomes are predominantly located randomly with respect to each other in interphase human cells. J. Cell Biol. 159:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer, T., and C. Cremer. 2001. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2:292–301. [DOI] [PubMed] [Google Scholar]
- Cremer, T., A. Kurz, R. Zirbel, S. Dietzel, B. Rinke, E. Schrock, M.R. Speicher, U. Mathieu, A. Jauch, P. Emmerich, et al. 1993. Role of chromosome territories in the functional compartmentalization of the cell nucleus. Cold Spring Harb. Symp. Quant. Biol. 58:777–792. [DOI] [PubMed] [Google Scholar]
- Cremer, T., G. Kreth, H. Koester, R.H. Fink, R. Heintzmann, M. Cremer, I. Solovei, D. Zink, and C. Cremer. 2000. Chromosome territories, interchromatin domain compartment, and nuclear matrix: an integrated view of the functional nuclear architecture. Crit. Rev. Eukaryot. Gene Expr. 10:179–212. [PubMed] [Google Scholar]
- Cremer, M., J. von Hase, T. Volm, A. Brero, G. Kreth, J. Walter, C. Fischer, I. Solovei, C. Cremer, and T. Cremer. 2001. Non-random radial higher-order chromatin arrangements in nuclei of diploid human cells. Chromosome Res. 9:541–567. [DOI] [PubMed] [Google Scholar]
- Cremer, M., L. Schermelleh, I. Solovei, and T. Cremer. 2003. Chromosomal arrangement during different phases of cell cycle. Encyclopedia of the Human Genome. Nature Publishing Group, London. In press.
- Croft, J.A., J.M. Bridger, S. Boyle, P. Perry, P. Teague, and W.A. Bickmore. 1999. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145:1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csink, A.K., and S. Henikoff. 1998. Large-scale chromosomal movements during interphase progression in Drosophila. J. Cell Biol. 143:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova, D.S., and R. Berezney. 2002. The spatio-temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J. Cell Sci. 115:4037–4051. [DOI] [PubMed] [Google Scholar]
- Edelmann, P., H. Bornfleth, D. Zink, T. Cremer, and C. Cremer. 2001. Morphology and dynamics of chromosome territories in living cells. Biochim. Biophys. Acta. 1551:M29–M39. [DOI] [PubMed] [Google Scholar]
- Gasser, S.M. 2002. Visualizing chromatin dynamics in interphase nuclei. Science. 296:1412–1416. [DOI] [PubMed] [Google Scholar]
- Habermann, F.A., M. Cremer, J. Walter, G. Kreth, J. von Hase, K. Bauer, J. Wienberg, C. Cremer, T. Cremer, and I. Solovei. 2001. Arrangements of macro- and microchromosomes in chicken cells. Chromosome Res. 9:569–584. [DOI] [PubMed] [Google Scholar]
- Heitz, E. 1932. Die herkunft der chromocentren. Planta. 18:571–635. [Google Scholar]
- Heun, P., T. Laroche, K. Shimada, P. Furrer, and S.M. Gasser. 2001. Chromosome dynamics in the yeast interphase nucleus. Science. 294:2181–2186. [DOI] [PubMed] [Google Scholar]
- Jähne, B. H. Haussecker, and P. Geissler. 1999. Handbook of Computer Vision and Applications. Academic Press, San Diego. 1500 pp.
- Kanda, T., K.F. Sullivan, and G.M. Wahl. 1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8:377–385. [DOI] [PubMed] [Google Scholar]
- Kimura, H., and P.R. Cook. 2001. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153:1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss, L.G. 1998. Characteristics of chromosomes in polarized normal human bronchial cells provide a blueprint for nuclear organization. Cytogenet. Cell Genet. 82:230–237. [DOI] [PubMed] [Google Scholar]
- Lamond, A.I., and W.C. Earnshaw. 1998. Structure and function in the nucleus. Science. 280:547–553. [DOI] [PubMed] [Google Scholar]
- Leitch, A.R. 2000. Higher levels of organization in the interphase nucleus of cycling and differentiated cells. Microbiol. Mol. Biol. Rev. 64:138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke, J., J. Claussen, S. Michel, I. Chudoba, P. Muhlig, M. Westermann, K. Sperling, N. Rubtsov, U.W. Grummt, P. Ullmann, et al. 2002. The DNA-based structure of human chromosome 5 in interphase. Am. J. Hum. Genet. 71:1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesko, S.A., D.E. Callahan, M.E. LaVilla, Z.P. Wang, and P.O. Ts'o. 1995. The experimental homologous and heterologous separation distance histograms for the centromeres of chromosomes 7, 11, and 17 in interphase human T-lymphocytes. Exp Cell Res. 219:499–506. [DOI] [PubMed] [Google Scholar]
- Lucas, J.N., and E. Cervantes. 2002. Significant large-scale chromosome territory movement occurs as a result of mitosis, but not during interphase. Int. J. Radiat. Biol. 78:449–455. [DOI] [PubMed] [Google Scholar]
- Ma, H., A.J. Siegel, and R. Berezney. 1999. Association of chromosome territories with the nuclear matrix. Disruption of human chromosome territories correlates with the release of a subset of nuclear matrix proteins. J. Cell Biol. 146:531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders, E.M., H. Kimura, and P.R. Cook. 1999. Direct imaging of DNA in living cells reveals the dynamics of chromosome formation. J. Cell Biol. 144:813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, W.F. 2002. Order and disorder in the nucleus. Curr. Biol. 12:R185–R192. [DOI] [PubMed] [Google Scholar]
- Marshall, W.F., A. Straight, J.F. Marko, J. Swedlow, A. Dernburg, A. Belmont, A.W. Murray, D.A. Agard, and J.W. Sedat. 1997. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr. Biol. 7:930–939. [DOI] [PubMed] [Google Scholar]
- Martou, G., and U. De Boni. 2000. Nuclear topology of murine, cerebellar Purkinje neurons: changes as a function of development. Exp. Cell Res. 256:131–139. [DOI] [PubMed] [Google Scholar]
- Nagele, R., T. Freeman, L. McMorrow, and H.Y. Lee. 1995. Precise spatial positioning of chromosomes during prometaphase: evidence for chromosomal order. Science. 270:1831–1835. [DOI] [PubMed] [Google Scholar]
- Nagele, R.G., T. Freeman, L. McMorrow, Z. Thomson, K. Kitson-Wind, and H. Lee. 1999. Chromosomes exhibit preferential positioning in nuclei of quiescent human cells. J. Cell Sci. 112:525–535. [DOI] [PubMed] [Google Scholar]
- Paddy, M.R., A.S. Belmont, H. Saumweber, D.A. Agard, and J.W. Sedat. 1990. Interphase nuclear envelope lamins form a discontinuous network that interacts with only a fraction of the chromatin in the nuclear periphery. Cell. 62:89–106. [DOI] [PubMed] [Google Scholar]
- Parada, L., and T. Misteli. 2002. Chromosome positioning in the interphase nucleus. Trends Cell Biol. 12:425–432. [DOI] [PubMed] [Google Scholar]
- Rabl, C. 1885. Über Zelltheilung. Morphologisches Jahrbuch. 10:214–330. [Google Scholar]
- Sadoni, N., S. Langer, C. Fauth, G. Bernardi, T. Cremer, B.M. Turner, and D. Zink. 1999. Nuclear organization of mammalian genomes. Polar chromosome territories build up functionally distinct higher order compartments. J. Cell Biol. 146:1211–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermelleh, L., I. Solovei, D. Zink, and T. Cremer. 2001. Two-color fluorescence labeling of early and mid-to-late replicating chromatin in living cells. Chromosome Res. 9:77–80. [DOI] [PubMed] [Google Scholar]
- Shelby, R.D., K.M. Hahn, and K.F. Sullivan. 1996. Dynamic elastic behavior of alpha-satellite DNA domains visualized in situ in living human cells. J. Cell Biol. 135:545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalnikova, M., S. Kozubek, E. Lukasova, E. Bartova, P. Jirsova, A. Cafourkova, I. Koutna, and M. Kozubek. 2000. Spatial arrangement of genes, centromeres and chromosomes in human blood cell nuclei and its changes during the cell cycle, differentiation and after irradiation. Chromosome Res. 8:487–499. [DOI] [PubMed] [Google Scholar]
- Solovei, I., J. Walter, M. Cremer, F. Habermann, L. Schermelleh, and T. Cremer. 2002. FISH on three-dimensionally preserved nuclei. FISH: A Practical Approach. J. Squire, B. Beatty, and S. Mai, editors. Oxford University Press, Oxford. 119–157.
- Sun, H.B., and H. Yokota. 1999. Correlated positioning of homologous chromosomes in daughter fibroblast cells. Chromosome Res. 7:603–610. [DOI] [PubMed] [Google Scholar]
- Sun, H.B., J. Shen, and H. Yokota. 2000. Size-dependent positioning of human chromosomes in interphase nuclei. Biophys. J. 79:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe, H., S. Muller, M. Neusser, J. von Hase, E. Calcagno, M. Cremer, I. Solovei, C. Cremer, and T. Cremer. 2002. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc. Natl. Acad. Sci. USA. 99:4424–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbar, T., and A. Belmont. 2001. Interphase movements of a DNA chromosome region modulated by VP16 transcriptional activator. Nat. Cell Biol. 3:134–139. [DOI] [PubMed] [Google Scholar]
- Vazquez, J., A.S. Belmont, and J.W. Sedat. 2001. Multiple regimes of constrained chromosome motion are regulated in the interphase Drosophila nucleus. Curr. Biol. 11:1227–1239. [DOI] [PubMed] [Google Scholar]
- Zink, D., and T. Cremer. 1998. Cell nucleus: chromosome dynamics in nuclei of living cells. Curr. Biol. 8:R321–R324. [DOI] [PubMed] [Google Scholar]
- Zink, D., T. Cremer, R. Saffrich, R. Fischer, M.F. Trendelenburg, W. Ansorge, and E.H. Stelzer. 1998. Structure and dynamics of human interphase chromosome territories in vivo. Hum. Genet. 102:241–251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}