

Abstract
RNA polymerase II transcribes most eukaryotic genes. Its catalytic subunit was tagged with green fluorescent protein and expressed in Chinese hamster cells bearing a mutation in the same subunit; it complemented the defect and so was functional. Photobleaching revealed two kinetic fractions of polymerase in living nuclei: ∼75% moved rapidly, but ∼25% was transiently immobile (association t1/2 ≈ 20 min) and transcriptionally active, as incubation with 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole eliminated it. No immobile but inactive fraction was detected, providing little support for the existence of a stable holoenzyme, or the slow stepwise assembly of a preinitiation complex on promoters or the nuclear substructure. Actinomycin D decreased the rapidly moving fraction, suggesting that engaged polymerases stall at intercalated molecules while others initiate. When wild-type cells containing only the endogenous enzyme were incubated with [3H]uridine, nascent transcripts became saturated with tritium with similar kinetics (t1/2 ≈ 14 min). These data are consistent with a polymerase being mobile for one half to five sixths of a transcription cycle, and rapid assembly into the preinitiation complex. Then, most expressed transcription units would spend significant times unassociated with engaged polymerases.
Keywords: green fluorescent protein; photobleaching; actinomycin D; DRB; kinetics
Introduction
RNA polymerase II is a multi-subunit enzyme responsible for transcription of most eukaryotic genes (Lee and Young, 2000). Exhaustive experiments performed over the last 30 yrs have given us detailed information on how this enzyme transcribes a naked DNA template in vitro, but we still know little about how it transcribes natural nucleosomal templates in vivo. Fortunately, protein dynamics in living cells can now be monitored using GFP; a hybrid gene encoding the protein of interest fused with GFP is expressed in a cell so the hybrid protein can be localized by its autofluorescence (Tsien, 1998). Fluorescence techniques like FRAP and fluorescence loss in photobleaching (FLIP)* then permit analysis of diffusion coefficients, rates of exchange of tagged proteins between different cellular compartments, and the proportions of mobile and immobile fractions (Ellenberg and Lippincott-Schwartz, 1999; White and Stelzer, 1999; Houtsmuller and Vermeulen, 2001; Phair and Misteli, 2001). Now, we have used these approaches to analyze the kinetics of the polymerase in living cells, using a cell line that we developed for the purpose.
The largest catalytic subunit of the polymerase bears a temperature-sensitive mutation in the CHO cell line, tsTM4. The wild-type subunit from human cells was tagged with GFP and expressed in tsTM4; this construct complemented the defect at the restrictive temperature, and enabled the mutant cells to grow normally (Sugaya et al., 2000). This indicates that the tagged polymerase must be functional. However, these cells contain both endogenous and tagged polymerases, and we can estimate their relative contributions to the total polymerizing activity as follows: during elongation, the COOH-terminal domain of the largest catalytic subunit becomes hyperphosphorylated and reactive to the H5 antibody. As a result, this hyperphosphorylated form (form IIO) is widely used as a marker for the active enzyme (Dahmus, 1996; Komarnitsky et al., 2000). Under the growth conditions used here, immunoblotting reveals that most of the H5-reactive form in the cell is the GFP-polymerase (GFP-pol) rather than the endogenous enzyme (Sugaya et al., 2000). We now use these cells to analyze the mobility of the GFP-pol, concentrating on changes occurring over the minutes required to complete a transcription cycle (including initiation, elongation, and termination). Determining whether GFP-pol diffuses as a core enzyme of ∼500 kD or a larger complex of 1,000–2,000 kD (Lee and Young, 2000) requires analysis over fractions of a second and the development of fluorescent standards of appropriate size. However, note that no larger complexes involved in repair have been detected (Houtsmuller et al., 1999). The kinetics are consistent with ∼75% of the GFP-pol being able to move quickly, with the remainder being transiently immobile (association t1/2 ≈ 20 min). No fraction immobilized in an inactive preinitiation complex could be detected. We also used a conventional biochemical approach (involving radiolabeling nascent transcripts with [3H]uridine) to confirm that the endogenous enzyme in wild-type cells completes a transcription cycle with roughly similar kinetics. Using current estimates of the length of a typical gene and the rate of elongation, we calculate that a polymerase would be engaged for only one half to five sixths of a transcription cycle; then, a typical expressed transcription unit would actually be transcribed for only a minority of the time.
Results
FRAP
For FRAP, GFP-pol in a square area in the nucleoplasm of a living nucleus was bleached, and the increase in fluorescence within the area was measured and expressed relative to the level seen in the whole nucleus (Fig. 1, A–C). As transcription occurs in many sites spread throughout the nucleoplasm (Sugaya et al., 2000), and as the irradiated volume is sufficiently large, many different molecules of GFP-pol active on many different transcription units will be bleached. Bleaching reduced the relative fluorescence in fixed cells from 1 to 0.07 (Fig. 1 B, curve 1) and did not affect “run-on” transcription in the bleached area (monitored using Br-UTP; see Materials and methods). After bleaching living cells, the intensity rose rapidly before the first image was collected, and then more slowly (Fig. 1 B, curve 2). Entry kinetics could be fitted (Kimura and Cook, 2001), assuming there were at least two populations in nuclei, with ∼75% entering quickly (t1/2 < 0.25 min) and the rest with “slower” first-order kinetics (t1/2 ≈ 20 min). 95% confidence intervals were 74–76% for the “fast” fraction, 18–32% for the “slow” fraction (t1/2 14–40 min). The fast fraction probably represents free GFP-pol that can diffuse throughout the nucleus. Its apparent rate of diffusion seems slightly slower than that of GFP (Fig. 1 E; unpublished data) and may reflect its larger size and/or its repeated association and dissociation from nuclear binding sites. Whatever governs its kinetics, its entry into the bleached zone is so fast that it makes essentially no contribution to the slower kinetics that we wish to study. These slow kinetics are probably dominated by the time taken for engaged and bleached polymerases to finish elongating and dissociate, as well as for unbleached ones to diffuse in and initiate (Fig. 1 C). This interpretation is supported by biochemical studies showing that ∼20% is engaged in a HeLa cell (Kimura et al., 1999). Moreover, pretreatment with 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), an inhibitor of elongation (Yamaguchi et al., 1998), eliminates the slow fraction (Fig. 1 B, curve 3); presumably, engaged polymerases became soluble and part of the fast fraction. We conclude that a complete transcription cycle has a half-life of ∼20 min in the absence of DRB. In addition, little inactive GFP-pol is bound stably to promoters or larger structures like “factories” (McCracken et al., 1997; Cook, 1999), as DRB eliminates essentially all the slow fraction. The slight shortfall in recovery of intensity after DRB treatment is probably due to the incomplete inhibition of transcription.
Figure 1.
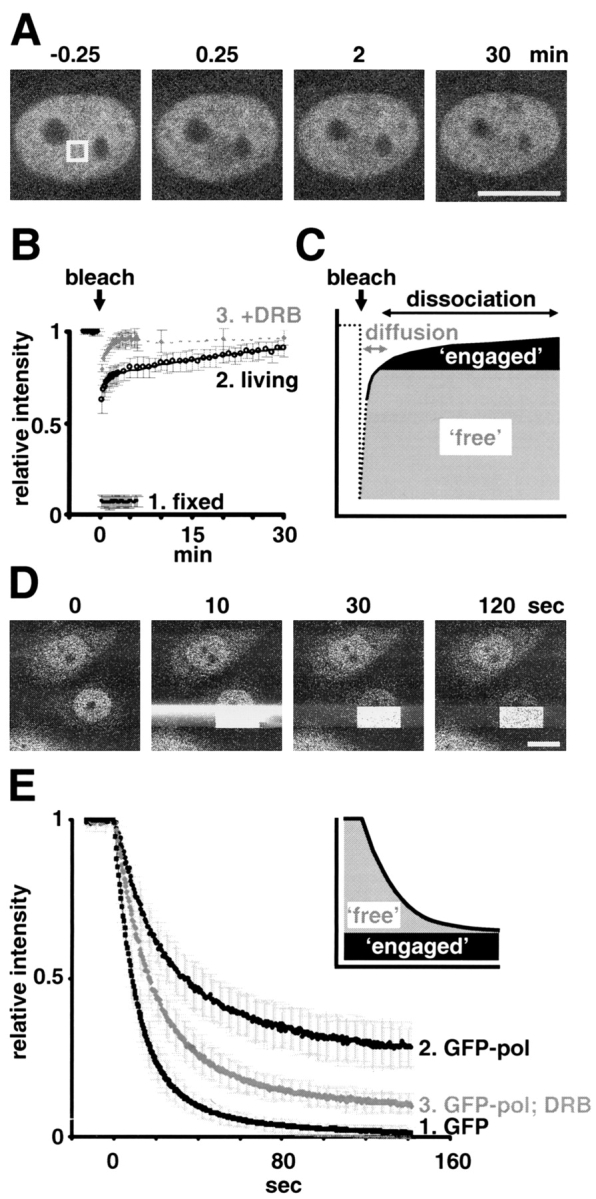
GFP-pol kinetics. (A) FRAP example. Images of typical equatorial sections before and after bleaching the square area. Bar, 10 μm. (B) FRAP results for fixed (4% PFA), living, and DRB-treated (100 μM for 0.5–2 h) cells (± SD, n ≥ 18). The curve for living cells fits the equation: relative intensity = 0.7464 + 0.2482· [1−exp(0.000568·t)] from 90 s. (C) FRAP interpretation. One kinetic population enters the bleached zone quickly (e.g., through diffusion), whereas the other can only initiate once engaged and bleached polymerases have terminated and dissociated. (D) FLIP example. Half the lower nucleus was bleached progressively as confocal images were collected approximately every 0.43 s. The intense signal to the left and right of the bleached rectangle is an artifact. Bar, 10 μm. (E) FLIP results (± SD, n ≥ 15). Inset; kinetics of curve 2 are consistent with there being two populations, one that rapidly enters the bleaching zone, the other being engaged and so remains unbleached.
FLIP
For FLIP, a field containing two nuclei was selected and raster scanned (Fig. 1 D). A low laser power sufficient for imaging was used for most of each scan, and then power was increased 25-fold to bleach a rectangle in the bottom half of the lower nucleus. This process was repeated until most fluorescence disappeared from the top half. Now, the intensity in the unbleached half was expressed relative to its original intensity, and values were further corrected for the slight effects of bleaching during imaging (using the reduction in fluorescence seen in the unbleached upper nucleus; Phair and Misteli, 2000). If all GFP-pol were freely diffusible, bleaching the bottom half should progressively reduce the relative intensity in the top half because unbleached molecules have plenty of time to diffuse into the target area and be bleached; this is the result obtained in control cells expressing GFP (Fig. 1 E, curve 1). If all were immobile (as in fixed cells; unpublished data), the relative intensity remains at unity because immobile molecules in the top half can never enter the bleaching zone. The results obtained lie between these extremes, and are again consistent with the existence of a large fast population and a smaller slow (engaged) one (Fig. 1 E, curve 2 and inset). The fast population decayed at a slightly lower rate than GFP, suggesting that it diffused more slowly and/or associated and dissociated from nuclear binding sites (Fig. 1 E, compare curves 1 and 2). DRB increased the fast fraction (Fig. 1 E, curve 3). Although it is difficult to distinguish the extent of the fast and slow fractions precisely from curves like these, the difference between curves 2 and 3 reflects the size of the DRB-sensitive fraction. It reaches 22% after 40 s and decreases thereafter, as the obscuring fast fraction is removed to reveal the engaged fraction, which then declines as polymerases terminate. Although 22% is an underestimate of the engaged fraction (as not all the obscuring pool will be bleached), it is reassuringly close to that obtained by FRAP.
Kinetics in wild-type cells determined by radiolabeling
We also examined the kinetics of the endogenous polymerase in wild-type cells using an independent technique (Fig. 2). Parental CHO-K1 cells were encapsulated in agarose beads to protect them, grown in [3H]uridine for different times, treated without and with sarkosyl, and the amount of radioactivity in RNA counted. Sarkosyl is a strong detergent that is widely used to extract completed transcripts while leaving nascent ones still associated with the polymerase engaged on its template (Kovelman and Roeder, 1990; Szentirmay and Sawadogo, 1994). Under our conditions, it removes completed transcripts, but leaves nascent ones in beads (Jackson et al., 1998; Kimura et al., 1999; Sugaya et al., 2000). In principle, radioactivity in all transcripts should increase, whereas that in nascent transcripts rises to a plateau (Fig. 2 A). In practice, there is a short lag of 2.5 min as internal pools become labeled; then, the maximum rate of incorporation is reached before turnover tempers the increase (Fig. 2 B, curve 1). Nascent (sarkosyl-resistant) transcripts initially become labeled with similar kinetics, but soon become saturated (Fig. 2 B, curve 2). Excluding the initial lag, it takes ∼14 min for label in nascent transcripts to reach half the maximum. As the increase in labeling is determined by rates of initiation and elongation, and as initiation and termination rates must be equal, this half-time is that of a complete transcription cycle. These results confirm those obtained by FRAP, and indicate that tagged and wild-type enzymes behave similarly.
Figure 2.
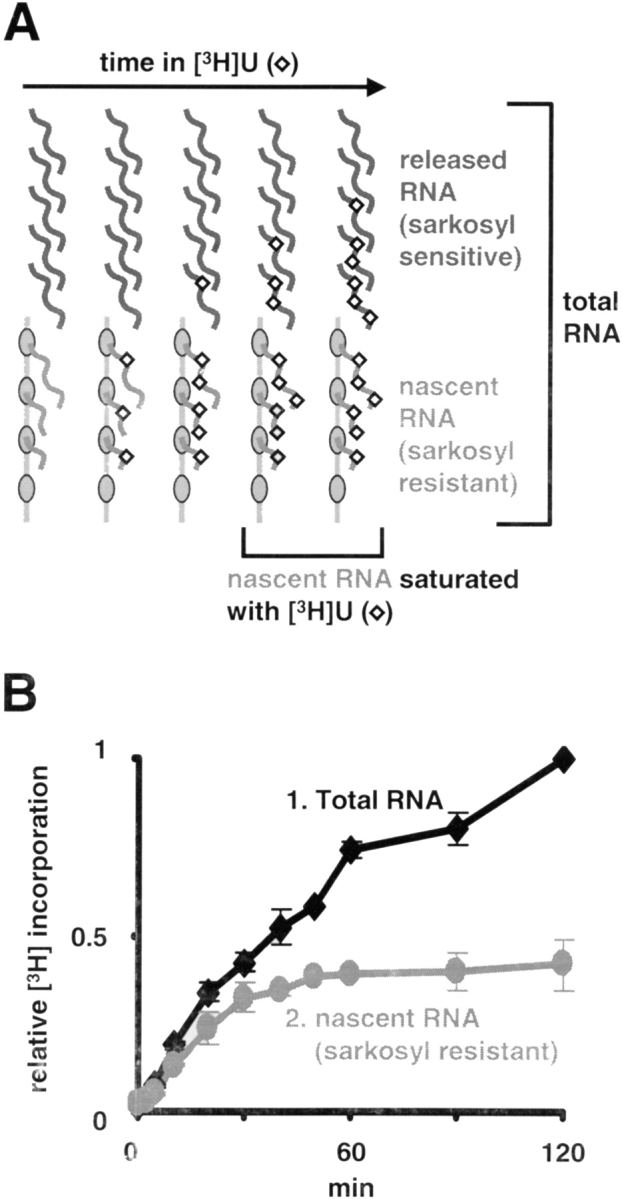
Labeling nascent transcripts. (A) Approach. Wild-type (CHO-K1) cells are grown in [3H]uridine, and treated ± sarkosyl to extract completed transcripts and leave nascent ones. Polymerases (ovals) at different stages of the transcription cycle incorporate 3H (diamonds) into RNA (wavy lines), as some terminate and others initiate. Radioactivity in all transcripts increases progressively, whereas that in nascent ones plateaus. (B) Incorporation (expressed as a fraction of the total at 120 min).
Effects of actinomycin D
Actinomycin D (actD) is a widely used transcriptional inhibitor, although its precise mode of action is unclear. It intercalates into DNA, and so could act like ethidium to promote dissociation of DNA-binding proteins (Lai and Herr, 1992; Kimura and Cook, 2001); alternatively, it could just stall the polymerase. We found it decreased the fast fraction seen by FRAP (Fig. 3 A, curve 2) and FLIP (Fig. 3 B, curve 2). This is consistent with the polymerase stalling at the intercalated drug, and with continuing initiation then increasing the engaged fraction. If so, incubation with the drug for longer amounts of time (3–4 h) should increase the engaged fraction, and it did (Fig. 3 A, curve 3). As DRB and actD had opposite effects, we pretreated cells with both and found that DRB prevented the actD-induced immobilization (Fig. 3 B, curve 3); however, the disengagement was not as great as was seen with DRB alone (Fig. 1 E, curve 3).
Figure 3.

Effect of actinomycin (actD; 5 μg/ml for 0.5–2.5 or 3–4 h) ± DRB (100 μM for 0.5–2.5 h) on polymerase mobility. (A) FRAP (n ≥ 8; 6 SD). Curve 1; data from Fig. 1 B, curve 2. (B) FLIP (n ≥ 15; ± SD). Curve 1; data from Fig. 1 E, curve 2.
Discussion
To what extent does GFP-pol behave like its natural counterpart?
This question arises in any study using a GFP-tagged protein. The best way of ensuring that a tagged protein behaves normally is to replace endogenous coding sequences with ones that specify the tagged molecule, establish a stable clone of cells expressing the modified genes, and confirm by genetic complementation that the cells grow and behave normally. This is rarely done in mammalian cells, as precise gene replacement is so difficult, and “transient” transfection and the study of the resulting populations of variably expressing cells so convenient; in addition, the mutants required to demonstrate complementation are usually unavailable. Here, we did not replace an existing gene, but added an extra tagged one under the control of a heterologous promoter, and this usually results in overexpression. However, we did both establish a stably expressing clone and demonstrate complementation using the appropriate mutant. The largest (catalytic) subunit of polymerase II was tagged with GFP, and expressed in CHO cells bearing a temperature-sensitive mutation in the same subunit; the tagged molecule allows the cells to grow much like the wild-type ones at the nonpermissive temperature, so it must be functional (Sugaya et al., 2000). Moreover, immunoblotting reveals that under our growth conditions, a significant fraction of the tagged subunit (but little of the endogenous temperature-sensitive subunit) is hyperphosphorylated, reactive with the H5 antibody, and resistant to sarkosyl; these are three characteristics of active subunits (Introduction).
Two properties of the tagged polymerase were analyzed in detail: (1) the engaged fraction; and (2) the time taken to complete half a transcription cycle. Given the successful genetic complementation, we might expect (but cannot formally prove) that the tagged polymerase behaves normally, simply because it is difficult to imagine how the cells could survive if the properties of such an important enzyme were changed significantly. Fortunately, the engaged fraction of the tagged polymerase in the nucleus (Fig. 1, B and E; see Materials and methods) turns out to be similar to that found previously for the untagged enzyme in HeLa cells (Kimura et al., 1999). Moreover, the half-life of GFP-pol in the complemented cells also proves to be similar to that of the untagged enzyme in wild-type CHO cells (Fig. 2). Taken at face value, this suggests that any overexpression has little influence on nuclear kinetics, and there is a simple explanation of why this may be so. The other subunits in the core enzyme will be expressed at the usual levels, and so excess GFP-tagged subunits cannot be incorporated into the core enzyme; therefore, we might expect them to remain in the cytoplasm, and this seems to be the case (Sugaya et al., 2000). But as with all studies using GFP as a tag, it remains possible that the tag and/or any overexpression influences the kinetics seen by FRAP and FLIP, and this should be borne in mind during the following discussion. Note that we did not study the diffusion of the GFP-pol for the reasons discussed in the Introduction, and because its apparent diffusion rate turns out to be sufficiently fast that it makes essentially no contribution to the slow kinetics analyzed (Fig. 1 E). However, the fast fraction appears to diffuse more slowly than GFP alone (Fig. 1 E; unpublished data), and future studies are needed to determine whether this reflects a larger size (perhaps of a core or holoenzyme) and/or repeated association and dissociation from different nuclear binding sites (e.g., factories, speckles, or Cajal bodies).
The kinetics of the polymerase
We can draw several conclusions from this work. First, results from both FRAP and FLIP (Fig. 1, B and E) are consistent with 20–25% GFP-pol being engaged. Biochemical analysis shows that a similar fraction of the untagged polymerase is engaged in HeLa cells (Kimura et al., 1999), but, as discussed above, this similarity could arise fortuitously. Second, little transcriptionally inactive GFP-pol is found in stable complexes, as DRB eliminates almost all the slow (engaged) fraction (Fig. 1 B). This result is not supported by additional data on the natural polymerase, but it implies that any unengaged enzyme on promoters or in larger structures like holoenzymes, preinitiation complexes, or factories (McCracken et al., 1997; Cook, 1999; Lee and Young, 2000) must exchange rapidly with soluble molecules (Misteli, 2001).
The third result surprised us. Data obtained using FRAP (on the tagged polymerase) and radiolabeling (on the natural enzyme) indicate that it takes 14–20 min to complete half a transcription cycle (Figs. 1 B and 2 B). How does this compare with other estimates (Jackson et al., 2000)? Although several highly active (polymerase II) transcription units like those encoding a heat shock protein (Lis and Wu, 1993), globin (Wijgerde et al., 1995), and actin (Femino et al., 1998) have been studied, no in vivo data exist for the time taken to initiate and terminate on a “typical” unit. Therefore, we determined it as follows: first, we calculated the average elongation time from the length of a transcription unit and the polymerization rate. Assuming a transcription unit in a CHO cell has the same length as a human gene (median length ∼14 kbp; International Human Genome Sequencing Consortium, 2001), and a polymerization rate of 1.1–2.5 × 103 nucleotides/min (Jackson et al., 2000), a typical transcription unit would be copied in 6–13 min. This complete elongation time is less than the half-times obtained by FRAP or radiolabeling, so elongating a typical gene probably occupies less than one half to one sixth of the transcription cycle. This fraction roughly equals that of all molecules that are transiently immobilized (i.e., one quarter to one fifth), as might be expected under steady-state conditions. These results are also consistent with the widely held assumption that initiation is rate-limiting. It follows that more than one engaged polymerase would rarely be found on a typical (expressed) transcription unit. This conclusion runs counter to the widespread belief that most polymerase II units are associated with many engaged enzymes. Although active units like the chorion and hsp70 genes in Drosophila, and the globin and actin genes in mammals can be associated with many polymerases (Osheim et al., 1985; O'Brien and Lis, 1991; Wijgerde et al., 1995; Femino et al., 1998), studies of typical (expressed) genes show that they are associated with one or less polymerase (Laird and Chooi, 1976; Fakan et al., 1986; Jackson et al., 1998; for review see Jackson et al., 2000). For example, the Drosophila genes encoding two household genes (tubulin B1 and glyceraldehyde phosphate dehydrogenase) are typically associated with <1 polymerase (Rougvie and Lis, 1990), and the mammalian actin gene under steady-state conditions with ∼2 (Femino et al., 1998). Even the major late unit of adenovirus, which is widely believed to be one of the most active units in a mammalian cell, has only one polymerase approximately every 7.5 kbp (Beyer et al., 1981; Wolgemuth and Hsu, 1981). These low numbers contrast with the ∼125 polymerases/transcripts seen on an active polymerase I unit (Miller, 1981).
Finally, actD decreases the fast fraction seen by FRAP (Fig. 3 A, curve 2) and FLIP (Fig. 3 B, curve 2), suggesting the polymerase stalls at the intercalated drug as continuing initiation increases the engaged fraction. Although this is unsupported by independent data on the natural enzyme, we have no reason to believe that the GFP tag influences the way the inhibitor acts.
Models for the initiation of transcription
Taken at face value, these data constrain current models for initiation. For example, the stepwise assembly model sees TBP binding to a promoter, followed successively by transcription factor (TF) IIB, polymerase/TFIIF, TFIIE, and TFIIH (Orphanides et al., 1996). However, little TFIIB-GFP (Chen et al., 2002) or unengaged GFP-pol is immobilized (Fig. 1), so assembly must be rapid relative to the other phases in the cycle. Another model sees the polymerase assembled into a stable transcription complex that repeatedly transcribes the same gene (Brown, 1984), but then the half-life obtained by radiolabeling should be much less than that obtained by FRAP. Note that our results do not exclude the possibility that a stable “scaffold” containing TFs facilitates recycling (Yudkovsky et al., 2000), but that scaffold would have to be free of polymerase.
Our data are consistent with the following parsimonious model for the transcription of a typical unit (Fig. 4). During most of the cycle, the polymerase, TFs, and promoter collide at random to bind and dissociate immediately. Only occasionally do collisions occur in the appropriate sequence that permit rapid assembly of a preinitiation complex (probably in the order described above), and this complex soon initiates. Next, elongation occupies one quarter of the cycle before the polymerase terminates and dissociates quickly. It follows that a typical “active” (i.e., expressed) transcription unit is misnamed, as it spends a significant time not being transcribed. Note that many newly made transcripts are nongenic, and it remains to be seen whether the transcription cycle of genic transcription units is shorter than that of nongenic units.
Figure 4.

A model for the transcription cycle. Polymerases (small ovals) and transcription factors (TFs; circles) spend most of their time (white segment) exchanging between nucleoplasm and a promoter and/or a transcription factory that contains one (McCracken et al., 1997) or more (Cook, 1999) polymerases (large ovals); when bound to the factory, the polymerase spends most of its time elongating (dark gray segment). Little (if any) bound but untranscribing polymerase is seen, so initiation and termination times must be short (light gray segments). The fraction of the cycle occupied by elongation (∼27%) is calculated from the average half-times obtained by FRAP and radiolabeling (i.e., t1/2 of 17 min) and an elongation t1/2 of 4.6 min (from an elongation rate of 1.8 × 103 nucleotides/min and a transcription unit that is 20% longer than a typical gene of 14 kbp; Dye and Proudfoot, 2001).
Materials and methods
FRAP and FLIP
A clone stably expressing GFP-pol (C23) was cultured at 39°C (Sugaya et al., 2000), and images were collected immediately as cells were grown on the microscope stage at 37°C. This clone expresses variable amounts of GFP-pol in the cytoplasm, but more constant amounts in the nucleus; cells expressing low cytoplasmic levels were selected for study to minimize any contribution of nuclear import to fluorescent recovery. However, note that such a contribution must be small, as repeated photobleaching of a small nuclear region over several seconds had little effect on cytoplasmic fluorescence. Fluorescence images were collected (Kimura and Cook, 2001) using a confocal microscope (Radiance2000 [Bio-Rad Laboratories]: 25-mW Ar laser at 4% power, 10× zoom, pinhole aperture 4, scan speed 600 lines/s, Kalman filtration 3, LP500 filter) fitted to a microscope (model TE300; Nikon) with a 60× PlanApo objective (N.A. 1.4). For FRAP, 10 equatorial images were collected, a small area was bleached (100% Ar laser power, 100× zoom, 3 scans), and images were collected using the original settings every 15 s for 3 min and every 1 min thereafter up to 30 min (or every 15 s for 6 min and at 10, 20, and 30 min after drug treatment). Relative intensities in the bleached area were measured and normalized using the average intensity before bleaching. For FLIP, a field with two cells was selected, imaged approximately every 0.43 s for 13 s, and the bottom half of a nucleus was bleached (100% laser power) during subsequent scans (Phair and Misteli, 2000). Curves were analyzed as described previously (Kimura and Cook, 2001). For example, curve 2 in Fig. 1 B could be fitted (using GraphPad Prism software v3.02; http://www.graphpad.com), assuming there were two populations, fast and slow. If recovery of the slow population occurs exponentially, kinetics are governed by R = C + P(1−exp−kt), where R is relative intensity, C is constant (equivalent to the relative intensity given by the fully equilibrated fast fraction), P is plateau value of the slow population, k is association constant, and t is time. Therefore, the half-time is t1/2 − ln(1/2)/k.
Transcriptional activity of bleached regions
The transcriptional activity of bleached regions of cells was assessed as follows: C23 cells grown on a glass-bottomed dish marked with a grid (MatTek) were bleached as for FRAP, incubated for 10–70 min, permeabilized, nascent transcripts were extended in Br-UTP, fixed, and immunolabeled (Pombo et al., 1999; Sugaya et al., 2000) using mouse anti-BrdU (1/200; Caltag) and donkey Cy3-conjugated anti–mouse immunoglobulin (1/200; Jackson ImmunoResearch Laboratories). Bleached cells were identified using the grid, and green and red confocal images were collected sequentially (4% power with Ar laser and 515/528 nm emission filter; 5% power with 1-mW HeNe laser and 560LP filter). The distribution and intensity of sites containing Br-RNA was similar in bleached and unbleached areas.
Radiolabeling
Wild-type CHO-K1 cells expressing only the endogenous polymerase were grown overnight in 1.85 kBq/ml methyl-[14C]thymidine to label DNA uniformly, encapsulated (Jackson and Cook, 1985) in agarose beads (107 cells/ml bead), regrown (4 h), and incubated in 3.7 Mbq/ml [3H]uridine. Incorporation was stopped by transferring 1-ml samples to 14 ml ice-cold PBS; beads were pelleted (3,000 rpm; 5 min; 4°C), washed two times in PBS, and radioactivity was measured by scintillation counting (Jackson and Cook, 1985). In some cases, beads were washed (5 min) in ice-cold 0.5% sarkosyl, spun, left overnight in ice-cold 0.5% sarkosyl, and washed two times before measuring radioactivity. 3H counts (in RNA) were normalized using 14C counts (in DNA).
Acknowledgments
We thank F. Iborra and N. White for help, and the Wellcome Trust for support.
H. Kimura's present address is Medical Research Institute, Tokyo Medical and Dental University, Yushima 1-5-45, Bunkyo-ku, Tokyo 113-8510 Japan.
K. Sugaya's present address is Research Center for Radiation Safety, National Institute of Radiological Sciences, 4-9-1, Anagawa, Chiba 263-8555 Japan.
Footnotes
Abbreviations used in this paper: actD, actinomycin D; DRB, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole; FLIP, fluorescence loss in photobleaching; GFP-pol, GFP-polymerase; TF, transcription factor.
References
- Beyer, A.L., A.H. Bouton, L.D. Hodge, and O.L. Miller. 1981. Visualization of the major late R strand transcription unit of adenovirus serotype 2. J. Mol. Biol. 147:269–295. [DOI] [PubMed] [Google Scholar]
- Brown, D.D. 1984. The role of stable complexes that repress and activate eucaryotic genes. Cell. 37:359–365. [DOI] [PubMed] [Google Scholar]
- Chen, D., C.S. Hinkley, R.W. Henry, and S. Huang. 2002. TBP dynamics in living human cells: sonstitutive association of TBP with mitotic chromosomes. Mol. Biol. Cell. 13:276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, P.R. 1999. The organization of replication and transcription. Science. 284:1790–1795. [DOI] [PubMed] [Google Scholar]
- Dahmus, M.E. 1996. Reversible phosphorylation of the C-terminal domain of RNA polymerase II. J. Biol. Chem. 271:19009–19012. [DOI] [PubMed] [Google Scholar]
- Dye, M.J., and N.J. Proudfoot. 2001. Multiple transcript cleavage precedes polymerase release in termination by RNA polymerase II. Cell. 105:669–681. [DOI] [PubMed] [Google Scholar]
- Ellenberg, J., and J. Lippincott-Schwartz. 1999. Dynamics and mobility of nuclear envelope proteins in interphase and mitotic cells revealed by green fluorescent protein chimeras. Methods. 19:362–372. [DOI] [PubMed] [Google Scholar]
- Fakan, S., G. Leser, and T.E. Martin. 1986. Immunoelectron microscope visualization of nuclear ribonucleoprotein antigens spread within transcription complexes. J. Cell Biol. 103:1153–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Femino, A.M., F.S. Fay, K. Fogarty, and R.H. Singer. 1998. Visualization of single RNA transcripts in situ. Science. 280:585–590. [DOI] [PubMed] [Google Scholar]
- Houtsmuller, A.B., and W. Vermeulen. 2001. Macromolecular dynamics in living cell nuclei revealed by fluorescence redistribution after photobleaching. Histochem. Cell Biol. 115:13–21. [DOI] [PubMed] [Google Scholar]
- Houtsmuller, A.B., S. Rademakers, A.L. Nigg, D. Hoogstraten, J.H. Hoeijmakers, and W. Vermeulen. 1999. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science. 284:958–961. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature. 409:860–921. [DOI] [PubMed] [Google Scholar]
- Jackson, D.A., and P.R. Cook. 1985. A general method for preparing chromatin containing intact DNA. EMBO J. 4:913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, D.A., F.J. Iborra, E.M.M. Manders, and P.R. Cook. 1998. Numbers and organization of RNA polymerases, nascent transcripts and transcription units in HeLa nuclei. Mol. Biol. Cell. 9:1523–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, D.A., A. Pombo, and F.J. Iborra. 2000. The balance sheet for transcription: an analysis of nuclear RNA metabolism in mammalian cells. FASEB J. 14:242–254. [PubMed] [Google Scholar]
- Kimura, H., and P.R. Cook. 2001. Kinetics of core histones in living cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153:1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, H., Y. Tao, R.G. Roeder, and P.R. Cook. 1999. Quantitation of RNA polymerase II and its transcription factors in an HeLa cell: little soluble holoenzyme but significant amounts of polymerases attached to the nuclear substructure. Mol. Cell. Biol. 19:5383–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarnitsky, P., E.J. Cho, and S. Buratowski. 2000. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14:2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovelman, R., and R.G. Roeder. 1990. Sarkosyl defines three intermediate steps in transcription initiation by RNA polymerase III: application to stimulation of transcription by E1A. Genes Dev. 4:646–658. [DOI] [PubMed] [Google Scholar]
- Laird, C.D., and W.Y. Chooi. 1976. Morphology of transcription units in Drosophila melanogaster. Chromosoma. 58:193–218. [DOI] [PubMed] [Google Scholar]
- Lai, J.-S., and W. Herr. 1992. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl. Acad. Sci. USA. 89:6958–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, T.I., and R.A. Young. 2000. Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 34:77–137. [DOI] [PubMed] [Google Scholar]
- Lis, J., and C. Wu. 1993. Protein traffic on the heat shock promoter: parking, stalling, and trucking along. Cell. 74:1–4. [DOI] [PubMed] [Google Scholar]
- McCracken, S., N. Fong, K. Yankulov, S. Ballantyne, G. Pan. J. Greenblatt, S.D. Patterson, M. Wickens, and D.L. Bentley. 1997. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature 385: 357–361. [DOI] [PubMed] [Google Scholar]
- Miller, O.L. 1981. The nucleolus, chromosomes, and visualization of genetic activity. J. Cell Biol. 91:15s–27s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T. 2001. Protein dynamics: implications for nuclear architecture and gene expression. Science. 291:843–847. [DOI] [PubMed] [Google Scholar]
- O'Brien, T., and J.T. Lis. 1991. RNA polymerase II pauses at the 5′ end of the transcriptionally induced Drosophila hsp70 gene. Mol. Cell Biol. 11:5285–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides, G., T. Lagrange, and D. Reinberg. 1996. The general transcription factors of RNA polymerase II. Genes Dev. 10:2657–2683. [DOI] [PubMed] [Google Scholar]
- Osheim, Y.N., O.L. Miller, and A.L. Beyer. 1985. RNP particles at splice junction sequences on Drosophila chorion transcripts. Cell. 43:143–151. [DOI] [PubMed] [Google Scholar]
- Phair, R.D., and T. Misteli. 2000. High mobility of proteins in the mammalian cell nucleus. Nature. 404:604–609. [DOI] [PubMed] [Google Scholar]
- Phair, R.D., and T. Misteli. 2001. Kinetic modelling approaches to in vivo imaging. Nat. Rev. Mol. Cell Biol. 2:898–907. [DOI] [PubMed] [Google Scholar]
- Pombo, A., D.A. Jackson, M. Hollinshead, Z. Wang. R.G. Roeder, and P.R. Cook. 1999. Regional specialization in human nuclei: visualization of discrete sites of transcription by RNA polymerase III. EMBO J. 18:2241–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougvie, A.E., and J.T. Lis. 1990. Postinitiation transcriptional control in Drosophila melanogaster. Mol. Cell. Biol. 10:6041–6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugaya, K., M. Vigneron, and P.R. Cook. 2000. Mammalian cell lines expressing functional RNA polymerase II tagged with the green fluorescent protein. J. Cell Sci. 113:2679–2683. [DOI] [PubMed] [Google Scholar]
- Szentirmay, M.N., and M. Sawadogo. 1994. Sarkosyl block of transcription reinitiation by RNA polymerase II as visualized by the colliding polymerases reinitiation assay. Nucleic Acids Res. 22:5341–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien, R.Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–544. [DOI] [PubMed] [Google Scholar]
- White, J., and E. Stelzer. 1999. Photobleaching GFP reveals protein dynamics inside live cells. Trends Cell Biol. 9:61–65. [DOI] [PubMed] [Google Scholar]
- Wijgerde, M., F. Grosveld, and P. Fraser. 1995. Transcription complex stability and chromatin dynamics in vivo. Nature. 377:209–213. [DOI] [PubMed] [Google Scholar]
- Wolgemuth, D.J., and M.T. Hsu. 1981. Visualization of nascent RNA transcripts and simultaneous transcription and replication in viral nucleoprotein complexes from adenovirus 2-infected HeLa cells. J. Mol. Biol. 147:247–268. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, Y., T. Wada, and H. Handa. 1998. Interplay between positive and negative elongation factors: drawing a new view of DRB. Genes Cells. 3:9–15. [DOI] [PubMed] [Google Scholar]
- Yudkovsky, N., J.A. Ranish, and S. Hahn. 2000. A transcription reinitiation intermediate that is stabilized by activator. Nature. 408:225–229. [DOI] [PubMed] [Google Scholar]