

Abstract
Collagen plays a critical role in hemostasis by promoting adhesion and activation of platelets at sites of vessel injury. In the present model of platelet–collagen interaction, adhesion is mediated via the inside-out regulation of integrin α2β1 and activation through the glycoprotein VI (GPVI)–Fc receptor (FcR) γ-chain complex. The present study extends this model by demonstrating that engagement of α2β1 by an integrin-specific sequence from within collagen or by collagen itself generates tyrosine kinase–based intracellular signals that lead to formation of filopodia and lamellipodia in the absence of the GPVI–FcR γ-chain complex. The same events do not occur in platelet suspensions. α2β1 activation of adherent platelets stimulates tyrosine phosphorylation of many of the proteins in the GPVI–FcR γ-chain cascade, including Src, Syk, SLP-76, and PLCγ2 as well as plasma membrane calcium ATPase and focal adhesion kinase. α2β1-mediated spreading is dramatically inhibited in the presence of the Src kinase inhibitor PP2 and in PLCγ2-deficient platelets. Spreading is abolished by chelation of intracellular Ca2+. Demonstration that adhesion of platelets to collagen via α2β1 generates intracellular signals provides a new insight into the mechanisms that control thrombus formation and may explain the unstable nature of β1-deficient thrombi and why loss of the GPVI–FcR γ-chain complex has a relatively minor effect on bleeding.
Keywords: integrin α2β1; blood platelets; cell spreading; PLCγ2; FAK
Introduction
Platelets play a critical role in the events that lead to thrombus formation and cessation of bleeding at sites of damage to the vasculature. They also play a major role in thrombotic diseases such as stroke and myocardial infarction. Platelet activation is an integrated process involving subendothelial matrix proteins and soluble agonists that support adhesion and activation. Collagen is unique among these stimuli in that it not only plays a key role in platelet tethering and adhesion, but it is also a powerful agonist that is capable of producing full activation. A full understanding of the role of collagen in hemostasis is essential toward an understanding of cardiovascular disease and new forms of therapeutic intervention.
Collagen interacts with platelets through direct and indirect mechanisms. At the medium and high shear rates found in arterioles and damaged vascular beds, von Willebrand factor (vWf)* bridges newly exposed collagen fibers to the GPIb–IX–V complex on the platelet surface. This interaction is facilitated by a fast on-rate of association between vWf and GPIbα and is essential for the initial tethering of platelets by collagen at medium to high rates of shear (Savage et al., 1996, 1998). The interaction is opposed by a rapid off-rate of dissociation such that platelets translocate (or roll) for several minutes on vWf in the absence of other proteins before forming stable adhesion through activated αIIbβ3 (Savage et al., 1996). The situation in vivo however is very different, as a number of extracellular matrix proteins work in tandem with vWf to promote stable adhesion, notably collagen.
Platelets interact with collagen through two major surface receptors, the integrin α2β1 and GPVI, as well as with a number of minor receptors of uncertain significance (for review see Watson and Gibbins, 1998). For many years, the interaction of collagen with α2β1 was thought to be essential for adhesion, whereas the role of GPVI was to mediate activation. The understanding of these events has changed considerably, however, after a number of recent observations using function blocking antibodies and genetically modified mice. In particular, the observation that platelets show dramatically impaired adhesion to collagen under static and shear conditions in the absence of functional GPVI demonstrates a critical role for the glycoprotein in platelet adhesion (Nieswandt et al., 2001a). This can be explained by the loss of inside-out signals from GPVI, which promote α2β1 activation and enable it to bind to collagen (Jung and Moroi, 1998, 2000). More recently, GPVI has been shown to play a critical role in the interaction of platelets with vWf in vitro (Goto et al., 2002) and with a denuded endothelial carotid artery in vivo (Massberg et al., 2003), demonstrating that it is required at the very initial stage of thrombus formation.
These observations highlight a central role for GPVI–Fc receptor (FcR) γ-chain in the interaction of platelets with collagen but do not explain why mice deficient in the glycoprotein show only a minor increase in bleeding times (Nieswandt et al., 2001b). It therefore seems prudent to suggest that other matrix proteins play a role in promoting thrombus formation and can thereby compensate for the absence of GPVI. In this context, it is pertinent to consider the role of α2β1 in greater detail, because not only is it a major receptor for collagen but it can also be activated by G protein–coupled receptor agonists independent of GPVI.
The role of α2β1 in platelet–collagen interactions is critically dependent on experimental conditions. A universal finding of α2β1 blockade is a delay in response to collagen, although in many cases the final extent of activation is not altered. However, under certain experimental conditions, blockade of the integrin can lead to an abolition of adhesion and activation. This is illustrated by the contrasting reports of Chen et al. (2002) and Holtkötter et al. (2002) on the adhesion of α2-deficient murine platelets to collagen under flow. Chen et al. (2002) reported a dramatic inhibition of adhesion to collagen, using washed platelets in a low Ca2+-containing buffer, conditions that favor the interaction with the integrin. In contrast, Holtkötter et al. (2002) reported a negligible effect of α2 ablation on adhesion, using plasma and a physiological concentration of Ca2+. A similar observation has also been reported in β1-deficient murine platelets in the presence of plasma (Nieswandt et al., 2001a). In a recent follow-up to this study, however, the same group described an increased tendency of the β1-deficient thrombi to fragment at later times in the experiment compared with those formed by wild-type platelets (Kuijpers et al., 2003). Careful examination of these thrombi revealed that they were more loosely packed than those found in control cells (Kuijpers et al., 2003). This observation demonstrates an unexpected role for α2β1 in the later stages of hemostasis that is critical for thrombus stability, even though it has no role in the initial events that underlie adhesion.
Suzuki-Inoue et al. (2001) have recently reported spreading of human platelets on Fab fragments of an α2β1-activating antibody, TS2/16. In light of this, we wondered whether the increased embolization of the β1-deficient thrombi was caused by a loss of integrin-mediated intracellular signals that mediate remodeling of the cytoskeleton and thereby contribute to thrombus stability. In the present study, we show that a collagen peptide that binds exclusively to α2β1 generates tyrosine kinase–based intracellular signals that underlie platelet spreading. Importantly, a similar set of observations are seen with collagen in murine platelets deficient in GPVI–FcR γ-chain. Both sets of responses are inhibited by α2β1 blockade. Strikingly, the intracellular signaling cascade used by α2β1 shares many of the features of the GPVI signaling cascade, including participation of Src kinases and PLCγ2. The observation that engagement of α2β1 can induce a similar set of signals to GPVI provides a new insight into the role of α2β1 in platelet activation by collagen and may explain the relatively minor effect of depletion of GPVI–FcR γ-chain on bleeding times (Nieswandt et al., 2001b) and the increased tendency of β1-deficient thrombi to embolize (Kuijpers et al., 2003).
Results
Spreading of human platelets on collagen-coated surfaces
Adhesion and spreading of washed human platelets on a collagen-coated surface was investigated in the presence of maximally effective concentrations of the cyclooxygenase inhibitor indomethacin, the P2Y1 and P2Y12 ADP receptor antagonists A3P5P and AR-C67085, and the αIIbβ3 antagonist lotrafiban. Confirmation that this cocktail of inhibitors was fully effective was shown by the abolition of shape change and αIIbβ3 aggregation to a high concentration of collagen (100 μg/ml) (unpublished data).
Platelets underwent time-dependent adhesion and formation of filopodia and lamellipodia on a collagen surface (Fig. 1, A and B) . The majority of platelets had spread fully after 30 min, although a number showed intermediate changes in morphology most likely because of a more recent contact with the collagen surface. Because the changes in morphology occurred in the presence of inhibitors, it is suggested that adhesion and spreading are directly mediated through collagen receptors. In contrast, very few platelets settled on a BSA-coated surface and those that did, did not undergo a change in morphology (Fig. 1 A).
Figure 1.
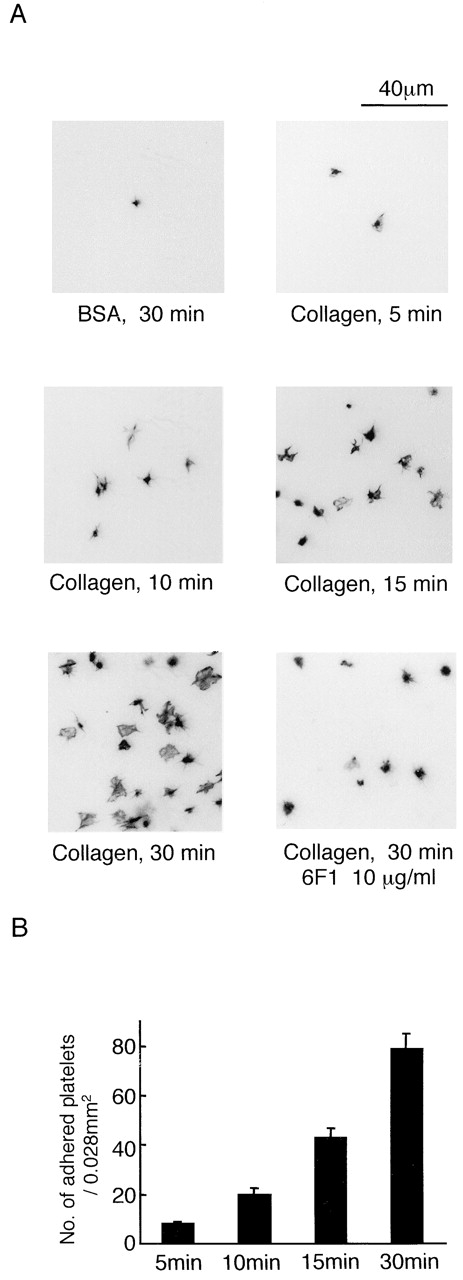
Spreading of human platelets on collagen. (A) Washed human platelets (2 × 107/ml) were pretreated with 100 μM EGTA, 10 μM indomethacin, 10 μM lotrafiban, 1 mM A3P5P, and 1 μM AR-C67085 (inhibitor cocktail) with or without 10 μg/ml 6F1 and 10 μg/ml VI.3. IV.3 was used to prevent platelet activation by the Fc portion of 6F1. Platelets were seeded on coverslips coated with 1% fatty acid–free BSA or 50 μg/ml collagen for the indicated times at 30°C. After unbound platelets were removed, the platelets were fixed by 3% paraformaldehyde, stained by TRITC-labeled phalloidin for actin fibers, and photographed with fluorescent microscopy. (B) The number of adhered platelets per 0.028 mm2. The graph illustrates the mean number of adhered platelets ± SEM per 0.028 mm2 from seven different images from three experiments. Photo images are representative of 5–10 different images from three experiments.
The role of the integrin α2β1 in adhesion and spreading on collagen was investigated using the α2β1-blocking mAb 6F1. The number of platelets that formed stable adhesions on collagen was reduced in the presence of 6F1 from 78.7 ± 6.2 per 0.028 mm2 to 31.5 ± 1.5 per 0.028 mm2 at 30 min (P < 0.01) (Fig. 1 A). In addition, the formation of filopodia and lamellipodia was impaired (Fig. 1 A). These results demonstrate a vital role for α2β1 in mediating adhesion and spreading on collagen. In contrast, adhesion and formation of filopodia and lamellipodia was readily seen in platelets exposed to a surface coated with a similar concentration of the GPVI-specific peptide CRP (unpublished data). This more marked response is likely to reflect the 10-fold greater density of the GPVI-specific binding motif, GPO, in CRP in comparison to collagen.
GPVI–FcR γ-chain–deficient murine platelets spread on collagen in the presence of ADP
Spreading of mouse platelets on a collagen-coated surface was investigated in the presence of the above cocktail of inhibitors. The platelets underwent time-dependent adhesion and formation of filopodia and lamellipodia on the collagen surface (Fig. 2 A). Murine platelets deficient in the GPVI–FcR γ-chain complex showed a marked reduction in adhesion to collagen of >60% and underwent only limited spreading (Fig. 2 B, 1 [i and ii]). The interaction of collagen with α2β1 is increased by inside-out activation of the integrin by stimuli such as ADP and PMA (Jung and Moroi, 1998, 2000). ADP (Fig. 2 B, 1 [iii]) or PMA (unpublished data) restored adhesion and spreading of FcR γ-chain–deficient platelets, although this was ablated in the presence of an α2β1-blocking antibody (Fig. 2 B, 1 [iv]) but not by an isotype-matched control (unpublished data). These findings demonstrate that α2β1 is able to support spreading on collagen in the absence of GPVI and that either the integrin and/or the stimulus used to promote inside-out signaling is able to mediate spreading.
Figure 2.
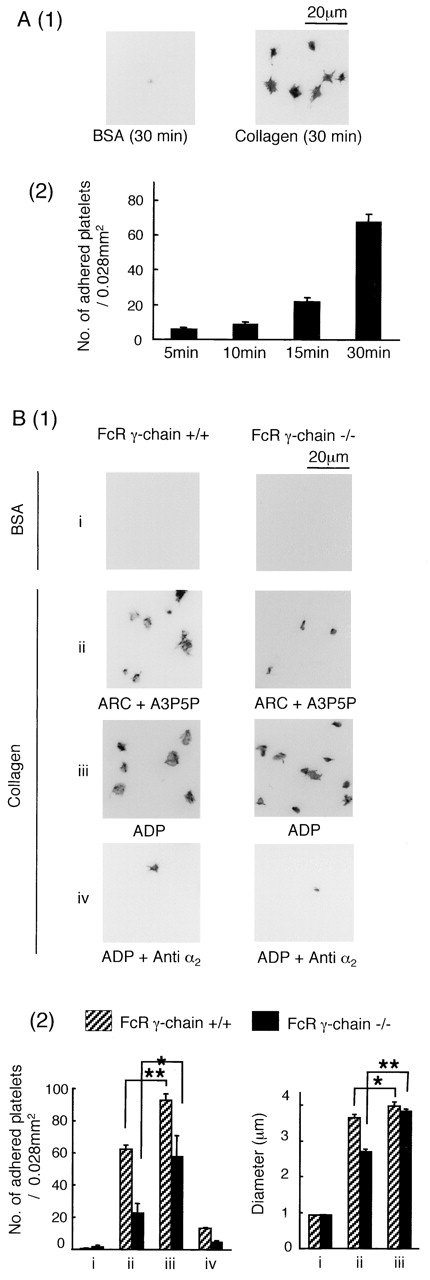
ADP stimulation promotes adhesion and spreading of FcR γ-chain–deficient mouse platelets on collagen. (A, 1) Washed murine platelets (3 × 107/ml) from wild-type mice (+/+) were pretreated with the inhibitor cocktail and seeded on BSA- or collagen-coated coverslips and stained as described in Fig. 1. Photo images are representative of 5–10 different images from three experiments. (A, 2) The graph illustrates the mean number of adhered platelets ± SEM per 0.028 mm2 from seven different images from at least two separate experiments. (B, 1) Washed platelets (3 × 107/ml) from FcR γ-chain (+/+) or (−/−) mice were pretreated with 100 μM EGTA, 10 μM indomethacin, and 10 μM lotrafiban, with or without 10 μg/ml anti–mouse integrin α2 antibody for 10 min. ADP (10 μM) was present in parts i, iii, and iv. The ADP receptor antagonists A3P5P (1 mM) plus AR-C67085 (1 μM) were present in part ii. Platelets were seeded on BSA- or collagen-coated coverslips and stained as described in Fig. 1. (B, 2) The mean number of adhered platelets ± SEM per 0.028 mm2 from 10 different images from three experiments is shown. One asterisk denotes P < 0.05, and two asterisks denote P < 0.01. The maximal diameter of between 100 and 200 adhered platelets was measured. For adhered platelets on BSA-coated surfaces, diameters of 10 platelets were counted because of a lack of cells.
Platelets spread on the α2β1-specific triple helical peptide GFOGER
We are presently unable to study spreading of human platelets on collagen in the absence of GPVI because of the lack of a GPVI-blocking antibody or access to individuals who are deficient in GPVI. To investigate the ability of α2β1 to stimulate spreading in human platelets, we therefore used an α2β1-specific, triple-helical peptide developed by Knight et al. (1998)(2000). GFOGER peptide contains one copy of an integrin α2β1 recognition motif, GFOGER, in a backbone of repeat GPP sequences and is cross-linked by NH2- and COOH-terminal cysteines. The GPP motif is inactive on α2β1 and GPVI but is required to confer a helical conformation on the peptide (Morton et al., 1995).
Human and murine platelets adhere and undergo extensive spreading on GFOGER-coated surfaces in the presence of indomethacin, ADP receptor antagonists, and lotrafiban (Fig. 3 A, 1 and 2, and B, 1). The formation of filopodia and lamellipodia can be clearly seen (Fig. 3 A, 3). Adhesion of human platelets to the GFOGER monolayer was ablated in the presence of the α2β1-blocking antibody 6F1 (Fig. 3 A, 1 and 2), but not in the presence of an isotype-matched control or the anti-GPIb mAb 6D1 (Fig. 3 A, 4). The latter provides evidence of specificity in the action of mAb 6F1, especially bearing in mind that GPIb is present at an ∼10 times greater level than the integrin. Adhesion of murine platelets to GFOGER is inhibited by an α2β1-blocking antibody (Fig. 3 B, 1 and 2) but not by an isotype-matched control (unpublished data). A similar number of platelets also undergo adhesion and spreading from wild-type, GPVI–FcR γ-chain–deficient, and αIIb-deficient mice, demonstrating that adhesion is not dependent on GPVI or the integrin αIIbβ3 (Fig. 3 B). Consistent with this, GFOGER peptide did not induce significant tyrosine phosphorylation of the FcR γ-chain in human platelets (Fig. 3 A, 5), as measured by precipitation with a GST–Syk SH2 domain fusion protein (Gibbins et al., 1996). Platelet adhesion to a monolayer of CRP served as a positive control for these studies (Fig. 3 A, 5). These findings are consistent with reports that GFOGER peptide is unable to bind GPVI (Knight et al., 1998, 2000) and demonstrate that engagement of α2β1 by a fibrillar collagen binding sequence mediates spreading.
Figure 3.
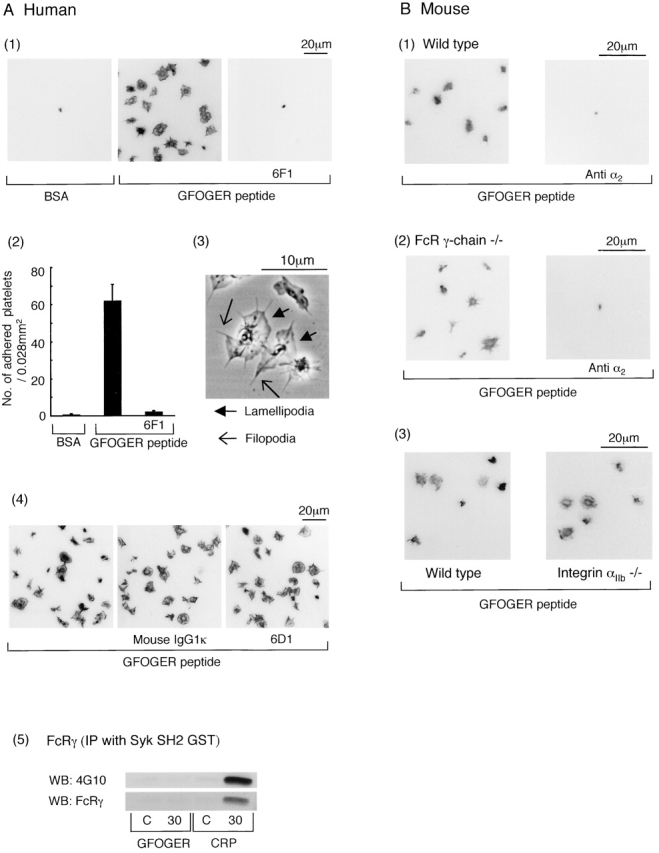
Spreading of human and mouse platelets on GFOGER peptide. (A, 1) Washed human platelets (2 × 107/ml) were pretreated with the inhibitor cocktail and then seeded on BSA- or GFOGER-coated coverslips in the presence or absence of 10 μg/ml 6F1 and 10 μg/ml F(ab')2 of IV.3. The platelets were fixed, stained, and photographed as described in Fig. 1. (A, 2) The mean number of adhered platelets ± SEM per 0.028 mm2 from between 10 and 20 images from six experiments was counted. (A, 3) Washed human platelets (2 × 107/ml) pretreated with the inhibitor cocktail were seeded on the GFOGER-coated glass plate for 30 min at 30°C. The picture was taken using phase contrast microscopy. (A, 4) Washed human platelets (2 × 107/ml) were pretreated with the inhibitor cocktail with or without 10 μg/ml 6D1 plus 10 μg/ml IV.3 or 10 μg/ml mouse IgG1κ plus 10 μg/ml IV.3 and then seeded on the BSA- or GFOGER-coated coverslips, stained, and photographed as described in Fig. 1. (A, 5) Washed human platelets (3 × 108/ml) were pretreated with 100 μM EGTA, 10 μM indomethacin, 10 μM lotrafiban, and 3 U/ml apyrase. 300 μl of washed platelets was seeded on the dishes coated with 1% BSA (C, control), 50 μg/ml GFOGER, or 50 μg/ml CRP for 30 min at 30°C. Platelets were dissolved by 150 μl of 3× lysis buffer. Unbound platelets that had been exposed to a BSA-coated surface served as the zero time point. FcR γ-chain was isolated by precipitation with GST–Syk SH2 fusion protein and blotted with 4G10 or anti–human FcR γ-chain antibody. Results are representative of two experiments. (B) Washed platelets (3 × 107/ml) from wild-type mice, FcR γ-chain (−/−) mice, and integrin αIIb (−/−) mice were pretreated with the inhibitor cocktail with or without 10 μg/ml anti–mouse integrin α2 antibody for 10 min. Platelets were seeded on GFOGER-coated coverslips and stained as described in Fig. 1.
α2β1 mediates platelet spreading through a Src kinase–dependent pathway
We considered it likely that the intracellular signals driven by α2β1 would be similar to those regulated by the platelet-specific integrin αIIbβ3. αIIbβ3 mediates spreading through a Src kinase–dependent pathway that lies upstream of the tyrosine kinase Syk (Obergfell et al., 2002). Consistent with this, the Src kinase inhibitor PP2 was observed to inhibit α2β1-mediated spreading (P < 0.01) but not adhesion of human platelets on GFOGER (Fig. 4 A). PP3, which is an inactive analogue of PP2, did not inhibit spreading on GFOGER (Fig. 4 A). PP2 also partially inhibited spreading but not adhesion of FcR γ-chain–deficient murine platelets on a collagen-coated surface treated with ADP (Fig. 4 B), suggesting that the Src kinase inhibitor does not interfere with the inside-out activation of α2β1 by the G protein receptor ligand. The structurally distinct Src kinase inhibitor SU6656 (20 μM) also inhibited spreading but not adhesion of human and FcR γ-chain–deficient murine platelets on GFOGER (unpublished data). These results demonstrate a key role for Src kinases in α2β1-mediated spreading but not adhesion.
Figure 4.
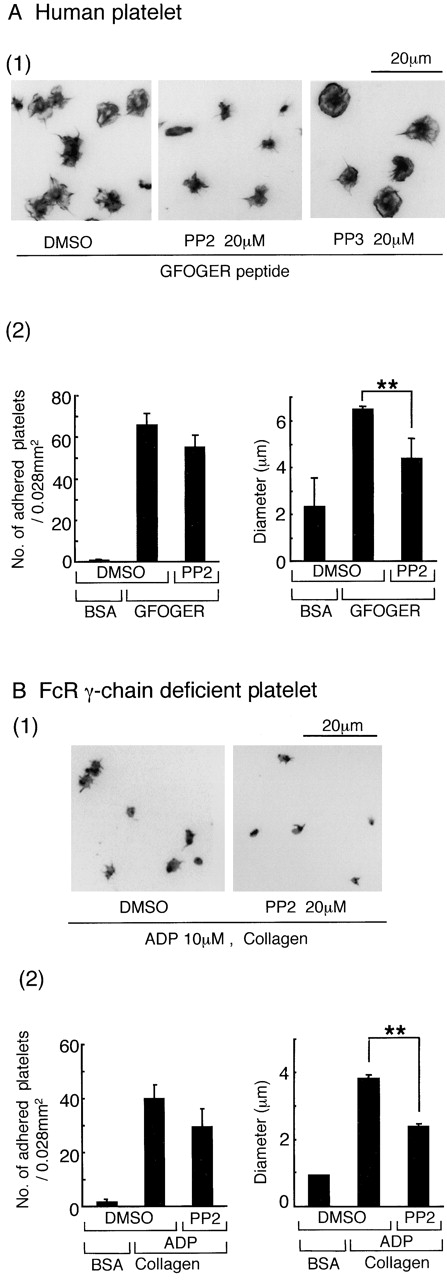
PP2 inhibits spreading, but not adhesion, mediated through integrin α2β1. (A, 1) Washed human platelets (2 × 107/ml) were pretreated with the inhibitor cocktail and 20 μM PP2, 20 μM PP3, or the equivalent volume of DMSO for 10 min before seeding on GFOGER-coated coverslips. The platelets were fixed, stained, and analyzed as described in Figs. 1 and 2. (A, 2) For the number of adhered platelets, data represent the mean number of adhered platelets ± SEM per 0.028 mm2 from 10 different images from three experiments. The mean diameter of between 150 and 400 platelets was measured. Two asterisks denote statistical significance of less than P < 0.01. (B, 1) Washed platelets (3 × 107/ml) from FcR γ-chain (−/−) mice were pretreated with 10 μM ADP, 100 μM EGTA, 10 μM indomethacin, 10 μM lotrafiban, and 20 μM PP2 or the equivalent volume of DMSO for 10 min before seeding on collagen-coated coverslips. (B, 2) For other conditions, see part A.
GFOGER peptide stimulates tyrosine phosphorylation of focal adhesion kinase (FAK) and PLCγ2
We investigated the ability of GFOGER to stimulate tyrosine phosphorylation when presented in suspension or as a monolayer. Tyrosine phosphorylation was measured by Western blotting using the anti-phosphotyrosine mAb 4G10. For the adhesion studies, unbound platelets were removed by aspiration and adhered cells were solubilized using Laemmli buffer. Unbound platelets that had been exposed to a surface of BSA or GFOGER served as controls for these studies. No significant difference was seen between the two sets of controls (unpublished data). The protein concentration of each lysate was determined using an assay that measures protein levels in the presence of SDS so that equal levels of protein could be added to each lane of the gel, making an appropriate correction for the contribution of GFOGER.
GFOGER stimulated a marked increase in tyrosine phosphorylation at 15 and 30 min when presented as a monolayer but not as a suspension (Fig. 5 A). The latter is in agreement with a previous study (Knight et al., 1999). These observations demonstrate a fundamental difference in the behavior of adherent and suspended platelets upon exposure to GFOGER. Tyrosine phosphorylation was dramatically inhibited in the presence of PP2, although increases in a major band of 60 kD and a more minor band of 125 kD were observed at later times of stimulation (Fig. 5 A).
Figure 5.
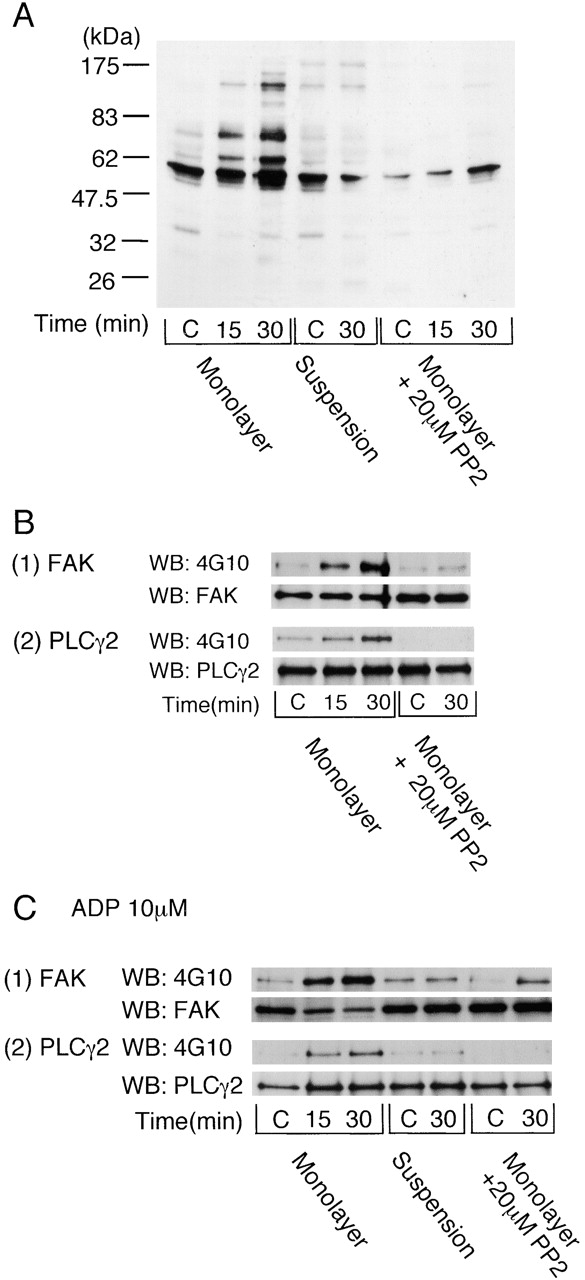
Integrin α2β1–mediated platelet spreading induces protein tyrosine phosphorylation. (A) Washed human platelets were pretreated with 100 μM EGTA, 10 μM indomethacin, 10 μM lotrafiban, and 3 U/ml apyrase. Where indicated, 10 μM ADP was added and apyrase omitted. Platelets were pretreated with 20 μM PP2 as shown. For adhesion studies, 500 μl washed platelets (4 × 108/ml) was seeded on the dishes coated with 1% BSA (C, control) or 50 μg/ml GFOGER at 30°C for the indicated times. After removal of unbound platelets, adhered platelets were dissolved by 500 μl Laemmli sample buffer. Unbound platelets that had been exposed to a BSA-coated surface served as the zero time point. For suspension studies, 500 μl washed platelets (4 × 108/ml) in a glass cuvette was stimulated with 10 μM ADP and then incubated with or without 50 μg/ml GFOGER for the indicated times with stirring at 30°C. After the protein concentrations of each sample were adjusted, proteins were separated by 8% SDS-PAGE, and protein tyrosine phosphorylation was blotted with 4G10. Results are representative of two separate experiments. (B) Washed human platelets (3 × 108/ml) were pretreated with the inhibitors and stimulated as described above. Reactions were stopped by the addition of an equal volume of 2× lysis buffer without removing unbound platelets. FAK (1) or PLCγ2 (2) proteins were isolated by immunoprecipitation with anti-FAK pAb or anti-PLCγ2 pAb and blotted with 4G10, anti-FAK mAb, or anti-PLCγ2 pAb. Results are representative of three separate experiments. (C) Washed human platelets (3 × 108/ml) were pretreated with 100 μM EGTA, 10 μM indomethacin, 10 μM lotrafiban, and 10 μM ADP. 20 μM PP2 was added as shown. Reactions were stopped, and FAK (1) or PLCγ2 (2) proteins were isolated by immunoprecipitation as described above. Results are representative of three separate experiments.
We sought to identify a number of the proteins that undergo an increase in tyrosine phosphorylation in adherent platelets through immunoprecipitation using specific antibodies and reprobing for phosphotyrosine. P125FAK is a nonreceptor tyrosine kinase that colocalizes with integrins at contact sites with the subendothelium and undergoes an increase in tyrosine phosphorylation on fibrinogen binding. Platelet adhesion to the GFOGER monolayer leads to tyrosine phosphorylation of focal adhesion kinase (FAK) (Fig. 5 B). The increase in tyrosine phosphorylation of FAK was substantially, but not completely, inhibited in the presence of a concentration of PP2 (20 μM) that causes complete inhibition of Src kinase activity in platelets (Fig. 5 B). PLCγ2 undergoes tyrosine phosphorylation in platelets exposed to a fibrinogen surface downstream of the integrin αIIbβ3 (Wonerow et al., 2002). PLCγ2 was also tyrosine phosphorylated in platelets that had adhered to GFOGER (Fig. 5 B). Tyrosine phosphorylation of PLCγ2 was completely inhibited by PP2 (20 μM).
We sought to further investigate the PP2-independent tyrosine phosphorylation of FAK by GFOGER. It has been previously shown that G protein–coupled receptor agonists are able to potentiate integrin-dependent tyrosine phosphorylation of FAK in platelets (Shattil et al., 1994). ADP potentiated tyrosine phosphorylation of FAK by a GFOGER monolayer in the presence of PP2, whereas it had no effect on phosphorylation of PLCγ2 (Fig. 5 C). Tyrosine phosphorylation of FAK by a GFOGER monolayer in the presence of ADP was maintained in the presence of a supramaximal concentration of PP2 (50 μM; unpublished data). On the other hand, tyrosine phosphorylation of FAK by GFOGER was not observed in suspension studies in the presence of ADP (Fig. 5 C).
These results demonstrate that adhesion to GFOGER leads to tyrosine phosphorylation of FAK and PLCγ2 through a Src kinase–dependent pathway. In addition, FAK is partially regulated by a Src kinase–independent pathway that is potentiated by ADP.
GFOGER peptide stimulates tyrosine phosphorylation of Src, Syk, SLP-76, and plasma membrane calcium ATPase (PMCA)
We sought to further characterize a number of the other tyrosine-phosphorylated proteins induced by the GFOGER monolayer and, in particular, to compare these with the sequence of events mediated by the major platelet integrin αIIbβ3. Obergfell et al. (2002) has recently shown a key role for Src in αIIbβ3-dependent spreading. This is consistent with the observation that PP2 and SU6656 partially inhibit spreading and protein tyrosine phosphorylation induced by GFOGER and suggests that the two integrins may signal in a similar way. Obergfell et al. (2002) used a phosphospecific antibody to Tyr-416 to demonstrate activation of Src during spreading on fibrinogen. GFOGER stimulated a 1.5-fold increase in phosphorylation on Tyr-416 at 5 and 10 min, the earliest time points that could be investigated because of the low number of adherent cells (Fig. 6 A). The smaller response to GFOGER relative to that induced by adhesion to fibrinogen (Obergfell et al., 2002) is likely to reflect the 50-fold lower density of α2β1 relative to that of αIIbβ3. Adhesion to fibrinogen induces tyrosine phosphorylation of Syk and SLP-76 but not the adaptor LAT, even though this is a prominent phosphorylated band in GPVI-activated platelets (Judd et al., 2000, 2002; Obergfell et al., 2001; Wonerow et al., 2002). GFOGER also stimulated marked tyrosine phosphorylation of Syk and SLP-76, whereas tyrosine phosphorylation of LAT was not detected (Fig. 6 B). Recently, it has been reported that the plasma membrane calcium ATPase (PMCA) on platelet membranes, which mediates calcium efflux from the cytoplasm, is phosphorylated on tyrosine by FAK and that this leads to its inhibition (Wan et al., 2003). PMCA is also phosphorylated after adhesion to GFOGER (Fig. 6 B, 4). These results demonstrate that activation of Src is an early event in platelet adhesion to GFOGER and that this is associated with tyrosine phosphorylation of Syk, SLP-76, and PMCA, but not LAT, as well as FAK and PLCγ2, as described in the previous section.
Figure 6.
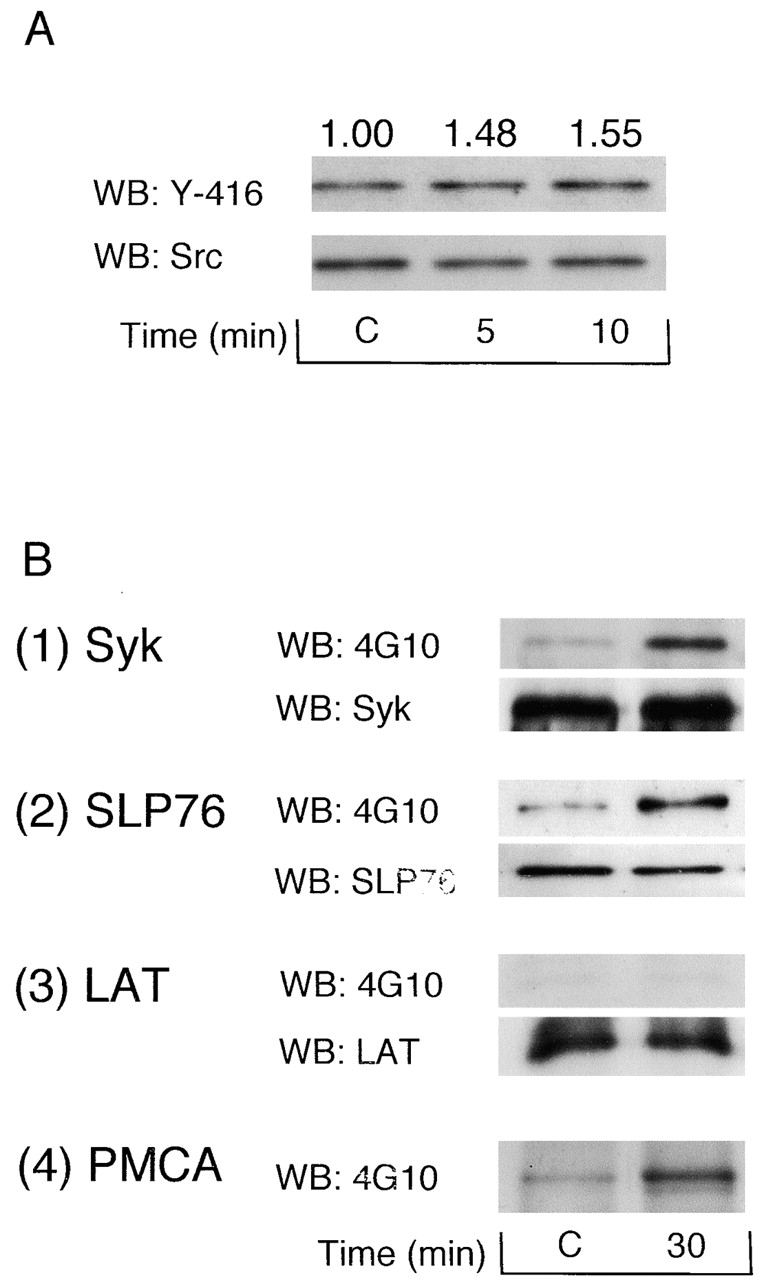
Src Tyr-416, Syk, SLP-76, and PMCA are tyrosine phosphorylated during spreading on GFOGER. (A) 300 μl of washed human platelets (3 × 108/ml) was pretreated with 100 μM EGTA, 10 μM indomethacin, 10 μM lotrafiban, and 3 U/ml apyrase and seeded on the dishes coated with 1% BSA (C, control) or 50 μg/ml GFOGER at 30°C for the indicated times. Reactions were stopped by the addition of 150 μl of 3× lysis buffer without removing unbound platelets. Src kinase was isolated by immunoprecipitation with anti-Src mAb, separated by 8% SDS-PAGE, and immunoblotted with anti–Src tyrosine-416 (Y-416) pAb or anti-Src mAb. Phosphorylation of Src Y-416 was quantified using Quantity One software for Macintosh. Optical density was standardized by the recruitment of Src. The optical density measurements are shown above the corresponding lane. (B) 300 μl washed human platelets (3 × 108/ml) was pretreated with the inhibitors and stimulated as described above. Reactions were stopped by the addition of 150 μl of 3× lysis buffer without removing unbound platelets. Syk, SLP-76, LAT, and PMCA were isolated by immunoprecipitation and Western blotted as described in the Materials and methods. Results are representative of two experiments.
GFOGER peptide stimulates spreading though PLCγ2 and mobilization of Ca2+
The above results demonstrate two pathways that have the potential to increase intracellular Ca2+ after adhesion to GFOGER, namely via activation of PLCγ2 downstream of Src, Syk, and SLP-76 and through inhibition of PMCA by FAK, which is partially regulated by Src. Dynamic video imaging of Fura2-loaded platelets confirmed that GFOGER stimulated an increase in Ca2+ in adherent cells (Fig. 7 A, 1). A record from a representative cell shows that the increase in Ca2+ consists of a series of oscillatory changes that decline over time (Fig. 7 A, 2). The increase in Ca2+ was inhibited completely in the presence of the intracellular Ca2+–chelating agent BAPTA-AM (Fig. 7 A, 1), which also reduced the number of adherent platelets and inhibited spreading (Fig. 7 B). In contrast, chelation of extracellular Ca2+ with 1 mM EGTA did not have a significant effect on adhesion or spreading on GFOGER (unpublished data). PLCγ2-deficient platelets were used to investigate a possible causal link between tyrosine phosphorylation of the lipase and spreading on GFOGER. A similar number of wild-type and PLCγ2-deficient platelets formed stable adhesions on GFOGER, whereas spreading of the lipase-deficient cells was significantly impaired (Fig. 8) . These results demonstrate that intracellular Ca2+ is essential for α2β1-mediated platelet spreading and that this contributes significantly to adhesion. The results also demonstrate a role for PLCγ2 in α2β1-mediated spreading.
Figure 7.
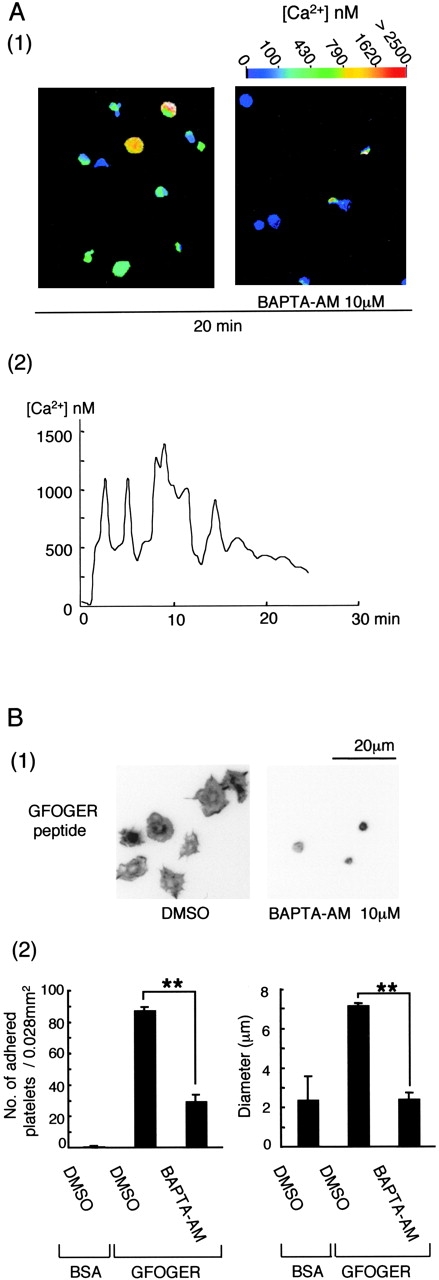
BAPTA-AM inhibits α2β1-mediated platelet adhesion and spreading on surfaces coated with GFOGER peptide. (A, 1) Washed human platelets (5 × 108/ml) were loaded with 10 μM Fura2-AM for 1 h at 30°C and then washed and resuspended to 2 × 107/ml in modified Tyrodes buffer with the inhibitor cocktail. 2 × 106 platelets were added to a GFOGER-coated coverslip. Fluorescence imaging and Ca2+ concentration measurements were performed with fluorescent microscopy using Openlab software for Macintosh. Results are representative of three experiments. (A, 2) The calcium concentration in a single platelet is shown. (B, 1) Washed human platelets (2 × 107/ml) were pretreated with the inhibitor cocktail, with or without 10 μM BAPTA-AM for 20 min. Platelets were seeded on BSA- or GFOGER-coated coverslips and stained and photographed as described in Fig. 1. (B, 2) The data represents the mean number of adhered platelets ± SEM per 0.028 mm2 from five different images from three experiments. The mean ± SEM diameter of 200 platelets from three experiments was measured. **, P < 0.01.
Figure 8.
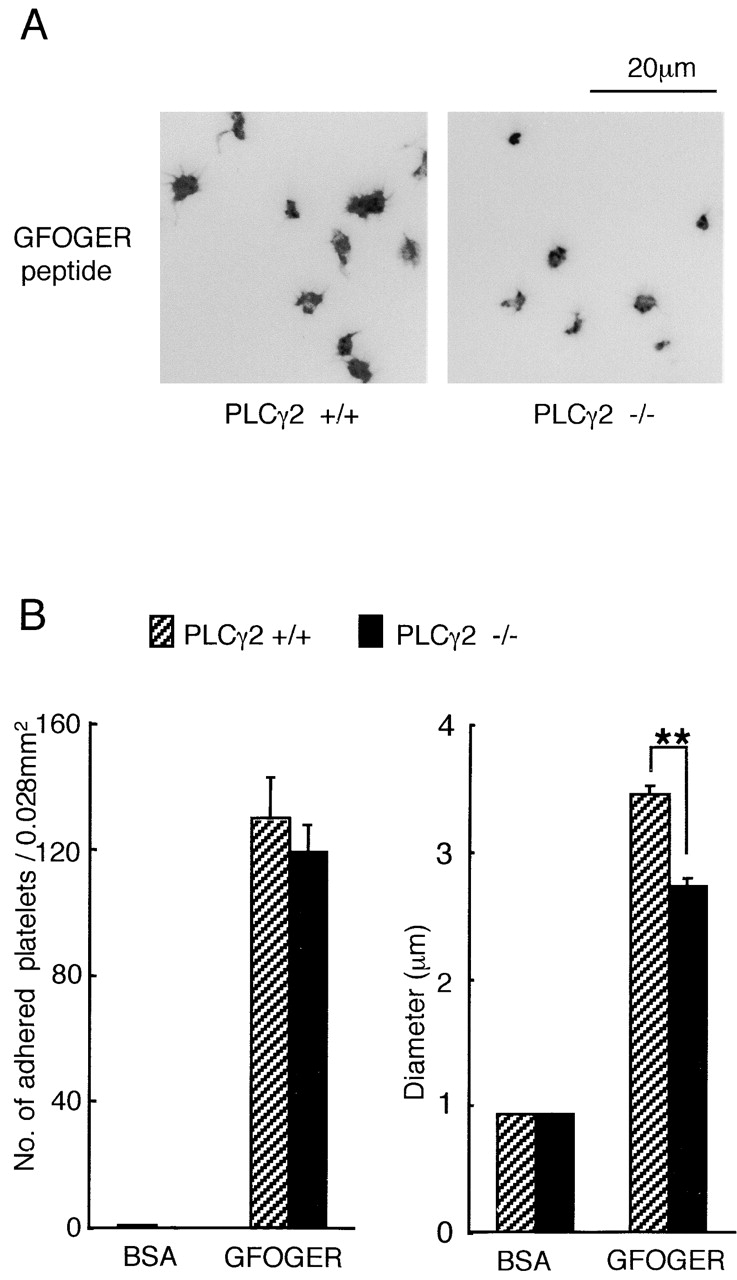
PLCγ2-deficient murine platelets exhibit limited spreading on GFOGER peptide. (A) Washed platelets (3 × 107/ml) from PLCγ2 (+/+) or (−/−) mice were pretreated with the inhibitor cocktail for 10 min. Platelets were seeded on BSA- or GFOGER-coated coverslips and stained as described in Fig. 1. (B) The number and diameter of adhered platelets were measured as described in Fig. 2. The data represent the mean number of adhered platelets ± SEM per 0.028 mm2 from 10 different images from two experiments. The mean ± SEM diameter of 200 platelets from two experiments was measured. **, P < 0.01.
Discussion
The present study provides evidence that activation of α2β1 induces spreading via a pathway that involves Src, Syk, SLP-76, PLCγ2, FAK, PMCA, and Ca2+ mobilization. This pathway was not detectable in platelet suspensions, explaining why previous studies have not reported signaling events through the integrin in platelets. The observation that α2β1 signals only in response to immobilized ligand demonstrates a unique mechanism to prevent platelet activation within blood vessels and yet promote activation upon damage to the subendothelial matrix. We speculate that α2β1-mediated spreading may have particular relevance in strengthening attachment of the thrombus to the subendothelial matrix through processes such as clot retraction. Such a role would account for the increased embolization of thrombi on collagen that is seen with β1-deficient platelets (Kuijpers et al., 2003). In addition, the observation that α2β1 and GPVI regulate a common set of proteins may enable the integrin to compensate for the absence of the glycoprotein, at least to a limited extent. This could explain the relatively small increase in bleeding time in mice that are deficient in the GPVI–FcR γ-chain complex.
Suzuki-Inoue et al. (2001) originally described spreading of human platelets on a surface coated with the F(ab')2 fragments of the anti–integrin α2β1 antibody TS2/16. More recently, Guidetti et al. (2002) have provided evidence that the small proteoglycan decorin induces tyrosine phosphorylation of Syk and PLCγ2 in platelets through integrin α2β1. In the present study, we have demonstrated spreading of human and murine platelets on an α2β1-specific peptide, GFOGER, that is unable to bind to GPVI or integrin αIIbβ3. Spreading on GFOGER is observed in the presence of a cyclooxygenase inhibitor and antagonists at αIIbβ3 and ADP receptors, demonstrating a direct role for α2β1 in mediating platelet activation. We also observed spreading of murine platelets deficient in GPVI–FcR γ-chain on collagen. These studies were performed in the presence of ADP or PMA to induce activation of α2β1 and thereby promote adhesion (Jung and Moroi, 1998, 2000).
Spreading on GFOGER is associated with a marked increase in tyrosine phosphorylation of Src, Syk, SLP-76, PLCγ2, FAK, and PMCA and elevation of intracellular Ca2+. Tyrosine phosphorylation of PLCγ2 is abolished in the presence of PP2 while phosphorylation of FAK is substantially inhibited. Spreading is reduced in the presence of Src kinase inhibitors and in PLCγ2-deficient platelets and abolished by chelation of intracellular Ca2+. These observations demonstrate that α2β1 mediates spreading through a Src kinase–dependent pathway that lies upstream of PLCγ2 and Ca2+. Syk and SLP-76 are likely to be downstream targets of Src by analogy with the signaling events mediated by αIIbβ3 (Judd et al., 2000; Obergfell et al., 2001, 2002). The limited degree of spreading observed in the presence of Src kinase inhibition may be mediated through phosphorylation of PMCA by FAK, leading to inhibition of Ca2+ extrusion (Wan et al., 2003) and a slow elevation in the intracellular levels of the cation. A residual increase in Ca2+ mobilization (unpublished data) is seen in the presence of PP2 on a GFOGER-coated surface and is therefore consistent with this possibility. These potential pathways are summarized in Fig. 9 .
Figure 9.
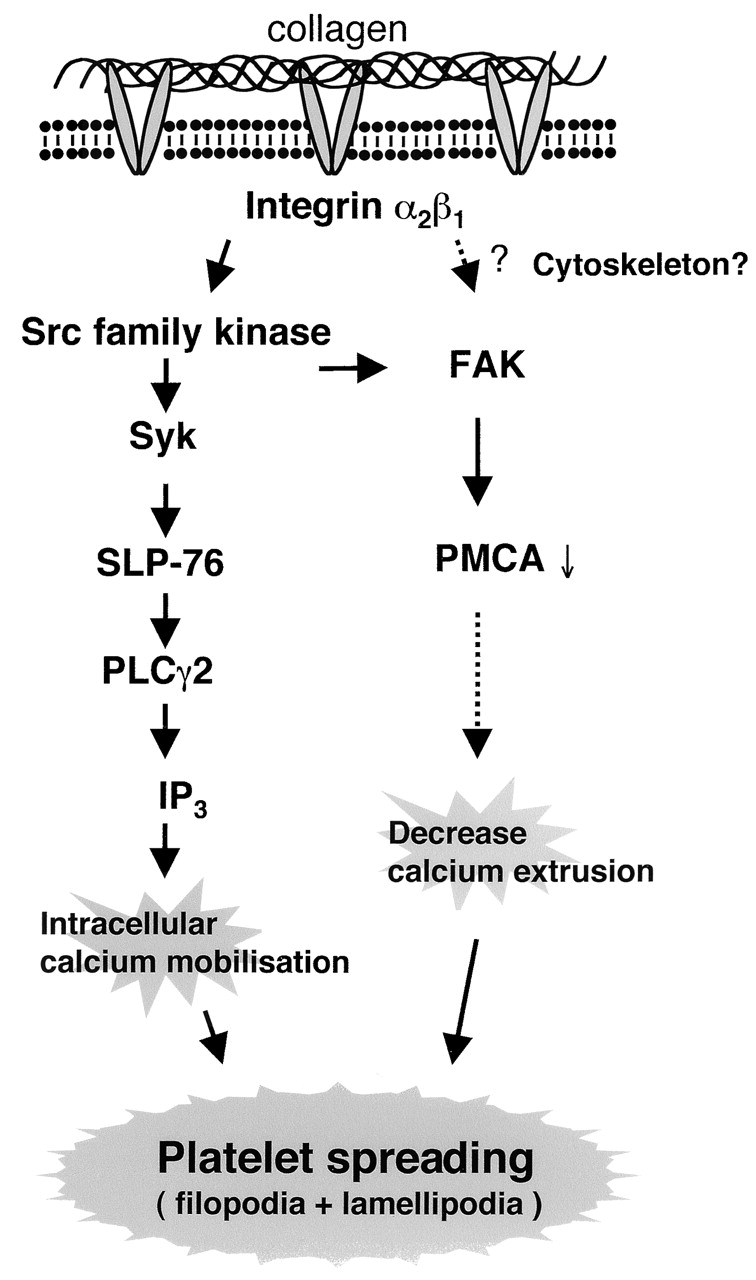
Model depicting the intracellular events during integrin α2β1–mediated platelet spreading. Integrin α2β1 mediates activation of a Src family kinase, Syk, and SLP-76 upstream of PLCγ2, Ca2+ mobilization, and platelet spreading. A second pathway can also give rise to Ca2+ mobilization and platelet spreading. This pathway may involve the tyrosine kinase FAK, whose phosphorylation is mediated, in part, through a Src kinase–independent pathway. A possible pathway of FAK-mediated increase in calcium is the inhibition of PMCA via tyrosine phosphorylation.
It is important to consider why binding of FcR γ-chain–deficient platelets to GFOGER does not require prior activation of α2β1, whereas this is not the case for collagen. This difference may be related to the frequency of the α2β1-specific binding sequence, GFOGER, in collagen. There are only two copies of GFOGER in a type I collagen molecule (a frequency of 0.4%), although it does contain a number of other GER-containing motifs that are likely to have a lower affinity for the integrin. In contrast, GFOGER is present at a frequency of 14% in a GFOGER peptide (one copy per 42 amino acids). This frequency may be sufficient to ensure stable adhesion to the low-affinity (or inactive) form of α2β1 through a combination of affinity and avidity. Moreover, once bound, the GFOGER peptide is likely able to convert the integrin to its high-affinity (or active) conformation through the generation of intracellular signals. On the other hand, we consider it unlikely that binding to GFOGER peptide can be explained by the presence of a significant population of platelets that express α2β1 in its active conformation, as adhesion of FcR γ-chain–deficient platelets to collagen is potentiated by stimuli that convert the integrin to a high-affinity conformation. Further, an antibody (7F6) developed by A. Schoolmeester and H. Deckmyn (Katholieke Universiteit Leuven Campus Kortrijk, Kortrijk, Belgium) that recognizes the active form of α2β1 demonstrated that the vast majority of the integrin is in an inactive conformation (unpublished data). It is also important to consider why platelets are activated by a monolayer of GFOGER peptide, whereas similar results are not seen in suspension studies. The same question applies to the interaction of platelets with fibrinogen. This is most likely brought about through enhanced avidity caused by movement of integrins to the exposed surface and potentiation of intracellular signaling (Hato et al., 1998).
It is of considerable interest that the mechanism of signaling by α2β1 is similar to that of αIIbβ3. Recently, Woodside et al. (2002) have reported that the NH2-terminal SH2 domain and linker region of Syk bind with high affinity to the tail of the β3 integrin subunit. In the same study, the authors demonstrated that Syk binds with a slightly lower affinity to the tail of the β1 integrin subunit. This gives rise to speculation that the signaling pathways between the two integrin β subunits are conserved, and that the sequence of events that have been established for αIIbβ3 may apply to other integrins, including α2β1. Consistent with this, both integrins are located outside of membrane rafts (Wonerow et al., 2002), and both regulate Syk, SLP-76, FAK, and PLCγ2, but not the raft-associated adaptor LAT.
The observation that α2β1 mediates spreading adds to the growing list of platelet receptors that are able to mediate this response, including GPVI, αIIbβ3, and G protein–coupled receptor stimuli. This apparent redundancy brings into question the significance of the present observations. Importantly, however, the role of soluble mediators during thrombus growth is compromised by their rapid removal in the flowing blood and by recruitment of further platelets into the developing thrombus. In this context, α2β1 may play an important role in the regulation of spreading, especially bearing in mind the high collagen content of the subendothelial matrix.
Additional work is required to establish the physiological and pathological significance of the α2β1-dependent signals reported in the present study. Although mice deficient in α2β1 do not exhibit a marked increase in tail bleeding times (Nieswandt et al., 2001a; Holtkötter et al., 2002), the integrin may play an important role in vascular beds that are high in collagen or that have a different type of lesion. In this context, the observation of increased embolization of β1-deficient thrombi is of particular interest (Kuijpers et al., 2003). The significance of α2β1-dependent signals may also be masked in the presence of GPVI. Indeed, the converse may also apply in that the relatively small increase in bleeding times observed in GPVI-deficient platelets (Nieswandt et al., 2001b) may be due to α2β1-mediated intracellular signals. Strikingly, the two receptors regulate a common set of proteins, including Src kinases and PLCγ2. Signaling through α2β1 may also have a significant role in pathological thrombosis, and this could explain the increase in thrombotic risk that is seen in individuals with polymorphisms that lead to an increase in expression of the integrin α2β1 (Kunicki, 2002). Agents that prevent signaling by α2β1 therefore represent potential novel anti-thrombotics. PLCγ2 also appears to be a particularly good target in that it lies downstream of the integrins α2β1 and αIIbβ3, as well as the major receptor for collagen, GPVI.
Materials and methods
Materials
Anti–integrin α2 mAb 6F1 and anti–glycoprotein Ib mAb 6D1 were gifts from B. Coller (Mt. Sinai Medical Center, New York, NY). Anti–SLP-76 pAb was donated by G. Koretzky (University of Pennsylvania, Philadelphia, PA). The anti-PMCA pAb was raised as previously described (Blankenship et al., 2000). 2-propylthio-d-fluoromethylene adenosine 5-trisphosphate (AR-C67085) was a gift from AstraZeneca. FcR γ-chain– (Snell et al., 2002), PLCγ2- (Wang et al., 2000), and αIIb-deficient mice (provided by J. Frampton and N.R. Emambokus, University of Birmingham) were bred on a B6 background. Mice were genotyped by PCR and wild-type litter mates used as controls. GFOGER peptide(GPC[GPP]5GFOGER[GPP]5GPC) was synthesized by TANA laboratories and cross-linked using N-succinimidyl 3-[2-pyridyldithio]propionate (SPDP) (Morton et al., 1995). 4-amino-5-(4-chlorophenyl)-7-(t-butil)pyrazolo[3,4-d]pyrimidine (PP2) and 4-amino-7-phenylpyrazolo[3,4-d]pyrimidine (PP3) were from Calbiochem. SU6656 was from SUGEN. Anti-phosphotyrosine mAb (4G10), anti–FcR γ-subunit pAb for Western blotting, anti-Src mAb (GD11), and anti-LAT pAb were from Upstate Biotechnology. Purified anti–mouse α2 integrin mAb (HMα2), hamster IgG1κ (A19-3), anti–SLP-76 mAb, and anti-FAK mAb (clone 8) for Western blotting were from Becton Dickinson. Hybridoma of anti–human FcR γRIIA (IV.3) was obtained from American Type Culture Collection, and F(ab')2 fragment of IV.3 was generated using ImmunoPure® F(ab')2 preparation kit. Anti-FAK pAb (C-903) and anti-Syk pAb (LR) for immunoprecipitation and anti-Syk mAb (4D10) for Western blotting were from Santa Cruz Biotechnology, Inc. An anti-PMCA mAb for Western blotting (5F10), mouse IgG1κ (MOPC 21), ADP, adenosine 3-phosphate 5-phosphate (A3P5P), and TRITC-conjugated phalloidin were from Sigma-Aldrich. Anti–phospho-Src family (Tyr416) pAb was from New England Biolabs, Inc. Fura2-AM was from Molecular Probes. RC DC protein assay kit II was from Bio-Rad Laboratories. All other reagents were from previously named sources (Wonerow et al., 2002).
Preparation of human and murine platelets
Venous blood from healthy drug-free volunteers was taken into 10% sodium citrate. Platelet-rich plasma was obtained after centrifugation at 200 g for 20 min with 15% acid–citrate–dextrose. EGTA (1 mM) was added, and the platelet-rich plasma was centrifuged at 640 g for 15 min. Murine blood was drawn from CO2 terminally anesthetized mice by cardiac puncture and taken into 100 μl of acid–citrate–dextrose, and washed platelets were prepared as previously described (Snell et al., 2002). Human and murine platelets were resuspended in modified Tyrodes buffer (137 mM NaCl, 11.9 mM NaHCO3, 0.4 mM Na2HPO4, 2.7 mM KCl, 1.1 mM MgCl2, 5.6 mM glucose, pH 7.3) at a cell density of 0.2–4 × 108/ml. Platelet suspensions were left for 30 min and then incubated with 10 μM lotrafiban, 10 μM indomethacin, 100 μM EGTA, and, where necessary, 1 mM A3P5P and 1 μM AR-C67085 for 10 min before experimentation. The concentration of DMSO in the incubation never exceeded 0.2%, and an equivalent volume of DMSO was present in controls.
Platelet spreading
For the spreading experiments, 50 μg/ml collagen or 50 μg/ml cross-linked GFOGER peptide was incubated on a coverslip overnight at 4°C. After washing twice with PBS, the coverslip was blocked with 1% fatty acid–free purified BSA in PBS for 2 h at room temperature and washed twice with modified Tyrodes. Washed murine platelets (3 × 107/ml) and washed human platelets (2 × 107/ml) were incubated on the coverslips at 30°C. After removal of unbound platelets by washing with modified Tyrodes buffer, adhered platelets were fixed, permeabilized, and stained by TRITC-conjugated phalloidin, as described previously (Suzuki-Inoue et al., 2001). Platelets were viewed on an inverted fluorescent microscope (Carl Zeiss MicroImaging, Inc.), and digital imaging (×400) was performed using Openlab software for Macintosh. The number and diameter of platelets were counted and measured by a ruler on the printed images (0.028 mm2/image). Statistical significance was evaluated by the t test using a software for statistics, T-Kentakun. In each case, P < 0.05 was taken as the minimum value to indicate statistical significance.
For phase contrast imaging, glass plates were incubated with 50 μg/ml of cross-linked GFOGER peptide overnight. The plates were then blocked with 1% BSA as described above. Platelets were viewed on a phase contrast microscope (Carl Zeiss MicroImaging, Inc.), and digital imaging (×600) was performed using LaserSharp 2000 software.
Protein precipitation studies
Six-well flat-bottomed plates were coated with cross-linked GFOGER peptide (50 μg/ml) overnight at 4°C. The plates were washed and blocked, as described above, before incubation with 500 μl washed platelets (4 × 108/ml) at 30°C for 15–30 min. Unbound platelets were removed by aspiration, and adhered cells were solubilized with 500 μl Laemmli sample buffer. A sample of the suspension was taken at the same time and used as a control. Protein concentrations were determined by RC DC protein assay kit II according to the manufacturer's instruction, and matched amounts of protein were separated by one-dimensional SDS-PAGE. Protein tyrosine phosphorylation was measured by Western blotting with 4G10. Protein loading was measured by Coomassie brilliant blue staining.
Immunoprecipitation studies were performed as previously described (Suzuki-Inoue et al., 2002). 500 μl of 2× ice-cold lysis buffer (2% vol/vol Nonidet P-40, 20 mM Tris, 300 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 2 mM Na3VO4, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 μg/ml pepstatin A, pH 7.3) was added to each well without removing unbound platelets. After removing debris and preclearing, the supernatants were incubated with 6 μl anti-FAK pAb, 2 μl anti-PLCγ2 pAb, 2 μl anti-Src mAb, 2 μl anti-Syk pAb, 2 μl anti–SLP-76 pAb, 4 μl anti-LAT pAb, or 20 μl anti-PMCA pAb and 30 μl protein A–Sepharose at 4°C overnight. FcR γ-chain was precipitated using glutathione-agarose conjugated with GST–Syk SH2 as previously described (Gibbins et al., 1996). After centrifugation, the pellet was washed and then solubilized by addition of 2× Laemmli sample buffer, and proteins were separated by SDS-PAGE on 8% gels. Protein tyrosine phosphorylation was detected by blotting with 4G10 or anti–Src Tyr416 pAb. Gels were reprobed with anti-FAK mAb, anti-PLCγ2 pAb, anti-Src mAb, anti-Syk mAb, anti–SLP-76 mAb, anti-LAT pAb, or anti-PMCA mAb. Phosphorylation of Src Tyr416 was quantified using Quantity One software for Macintosh.
Intracellular Ca2+ measurement
Platelet suspensions (5 × 108/ml) were loaded with 10 μM Fura2-AM at 30°C for 1 h. Platelets were washed and resuspended to 2 × 107/ml in modified Tyrodes buffer with 1 mM EGTA, 10 μM indomethacin, 10 μM lotrafiban, 1 mM A3P5P, and 1 μM AR-C67085. 2 × 106 platelets were added to a GFOGER-coated coverslip, and fluorescence imaging was performed using Openlab software for Macintosh as described previously (Mountford et al., 1999).
Acknowledgments
We are grateful to Dr. B. Coller, Dr. M. Tomlinson, and Dr. G. Koretzky for donating the antibodies and to Dr. H. Deckmyn and Ms. A. Schoolmeester for supplying anti–integrin α2 antibody (7F6). Gratitude is expressed to Peter Busby, Ben Atkinson, Gavin Jarvis, and Andrew Pearce for their kind help with these studies.
This work was supported by grants from the British Heart Foundation, Japan Clinical Pathology Foundation for International Exchange, and Mochida Memorial Foundation for Medical and Pharmaceutical Research, Japan. S.P. Watson holds a British Heart Foundation Chair in Cardiovascular Sciences.
Footnotes
Abbreviations used in this paper: FAK, focal adhesion kinase; FcR, Fc receptor; GPVI, glycoprotein VI; PMCA, plasma membrane calcium ATPase; vWf, von Willebrand factor.
References
- Blankenship, K.A., C.B. Dawson, G.R. Aronoff, and W.L. Dean. 2000. Tyrosine phosphorylation of human platelet plasma membrane Ca2+-ATPase. Hypertension. 35:103–107. [DOI] [PubMed] [Google Scholar]
- Chen, J., T.G. Diacovo, D.G. Grenache, S.A. Santoro, and M.M. Zutter. 2002. The α2 integrin subunit-deficient mouse. Am. J. Pathol. 161:337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbins, J.M., J. Asselin, R. Farndale, M. Barnes, C. Law, and S.P. Watson. 1996. Tyrosine phosphorylaion of the Fc receptor γ-chain in collagen-stimulated platelets. J. Biol. Chem. 271:18095–18099. [DOI] [PubMed] [Google Scholar]
- Goto, S., N. Tamura, S. Handa, M. Arai, K. Kodama, and H. Takayama. 2002. Involvement of glycoprotein VI in platelet thrombus formation on both collagen and von Willebrand factor surfaces under flow conditions. Circulation. 106(2):266–272. [DOI] [PubMed] [Google Scholar]
- Guidetti, G., A. Bertoni, M. Viola, E. Tira, C. Balduini, and M. Torti. 2002. The small proteoglycan decorin supports adhesion and activation of human platelets. Blood. 100:1707–1714. [PubMed] [Google Scholar]
- Hato, T., N. Pampori, and S.J. Shattil. 1998. Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin αIIbβ3. J. Cell Biol. 141:1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtkötter, O., B. Nieswandt, N. Smyth, W. Muller, M. Hafner, V. Schulte, T. Krieg, and B. Eckes. 2002. Integrin α2-deficient mice develop normally, are fertile, but display partially defective platelet interaction with collagen. J. Biol. Chem. 277:10789–10794. [DOI] [PubMed] [Google Scholar]
- Judd, B.A., P.S. Myung, L. Leng, A. Obergfell, W.S. Pear, S.J. Shattil, and G.A. Koretzky. 2000. Hematopoietic reconstitution of SLP-76 corrects hemostasis and platelet signaling through αIIb β3 and collagen receptors. Proc. Natl. Acad. Sci. USA. 97:12056–12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judd, B.A., P.S. Myung, A. Obergfell, E.E. Myers, A.M. Cheng, S.P. Watson, W.S. Pear, D. Allman, S.J. Shattil, and G.A. Koretzky. 2002. Differential requirement for LAT and SLP-76 in GPVI versus T cell receptor signaling. J. Exp. Med. 195:705–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, S.M., and M. Moroi. 1998. Platelets interact with soluble and insoluble collagens through characteristically different reactions. J. Biol. Chem. 273:14827–14837. [DOI] [PubMed] [Google Scholar]
- Jung, S.M., and M. Moroi. 2000. Signal-transducing mechanisms involved in activation of the platelet collagen receptor integrin α2β1. J. Biol. Chem. 275:8016–8026. [DOI] [PubMed] [Google Scholar]
- Knight, C.G., L.F. Morton, D.J. Onley, A.R. Peachey, A.J. Messent, P.A. Smethurst, D.S. Tuckwell, R.W. Farndale, and M.J. Barnes. 1998. Identification in collagen type I of an integrin α2β1-binding site containing an essential GER sequence. J. Biol. Chem. 273:33287–33294. [DOI] [PubMed] [Google Scholar]
- Knight, C.G., L.F. Morton, D.J. Onley, A.R. Peachey, T. Ichinohe, M. Okuma, R.W. Farndale, and M.J. Barnes. 1999. Collagen-platelet interaction: Gly-Pro-Hyp is uniquely specific for platelet Gp VI and mediates platelet activation by collagen. Cardiovasc. Res. 41:450–457. [DOI] [PubMed] [Google Scholar]
- Knight, C.G., L.F. Morton, A.R. Peachey, D.S. Tuckwell, R.W. Farndale, and M.J. Barnes. 2000. The collagen-binding A-domains of integrins α1β1 and α2β1 recognize the same specific amino acid sequence, GFOGER, in native (triple-helical) collagens. J. Biol. Chem. 275:35–40. [DOI] [PubMed] [Google Scholar]
- Kuijpers, M.J.E., V. Schulte, W. Bergmeier, T. Lindhout, C. Brakebusch, S. Offermanns, R. Fässler, J.W.M. Heemskerk, and B. Nieswandt. 2003. Complementaly roles of platelet glycoprotein VI and α2β1 integrin in collagen-induced thrombus formation in flowing whole blood ex vivo. FASEB J. In press. [DOI] [PubMed] [Google Scholar]
- Kunicki, T.J. 2002. The influence of platelet collagen receptor polymorphisms in hemostasis and thrombotic disease. Arterioscler. Thromb. Vasc. Biol. 22:14–20. [DOI] [PubMed] [Google Scholar]
- Massberg, S., M. Gawaz, S. Gruner, V. Schulte, I. Konrad, D. Zohlnhofer, U. Heinzmann, and B. Nieswandt. 2003. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J. Exp. Med. 197:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, L.F., P.G. Hargreaves, R.W. Farndale, R.D. Young, and M.J. Barnes. 1995. Integrin α2β1-independent activation of platelets by simple collagen-like peptides: collagen tertiary (triple-helical) and quaternary (polymeric) structures are sufficient alone for α2β1-independent platelet reactivity. Biochem. J. 306:337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountford, J.C., S.K. Melford, C.M. Bunce, J. Gibbins, and S.P. Watson. 1999. Collagen or collagen-related peptide cause (Ca2+)I elevation and increased tyrosine phosphorylation in human megakaryocytes. Thromb. Haemost. 82:1153–1159. [PubMed] [Google Scholar]
- Nieswandt, B., C. Brakebusch, W. Bergmeier, V. Schulte, D. Bouvard, R. Mokhtari-Nejad, T. Lindhout, J.W. Heemskerk, H. Zirngibl, and R. Fassler. 2001. a. Glycoprotein VI but not α2β1 integrin is essential for platelet interaction with collagen. EMBO J. 20:2120–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieswandt, B., V. Schulte, W. Bergmeier, R. Mokhtari-Naja, K. Rackebrandt, J.P. Cazenave, P. Ohlmann, C. Gachet, and H. Zirngibl. 2001. b. Long-term antithrombotic protection by in vivo depletion of platelet glycoprotein VI in mice. J. Exp. Med. 193:459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obergfell, A., B.A. Judd, M.A. del Pozo, M.A. Schwartz, G.A. Koretzky, and S.J. Shattil. 2001. The molecular adapter SLP-76 relays signals from platelet integrin αIIbβ3 to the actin cytoskeleton. J. Biol. Chem. 276:5916–5923. [DOI] [PubMed] [Google Scholar]
- Obergfell, A., K. Eto, A. Mocsai, C. Buensuceso, S.L. Moores, J.S. Brugge, C.A. Lowell, and S.J. Shattil. 2002. Coordinate interactions of Csk, Src, and Syk kinases with αIIbβ3 initiate integrin signaling to the cytoskeleton. J. Cell Biol. 157:265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage, B., E. Saldivar, and Z.M. Ruggeri. 1996. Initiation of platelet adhesion by arrest on fibrinogen or tranlocation on von Willebrand factor. Cell. 84:289–297. [DOI] [PubMed] [Google Scholar]
- Savage, B., F. Almus-Jacobs, and Z.M. Ruggeri. 1998. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 94:657–666. [DOI] [PubMed] [Google Scholar]
- Shattil, S.J., B. Haimovich, M. Cunningham, L. Lipfert, J.T. Parsons, M.H. Ginsberg, and J.S. Brugge. 1994. Tyrosine phosphorylation of pp125FAK in platelets requires coordinated signaling through integrin and agonist receptors. J. Biol. Chem. 269:14738–14745. [PubMed] [Google Scholar]
- Snell, D.C., V. Schulte, G.E. Jarvis, K. Arase, D. Sakurai, T. Saito, S.P. Watson, and B. Nieswandt. 2002. Differential effect of reduced glycoprotein VI levels on activation of murine platelets by glycoprotein VI ligands. Biochem. J. 368:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki-Inoue, K., Y. Yatomi, N. Asazuma, M. Kainoh, T. Tanaka, K. Satoh, and Y. Ozaki. 2001. Rac, a small guanosine triphosphate-binding protein, and p21-activated kinase are activated during platelet spreading on collagen-coated surfaces: roles of integrin α2β1. Blood. 98:3708–3716. [DOI] [PubMed] [Google Scholar]
- Suzuki-Inoue, K., D. Tulasne, Y. Shen, T. Bori-Sanz, O. Inoue, S.M. Jung, M. Moroi, R.K. Andrews, M.C. Berndt, and S.P. Watson. 2002. Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J. Biol. Chem. 277:21561–21566. [DOI] [PubMed] [Google Scholar]
- Wan, T.C., M. Zabe, and W.L. Dean. 2003. Plasma membrane Ca2+-ATPase isoform 4b is phosphorylated on tyrosine 1176 in activated human platelets. Thromb. Haemost. 89:122–131. [PubMed] [Google Scholar]
- Wang, D., J. Feng, R. Wen, J.C. Marine, M.Y. Sangster, E. Parganas, A. Hoffmeyer, C.W. Jackson, J.L. Cleveland, P.J. Murray, and J.N. Ihle. 2000. Phospholipase Cγ2 is essential in the functions of B cell and several Fc receptors. Immunity. 13:25–35. [DOI] [PubMed] [Google Scholar]
- Watson, S.P., and J. Gibbins. 1998. Collagen receptor signalling in platelets: extending the role of the ITAM. Immunol. Today. 19:260–264. [DOI] [PubMed] [Google Scholar]
- Wonerow, P., A. Obergfell, J.I. Wilde, R. Bobe, N. Asazuma, T. Brdicka, A. Leo, B. Schraven, V. Horejsi, S.J. Shattil, and S.P. Watson. 2002. Differential role of glycolipid-enriched membrane domains in glycoprotein VI- and integrin-mediated phospholipase Cγ2 regulation in platelets. Biochem. J. 364:755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodside, D.G., A. Obergfell, A. Talapatra, D.A. Calderwood, S.J. Shattil, and M.H. Ginsberg. 2002. The N-terminal SH2 domains of Syk and ZAP-70 mediate phosphotyrosine-independent binding to integrin β cytoplasmic domains. J. Biol. Chem. 277:39401–39408. [DOI] [PubMed] [Google Scholar]