

Abstract
The adaptor protein complex-1 (AP-1) sorts and packages membrane proteins into clathrin-coated vesicles (CCVs) at the TGN and endosomes. Here we show that this process is highly regulated by phosphorylation of AP-1 subunits. Cell fractionation studies revealed that membrane-associated AP-1 differs from cytosolic AP-1 in the phosphorylation status of its β1 and μ1 subunits. AP-1 recruitment onto the membrane is associated with protein phosphatase 2A (PP2A)–mediated dephosphorylation of its β1 subunit, which enables clathrin assembly. This Golgi-associated isoform of PP2A exhibits specificity for phosphorylated β1 compared with phosphorylated μ1. Once on the membrane, the μ1 subunit undergoes phosphorylation, which results in a conformation change, as revealed by increased sensitivity to trypsin. This conformational change is associated with increased binding to sorting signals on the cytoplasmic tails of cargo molecules. Dephosphorylation of μ1 (and μ2) by another PP2A-like phosphatase reversed the effect and resulted in adaptor release from CCVs. Immunodepletion and okadaic acid inhibition studies demonstrate that PP2A is the cytosolic cofactor for Hsc-70–mediated adaptor uncoating. A model is proposed where cyclical phosphorylation/dephosphorylation of the subunits of AP-1 regulate its function from membrane recruitment until its release into cytosol.
Keywords: adaptor protein-1; phosphoregulation; protein phosphatase 2A; clathrin-coated vesicle; uncoating
Introduction
Adaptor protein complex-1 (AP-1)* plays a central role in the sorting and packaging of membrane proteins into clathrin-coated vesicles (CCVs) at the TGN and endosomes (Klumperman et al., 1993; Meyer et al., 2000, 2001). These vesicles transport mannose 6-phosphate receptors (MPRs) with their bound lysosomal enzymes from the TGN to endosomes and then retrieve the receptors and other membrane proteins from endosomes back to the TGN. The formation of CCVs is a complex process involving interactions of AP-1 with cargo molecules, clathrin, and a variety of accessory proteins. Once the CCVs bud from the donor compartment, the clathrin is disassembled and the AP-1 is subsequently released into the cytoplasm (Huang et al., 2001). This “uncoating” process is known to involve Hsc-70, cyclin G–associated kinase (GAK/auxilin 2), and an unidentified cytoplasmic factor (Hannan et al., 1998; Greener et al., 2000; Umeda et al., 2000).
AP-1 is a heterotetrameric complex composed of two ∼100-kD subunits (termed γ and β1), an ∼47-kD subunit (μ1), and an ∼20-kD subunit (σ1). The hinge regions of the β1 and γ subunits bind clathrin via clathrin box motifs (Shih et al., 1995; Doray and Kornfeld, 2001), and the appendage of the γ subunit binds a number of accessory proteins, including γ synergin (Page et al., 1999); Rabaptin-5 (Shiba et al., 2002); Golgi-localized, γ ear–containing, adenosine 5′ diphosphate-ribosylation factor–binding proteins (GGAs) (Doray et al., 2002b); and enthoprotin/clint (Kalthoff et al., 2002; Wasiak et al., 2002). The μ1 subunit interacts with tyrosine-based sorting motifs present on the cytoplasmic tails of the MPRs and other cargo molecules (Bremnes et al., 1998; Ohno et al., 1998; Owen and Evans, 1998), and either it or the β1 subunit binds dileucine-based sorting motifs (Bremnes et al., 1998; Rapoport et al., 1998). These various protein–protein interactions must be highly coordinated if the assembly/disassembly of AP-1–containing CCVs is to proceed efficiently. Several reports have indicated that this process may be regulated by phosphorylation/dephosphorylation events. To date, clathrin (Hill et al., 1988), β1 and μ1 subunits of AP-1(Wilde and Brodsky, 1996), GGAs 1 and 3 (Doray et al., 2002a), and the cytoplasmic tails of MPRs (Meresse et al., 1990) have been shown to be phosphorylated. Further, it has been established that phosphorylation of the β1 hinge impairs clathrin binding and assembly (Wilde and Brodsky, 1996). Although the effect of μ1 phosphorylation on ligand binding has not been examined, it has been found that phosphorylation of the μ2 subunit of AP-2 greatly enhances the binding of sorting signals at the plasma membrane (Fingerhut et al., 2001; Ricotta et al., 2002). Finally, the phosphorylation of casein kinase-2 (CK2) sites in the cytoplasmic tails of the MPRs enhances binding to AP-1 (Le Borgne et al., 1993; Mauxion et al., 1996; Dittie et al., 1997).
We now report that phosphorylation of μ1 and β1 is differentially regulated and that phosphorylation of μ1 strongly enhances ligand binding, whereas dephosphorylation of this subunit by protein phosphatase 2A (PP2A) is an essential step in the CCV uncoating process. These data, together with the previous findings of others, are incorporated into a model whereby cycles of AP-1 phosphorylation/dephosphorylation regulate CCV assembly and disassembly.
Results
In vivo phosphorylation of AP-1
Wilde and Brodsky (1996) have reported that the μ1 and β1 subunits of AP-1 are phosphoproteins, and that the phosphorylated form of β1 is predominantly cytosolic. The subcellular distribution of phosphorylated μ1 was not examined. To pursue this point, membrane and cytosolic fractions were prepared from mouse L cells (LSS) that had been incubated with [32P]orthophosphate for 4 h. Then the AP-1 in each fraction was immunoprecipitated and analyzed by SDS-PAGE. As shown in Fig. 1 A, β1 was the only subunit labeled in the cytosolic AP-1, whereas μ1 was selectively labeled on the membrane-associated AP-1. Western blotting of the fractions showed an equal distribution of AP-1 between the membranes and the cytosol. The addition of 10 nM okadaic acid to the labeling media resulted in phosphorylation of the β1 subunit isolated from both locations as well as a shift of AP-1 distribution toward the membranes. Because this concentration of okadaic acid has been reported to selectively inhibit PP2A (Mumby and Walter, 1993; Bertrand et al., 1997), this result suggests that the β1 subunit is dephosphorylated by PP2A upon recruitment of AP-1 onto the membrane, whereas μ1 becomes phosphorylated.
Figure 1.

In vivo phosphorylation of AP-1 subunits. Confluent P100 dishes of mouse L cells (LSS) were labeled with 1 mCi/ml of [32P]orthophosphoric acid for 4 h in the presence or absence of 10 nM okadaic acid in the labeling medium. Membrane–cytosol fractionation was performed after harvesting the cells, and each fraction was immunoprecipitated with 2 μg of anti–AP-1 γ monoclonal antibody. The immunoprecipitated proteins were eluted in sample buffer and subjected to SDS-PAGE for subsequent autoradiography (A) and Western blotting (B). The band corresponding to the AP-1 γ subunit in the Western blots was quantitated using a densitometer and expressed as a ratio with the first lane as the point of reference. M, membrane; C, cytosol.
Phosphorylation of μ1 enhances ligand binding
AP-2 with a phosphorylated μ2 subunit exhibits a 15–30-fold enhanced avidity for tyrosine- and dileucine-based sorting motifs as compared with the nonphosphorylated species in binding studies (Fingerhut et al., 2001; Ricotta et al., 2002). It has also been established that the phosphorylation of the μ2 subunit is essential for endocytosis (Olusanya et al., 2001; Conner and Schmid, 2002). To determine whether phosphorylation of μ1 also enhances ligand binding, AP-1 was purified from cytosol that contained dephosphorylated μ1 and from CCVs that contained AP-1 with phosphorylated μ1 (Wilde and Brodsky, 1996). The ability of these AP-1s to bind to GST fusion peptides encoding wild-type and mutant forms of the cytoplasmic tails of the MPRs was then examined. As shown in Fig. 2 A, the CCV-derived AP-1 bound much better to the cation-dependent MPR (CD-MPR) full-length tail, and this binding was further enhanced when the CK2 site in the tail was phosphorylated. The CCV-derived AP-1 also exhibited strong binding to a GST fusion peptide containing the COOH-terminal 18 residues, which includes the acidic cluster CK2 site. However, high-avidity binding only occurred when the CK2 site was phosphorylated, demonstrating that phosphorylation of both the μ1 subunit and the CK2 site is required for optimal binding in this pull-down assay. This is consistent with the reports that phosphorylation of CK2 sites on the full-length MPR cytoplasmic tails stimulates AP-1 binding (Mauxion et al., 1996; Dittie et al., 1997).
Figure 2.
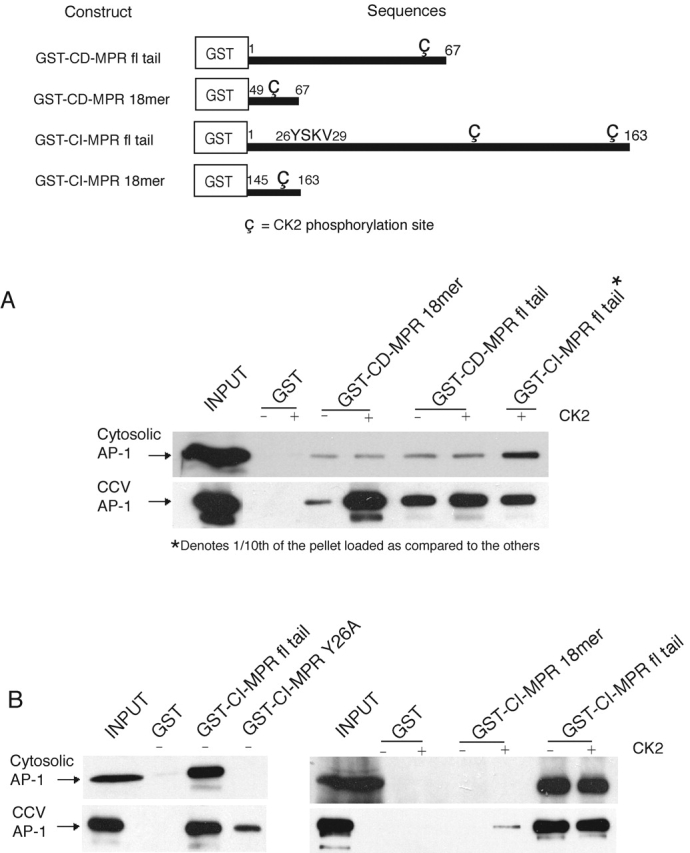
AP-1 from cytosolic versus CCV source display differential ligand binding. A schematic representation of the GST-fused MPR cytoplasmic tail constructs that were expressed and purified from E. coli is shown in the top panel. The CK2 sites are represented symbolically. (A and B) 100 μg of GST-fused CD-MPR or CI-MPR cytoplasmic tail constructs was incubated at 37°C for 3 h in the presence or absence of 500 U of recombinant human CK2 (Calbiochem) in buffer A supplemented with 1.5 mM ATP and 2 μM polylysine. Efficacy of phosphorylation achieved by this method was 75–80%. The CK2-treated ligands were then incubated with GSH-beads at room temperature for 2 h in 350 μl of buffer B. The beads were sedimented and washed with 1 ml of buffer B to remove unbound ligand and CK2. The glutathione-Sepharose beads with the various bound ligands were used in pull-down assays to assess their binding avidity toward AP-1 immunopurified from cytosol or CCVs. 10% of the input and 25% of the pellets (except as noted) were loaded onto a 10% SDS-PAGE gel and transferred onto nitrocellulose for immunoblotting with anti–AP-1γ (100/3) monoclonal antibody. The figures represent one of two to four independent experiments. fl, full length.
The difference in the ligand binding avidity between the two sources of AP-1 was less evident in the case of the cation-independent MPR (CI-MPR), which binds with much higher avidity to AP-1, as shown in the last lane of Fig. 2 A. Both forms of AP-1 bound similarly to the full-length wild-type cytoplasmic tail, and phosphorylation of the tail by CK2 did not enhance binding to any significant extent (Fig. 2 B, right). However, replacement of the essential tyrosine within the 26YSKV29 motif with an alanine resulted in a complete loss of binding by the cytosolic AP-1, whereas the CCV-derived AP-1 still bound, albeit to a much lesser extent compared with the wild-type tail (Fig. 2 B, left). This shows that both forms of AP-1 interact well with the tyrosine-based motif, whereas the AP-1 with phosphorylated μ1 binds better to the other sorting motifs. This conclusion is supported by the finding that the CCV-derived AP-1, but not the cytosolic AP-1, also bound to a GST fusion peptide containing the CK2 site present in the last 18 residues of the tail, but only when the site was phosphorylated (Fig. 2 B, right).
Taken together, these findings demonstrate that phosphorylation of the μ1 subunit enhances binding to sorting signals in the cytoplasmic tails of the MPRs, especially to the CK2 acidic cluster sites. This effect is most striking in the case of the CD-MPR; perhaps because it has a tyrosine-based sorting motif that is poorly recognized by AP-1 (Honing et al., 1997), and the other sorting motifs assume greater importance. One possible explanation for the enhanced binding to the CK2 acidic sorting motifs is that phosphorylation of μ1 induces a conformational change that exposes basic surfaces that are buried in the nonphosphorylated state. It is also apparent that μ1 phosphorylation is not the only factor that regulates the interaction of AP-1 with the MPR cytoplasmic tails, as phosphorylation of the CK2 sites enhances binding as well.
We next tested whether dephosphorylation of CCV-derived AP-1 impaired ligand binding. In a preliminary experiment, we showed that in vitro–phosphorylated μ1 (and μ2) was dephosphorylated by low nanogram amounts of PP2A, and this dephosphorylation was completely prevented by okadaic acid (Fig. 3 A). As seen in Fig. 3 B, the PP2A treatment decreased the ability of the CCV-derived AP-1 to bind to the CD-MPR tail. Because μ1 is the only subunit of the CCV-derived AP-1 that is phosphorylated, this finding confirms that μ1 phosphorylation enhances ligand binding.
Figure 3.
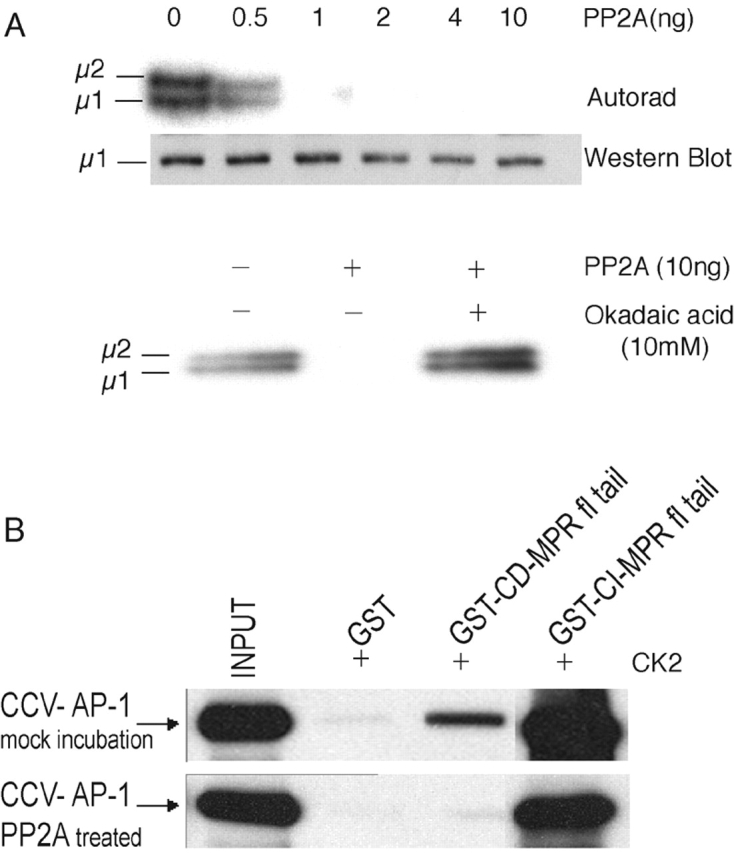
Dephosphorylation of the μ1 subunit of AP-1 decreases ligand binding avidity. (A, top) 25 μg of purified CCVs was incubated at 30°C for 4 h in the presence of ATP (γ32P) to phosphorylate μ1 (and μ2) in vitro using endogenous GAK as the kinase, as described in the Materials and methods. The CCVs were then incubated with purified bovine kidney–derived PP2A at the indicated amounts for an additional 15 min. The complete reaction was subjected to SDS-PAGE on a 12% gel, dried, and filmed. Another identical set was subjected to Western blotting with an antibody toward μ1. (A, bottom) An autorad of a similar in vitro dephosphorylation reaction that was performed in the presence or absence of 10 mM okadaic acid. (B) 50-μg aliquots of immunopurified AP-1 derived from bovine liver CCVs were incubated with or without 50 ng of PP2A in buffer A at 37°C for 1 h. Aliquots were then analyzed for binding to phosphorylated GST-fused MPR ligands as in Fig. 2. 10% of the input and 25% of the pellets were subjected to SDS-PAGE on a 10% gel and immunoblotted with anti–AP-1γ (100/3) monoclonal antibody.
One mechanism whereby the phosphorylation of μ1 could affect ligand binding is by inducing a conformation change in this subunit that favors interaction with the ligand. As a way to explore this, we looked for an alteration in trypsin sensitivity of phosphorylated and nonphosphorylated μ1. Fig. 4 A shows that the μ1 subunit of cytosol-derived AP-1 is completely resistant to concentrations of trypsin that totally cleave the β1 subunit. In contrast, the μ1 subunit of CCV-derived AP-1 is readily cleaved by low concentrations of trypsin (Fig. 4 B). This finding suggests that upon phosphorylation, the μ1 subunit undergoes a conformational change that exposes a trypsin cleavage site, as observed in the case of μ2 (Matsui and Kirchhausen, 1990). This change in conformation could expose regions of μ1 that are involved in ligand binding, perhaps similar to the “jack knife” conformational change proposed by Collins et al. (2002) for μ2. It should also be noted that the β1 subunit of AP-1 derived from the two sources showed a modest difference in susceptibility to trypsin. This suggests that phosphorylation of this subunit also results in a conformational change or that phosphorylation of μ1 induces a change in β1 conformation.
Figure 4.
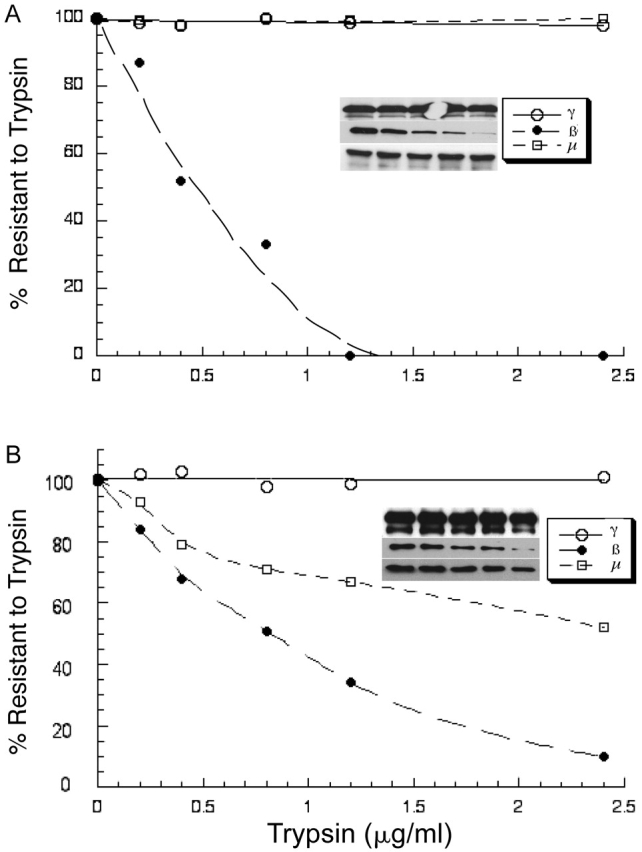
Phosphorylated AP-1 μ1 has increased sensitivity to trypsin. 2.5 μg of immunopurified AP-1 from bovine liver cytosol (A) or CCVs (B) in 100 μl buffer E was incubated with varying amounts of trypsin as indicated. The reaction was performed at 37°C for 8 min and immediately terminated by the addition of an excess of soybean trypsin inhibitor at 4°C. 25% of the reaction was subjected to SDS-PAGE and Western blotting for the γ, β1, and μ1 subunits of AP-1 with their respective antibodies, as mentioned under the Materials and methods. The bands were quantitated using a densitometer. The amount of uncleaved (trypsin resistant) subunits expressed as a percentage of the starting amount was plotted against trypsin concentration using KaleidaGraph. The inset shows the blots of the three subunits, γ, β1, and μ1.
PP2A is the cytosolic cofactor for adaptor release from CCVs
Hannan et al. (1998) have reported that AP-1 and AP-2 release from CCVs requires ATP, Hsc-70, and a cytosolic factor with an apparent size of ∼100 kD on gel filtration. Because PP2A can exist as a heterotrimer (or dimer) of ∼100–125 kD, we hypothesized that it could be the cytosolic factor of Hannan et al. (1998) and function via its ability to dephosphorylate μ1. This would be predicted to impair AP-1 binding to its membrane ligands and thereby facilitate its release into the cytosol. To test this hypothesis, CCVs were treated with cytosol, or with Hsc-70 and PP2A, either alone or together, in the presence of an ATP-regenerating system, and the release of AP-1 was determined. As shown in Fig. 5 A, cytosol plus ATP induced AP-1 release as expected. Importantly, the combination of recombinant Hsc-70 and purified PP2A in the same conditions was even more effective in stimulating AP-1 release. No enhancement of AP-1 release was noted when these proteins were tested alone. The combination of Hsc-70 and PP2A also induced AP-2 release from the CCVs (Fig. 5 B). Half-maximal release of AP-1 occurred at a PP2A concentration of 2.2 ng/reaction (∼44 ng/ml) (Fig. 5 C).
Figure 5.
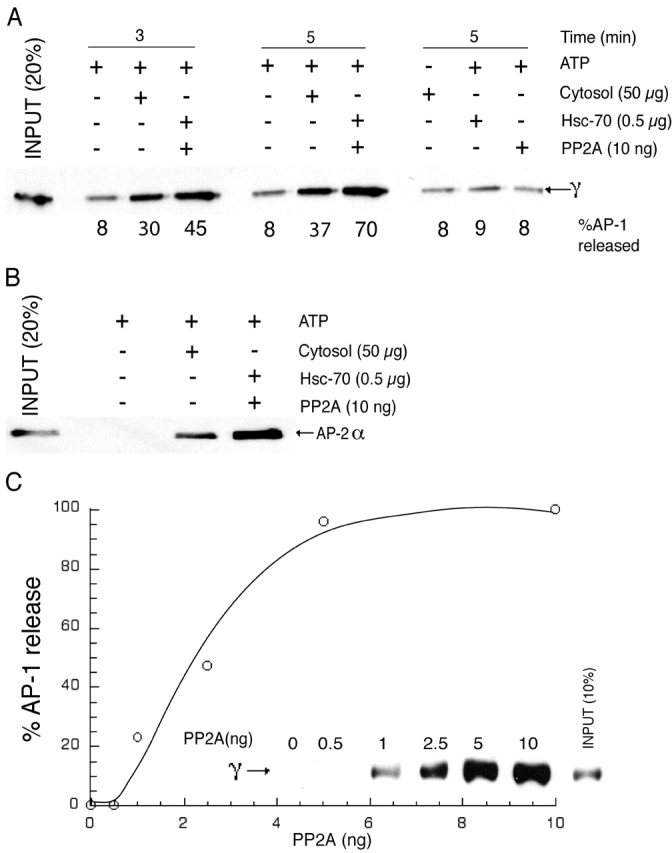
PP2A mediates adaptor release in the presence of Hsc-70 and an ATP-regenerating system. (A and B) 5 μg of purified bovine liver CCVs was incubated with varying combinations of bovine brain cytosol, Hsc-70, PP2A, and ATP-regenerating system at 25°C for 3 min (A and B) or 5 min (A) in a final volume of 50 μl. At the indicated time, the reaction was centrifuged at 100,000 g in a Beckman Coulter ultracentrifuge. 40 μl of the supernatant was collected and subjected to SDS-PAGE, followed by immunoblotting for released AP-1 (A) and AP-2 (B). (C) 5 μg of CCVs and 0.5 μg Hsc-70 were incubated with varying concentrations of PP2A in the presence of the ATP-regenerating system for 5 min and worked up as above. The bands corresponding to the released AP-1 were quantitated using a densitometer and expressed as a percentage of the input. The figures represent 1 of 5–10 independent experiments.
Two additional experiments were performed to confirm that PP2A was indeed the cytosolic factor involved in AP-1 uncoating from CCVs. First, we established that okadaic acid inhibited the cytosol-mediated AP-1 release from CCVs as well as release catalyzed by the Hsc-70–PP2A combination (Fig. 6 A). In a second experiment, it was shown that immunodepletion of PP2A from cytosol (Fig. 6 B) resulted in the loss of AP-1–releasing activity, and the addition of PP2A restored this function (Fig. 6 C). These experiments indicate that PP2A is the cytosolic factor that acts in conjunction with Hsc-70 to catalyze adaptor release from CCVs. The observation that okadaic acid causes increased, nonspecific membrane localization of AP-2 (Lauritsen et al., 2000) supports the notion that μ2 dephosphorylation is also essential for release from membranes. In addition, overexpression of GAK, the kinase implicated in μ1/2 phosphorylation and clathrin uncoating, resulted in the sustained association of AP-1 with TGN budding profiles and of AP-2 with plasma membrane pits independent of clathrin (Umeda et al., 2000; Zhao et al., 2001).
Figure 6.
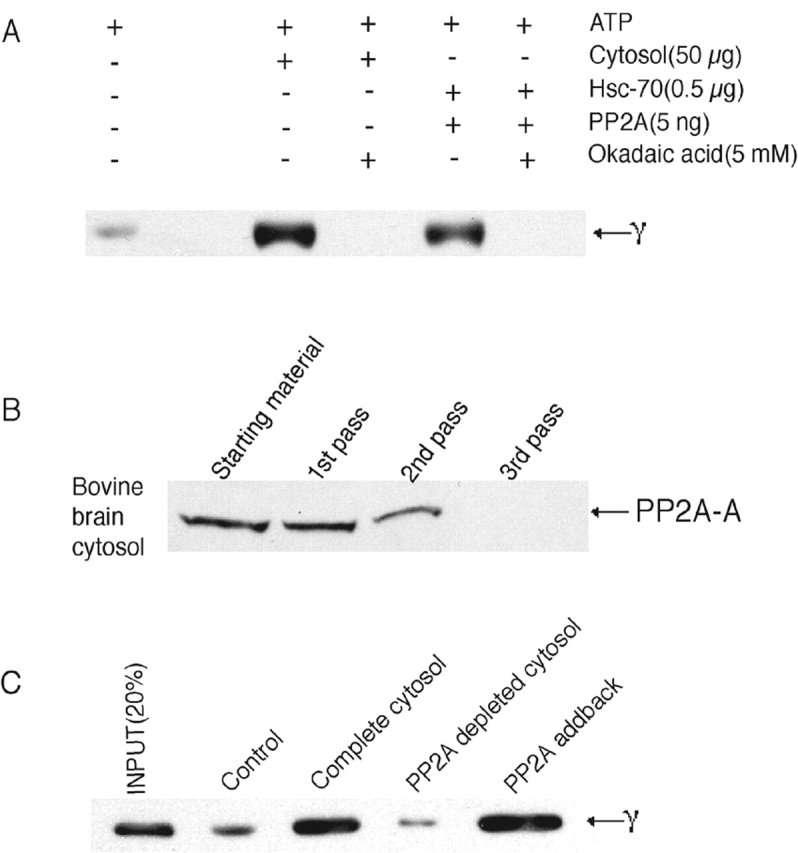
PP2A is the cytosolic cofactor that mediates AP-1 uncoating. (A) Coat release assays were performed as described in the Materials and methods and in Fig. 5 in the presence or absence of 5 mM okadaic acid. The duration of incubation was 4 min, and the released coat components were subjected to electrophoresis followed by immunoblotting as before. (B) PP2A was immunodepleted from bovine brain cytosol by repeated passage through an anti-PP2A affinity column as described in the Materials and methods. The depletion was confirmed by immunoblotting the runthrough fraction after each passage through the affinity column. (C) Whole cytosol and depleted cytosol were used in the coat release assay as described before. 10 ng of bovine kidney PP2A (Calbiochem) was added back to the depleted cytosol in the “addback” lane.
Hsc-70 is known to be recruited onto CCVs via binding to the DnaJ homologues auxilin 2 (GAK) and auxilin 1, which are present on the vesicles (Jiang et al., 1997; Ungewickell et al., 1997). Because phosphatases have been reported to associate with heat shock proteins (Gear et al., 1997; Polanowska-Grabowska et al., 1997), we tested whether PP2A interacts with Hsc-70. A GST fusion protein containing the common A subunit of the PP2A family was immobilized on glutathione-Sepharose beads and incubated with bovine liver cytosol or purified Hsc-70. Bound proteins were eluted and subjected to SDS-PAGE followed by Western blotting using a monoclonal antibody to Hsc-70. Fig. 7 (A and B) shows that Hsc-70 from both sources bound to GST–PP2A-A subunit, but not to GST. This interaction was independent of ATP. To determine whether PP2A interacts with Hsc-70 in cytosol, the PP2A was immunoprecipitated from bovine liver cytosol and analyzed for the presence of Hsc-70 (Fig. 7 C). Hsc-70 was readily detected in the anti-PP2A immunoprecipitate but not in the preimmune control. Together, these experiments show that PP2A and Hsc-70 interact with each other. This could serve as a mechanism for cytosolic PP2A recruitment onto CCVs, where it functions in adaptor release. Further, it has been reported that Hsc-70 activates phosphoprotein phosphatases (Mivechi et al., 1993), possibly providing another means for cooperative interactions in the uncoating process.
Figure 7.
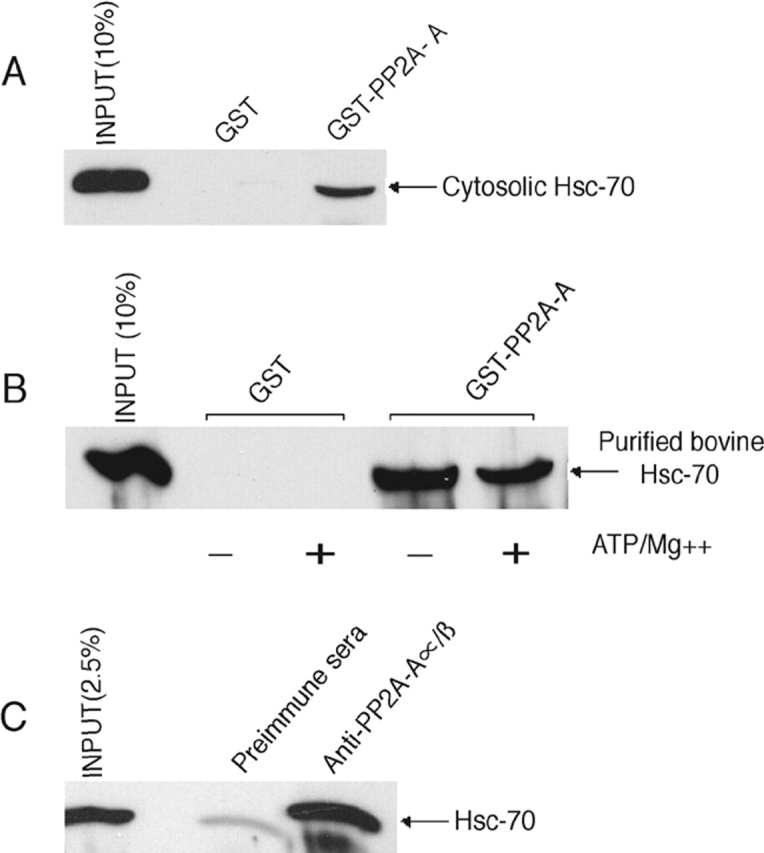
PP2A interacts with Hsc-70 via its regulatory A subunit. (A) 100 μg of GST or GST fusion peptide corresponding to the PP2A A subunit protein was bound to glutathione-Sepharose beads and used to pull down Hsc-70 from 2 mg of bovine liver cytosol. 20% of the pellet was subjected to SDS-PAGE and subsequent immunoblotting with anti–Hsc-70 mAb. The “smile” in the bound Hsc-70 is caused by the GST–PP2A-A fusion protein, which runs at a similar molecular weight as Hsc-70, i.e., ∼70 kD. (B) Aliquots (2.5 μg) of recombinant bovine Hsc-70 were incubated at 25°C for 15 min in the presence or absence of 800 μM ATP and 5 mM magnesium acetate in 25 μl of buffer A. The reactions were then used for binding to 100 μg of GST or GST–PP2A-A fusion proteins prebound to glutathione-Sepharose beads at 4°C for 4 h. 20% of the pellet was subjected to SDS-PAGE and subsequent immunoblotting with anti–Hsc-70 mAb. (C) Bovine liver cytosol (2.5 mg) was subjected to immunoprecipitation with either rabbit preimmune sera or rabbit polyclonal antibody toward PP2A-Aα/β. The immune complexes were captured onto BSA-blocked protein A–agarose beads (Sigma-Aldrich). After four washes with PBS, the bound proteins were eluted by boiling in SDS sample buffer and subjected to SDS-PAGE. Immunoblotting was performed using mAb against Hsc-70.
AP-1 released from CCVs by PP2A and Hsc-70 displays low-affinity ligand binding
If PP2A dephosphorylates the μ1 subunit of AP-1 during the uncoating process, then the released AP-1 should exhibit lower-avidity binding to sorting signals relative to AP-1 with a phosphorylated μ1 subunit. This was tested by comparing ligand binding of AP-1 released from CCVs by Tris treatment versus PP2A–Hsc-70 (Fig. 8) . Whereas AP-1 recovered by both procedures bound GST–CI-MPR full-length cytoplasmic tail approximately equally, the AP-1 released by PP2A–Hsc-70 bound much less well to the CD-MPR cytoplasmic tail. This result provides further evidence that PP2A-catalyzed release of AP-1 from CCVs is associated with dephosphorylation of μ1.
Figure 8.

AP-1 released from CCVs by PP2A and Hsc-70 displays low-avidity ligand binding. AP-1 was extracted from CCVs either with 1.0 M Tris at 4°C or by incubation at 25°C in the presence of PP2A, Hsc-70, and ATP. The supernatants from these reactions were collected as sources of AP-1. GST-fused CK-phosphorylated MPR tails bound to glutathione-Sepharose beads were used as ligands for binding to AP-1 from the two sources. The beads were washed, eluted by boiling in SDS sample buffer, and subjected to SDS-PAGE and immunoblotting with mAb100/3 to detect bound AP-1.
Golgi-associated phosphatase has a preference for phosphorylated β1 versus phosphorylated μ1
The in vivo 32P labeling experiment showed that the β1 subunit of AP-1 is phosphorylated in the cytosol but not on the membrane, whereas the reverse was true for μ1 (Fig. 1). This indicated that the Golgi apparatus contains a phosphatase that acts efficiently on β1 and less well on μ1. On the other hand, the PP2A recruited onto CCVs would be predicted to act efficiently on μ1 while sparing β1 from dephosphorylation. To test this directly, we compared dephosphorylation of β1 and μ1 mediated by Golgi membranes versus purified PP2A from bovine kidney (Fig. 9) . The kidney PP2A dephosphorylated μ1 much more rapidly than β1, whereas the Golgi membrane extract acted on β1 more readily than μ1. Dephosphorylation catalyzed by Golgi membranes was inhibited by 50 nM okadaic acid, indicating that it is mediated by a member of the PP2A family (unpublished data). Thus, we conclude that different isoforms of PP2A residing in specific subcellular compartments display differential substrate specificity toward β1 and μ1.
Figure 9.

Golgi-associated PP2A has a preference for phosphorylated β1 versus μ1. (A and B) Confluent P60 dishes of LSS cells were labeled with 1 mCi/ml of [32P]orthophosphoric acid for 4 h in the presence of 10 nM okadaic acid in the labeling media. The cells were harvested and whole cell lysates were subjected to immunoprecipitation with 2 μg of anti–AP-1 γ monoclonal antibody. The immune complexes were captured on BSA-blocked protein G–agarose beads, washed, and thereafter treated with or without 10 ng of PP2A (A) or 50 μg of rat liver Golgi-enriched membranes in buffer B (B) for the times indicated in the figures. The complete reaction was subjected to SDS-PAGE followed by autoradiography.
Discussion
These results, combined with previous findings of others, lead us to propose a model whereby cycles of phosphorylation and dephosphorylation regulate AP-1 function (Fig. 10) . AP-1 in the cytosol is selectively phosphorylated on the hinge of its β1 subunit, which impairs its binding to clathrin (Wilde and Brodsky, 1996). Upon recruitment of AP-1 onto Golgi membranes, the β1 subunit becomes dephosphorylated, most likely by a PP2A family member. The μ1 subunit, in contrast, becomes phosphorylated at this time. This phosphorylation is likely to be catalyzed by GAK (auxilin-2) (Kanaoka et al., 1997; Kimura et al., 1997), which binds to the γ-appendage of AP-1 (Umeda et al., 2000) and copurifies with AP-1 on a hydroxyapatite column (Ricotta et al., 2002). Although adaptor-associated kinase-1 (AAK-1) has also been shown to phosphorylate μ1 in vitro, it is primarily associated with AP-2 at the plasma membrane (Conner and Schmid, 2002) and copurifies with AP-2 on a hydroxyapatite column (Ricotta et al., 2002). Clearly, the rate of μ1 phosphorylation must exceed the rate of dephosphorylation, because at steady-state, the μ1 subunit of membrane-associated AP-1 is phosphorylated (Fig. 1). In this regard, it is relevant that the Golgi-associated PP2A has a strong preference for the β1 subunit over the μ1 subunit (Fig. 9). Phosphorylation of the μ1 subunit enhances binding to sorting signals on the cytoplasmic tails of the MPR cargo molecules, which should facilitate their capture and retention in the forming CCVs. This was most evident with the CD-MPR, but was also observed with the COOH-terminal CK2 site on the CI-MPR. This is similar to the effect of μ2 phosphorylation on binding to sorting signals on cargo proteins at the plasma membrane (Fingerhut et al., 2001; Olusanya et al., 2001; Ricotta et al., 2002).
Figure 10.
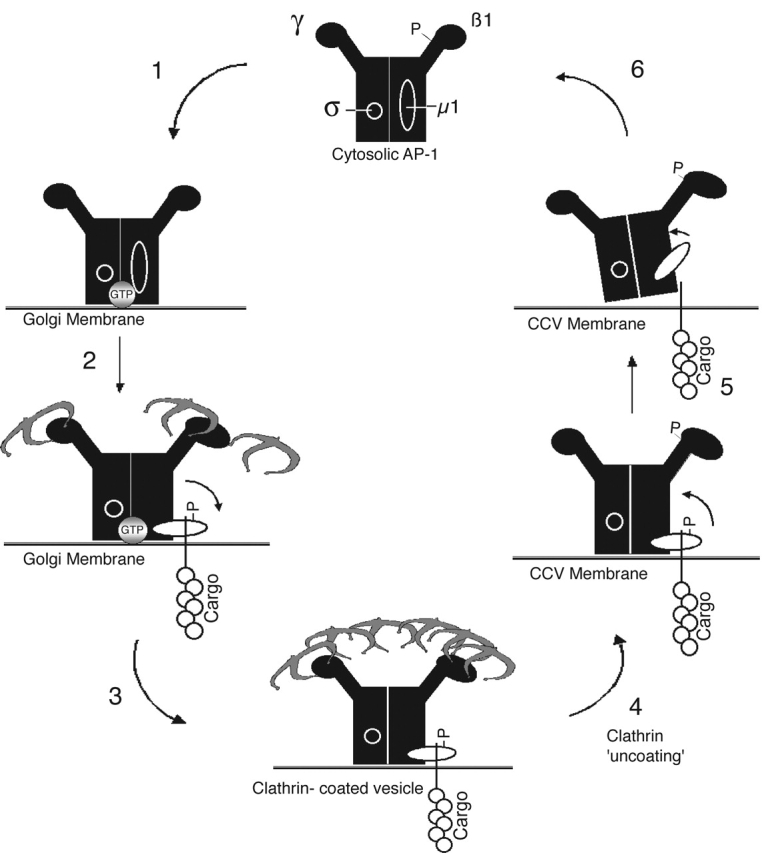
Working model for phosphoregulation of AP-1. (1) Cytosolic AP-1 has a phosphorylated β1 hinge that prevents its association with clathrin and a dephosphorylated μ1 subunit, which presumably is in its inactive/ “closed” conformation. AP-1 is recruited onto the Golgi membranes via ARF-GTP (gray circle) where the β1 hinge gets dephosphorylated by a Golgi-associated isoform of PP2A. (2) Dephosphorylation of the β1 hinge allows AP-1 to initiate clathrin assembly. Concurrently, μ1 is phosphorylated, most likely by GAK (auxilin 2) (Umeda et al., 2000). This results in a conformational change that exposes ligand binding sites on μ1, thereby enhancing interactions with cargo sorting signals at the TGN. (3) During the maturation of the clathrin-coated bud, the β1 is maintained in a dephosphorylated state and μ1 in a phosphorylated state, presumably by GAK, which is incorporated into the forming CCV as an active enzyme (Korolchuk and Banting, 2002). (4) After budding, clathrin dissociation is mediated by GAK and Hsc-70. β1 phosphorylation could occur at this point, contributing to clathrin dissociation. The AP-1 remains on the vesicle membrane, possibly performing postbudding roles. (5) At some point before vesicle fusion with the acceptor compartment, AP-1 is released from the vesicle membrane. Hsc-70 may recruit PP2A from the cytosol and, in concert, mediate AP-1 release. This is achieved by dephosphorylation of μ1, which results in decreased avidity for cargo sorting signals due to reversion to its inactive conformation. (6) Released AP-1 returns to the cytosolic pool with phosphorylated β1 subunit and dephosphorylated μ1 subunit, both in their functionally inactive states.
Recently, Collins et al. (2002) have determined the structure of the AP-2 “core” complex and find that the binding site for the YXX∅ (where ∅ is a bulky hydrophobic residue) endocytic motif in μ2 is blocked by portions of the β2 subunit. These authors propose that a conformational change must occur to allow binding to this sorting signal, and they postulate that this is triggered by phosphoregulation of μ2. Phosphorylation of μ1 at the Golgi membrane may also induce a conformational change that exposes the binding site for phosphorylated CK2 sites and possibly other binding motifs present in the MPR cytoplasmic tails. Our finding that μ1 phosphorylation increases sensitivity to trypsin digestion is consistent with this hypothesis. The likely source of the kinase that phosphorylates the MPRs is the CK2 associated with AP-1 (Meresse et al., 1990, Doray et al., 2002b).
It has recently been demonstrated that the MPRs also bind to the GGA family of proteins (Puertollano et al., 2001; Takatsu et al., 2001; Zhu et al., 2001) and that this interaction is required for packaging of the MPRs into AP-1 CCVs (Doray et al., 2002b). We found that the GGAs with their bound MPRs interact with AP-1 and that the CK2 associated with AP-1 phosphorylates GGAs 1 and 3 (Doray et al., 2002b). This results in autoinhibition of cargo binding, providing a mechanism for the transfer of the MPRs from these GGAs to AP-1. The CK2 would also be expected to phosphorylate the CK2 sites in the cytoplasmic tails of the MPRs, thereby enhancing their binding to AP-1.
The membrane-associated AP-1 also binds clathrin and facilitates clathrin assembly, which proceeds to the formation of a CCV that eventually buds off from the membrane. The vesicle carries within itself a dormant CK2 (associated with AP-1) and an active GAK (Korolchuk and Banting, 2002). After budding, clathrin disassembly occurs, most likely triggered by GAK and Hsc-70 (Braell et al., 1984; Schlossman et al., 1984; Prasad et al., 1993; Barouch et al., 1994; Ungewickell et al., 1995; Umeda et al., 2000; Morgan et al., 2001). During this process, the β1 subunit of AP-1 becomes phosphorylated, but the kinase that mediates this event is currently unknown. However, it is tempting to speculate that the phosphorylation of the β1 hinge could additionally contribute to clathrin dissociation.
After clathrin release, the adaptors remain on the membrane, possibly to mediate “postbudding” roles (Huang et al., 2001), such as the interaction with motor kinesins like KIF13A (Nakagawa et al., 2000) or with Rabaptin-5 (Shiba et al., 2002). At some point, presumably before vesicle fusion with the acceptor compartment, the adaptors are released back to the cytosol. This event, in our model, is mediated by Hsc-70 in concert with PP2A, which is recruited via its interaction with Hsc-70 (Figs. 5–7). PP2A-mediated dephosphorylation would reverse the conformation change in the μ1 subunit that was induced by its phosphorylation. This would be expected to decrease the avidity for cargo sorting signals, thereby facilitating dissociation from the membrane. AP-1 dissociation may be further stimulated by PP2A-mediated dephosphorylation of the CK2 sites in the cytoplasmic tails of the cargo molecules (Molloy et al., 1998). In addition, dephosphorylation of μ1 could decrease surface charge interactions between the μ1 subunit and the lipid bilayer of the membrane, as speculated for μ2 (Collins et al., 2002). The released AP-1 will have a phosphorylated β1 subunit and a dephosphorylated μ1 subunit, as found in the cytosol of mouse L cells (Fig. 1). A similar mechanism appears to account for AP-2 release from CCVs (Fig. 5 B). This requirement for μ2 dephosphorylation in CCV recycling could explain why maintaining μ2 in a phosphorylated state eventually results in inhibition of CCV-mediated transferrin uptake (Conner and Schmid, 2002).
This model explains the critical role of cycles of phosphorylation and dephosphorylation in regulating clathrin-mediated cargo transport at the TGN. In future studies, it will be necessary to identify the PP2A family member associated with the TGN and the kinase that phosphorylates the β1 subunit. In addition, it will be important to gain an understanding of how the kinases and phosphatases are themselves regulated so that they act at the correct time to coordinate this complex series of events.
Materials and methods
Materials
Rabbit skeletal muscle creatine phosphokinase, recombinant bovine Hsc-70 expressed in Escherichia coli, and PP2A1 from bovine kidney were obtained from Calbiochem. Phosphocreatine, okadaic acid, protein G–agarose beads, and adenosine 5′ triphosphate were from Sigma-Aldrich. [32P]orthophosphoric acid (Amersham Biosciences) for in vivo labeling and glutathione–Sepharose 4B beads were from Amersham Biosciences. ATP (γ32P) was from ICN Biomedicals. Tissue culture medium was purchased from Invitrogen, and protease inhibitor cocktail tablets were from Roche Diagnostics.
Buffers.
The following buffers were used: buffer A (10 mM [NH4]2 SO4, 20 mM Hepes, pH 7.0, 2 mM magnesium acetate, 25 mM KCl, 1 mM PMSF); buffer B (20 mM Hepes, pH 7.2, 5 mM magnesium acetate, 125 mM potassium acetate, 0.1% Triton X-100, 1 mM DTT); buffer C (1.0 M Tris-HCl, pH 7.0, 1 mM EDTA, 1 mM PMSF); buffer D (0.1 M Tris, pH 8.0, 0.1 M NaCl, 0.5% sodium deoxycholate, 0.2% SDS, 1% Triton X-100, 10 nM okadaic acid); and buffer E (25 mM Tris-Cl, pH 7.5, 125 mM potassium acetate, 5 mM magnesium acetate, 1 mM DTT). All chemicals were reagent grade and obtained from Sigma-Aldrich.
Antibodies.
Rat anti–Hsc-70 monoclonal antibody was obtained from Calbiochem. Rabbit anti–PP2A-Aα/β was from Santa Cruz Biotechnology, Inc. AP-1 was probed with anti–AP-1 γ-hinge monoclonal 100/3 antibody and anti-β1/2 monoclonal 100/1 antibody; AP-2 with anti–AP-2α monoclonal 100/2 antibody from Sigma-Aldrich. Rabbit anti-μ1 (RY/1) was prepared as described earlier (Traub et al., 1995). Anti-clathrin monoclonal antibody TD.1 was from BAbCo. Anti–AP-1 γ monoclonal antibody from Transduction Laboratories was used for immunoprecipitation of AP-1. Species-specific HRP-conjugated secondary antibodies used for Western blotting were from Amersham Biosciences.
Methods
Bovine liver was harvested immediately after slaughter and was used for CCV preparation as previously described (Meresse et al., 1990). Sucrose gradient–purified CCVs were stored at −80°C until usage. Bovine brain cytosol was prepared as before (Drake et al., 2000), and bovine liver cytosol was the post–70,000 g centrifugation byproduct during the CCV preparation. Tris extraction of the bovine liver CCVs was performed at 4°C overnight in buffer C. After centrifugation at 100,000 g for 1 h, the supernatant containing the released AP-1 was collected and dialyzed against PBS. AP-1 was immunopurified from the cytosol and Tris extracts of CCVs using affinity chromatography and peptide elution as described earlier (Zhu et al., 1998).
Coat release assay.
5 μg of bovine liver CCVs was incubated with or without 0.5 μg of Hsc-70/10 ng PP2A/50 μg bovine brain cytosol, as indicated in the figure legends for Figs. 5 and 6, in the presence of an ATP-regenerating system (containing 800 μM ATP, creatine phosphokinase, and 5 mM creatine phosphate) in a final volume 50 μl of buffer A. The reactions were performed in thick-walled ultracentrifuge Eppendorf tubes at 25°C. At the indicated times, the reactions were centrifuged in a Beckman TLA 100.3 rotor at 100,000 g for 10 min at 4°C. 40 μl of the supernatant was removed without disturbing the pellet and analyzed by SDS-PAGE and Western blotting for the presence of released coat components.
Pull-down assays.
GST–CI-MPR and CD-MPR peptides, wild-type and mutants, were expressed and purified as before (Doray et al., 2002a). GST–PP2A-A plasmid was a generous gift from Marc C. Mumby (University of Texas Southwestern Medical Center, Dallas, TX). The reactions contained 100 μg of purified GST fusion ligand that had been prebound to glutathione-Sepharose beads at room temperature for 2 h and 4 μg of immunopurified AP-1 from either bovine liver cytosol or Tris extracts of bovine liver CCVs in a final volume of 350 μl of buffer B. The reactions were incubated at 4°C for 4 h with constant tumbling. The beads were then collected by centrifugation at 3,000 rpm and given four washes, each with 1 ml of buffer B. The pellet was boiled in SDS sample buffer for Western blotting.
Western blotting.
Samples boiled in SDS sample buffer were subjected to SDS-PAGE on a 10% gel, transferred onto nitrocellulose, and immunoblotted with the antibodies mentioned before. The blot was developed using an ECL detection kit from Amersham Biosciences and filmed with KODAK X-OMAT K (Eastman Kodak Co.).
Immunodepletion of cytosolic PP2A.
An affinity column was prepared by coupling 200 μg of anti–PP2A-Aα/β to 250 mg of CNBr-activated Sepharose beads (Sigma-Aldrich). Bovine brain cytosol was subjected to repeated passes through this column until the PP2A was depleted, as determined by immunoblotting. The beads were regenerated by washing with glycine, pH 2.5.
In vivo phosphorylation, membrane–cytosol fractionation, and immunoprecipitation of AP-1.
Mouse L cells (LSS) were grown in α-MEM (supplemented with 10% FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin) at 37°C and 5% CO2. Four confluent P100 dishes were labeled with 1 mCi/ml [32P]orthophosphoric acid/phosphate-free α-MEM/10% FBS/20 mM Hepes, pH 7.4, with or without 10 nM okadaic acid for 4 h at 37°C and 5% CO2. The medium was removed and the dishes washed with ice cold PBS. The cells were scraped in 1 ml of homogenization buffer (100 mM MES, pH 6.5, 0.25% sucrose, 1 mM EGTA, 10 mM okadaic acid, and protease inhibitor cocktail). The cell suspension was sonicated three times, 10 s each, at intensity 3.5 using a Fisher Scientific sonic dismembrator (model no. 550), followed by centrifugation at 650 g for 15 min to remove intact cells and nuclei. The postnuclear supernatant was collected and subjected to centrifugation at 100,000 g in a Beckman TLA 100.3 rotor for 60 min. The supernatant was collected (cytosol) and the pellet was resuspended in buffer B (supplemented with 10 mM okadaic acid and 0.4% Triton X-100), sonicated, and centrifuged at 14,000 g for an additional 15 min to remove insoluble membrane proteins. The two supernatant fractions were incubated with 10 μl of anti–AP-1γ antibody overnight with tumbling. The next morning, BSA-blocked protein G–agarose beads were added to the tubes and incubated for an additional hour. The beads were then pelleted and washed repeatedly with buffer D until no further radioactivity was released. The washed pellets were boiled in sample buffer and subjected to SDS-PAGE using a 10% gel. The gel was dried and filmed with KODAK X-OMAT MR. Duplicate sets were subjected to Western blotting to confirm the specificity of the immunoprecipitation.
Dephosphorylation assays.
Dephosphorylation assays were performed on phosphorylated AP-1 prepared both in vivo and in vitro. AP-1 was phosphorylated in vivo in the presence of okadaic acid and immunoprecipitated from whole cell lysates as above. The washed beads were then subjected to dephosphorylation in buffer A with either commercially obtained bovine kidney PP2A or Golgi-enriched membranes. The duration of incubations and the amount of PP2A and Golgi membrane used are indicated in the figure legends. CCVs were used to phosphorylate the μ1 subunit of AP-1 in vitro because they contain AP-1 as well as GAK kinase, which is known to act on μ1 (Umeda et al., 2000). 25 μg of CCVs was incubated for 4 h at 30°C in buffer A supplemented with 1.5 mM ATP γ32P (ICN Biomedicals; specific activity 12.5 μCi/mmol ATP) in a final volume of 40 μl. After incubation, PP2A was added, as indicated, for an additional 15 min, and the entire mixture was subjected to SDS-PAGE on a 12% gel and autoradiography after drying. The phosphorylated 50-kD and 47-kD bands were the only detectable products of the reaction. The use of a 12% gel was required to resolve the μ1 and μ2 subunits (Loeb et al., 1989), which otherwise run together (Korolchuk and Banting, 2002).
Limited proteolysis of purified AP-1.
Aliqouts (2.5 μg) of purified AP-1, of either cytosolic or CCV source, were incubated with various amounts of bovine pancreatic trypsin (Sigma-Aldrich), as indicated in the figure legend for Fig. 4, in a final volume of 100 μl of buffer E at 37°C for 8 min. The tubes were then placed on ice, and the reactions were arrested by the addition of an excess of soybean trypsin inhibitor (Sigma-Aldrich). Aliquots of the reactions were subjected to SDS-PAGE and immunoblotting. The bands were quantitated using an Eastman Kodak Co. densitometer and plotted using KaleidaGraph.
Acknowledgments
We thank Rosalind Kornfeld, Alexander Ungewickell, Wang Sik Lee, and Hongdong Bai for critical reading of the manuscript and helpful comments.
This work was supported by National Institutes of Health grant RO1 CA-08759 to S. Kornfeld.
Footnotes
Abbreviations used in this paper: AP-1, adaptor protein complex-1; CCV, clathrin-coated vesicle; CD-MPR, cation-dependent MPR; CI-MPR, cation-independent MPR; CK2, casein kinase-2; GAK, cyclin G–associated kinase; GGA, Golgi-localized, γ ear–containing, adenosine 5′ diphosphate-ribosylation factor–binding protein; MPR, mannose 6-phosphate receptor; PP2A, protein phosphatase 2A.
References
- Barouch, W., K. Prasad, L.E. Greene, and E. Eisenberg. 1994. ATPase activity associated with the uncoating of clathrin baskets by Hsp70. J. Biol. Chem. 269:28563–28568. [PubMed] [Google Scholar]
- Bertrand, F., P. Turowski, and B.A. Hemmings. 1997. Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid and tautomycin. J. Biol. Chem. 272:13856–13863. [DOI] [PubMed] [Google Scholar]
- Braell, W.A., D.M. Schlossman, S.L. Schmid, and J.E. Rothman. 1984. Dissociation of clathrin coats coupled to the hydrolysis of ATP: role of an uncoating ATPase. J. Cell Biol. 99:734–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremnes, T., V. Lauvrak, B. Lindqvist, and O. Bakke. 1998. A region from the medium chain adaptor subunit (μ) recognizes leucine- and tyrosine-based sorting signals. J. Biol. Chem. 273:8638–8645. [DOI] [PubMed] [Google Scholar]
- Collins, B.M., A.J. McCoy, H.M. Kent, P.R. Evans, and D.J. Owen. 2002. Molecular architecture and functional model of the endocytic AP2 complex. Cell. 109:523–535. [DOI] [PubMed] [Google Scholar]
- Conner, S.D., and S.L. Schmid. 2002. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J. Cell Biol. 156:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittie, A.S., L. Thomas, G. Thomas, and S.A. Tooze. 1997. Interaction of furin in immature secretory granules from neuroendocrine cells with the AP-1 adaptor complex is modulated by casein kinase II phosphorylation. EMBO J. 16:4859–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doray, B., and S. Kornfeld. 2001. γ subunit of the AP-1 adaptor complex binds clathrin: implications for cooperative binding in clathrin vesicle assembly. Mol. Biol. Cell. 12:1925–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doray, B., K. Bruns, P. Ghosh, and S.A. Kornfeld. 2002. a. Autoinhibition of the ligand-binding site of GGA1/3 VHS domains by an internal acidic cluster-dileucine motif. Proc. Natl. Acad. Sci. USA. 99:8072–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doray, B., P. Ghosh, J. Griffith, H. Geuze, and S. Kornfeld. 2002. b. Cooperation of GGAs and AP-1 in packaging MPRs at the trans-Golgi network. Science. 297:1700–1703. [DOI] [PubMed] [Google Scholar]
- Drake, M.T., Y. Zhu, and S. Kornfeld. 2000. The assembly of AP-3 adaptor complex-containing clathrin-coated vesicles on synthetic liposomes. Mol. Biol. Cell. 11:3723–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingerhut, A., K. von Figura, and S. Honing. 2001. Binding of AP2 to sorting signals is modulated by AP2 phosphorylation. J. Biol. Chem. 276:5476–5482. [DOI] [PubMed] [Google Scholar]
- Gear, A.R., C.G. Simon, and R. Polanowska-Grabowska. 1997. Platelet adhesion to collagen activates a phosphoprotein complex of heat-shock proteins and protein phosphatase 1. J. Neural Transm. 104:1037–1047. [DOI] [PubMed] [Google Scholar]
- Greener, T., X. Xhao, H. Nojima, E. Eisenberg, and L.E. Greene. 2000. Role of cyclin G-associated kinase in uncoating clathrin-coated vesicles from non-neuronal cells. J. Biol. Chem. 275:1365–1370. [DOI] [PubMed] [Google Scholar]
- Hannan, L.A., S.L. Newmyer, and S.L. Schmid. 1998. ATP- and cytosol-dependent release of adaptor proteins from clathrin-coated vesicles: a dual role for Hsc70. Mol. Biol. Cell. 9:2217–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, B.L., K. Drickamer, F.M. Brodsky, and P. Parham. 1988. Identification of the phosphorylation sites of clathrin light chain LCb. J. Biol. Chem. 263:5499–5501. [PubMed] [Google Scholar]
- Honing, S., M. Sosa, A. Hille-Rehfeld, and K. von Figura. 1997. The 46-kDa mannose 6-phosphate receptor contains multiple binding sites for clathrin adaptors. J. Biol. Chem. 272:19884–19890. [DOI] [PubMed] [Google Scholar]
- Huang, F., A. Nesterov, R.E. Carter, and A. Sorkin. 2001. Trafficking of yellow-fluorescent-protein-tagged μ1 subunit of clathrin adaptor AP-1 complex in living cells. Traffic. 2:345–357. [DOI] [PubMed] [Google Scholar]
- Jiang, R.F., T. Greener, W. Barouch, L. Greene, and E. Eisenberg. 1997. Interaction of auxilin with the molecular chaperone, Hsc70. J. Biol. Chem. 272:6141–6145. [DOI] [PubMed] [Google Scholar]
- Kalthoff, C., S. Groos, R. Kohl, S. Mahrhold, and E.J. Ungewickell. 2002. Clint: a novel clathrin-binding ENTH-domain protein at the Golgi. Mol. Biol. Cell. 13:4060–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaoka, Y., S.H. Kimura, I. Okazaki, M. Ikeda, and H. Nojima. 1997. GAK: a cyclin G associated kinase contains a tensin/auxilin-like domain. FEBS Lett. 402:73–80. [DOI] [PubMed] [Google Scholar]
- Kimura, S.H., H. Tsuruga, N. Yabuta, Y. Endo, and H. Nojima. 1997. Structure, expression, and chromosomal localization of human GAK. Genomics. 44:179–187. [DOI] [PubMed] [Google Scholar]
- Klumperman, J., A. Hille, T. Veenendaal, V. Oorschot, W. Stoorvogel, K. von Figura, and H.J. Geuze. 1993. Differences in the endosomal distributions of the two mannose 6-phosphate receptors. J. Cell Biol. 121:997–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk, V.I., and G. Banting. 2002. CK2 and GAK/auxilin2 are major protein kinases in clathrin-coated vesicles. Traffic. 3:428–439. [DOI] [PubMed] [Google Scholar]
- Lauritsen, J.P., C. Menne, J. Kastrup, J. Dietrich, N. Odum, and C. Geisler. 2000. β2-adaptin is constitutively de-phosphorylated by serine/threonine protein phosphatase PP2A and phosphorylated by a staurosporine-sensitive kinase. Biochim. Biophys. Acta. 1497:297–307. [DOI] [PubMed] [Google Scholar]
- Le Borgne, R., A. Schmidt, F. Mauxion, G. Griffiths, and B. Hoflack. 1993. Binding of AP-1 Golgi adaptors to membranes requires phosphorylated cytoplasmic domains of the mannose 6-phosphate/insulin-like growth factor II receptor. J. Biol. Chem. 268:22552–22556. [PubMed] [Google Scholar]
- Loeb, J.E., B. Cantournet, J.P. Vartanian, J. Goris, and W. Merlevede. 1989. Phosphorylation/dephosphorylation of the β light chain of clathrin from rat liver coated vesicles. Eur. J. Biochem. 182:195–202. [DOI] [PubMed] [Google Scholar]
- Matsui, W., and T. Kirchhausen. 1990. Stabilization of clathrin coats by the core of the clathrin-associated protein complex AP-2. Biochemistry. 29:10791–10798. [DOI] [PubMed] [Google Scholar]
- Mauxion, F., R. Le Borgne, H. Munier-Lehmann, and B. Hoflack. 1996. A casein kinase II phosphorylation site in the cytoplasmic domain of the cation-dependent mannose 6-phosphate receptor determines the high affinity interaction of the AP-1 Golgi assembly proteins with membranes. J. Biol. Chem. 271:2171–2178. [DOI] [PubMed] [Google Scholar]
- Meresse, S., T. Ludwig, R. Frank, and B. Hoflack. 1990. Phosphorylation of the cytoplasmic domain of the bovine cation-independent mannose 6-phosphate receptor. Serines 2421 and 2492 are the targets of a casein kinase II associated to the Golgi-derived HAI adaptor complex. J. Biol. Chem. 265:18833–18842. [PubMed] [Google Scholar]
- Meyer, C., D. Zizioli, S. Lausmann, E.L. Eskelinen, J. Hamann, P. Saftig, K. von Kigura, and P. Schu. 2000. μ1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 19:2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, C., E.L. Eskelinen, M.R. Guruprasad, K. von Figura, and P. Schu. 2001. μ1A deficiency induces a profound increase in MPR300/IGF-II receptor internalization rate. J. Cell Sci. 114:4469–4476. [DOI] [PubMed] [Google Scholar]
- Mivechi, N.F., L.D. Trainor, and G.M. Hahn. 1993. Purified mammalian HSP-70 KDA activates phosphoprotein phosphatases in vitro. Biochem. Biophys. Res. Commun. 192:954–963. [DOI] [PubMed] [Google Scholar]
- Molloy, S.S., L. Thomas, C. Kamibayashi, M.C. Mumby, and G. Thomas. 1998. Regulation of endosome sorting by a specific PP2A isoform. J. Cell Biol. 142:1399–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, J.R., K. Prasad, S. Jin, G.J. Augustine, and E.M. Lafer. 2001. Uncoating of clathrin-coated vesicles in presynaptic terminals: roles for Hsc70 and auxilin. Neuron. 32:289–300. [DOI] [PubMed] [Google Scholar]
- Mumby, M.C., and G. Walter. 1993. Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol. Rev. 73:673–699. [DOI] [PubMed] [Google Scholar]
- Nakagawa, T., M. Setou, D. Seog, K. Ogasawara, N. Dohmae, K. Takio, and N. Hirokawa. 2000. A novel motor, KIF13A, transports mannose-6-phosphate receptor to plasma membrane through direct interaction with AP-1 complex. Cell. 103:569–581. [DOI] [PubMed] [Google Scholar]
- Ohno, H., R.C. Aguilar, D. Yeh, D. Taura, T. Saito, and J.S. Bonifacino. 1998. The medium subunits of adaptor complexes recognize distinct but overlapping sets of tyrosine-based sorting signals. J. Biol. Chem. 273:25915–25921. [DOI] [PubMed] [Google Scholar]
- Olusanya, O., P.D. Andrews, J.R. Swedlow, and E. Smythe. 2001. Phosphorylation of threonine 156 of the μ2 subunit of the AP2 complex is essential for endocytosis in vitro and in vivo. Curr. Biol. 11:896–900. [DOI] [PubMed] [Google Scholar]
- Owen, D.J., and P.R. Evans. 1998. A structural explanation for the recognition of tyrosine-based endocytic signals. Science. 282:1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, L.J., P.J. Sowerby, W.W. Lui, and M.S. Robinson. 1999. γ-Synergin: an EH domain–containing protein that interacts with γ-adaptin. J. Cell Biol. 146:993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanowska-Grabowska, R., C.G. Simon, Jr., R. Falchetto, J. Shabanowitz, D.F. Hunt, and A.R. Gear. 1997. Platelet adhesion to collagen under flow causes dissociation of a phosphoprotein complex of heat-shock proteins and protein phosphatase 1. Blood. 90:1516–1526. [PubMed] [Google Scholar]
- Prasad, K., W. Barouch, L. Greene, and E. Eisenberg. 1993. A protein cofactor is required for uncoating of clathrin baskets by uncoating ATPase. J. Biol. Chem. 268:23758–23761. [PubMed] [Google Scholar]
- Puertollano, R., R.C. Aguilar, I. Gorshkova, R.J. Crouch, and J.S. Bonifacino. 2001. Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science. 292:1712–1716. [DOI] [PubMed] [Google Scholar]
- Rapoport, I., Y.C. Chen, P. Cupers, S.E. Shoelson, and T. Kirchhausen. 1998. Dileucine-based sorting signals bind to the β chain of AP-1 at a site distinct and regulated differently from the tyrosine-based motif-binding site. EMBO J. 17:2148–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricotta, D., S.D. Conner, S.L. Schmid, K. von Figura, and S. Honing. 2002. Phosphorylation of the AP2 μ subunit by AAK1 mediates high affinity binding to membrane protein sorting signals. J. Cell Biol. 156:791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlossman, D.M., S.L. Schmid, W.A. Braell, and J.E. Rothman. 1984. An enzyme that removes clathrin coats: purification of an uncoating ATPase. J. Cell Biol. 99:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba, Y., H. Takatsu, H.W. Shin, and K. Nakayama. 2002. γ-Adaptin interacts directly with Rabaptin-5 through its ear domain. J. Biochem. (Tokyo). 131:327–336. [DOI] [PubMed] [Google Scholar]
- Shih, W., A. Gallusser, and T. Kirchhausen. 1995. A clathrin-binding site in the hinge of the β2 chain of mammalian AP-2 complexes. J. Biol. Chem. 270:31083–31090. [DOI] [PubMed] [Google Scholar]
- Takatsu, H., Y. Katoh, Y. Shiba, and K. Nakayama. 2001. Golgi-localizing, γ-adaptin ear homology domain, ADP-ribosylation factor-binding (GGA) proteins interact with acidic dileucine sequences within the cytoplasmic domains of sorting receptors through their Vps27p/Hrs/STAM (VHS) domains. J. Biol. Chem. 276:28541–28545. [DOI] [PubMed] [Google Scholar]
- Traub, L.M., S. Kornfeld, and E. Ungewickell. 1995. Different domains of the adaptor protein complex are required for Golgi membrane binding and clathrin recruitment. J. Biol. Chem. 270:4933–4942. [DOI] [PubMed] [Google Scholar]
- Umeda, A., A. Meyerholz, and E. Ungewickell. 2000. Identification of the universal cofactor (auxilin 2) in clathrin coat dissociation. Eur. J. Cell Biol. 79:336–342. [DOI] [PubMed] [Google Scholar]
- Ungewickell, E., H. Ungewickell, S.E. Holstein, R. Lindner, K. Prasad, W. Barouch, B. Martin, L.E. Greene, and E. Eisenberg. 1995. Role of auxilin in uncoating clathrin-coated vesicles. Nature. 378:632–635. [DOI] [PubMed] [Google Scholar]
- Ungewickell, E., H. Ungewickell, and S.E. Holstein. 1997. Functional interaction of the auxilin J domain with the nucleotide and substrate-binding modules of Hsc70. J. Biol. Chem. 272:19594–19600. [DOI] [PubMed] [Google Scholar]
- Wasiak, S., V. Legendre-Guillemin, R. Puertollano, F. Blondeau, M. Girard, E. de Heuvel, D. Boismenu, A.W. Bell, J.S. Bonifacino, and P.S. McPherson. 2002. Enthoprotin: a novel clathrin-associated protein identified through subcellular proteomics. J. Cell Biol. 158:855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde, A., and F.M. Brodsky. 1996. In vivo phosphorylation of adaptors regulates their interaction with clathrin. J. Cell Biol. 135:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X., T. Greener, H. Al-Hasani, S.W. Cushman, E. Eisenberg, and L.E. Greene. 2001. Expression of auxilin or AP180 inhibits endocytosis by mislocalizing clathrin: evidence for formation of nascent pits containing AP1 or AP2 but not clathrin. J. Cell Sci. 114:353–365. [DOI] [PubMed] [Google Scholar]
- Zhu, Y., L.M. Traub, and S. Kornfeld. 1998. ADP-ribosylation factor 1 transiently activates high affinity adaptor protein complex AP-1 binding sites on Golgi membranes. Mol. Biol. Cell. 9:1323–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Y., B. Doray, A. Poussu, V.P. Lehto, and S. Kornfeld. 2001. Binding of GGA2 to the lysosomal enzyme sorting motif of the mannose 6-phosphate receptor. Science. 292:1716–1718. [DOI] [PubMed] [Google Scholar]