

Abstract
Clustering of acetylcholine receptors (AChRs) is a critical step in neuromuscular synaptogenesis, and is induced by agrin and laminin which are thought to act through different signaling mechanisms. We addressed whether laminin redistributes postsynaptic proteins and requires key elements of the agrin signaling pathway to cause AChR aggregation. In myotubes, laminin-1 rearranged dystroglycans and syntrophins into a laminin-like network, whereas inducing AChR-containing clusters of dystrobrevin, utrophin, and, to a marginal degree, MuSK. Laminin-1 also caused extensive coclustering of rapsyn and phosphotyrosine with AChRs, but none of these clusters were observed in rapsyn −/− myotubes. In parallel with clustering, laminin-1 induced tyrosine phosphorylation of AChR β and δ subunits. Staurosporine and herbimycin, inhibitors of tyrosine kinases, prevented laminin-induced AChR phosphorylation and AChR and phosphotyrosine clustering, and caused rapid dispersal of clusters previously induced by laminin-1. Finally, laminin-1 caused normal aggregation of AChRs and phosphotyrosine in myotubes lacking both Src and Fyn kinases, but these clusters dispersed rapidly after laminin withdrawal. Thus, laminin-1 redistributes postsynaptic proteins and, like agrin, requires tyrosine kinases for AChR phosphorylation and clustering, and rapsyn for AChR cluster formation, whereas cluster stabilization depends on Src and Fyn. Therefore, the laminin and agrin signaling pathways overlap intracellularly, which may be important for neuromuscular synapse formation.
Keywords: laminin; tyrosine phosphorylation; neuromuscular junction; clustering; acetylcholine receptors
Introduction
The synaptic basal lamina at the neuromuscular junction (NMJ)* contains proteins that direct a central aspect of synaptogenesis, the clustering of acetylcholine receptors (AChRs). Although initially AChR clusters of somewhat lower receptor density can be formed in the absence of motor neurons (Yang et al., 2000, 2001; Lin et al., 2001), nerve-derived agrin is required at later stages as a key player in the synaptic basal lamina in order to maintain synapses and localize tightly packed AChR clusters precisely underneath the nerve terminal (Gautam et al., 1996). Aggregation of AChRs is also induced by some forms of laminin (Sugiyama et al., 1997; Montanaro et al., 1998; Burkin et al., 2000).
Nerve-independent and neural agrin–induced clustering of AChRs both require the receptor tyrosine kinase MuSK (Glass et al., 1996; Lin et al., 2001; Yang et al., 2001). Agrin activates MuSK, which initiates a signaling cascade that requires the AChR-associated protein rapsyn to cause clustering of AChRs (Gautam et al., 1995; for review see Sanes and Lichtman, 2001; Huh and Fuhrer, 2002). Via this pathway, agrin also induces clustering of many other postsynaptic proteins, leading to coextensive aggregates of these proteins with AChRs (Wallace, 1989; Meier et al., 1997; Marangi et al., 2001). Within the agrin signaling pathway, AChRs play an active role in the clustering of several postsynaptic proteins, including rapsyn (Marangi et al., 2001). Furthermore, agrin causes rapid activation of Src-related kinases and tyrosine phosphorylation of AChR β and δ subunits (Mittaud et al., 2001); phosphorylation of β is required for efficient AChR clustering (Borges and Ferns, 2001). Finally, after AChR clusters are formed, tyrosine phosphorylation and Src and Fyn kinases are necessary for cluster stabilization (Ferns et al., 1996; Smith et al., 2001).
AChR clustering in myotubes is also induced by laminin-1 and laminin-2/4 (merosin) (Sugiyama et al., 1997; Montanaro et al., 1998; Burkin et al., 2000). The distribution of laminins in the muscle basal lamina is under tight developmental control (Patton et al., 1997). Laminin-1 is expressed early in development and, although hardly detectable at the NMJ itself, at the time of neuromuscular synaptogenesis (E15) before it disappears from muscles later on. Laminin-2 is expressed throughout development, whereas expression of laminin-4 starts at the synapse during synaptogenesis, resulting in a strong expression of merosin (laminin-2/4) at the earliest synapses (E15). Laminin-4 expression persists mainly synaptically at later stages (Patton et al., 1997). Thus, all laminins capable of inducing AChR clusters are expressed during NMJ formation, and NMJs of all developmental stages always contain at least one of these laminin isoforms. These observations are consistent with an important role of laminin-1 and laminin-2/4 in AChR clustering during NMJ formation. Synaptic laminins also direct other aspects of NMJ development. In the absence of the β2 chain, NMJs display less postjunctional folds, poor presynaptic differentiation, and Schwann cell invasion into the synaptic cleft (Noakes et al., 1995), whereas without the α4 chain, active zones are not aligned with junctional folds (Patton et al., 2001).
The actual mechanism of AChR clustering induced by laminin-1 and laminin-2/4 remains poorly characterized, and it is likewise unknown whether these laminins cause clustering of many other proteins in addition to the AChR. However, laminin-1 and laminin-2/4 appear to activate the same pathway (Burkin et al., 2000), which is known to differ from the agrin pathway in several ways. First, some laminin-induced AChR clustering can occur independently of MuSK in MuSK−/− myotubes, although at lower levels (at least 20 times) than in wild-type cells (Sugiyama et al., 1997). Second, short laminin treatments (15–45 min) do not cause phosphorylation of the AChR β subunit (Sugiyama et al., 1997; Montanaro et al., 1998). These observations suggest that laminin may act independently of tyrosine kinases. Finally, laminin-induced AChR clustering shows slower kinetics and leads to larger AChR clusters with higher AChR densities than in the case of agrin (Sugiyama et al., 1997; Montanaro et al., 1998).
Nevertheless, the agrin and laminin pathways share some common principles. Both agrin and laminin are known to require dystroglycan to induce AChR clusters of normal morphology and stability in myotubes (Jacobson et al., 1998, 2001; Montanaro et al., 1998; Grady et al., 2000). Agrin and laminin act synergistically in AChR clustering and laminin-pretreated myotubes require much less agrin for subsequent clustering, suggesting that laminin primes myotubes to be more sensitive towards agrin (Sugiyama et al., 1997; Burkin et al., 2000). Furthermore, integrins such as α7β1 are implied as receptors in both laminin- and agrin-induced AChR clustering, because in myotubes antibodies against β1 or α7 integrin modulate both clustering processes (Martin and Sanes, 1997; Burkin et al., 1998). α7β1 integrin is more frequently found at AChR clusters induced by laminin than by agrin, but the integrin is also prominent outside of such clusters (Burkin et al., 1998). These data suggest that agrin and laminin act together to achieve maximal AChR clustering. In support of this idea, agrin binds directly to laminin-1, -2, and particularly laminin-4 (Denzer et al., 1997), which may contribute to the synaptic concentration of agrin.
An intrinsic property of laminins is to build extracellular matrices by forming an organized network with other proteins, which stabilizes cells by interaction with cytoskeleton-linked surface receptors (Henry and Campbell, 1999). Furthermore, laminins 1–4 polymerize while bound to muscle receptors like integrins and dystroglycan, leading to reorganization of these receptors into a network-like structure or small clusters that colocalize with laminin (Cohen et al., 1997; Colognato et al., 1999). Thus, laminin-induced AChR clustering could, in principle, occur by extracellular protein interactions that redistribute dystroglycan and other members of the dystrophin/utrophin glycoprotein complex (D/UGC), followed by redistribution of AChRs, which are known to interact with the D/UGC (Fuhrer et al., 1999). Together, these various lines of evidence imply that laminin-induced AChR clustering may not require intracellular signaling, in particular tyrosine kinases.
Therefore, to unravel the mechanism by which laminin causes AChR clustering, we have analyzed whether laminin redistributes postsynaptic proteins and requires an intracellular signaling cascade similar to that of agrin. Because laminin-1 is best characterized in terms of inducing AChR clustering, we concentrated on this form of laminin. We find that laminin-1 differentially redistributes postsynaptic proteins into a network or AChR-containing clusters, in contrast to agrin, which is known to cause co-clustering of all these proteins with AChRs. Furthermore, laminin-1 initiates phosphotyrosine signaling that, similar to agrin, requires rapsyn and tyrosine kinases for formation of AChR clusters, whereas Src and Fyn are necessary for stabilization of clusters.
Results
Laminin-1 differentially redistributes postsynaptic proteins
To analyze whether laminin affects the distribution of postsynaptic proteins in a different way than agrin does, we first incubated C2 myotubes with laminin-1 and examined the distribution of AChRs. No morphological changes of the myotubes were observed when compared to agrin-treated or untreated myotubes (Fig. 1 A). Staining for AChRs with rhodamine-conjugated α-bungarotoxin (α-btx) revealed that a 16-h incubation with 100 nM laminin-1 increased the number of AChR clusters by 5.8 ± 2.6-fold (mean ± SD; 12 experiments). Furthermore, laminin-induced aggregates were more elongated, larger, and showed a higher fluorescence intensity than their counterparts induced by agrin (Fig. 1 A), similar to previous reports (Sugiyama et al., 1997; Montanaro et al., 1998).
Figure 1.
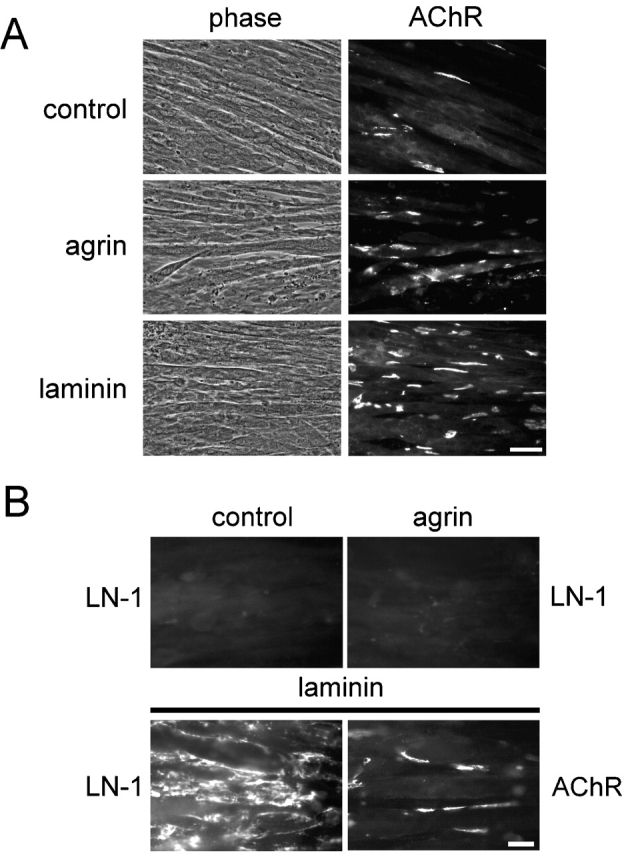
Laminin-1 adheres to C2 myotubes and induces AChR cluster formation. (A) Cells were incubated with or without 0.1 nM neural agrin or 100 nM laminin-1 for 16 h and stained with rhodamine-conjugated α-btx. Bar, 50 μm. (B) Myotubes were treated with laminin-1 or agrin and double labeled with rhodamine-α-btx (AChR) and antiserum against laminin-1 followed by Alexa350-conjugated secondary antibodies (LN-1). Rhodamine channels of panels in top row are not shown. Bar, 20 μm.
We examined the distribution of laminin added to our myotubes and found that laminin-1 formed large deposits resembling a coarse network on the cell surface (Fig. 1 B). Higher magnification also occasionally revealed areas where laminin was arranged in a more regular, polygonal fashion (unpublished data), resembling the polygonal organization of laminin-1 when added to myotubes at lower concentrations (Colognato et al., 1999).
Interestingly, the localization of AChR clusters induced by laminin-1 showed no obvious correlation with the distribution of laminin-1 itself (Fig. 1 B; see Fig. 3). Therefore, we systematically analyzed how other proteins were distributed after laminin treatment. By assessing the distribution of α- and β-dystroglycan, syntrophin isoforms, rapsyn, tyrosine-phosphorylated proteins, MuSK, utrophin, and α-dystrobrevin-1, we observed three different classes of proteins. The first class, including α- and β-dystroglycan and syntrophins, was rearranged into network-like patches by laminin-1 and showed no obvious colocalization with AChR clusters (Fig. 2 A). These patches resembled the distribution of laminin-1 itself (Fig. 1 B). Indeed, pairwise double staining revealed partial colocalization of laminin-1 with α-dystroglycan, and, to a lesser degree, with β-dystroglycan and syntrophin (unpublished data).
Figure 3.
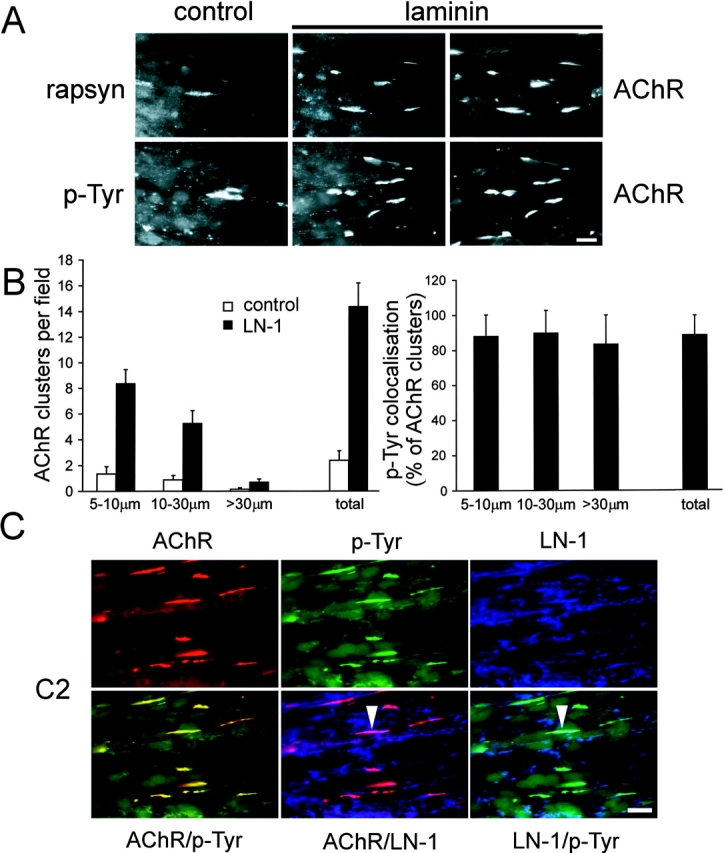
Laminin-1 induces clusters of rapsyn and phosphotyrosine-containing proteins that highly colocalize with AChRs but not with surface-bound laminin. (A) Myotubes cultures were treated with or without laminin-1 (100 nM for 16 h) and double labeled with rhodamine-α-btx (right) and antisera against phosphotyrosine and rapsyn followed by fluorescein-conjugated secondary and tertiary antibodies (middle and left). Bar, 20 μm. (B) Quantitation of AChR clustering after laminin treatment and colocalization with laminin-induced phosphotyrosine clusters. Three subgroups of AChR clusters (size, 5–10 μm, 10–30 μm, and >30 μm) or total AChR clusters of all sizes were quantitated (left graph). A high percentage of AChR clusters of all sizes colocalizes with phosphotyrosine clusters (right graph). Data represent mean ± SD of eight experiments. (C) Laminin-treated C2 myotubes were triple stained for AChR, phosphotyrosine, and laminin-1 (LN-1; top). Combinations of two markers were merged (bottom). Whereas AChRs and phosphotyrosine clusters largely colocalize (yellow, left), only very few clusters of AChRs or phosphotyrosine colocalize with laminin (arrowheads). Bar, 20 μm.
Figure 2.
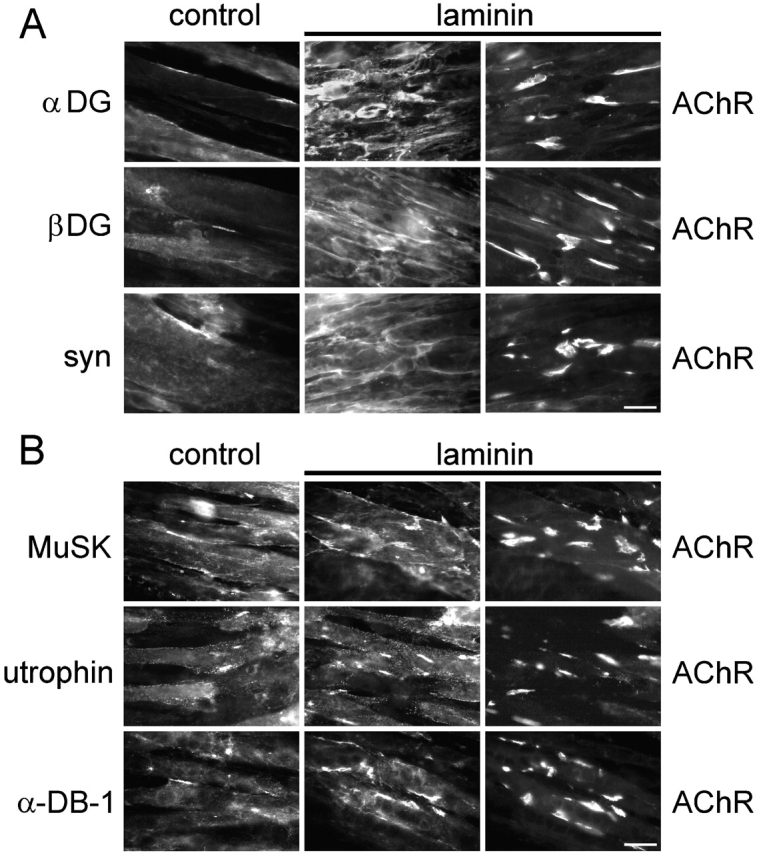
Laminin-1 redistributes α- and β-dystroglycan and syntrophins, hardly affects the distribution of MuSK, and induces clustering of utrophin and α-dystrobrevin-1. C2 myotubes were treated with or without laminin-1 (100 nM for 16 h) and double labeled with rhodamine-α-btx (right) and (A) antisera against α-dystroglycan, β-dystroglycan and syntrophins or (B) antisera against MuSK, utrophin, and α-dystrobrevin-1 followed by fluorescein-conjugated secondary and tertiary antibodies (middle and left). Bars, 20 μm.
The second class of proteins comprised MuSK, α-dystrobrevin-1, and utrophin. These proteins formed laminin-induced clusters but were also detectable in a diffuse distribution along myotubes (Fig. 2 B). Clustering was most pronounced for α-dystrobrevin-1, where laminin caused a 4.8 ± 1.4-fold (mean ± SD; four experiments) increase in the number of aggregates. Laminin-induced clustering was less pronounced for utrophin (2.7 ± 0.7-fold increase; mean ± SD; four experiments) and hardly detectable for MuSK (1.4 ± 0.5-fold increase; mean ± SD; four experiments). In addition, clusters of all three proteins colocalized with AChR clusters to a high degree. Whereas after laminin treatment 64.4 ± 0.1% of α-dystrobrevin-1 clusters colocalized with AChRs, this degree of colocalization was 83.2 ± 3.9% and 64.1 ± 12.1% for utrophin and MuSK, respectively (mean ± SD; four experiments; unpublished data).
Finally, the third class of proteins, comprising rapsyn- and tyrosine-phosphorylated proteins, formed laminin-induced clusters as efficiently as AChRs and strictly colocalized with AChRs in clusters (Fig. 3 A). The intensity of laminin-induced phosphotyrosine aggregates was high and appeared brighter than in the case of agrin (Fig. 3 A; unpublished data). For quantitation, we analyzed three different AChR cluster populations (5–10 μm, 10–30 μm, and clusters >30 μm in length; Fig. 3 B, left) and their corresponding phosphotyrosine aggregates (unpublished data). Laminin-1 increased the number of AChR and phosphotyrosine clusters of all sizes, with the total increase of all AChR clusters being 5.8 ± 2.6-fold (mean ± SD; 12 experiments; Fig. 3 B) and of phosphotyrosine clusters being 5.8 ± 1.6-fold (mean ± SD; seven experiments; unpublished data). Furthermore, all AChR clusters colocalized efficiently with phosphotyrosine-containing proteins showing an overall colocalization of 88% (Fig. 3 B, right). Triple staining for laminin (blue), AChR (red), and phosphotyrosine (green), and subsequent merging of digitized pictures confirmed the high colocalization between AChR and phosphotyrosine clusters (Fig. 3 C, yellow clusters). In contrast, when pictures of surface-bound laminin were merged to AChRs or phosphotyrosine, almost no overlap could be detected.
Together, these data show that postsynaptic proteins are differentially distributed by laminin-1 (Table I). Most strikingly, phosphotyrosine and rapsyn cluster very efficiently in response to laminin, and strictly colocalize with AChRs. This reveals a difference to agrin, which induces significant coclustering with AChRs of all the postsynaptic proteins examined here, including dystroglycans, syntrophins, and MuSK (Table I; Marangi et al., 2001).
Table I. Redistribution of postsynaptic proteins in response to laminin or agrin treatment of cultured myotubes.
| Protein | Laminin treatment | Agrin treatment |
|---|---|---|
| α-dystroglycan | Laminin-like network | Clusters colocalizing with AChRs; some diffuse |
| β-dystroglycan | Laminin-like network | Clusters colocalizing with AChRs; some diffuse |
| Syntrophins | Laminin-like network | Clusters colocalizing with AChRs; some diffuse |
| α-dystrobrevin-1 | Clusters colocalizing with AChRs; some diffuse | Clusters colocalizing with AChRs |
| Utrophin | Clusters colocalizing with AChRs; some diffuse | Clusters colocalizing with AChRs |
| MuSK | Marginal clustering; colocalizes with AChRs; some diffuse | Clusters colocalizing with AChRs |
| Rapsyn | Clusters strictly colocalizing with AChRs | Clusters strictly colocalizing with AChRs |
| Phosphotyrosine | Intense clusters strictly colocalizing with AChRs | Clusters strictly colocalizing with AChRs |
C2 myotubes were treated with laminin and analyzed by double labeling for AChRs and the indicated proteins as described in Materials and methods. Results of agrin-treated cells are from Marangi et al. (2001).
Laminin-induced AChR clustering requires rapsyn
Based on its colocalization with laminin-induced AChR clusters, we analyzed whether rapsyn is necessary for laminin-induced clustering of AChRs and phosphotyrosine. For this purpose, we used myotubes derived from rapsyn −/− mice (Fuhrer et al., 1999) and treated them with laminin-1 for prolonged periods. Although the corresponding control wild-type myotubes formed laminin-induced clusters of slightly smaller sizes than in C2, the difference to rapsyn −/− cells was striking, because no laminin-induced clusters of AChRs and phosphotyrosine could be detected in rapsyn −/− myotubes (Fig. 4). Thus, like agrin, laminin-1 requires rapsyn for the formation of AChR and phosphotyrosine clusters.
Figure 4.

Laminin-induced clustering of AChRs and phosphotyrosine requires rapsyn. Rapsyn −/− myotubes (clones 11–4 and 11–7) were treated with 100 nM laminin-1 for 16 h and double labeled for AChRs and phosphotyrosine using rhodamine-conjugated α-btx and antiserum against phosphotyrosine followed by secondary FITC-conjugated antibodies. No clusters are visible, unlike wild-type cells (clone 12–10), which show many clusters of AChRs and phosphotyrosine. Bar, 20 μm.
Laminin-1 induces tyrosine phosphorylation of AChR β and δ subunits
Based on the high degree of colocalization between phosphotyrosine and AChR clusters in laminin-treated wild-type myotubes (Fig. 3), we analyzed whether AChRs are tyrosine phosphorylated in response to laminin-1. In 1R- cells, mutant derivatives of C2 that lack functional AChRs and clusters of some other postsynaptic proteins (Black et al., 1987; Marangi et al., 2001), no phosphotyrosine clusters could be detected after laminin treatment (Fig. 5 A). This suggests that AChRs or other AChR-associated proteins are tyrosine phosphorylated in the wild-type (Fig. 5 A). We analyzed AChR phosphorylation in C2 myotubes directly, by precipitation of AChRs using biotinylated α-btx followed by phosphotyrosine immunoblotting. When myotubes were treated with laminin-1 for increasing times (Fig. 5 B), tyrosine phosphorylation of the AChR β subunit was first visible after 9 h of treatment and significantly increased by 2.4-fold after 24 h (Fig. 5 C). We also detected tyrosine phosphorylation of the AChR δ subunit induced by laminin-1. The AChR β phosphorylation induced by laminin-1 showed striking parallels to AChR clustering, as clusters are first seen after 8 h and maximal after 24 h of laminin treatment (Sugiyama et al., 1997). In contrast, agrin-induced AChR β phosphorylation is stronger and peaks around 1 h of agrin treatment, i.e., several hours before clustering (Fig. 5 C; Ferns et al., 1996).
Figure 5.
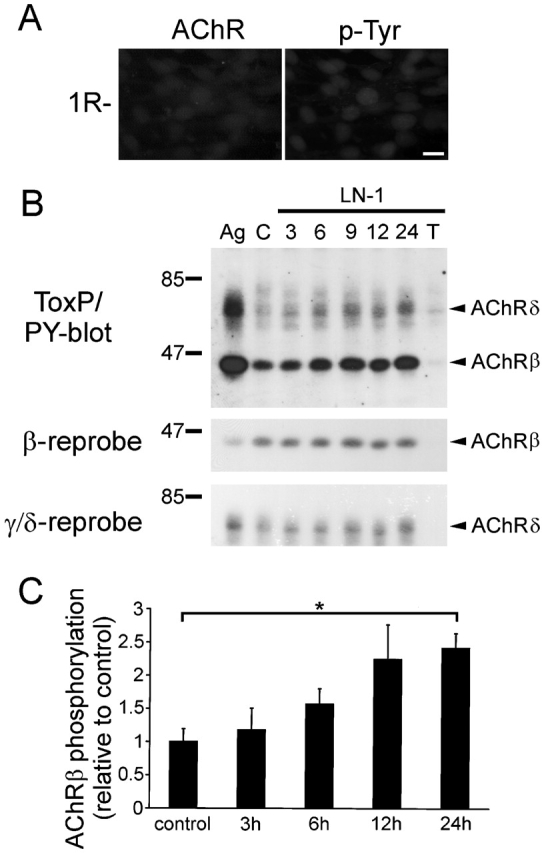
Laminin-1 induces tyrosine phosphorylation of AChR β and δ subunits. (A) 1R- cells were treated with 100 nM laminin-1 for 16 h and double labeled for AChRs and phosphotyrosine using rhodamine-conjugated α-btx and anti-phosphotyrosine antiserum followed by secondary FITC-conjugated antibodies. Bar, 20 μm. (B) C2 myotubes were treated with 80 nM laminin-1 for 3, 6, 9, 12, and 24 h, and lysed. AChRs were precipitated with biotinylated α-btx followed by streptavidin-conjugated Sepharose beads and analyzed by phosphotyrosine immunoblotting. As controls, cells were not treated with laminin (C), treated with 0.25 nM neural agrin for 1 h (Ag), or treated with 24 h laminin-1 and excess free toxin before precipitation (T). Blots were stripped and reprobed for AChR β and γ/δ subunits. (C) Quantitation of phosphotyrosine immunoblots (AChR β subunit, as shown in B, top) by densitometric scanning. Values for untreated cells were set to 1 (control). Data represent mean ± SEM of at least three experiments. *Differs significantly from control (P < 0.0007, by two-sampled t test assuming unequal variances).
Tyrosine phosphorylation is required for laminin-induced AChR clustering and stabilization of clusters
Agrin-induced clustering of AChRs requires tyrosine kinase activity, as shown by the inhibition of clustering by two kinase inhibitors, herbimycin and staurosporine (Wallace, 1994; Ferns et al., 1996). Therefore, we asked whether laminin-induced clustering of AChRs and phosphotyrosine proteins is affected in a similar way. When C2 myotubes were treated with laminin-1 together with staurosporine or herbimycin, no clusters of AChRs or phosphotyrosine could be induced (Fig. 6 A). In addition, in the presence of laminin-1 and inhibitors, no difference to nonlaminin control myotubes was visible, even when AChR clusters of different sizes were quantitated (Fig. 6 B). This indicates that the inhibitors do not act on already existing clusters or on growth of clusters, but preferentially inhibit formation of new AChR clusters of all sizes. Herbimycin and staurosporine did not affect the distribution of laminin-1 on the surface of myotubes (Fig. 6 A). Thus, staurosporine and herbimycin inhibit induction of AChR and phosphotyrosine clustering without affecting spontaneous clustering and without altering the network-like deposition of laminin on the surface.
Figure 6.
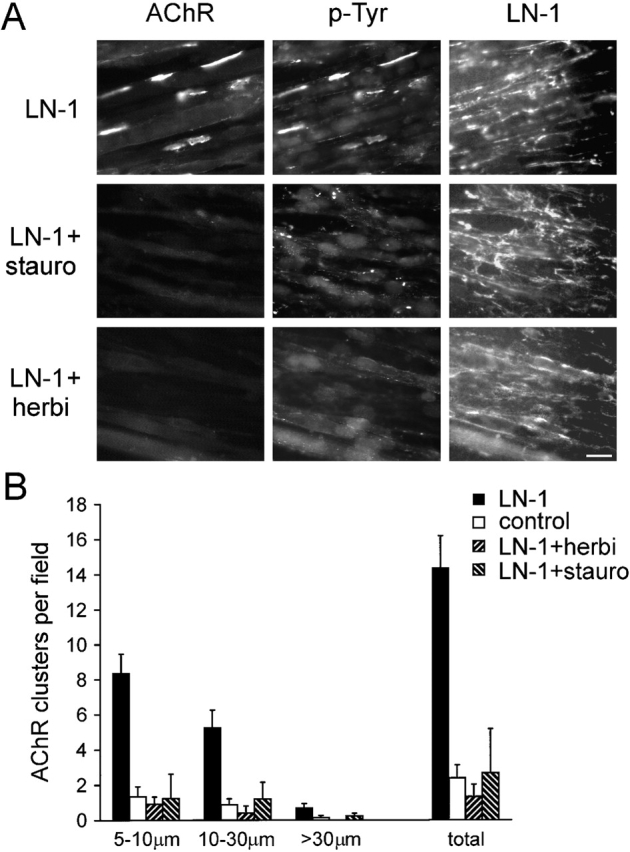
Staurosporine and herbimycin inhibit AChR and phosphotyrosine cluster formation, but not binding and distribution of laminin-1 on myotube surfaces. (A) C2 myotubes were treated with 120 nM laminin-1 for 7 h in the presence of 10 nM staurosporine or 1 μM herbimycin and triple stained for AChRs, phosphotyrosine, and laminin. AChRs were visualized with rhodamine-α-btx; phosphotyrosine and laminin-1 were visualized with primary antisera followed by FITC- and Alexa350-conjugated secondary antibodies, respectively. Bar, 20 μm. (B) Total AChR clusters or three subgroups of AChR clusters (size, 5–10 μm, 10–30 μm, and >30 μm) were analyzed after treatment with laminin-1 alone or together with 10 nM staurosporine or 1 μM herbimycin. Both inhibitors prevent formation of laminin-induced AChR clusters of all sizes. Data represent mean ± SD of at least three experiments.
Next, we analyzed whether inhibition of AChR and phosphotyrosine clustering by staurosporine and herbimycin was paralleled by a reduction of tyrosine phosphorylation of the AChR itself. α-btx-AChR precipitation and phosphotyrosine immunoblotting revealed that indeed herbimycin and staurosporine reduced the level of laminin-induced AChR β subunit phosphorylation (Fig. 7 A). As judged from multiple experiments, this reduction occurred to at least the level of spontaneous β phosphorylation observed in nonlaminin-treated control cells (Fig. 7 B). Thus, apart from clustering of AChRs and phosphotyrosine, staurosporine and herbimycin also inhibit tyrosine phosphorylation of AChR β subunits.
Figure 7.
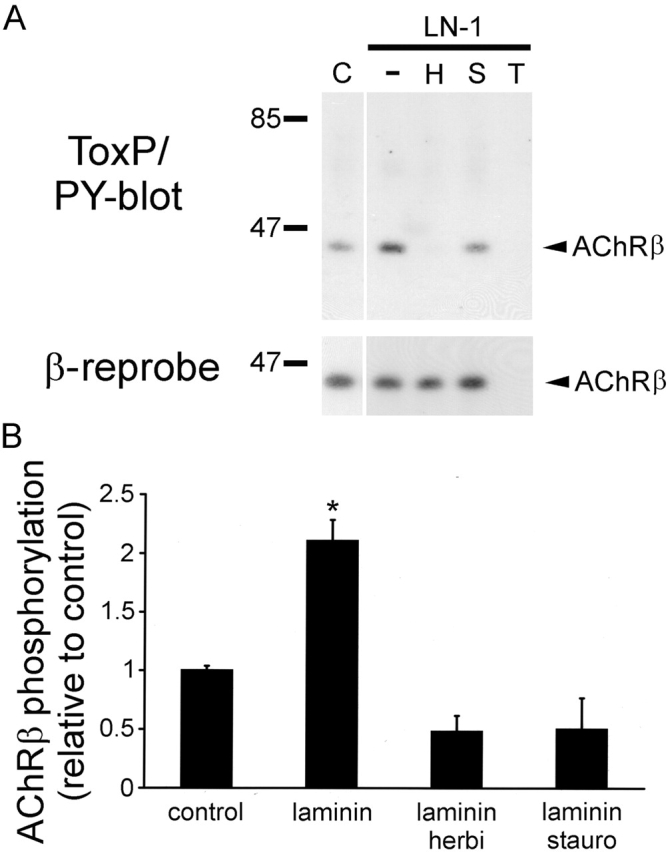
Staurosporine and herbimycin inhibit laminin-induced tyrosine phosphorylation of AChR β subunits. (A) C2 myotubes were treated for 8 h with 120 nM laminin alone (−) or in the presence of 1 μM herbimycin (H) or 10 nM staurosporine (S), lysed, and precipitated using biotinylated α-btx followed by streptavidin-conjugated Sepharose. Precipitates were analyzed by phosphotyrosine immunoblotting. Untreated cells (C) and excess free toxin (T) were used as controls. Blots were stripped and reprobed for AChR β subunits. Both inhibitors reduce laminin-induced AChR β phosphorylation to at least the levels observed in untreated cells. (B) Quantitation of immunoblots by densitometric scanning. Values for untreated cells were set to 1 (control). Data represent mean ± SEM of at least three experiments. *Differs significantly from the other values (P < 0.026, by two-sampled t test assuming unequal variances).
Tyrosine kinases are also required for stabilization of agrin-induced AChR clusters, because after withdrawal of agrin, herbimycin and staurosporine cause rapid dispersal of preformed clusters (Ferns et al., 1996). We asked if stabilization of laminin-induced clusters is governed by a similar mechanism involving tyrosine kinases. We induced AChR clusters with laminin, withdrew laminin, and treated the myotubes with staurosporine or herbimycin. Preformed AChR clusters disappeared after 8 h of treatment with either inhibitor, but not when cells were incubated in inhibitor-free medium (Fig. 8). Together, these data demonstrate that tyrosine kinases are involved in laminin-induced AChR phosphorylation, cluster formation, and cluster stabilization, similar to their action in agrin signaling.
Figure 8.

Laminin-induced AChR clusters are dispersed by herbimycin and staurosporine. C2 myotubes were treated with 100 nM laminin-1 for 16 h and washed. Cells were reincubated without laminin and with or without inhibitors for 8 h as indicated. AChRs were visualized with rhodamine-α-btx and clusters quantitated. Data represent mean ± SD of 20 visual fields. Bar, 20 μm.
Src and Fyn are necessary for stabilization of laminin-induced AChR clusters
Recent findings have shown that Src and Fyn are together necessary for AChR cluster maintenance after agrin treatment (Smith et al., 2001). To analyze whether Src and Fyn also play a role in laminin signaling, we examined laminin-induced AChR clustering in myotubes derived from mice lacking both Src and Fyn (src −/−;fyn −/− cells; Smith et al., 2001). Src −/−;fyn −/− cells formed myotubes of normal morphology indistinguishable from control wild-type or C2 myotubes (Fig. 9 A), as reported previously (Smith et al., 2001). Laminin-1 induced identical AChR clustering in src −/−;fyn −/− cells as in C2 and control wild-type cells, indicating that the absence of Src and Fyn does not affect cluster formation (Fig. 9). Furthermore, clustering of phosphotyrosine was also normal in laminin-treated src −/−;fyn −/− cells (unpublished data). Thus, similar to agrin, Src and Fyn are not required for laminin-induced formation of AChR and phosphotyrosine clusters.
Figure 9.

Laminin-1 induces normal AChR clustering in src −/−;fyn −/− myotubes. C2, src −/−;fyn −/− and control wild-type cells were treated with 100 nM laminin-1 for 16 h and stained for AChRs with rhodamine-α-btx. Bar, 20 μm. Quantitation shows that laminin-1 induces similar clustering of AChRs in C2 and src −/−;fyn −/− cells. We analyzed two clones each of src −/−;fyn −/− and control wild-type cells, which all gave identical results to C2 (unpublished data). Data represent mean ± SEM of at least five experiments.
We next studied the role of Src and Fyn in stabilization of laminin-induced AChR and phosphotyrosine clusters. For this purpose, we stimulated cells with laminin-1 to induce clusters, withdrew laminin, and incubated cells in laminin-free medium for increasing times. In src −/−;fyn −/− cells, preformed clusters of AChRs and phosphotyrosine dispersed rapidly after laminin withdrawal and reached control levels already after 4 h (Fig. 10 A). In contrast, in C2 and wild-type control cells, clusters were very stable after laminin withdrawal and showed no detectable dispersal within 4 h (Fig. 10; unpublished data). Thus, the combination of Src and Fyn is required for maintenance of laminin-induced AChR and phosphotyrosine clusters, similarly to the role of these kinases in stabilization of agrin-induced AChR clusters.
Figure 10.

The stability of laminin-induced clusters of AChRs and phosphotyrosine is reduced in myotubes lacking Src and Fyn. (A) We treated src −/−;fyn −/− and C2 myotubes with or without 100 nM laminin-1 for 16 h and withdrew laminin for 4 h. Remaining clusters of AChRs and phosphotyrosine were then visualized by rhodamine-conjugated α-btx and phosphotyrosine antibodies followed by secondary FITC-conjugated antiserum. Bar, 20 μm. (B) Cells were treated with laminin-1 as above, laminin was withdrawn for 1–4 h, and remaining clusters of AChRs and phosphotyrosine quantitated. As controls, cells were not treated with laminin (C) or laminin was not withdrawn (0 h). Data represent mean ± SEM of at least five experiments. Two clones of src −/−;fyn −/− cells gave identical results, and control wild-type cells were similar to C2 (unpublished data).
Discussion
In this study, we show that laminin-1 redistributes dystroglycans and syntrophins into a coarse network and induces clusters of AChRs, rapsyn, α-dystrobrevin-1, utrophin and, less efficiently, MuSK. Laminin-1 also induced clusters containing phosphotyrosine that strictly colocalized with AChRs clusters, and AChR phosphorylation which strongly correlated with AChR clustering. This is surprising because in earlier studies that used short laminin treatments, no phosphorylation of AChRs or MuSK was observed (Sugiyama et al., 1997; Montanaro et al., 1998). Furthermore, clustering of AChRs in response to laminin required rapsyn and the activity of tyrosine kinases, and maintenance of laminin-induced AChR clusters was dependent on Src and Fyn. Although our experiments were done with laminin-1, we think that the results are also important for laminin-2/4, because all these laminin isoforms cause AChR clustering by a very similar mechanism in cultured myotubes (Burkin et al., 2000).
Laminin-1 redistributes dystroglycans and syntrophin
Laminin-1, as well as laminin-2/4, can bind to myotube surface receptors and form cell-associated polygonal networks (Colognato et al., 1999; Montanaro et al., 1999). Furthermore, α-dystroglycan is a binding partner for laminins (Henry and Campbell, 1999). Our observed laminin network differs from that described by Colognato et al. (1999), in that although we occasionally see exogenous laminin-1 in a regular polygonal arrangement (PAM and STW, unpublished data), most of our laminin-1 appears as bright and bulky deposits forming a rather coarse network. This most likely originates from the higher concentration of our laminin used, which is required in order to cause AChR clustering.
In our experiments, α- and β-dystroglycan and syntrophins codistribute with exogenously applied laminin-1. This codistribution of dystroglycans is consistent with the known tight binding of laminin directly to α-dystroglycan (Henry and Campbell, 1999). Our antibody against syntrophin detects all muscle syntrophin isoforms, including extrasynaptic ones. Others have found that dystrophin codistributes with laminin, at least in a polygonal network when low concentrations of laminin-1 are added to myotubes (Colognato et al., 1999). Together, these data indicate that components of the dystrophin–glycoprotein complex are redistributed by laminin, and laminin-1 appears able to organize this complex from the extracellular matrix to the underlying cytoskeleton. Laminin-1 may thereby contribute to muscle stability extrasynaptically.
Laminin-1 clusters postsynaptic proteins
The fact that laminin-1 redistributes and colocalizes with some muscle proteins (as described above; Table I) raised the possibility that laminin may drive clustering of AChRs merely by extracellular protein rearrangements, because AChRs indirectly interact with dystroglycan (Fuhrer et al., 1999). However, we found that laminin-induced AChR clusters hardly colocalize with exogenously applied laminin itself, nor with dystroglycans or syntrophins. Rather, laminin-1 caused clustering of the synaptic proteins utrophin and α-dystrobrevin-1, and these clusters colocalized strongly with AChRs. In the case of MuSK, laminin-1 induced a marginal clustering. Furthermore, laminin caused aggregation of rapsyn and phosphotyrosine, which colocalized precisely with AChR clusters but showed very little overlap with the laminin network distribution (Table I). The regular hexagonal network that is formed by applying low laminin concentrations to myotubes can be disassembled by genistein, showing that its formation requires tyrosine phosphorylation (Colognato et al., 1999). Interestingly, we found that staurosporine and herbimycin did not affect the distribution of laminin-1 yet abolished AChR clustering. Similarly, genistein did not affect the distribution of laminin-1 in our conditions (STW, PAM, and CF, unpublished data). These observations show that laminin-1 does not act in AChR clustering merely by protein redistribution based on extracellular protein interactions. If involved at all, the laminin network clearly is not sufficient for clustering. Rather, clustering involves intracellular signaling mechanisms.
Laminin-induced AChR clustering requires rapsyn and tyrosine phosphorylation
Based on the coclustering of rapsyn with AChRs induced by laminin-1, we investigated the laminin signaling pathway by examining the role of rapsyn. Use of rapsyn −/− myotubes showed that indeed rapsyn is required for clustering of AChR and phosphotyrosine, very similar to its role in agrin signaling. Rapsyn acts in agrin signaling downstream of MuSK (Apel et al., 1997). As laminin-induced AChR clustering can occur in the absence of MuSK (Sugiyama et al., 1997), the agrin and laminin pathways thus converge at a step downstream of MuSK involving rapsyn. This role of rapsyn reconfirms that laminin requires intracellular signaling mechanisms for AChR clustering.
We investigated this signaling mechanism in more detail by analyzing the role of tyrosine phosphorylation. Laminin induced phosphorylation of AChR subunits, mostly on the β subunit. These results extend earlier findings that short (15–45 min) laminin treatments do not induce phosphorylation of the AChR β subunit and of MuSK (Sugiyama et al., 1997; Montanaro et al., 1998). In our hands, AChR β subunit phosphorylation paralleled clustering, as it was detectable after 9 h and strongest after 24 h. In contrast, in the case of agrin, AChR β phosphorylation precedes cluster formation and peaks after about 1 h (Ferns et al., 1996). Thus, the mechanisms that trigger β phosphorylation are different between agrin and laminin. Furthermore, although in our biochemical experiments β phosphorylation is relatively weak after laminin treatment, phosphotyrosine clustering is very intense, implying that other proteins are tyrosine-phosphorylated and co-clustered with AChRs.
However, most importantly, the same kinase inhibitors that block AChR phosphorylation and clustering by agrin, herbimycin and staurosporine, also inhibit laminin-induced aggregation of AChRs and block phosphorylation of AChR β subunits. These results raise the possibility that phosphorylation of the AChR itself, on its β subunit, may be important for clustering, in analogy to the role of β phosphorylation in receptor clustering induced by agrin (Borges and Ferns, 2001). Several control experiments indicated that the effects of herbimycin and staurosporine during their 8–9 h incubation are specific and not due to general toxicity of these chemicals. First, the inhibitors did not affect the general morphology of C2 (Fig. 6 A). Second, when treated with inhibitor for 8 h, followed by extensive washing and incubation with laminin or agrin in the absence of inhibitor, C2 cells formed normal AChR clusters (STW and CF, unpublished data). Finally, when adding agrin and inhibitors simultaneously for 8 h (i.e., leaving out the normal 1-h preincubation with inhibitor), C2 formed normal clusters (STW and CF, unpublished data).
These data reveal that laminin activates one or several tyrosine kinases, and that this activity is required for laminin-, as well as for agrin-induced AChR clustering. This kinase(s) is downstream of MuSK, because staurosporine does not affect MuSK activation (Fuhrer et al., 1997), and may be a member of the Src family, because such kinases are activated in response to agrin (Mittaud et al., 2001). Src family kinases, together with focal adhesion kinase FAK, are activated in integrin signaling, which leads to tyrosine phosphorylation and rearrangement of cytoskeletal proteins such as paxillin (Thomas and Brugge, 1997; Thomas et al., 1998). Paxillin, talin, and α-fodrin are localized postsynaptically at the NMJ and are substrates for Src family kinases (Hall and Sanes, 1993; Thomas and Brugge, 1997). Thus, given the role of α7 and β1 integrins in both laminin and agrin-induced AChR clustering, signaling mediators and cytoskeletal substrates downstream of integrins, in particular tyrosine kinases, are good candidates to be involved as a common element in laminin as well as agrin signaling.
Src and Fyn are required for stabilization of laminin-induced AChR clusters
Similar to agrin, Src and Fyn are not necessary for laminin-induced AChR clustering. However, the Src-related kinase Yes is upregulated in src −/−;fyn −/− myotubes, and might thus compensate (Smith et al., 2001). In contrast, these mutant cells show that Src and Fyn are required for maintaining AChR clusters after laminin-withdrawal. Furthermore, tyrosine phosphorylation as such is required for cluster stabilization, as shown by herbimycin and staurosporine. These findings reveal strong parallels to agrin, where tyrosine phosphorylation, Src, and Fyn are all required for stabilizing AChR clusters (Ferns et al., 1996; Smith et al., 2001). Src and Fyn may act in stabilization of laminin- or agrin-induced AChR clusters through their adaptor activities, by mediating protein interactions using their SH2 or SH3 domains (Smith et al., 2001). Such kinase activity–independent roles are known from focal adhesion sites, where kinase-inactive Src can recruit FAK (Kaplan et al., 1995; Thomas and Brugge, 1997). Alternatively and/or in parallel, Src and Fyn may act by phosphorylating other proteins. A good candidate is dystrobrevin, which is highly phosphorylated in Torpedo electric organ (Wagner et al., 1993). Interestingly, laminin-1 clusters α-dystrobrevin-1 in a similar fashion as agrin does, and agrin-induced aggregates of AChRs are unstable in the absence of α-dystrobrevin (Grady et al., 2000). Thus, these data raise the possibility that Src and Fyn act in stabilization of AChR clusters (induced by agrin or laminin) by linking the receptor to components of the D/UGC such as dystrobrevin.
The laminin signaling pathway activates the agrin signaling pathway
Previous studies suggested that laminin-1 acts through a different pathway than agrin (Sugiyama et al., 1997; Montanaro et al., 1998), largely based on the differential role of MuSK and rapid AChR β phosphorylation. However, our results demonstrate that laminin-1 activates an intracellular signaling pathway that strongly overlaps with that of agrin at an early step, downstream of MuSK, at the level of tyrosine kinases and rapsyn. Thus, rapsyn and tyrosine phosphorylation (possibly also of AChR β) are required for both agrin- and laminin-induced AChR clustering. Furthermore, laminin and agrin induce comparable coclustering of utrophin and α-dystrobrevin-1 with AChRs. Nonetheless, other findings argue against a full convergence of both pathways, because agrin and laminin have additive effects when added together to myotubes, even when agrin is applied at a saturating concentration (Sugiyama et al., 1997; Burkin et al., 2000). Thus, neither molecule alone seems able to fully activate the signal transduction pathway leading to clustering, and only combined application allows maximal aggregation. Therefore, we think that the laminin and agrin pathways share rapsyn and one or several kinases but also each have unique signaling steps.
This cooperation, particularly on the level of tyrosine kinases, may be important for postsynaptic organization in vivo. Myotubes are known to contain ample tyrosine phosphatase activity that continuously dephosphorylates proteins such as the AChR (Wallace, 1995). Upon blockage by pervanadate (Wallace, 1995), or treatment with agrin (Borges and Ferns, 2001), AChRs become stably phosphorylated and linked to the cytoskeleton, showing that agrin, by activating tyrosine kinases, overcomes the phosphatase activity, thereby driving clustering. By activating the same downstream kinases as agrin, laminin may create an intracellular signaling environment in the muscle that primes myofibers to be more receptive towards a clustering signal like agrin. Consistent with this, laminin-pretreated myotubes require 20-fold less agrin to trigger subsequent AChR clustering (Burkin et al., 2000). Because activated kinases can diffuse intracellularly, such a priming mechanism would operate along considerable distances within muscle fibers and not be restricted to the location of laminin, in agreement with our observation that laminin deposits hardly colocalize with AChR clusters. Based on expression patterns (Patton et al., 1997), laminin-1 may cause widespread signaling throughout the muscle and render muscle fibers (which grow from the middle extending longitudinally) receptive for a clustering signal early in development and around the period of synapse formation, as laminin-1 is lost later. Similarly, laminin-2/4 (which induces AChR clusters indistinguishably from laminin-1 in cultured myotubes; Burkin et al., 2000) may prime the muscle, particularly local synaptic areas, to be receptive around synaptogenesis and later. Neural agrin may enhance such local laminin-2/4-signaling, and the fine tuning of this signaling with the more widespread effects of laminin-1 may be an underlying mechanism for AChR clustering in the middle of myofibers.
It is possible that at certain developmental stages, depending on the level of laminin expression, such a laminin priming mechanism may actually lead to AChR clusters independently of agrin. This may be the case at the earliest stages of NMJ formation, where muscle prepatterning leads to AChR clustering independently of neural agrin and even motorneurons (Yang et al., 2000, 2001; Lin et al., 2001). At this stage, laminin-induced AChR clustering may involve MuSK, given the absence of early clusters in MuSK −/− mice (Lin et al., 2001; Yang et al., 2001), and may occur by phosphorylation and activation of MuSK not through agrin-like ligands but through the downstream kinases activated by laminin. In agreement with this, MuSK can be phosphorylated by other kinases such as Src or Fyn in myotubes (Mohamed et al., 2001), and laminin-induced AChR clustering occurs to a reduced degree in MuSK −/− myotubes in vitro (Sugiyama et al., 1997). Furthermore, upon ectopic injection into myofibers in vivo, MuSK causes AChR clustering independent of the MuSK ectodomain (which would interact with agrin-like ligands) but dependent on its kinase domain (Sander et al., 2001).
However, as development proceeds, laminin does not seem able to cause efficient nerve-bound clustering of AChR in the absence of agrin, because no such clusters are seen at birth in agrin −/− mice (Gautam et al., 1996). However, the remaining few clusters seen in these animals at birth, could raise from laminin. It remains to be investigated how cooperation between agrin and laminin is regulated in vivo and how their signaling pathways mediate AChR clustering throughout development.
Materials and methods
Cell culture
Cell culture reagents were purchased from Life Technologies (Basel). C2C12 (C2), src −/−;fyn −/− cells (clones DM11 and DM15), and the corresponding wild-type cells (clones SW5 and SW10) were grown and fused to form myotubes as previously described (Fuhrer et al., 1997; Smith et al., 2001). Rapsyn −/− cells (clones 11-4 and 11-7) and their corresponding wild-type cells (clone 12-10) were grown and fused as src −/−;fyn −/− cells.
Agrin, laminin, and tyrosine kinase inhibitors
Laminin-1, purified from basement membranes of Engelbreth-Holm-Swarm Mouse sarcoma, was purchased from Sigma-Aldrich (L-2020; Fluka Chemie AG, Buchs; Sugiyama et al., 1997). To induce clustering, 100 nM laminin-1 was added for 16 h to C2, src −/−;fyn −/−, and rapsyn −/− cells on day 2, day 3–4, and day 2–3 in fusion medium, respectively. As control, agrin-induced clustering and phosphorylation was examined in parallel. COOH-terminal constructs of neurally derived agrin (C-Ag12,4,8) were expressed and quantitated as described earlier (Fuhrer et al., 1997). To study the effects of kinase inhibitors on laminin-induced clustering of AChRs and phosphotyrosine, staurosporine (S-5921; Sigma-Aldrich) and herbimycin A (Life Technologies) were used as described below.
Precipitation assays and immunoblot analysis
To examine laminin-induced tyrosine phosphorylation of AChRs in C2 myotubes, cells were treated with 80 nM laminin for 3–24 h, and lysates were subjected to AChR precipitation using biotin-α-btx (Molecular Probes) (Mittaud et al., 2001). As controls, cells were treated for 1 h with 0.25 nM agrin, or an excess (10 μM) of free α-btx was added after a 24-h laminin treatment before AChR precipitation. To inhibit laminin-induced AChR phosphorylation, C2 cells were pretreated with 1 μM herbimycin or 10 nM staurosporine for 1 h, followed by 7–8 h treatment with 120 nM laminin in the presence of the inhibitors. After biotin-α-btx precipitation, tyrosine-phosphorylated proteins were detected by immunoblotting using a mixture of 4G10 (Upstate Biotech) and PY-20 antibodies (Transduction Labs). To visualize precipitated AChRs, blots were stripped and reprobed for the AChR β subunit with mAb124 antibodies or for the AChR γ/δ subunits with mAb88B, provided by Dr. S.C. Froehner (University of Washington, Seattle, WA). Quantitation was performed by densitometric scanning as described previously (Marangi et al., 2001).
Immunocytochemical staining procedures
AChRs were visualized by incubating cells with 100 nM tetramethylrhodamine-conjugated α-btx (Molecular Probes) in fusion medium for 1 h at 37°C before fixation and double staining for other postsynaptic markers. Staining procedures for α-dystroglycan, β-dystroglycan, syntrophins (α-, β1- and β2-isoforms), rapsyn, MuSK, α-dystrobrevin-1, utrophin, and tyrosine-phosphorylated proteins were all done as described previously, using fluorescein-coupled secondary antisera (Marangi et al., 2001). To detect exogenously added laminin-1 in double or triple stains, purified laminin antibodies mAb1905 (Chemicon) were added for 1 h at room temperature together with α-btx before fixation and addition of other primary antibodies. Cells were then treated with secondary Alexa Fluor 350–conjugated goat anti–rat antibodies (Molecular Probes) to detect mAb1905 and with fluorescein-coupled secondary or tertiary antibodies, to detect the other primary antibodies. Although mAb1905 is able to detect endogenous laminin-1 in C2 myotubes, this signal is neglectable in comparison to the much stronger intensity seen in laminin-1–treated cells stained with mAb1905 (Fig. 1 B). Finally, in all cases, cells were mounted in a medium containing glycerol and p-phenylenediamine (Sigma-Aldrich), to reduce fluorescence fading.
Fluorescence microscopy and quantitation of clusters
Stained myotubes were examined with a fluorescence microscope and signals on pictures were counted as clusters as previously described (Marangi et al., 2001). Such signals had to be at least 10 μm in length to score as a cluster. However, in Figs. 3 and 6, we also counted clusters of 5–10 μm in length. For laminin-induced clustering of MuSK, α-dystrobrevin, and utrophin, at least 10 pictures per treatment were counted in each experiment and the number of clusters averaged. Data from four such experiments were used to calculate mean ± SD. Clusters of AChRs and phosphotyrosine were quantitated in one of several ways, which all gave similar results. In Figs. 3 and 6, pictures taken at 200× magnification (from at least three experiments) were evaluated as described above for MuSK, α-dystrobrevin, and utrophin. Similarly, in Figs. 9 and 10, pictures taken at 400× magnification (from at least five experiments) were evaluated to calculate mean ± SEM. Finally, in Fig. 8, a total of 20 pictures (400× magnification) per treatment were counted to yield mean ± SD of AChR clusters.
Acknowledgments
We are very grateful to Drs. S.J. Burden, C.L. Smith, and M. Gautam for preparing, and to S. Erb-Vögtli for culturing knockout muscle cells. We also thank Drs. M. Gesemann and S.J. Burden for comments on the manuscript and members of the Fuhrer laboratory for helpful discussions.
This work was supported by the Dr. Eric Slack-Gyr Foundation, the National Center of Competence in Research “Neural Plasticity and Repair,” and by grants from the Swiss National Science Foundation and the Swiss Foundation for Research on Muscle Diseases (to C. Fuhrer).
Footnotes
Abbreviations used in this paper: α-btx, α-bungarotoxin; AChR, acetylcholine receptor; D/UGC, dystrophin/utrophin glycoprotein complex; FAK, focal adhesion kinase; NMJ, neuromuscular junction.
References
- Apel, E.D., D.J. Glass, L.M. Moscoso, G.D. Yancopoulos, and J.R. Sanes. 1997. Rapsyn is required for MuSK signaling and recruits synaptic components to a MuSK-containing scaffold. Neuron. 18:623–635. [DOI] [PubMed] [Google Scholar]
- Black, R., D. Goldman, S. Hochschwender, J. Lindstrom, and Z.W. Hall. 1987. Genetic variants of C2 muscle cells that are defective in synthesis of the α-subunit of the acetylcholine receptor. J. Cell Biol. 105:1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges, L.S., and M. Ferns. 2001. Agrin-induced phosphorylation of the acetylcholine receptor regulates cytoskeletal anchoring and clustering. J. Cell Biol. 153:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkin, D.J., M. Gu, B.L. Hodges, J.T. Campanelli, and S.J. Kaufman. 1998. A functional role for specific spliced variants of the α7β1 integrin in acetylcholine receptor clustering. J. Cell Biol. 143:1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkin, D.J., J.E. Kim, M. Gu, and S.J. Kaufman. 2000. Laminin and α7β1 integrin regulate agrin-induced clustering of acetylcholine receptors. J. Cell Sci. 113:2877–2886. [DOI] [PubMed] [Google Scholar]
- Cohen, M.W., C. Jacobson, P.D. Yurchenco, G.E. Morris, and S. Carbonetto. 1997. Laminin-induced clustering of dystroglycan on embryonic muscle cells: comparison with agrin-induced clustering. J. Cell Biol. 136:1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognato, H., D.A. Winkelmann, and P.D. Yurchenco. 1999. Laminin polymerization induces a receptor–cytoskeleton network. J. Cell Biol. 145:619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denzer, A.J., R. Brandenberger, M. Gesemann, M. Chiquet, and M.A. Ruegg. 1997. Agrin binds to the nerve-muscle basal lamina via laminin. J. Cell Biol. 137:671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferns, M., M. Deiner, and Z. Hall. 1996. Agrin-induced acetylcholine receptor clustering in mammalian muscle requires tyrosine phosphorylation. J. Cell Biol. 132:937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer, C., J.E. Sugiyama, R.G. Taylor, and Z.W. Hall. 1997. Association of muscle-specific kinase MuSK with the acetylcholine receptor in mammalian muscle. EMBO J. 16:4951–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer, C., M. Gautam, J.E. Sugiyama, and Z.W. Hall. 1999. Roles of rapsyn and agrin in interaction of postsynaptic proteins with acetylcholine receptors. J. Neurosci. 19:6405–6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam, M., P.G. Noakes, J. Mudd, M. Nichol, G.C. Chu, J.R. Sanes, and J.P. Merlie. 1995. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 377:232–236. [DOI] [PubMed] [Google Scholar]
- Gautam, M., P.G. Noakes, L. Moscoso, F. Rupp, R.H. Scheller, J.P. Merlie, and J.R. Sanes. 1996. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 85:525–535. [DOI] [PubMed] [Google Scholar]
- Glass, D.J., D.C. Bowen, T.N. Stitt, C. Radziejewski, J. Bruno, T.E. Ryan, D.R. Gies, S. Shah, K. Mattsson, S.J. Burden, et al. 1996. Agrin acts via a MuSK receptor complex. Cell. 85:513–523. [DOI] [PubMed] [Google Scholar]
- Grady, R.M., H. Zhou, J.M. Cunningham, M.D. Henry, K.P. Campbell, and J.R. Sanes. 2000. Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin-glycoprotein complex. Neuron. 25:279–293. [DOI] [PubMed] [Google Scholar]
- Hall, Z.W., and J.R. Sanes. 1993. Synaptic structure and development: the neuromuscular junction. Cell. 72:99–121. [DOI] [PubMed] [Google Scholar]
- Henry, M.D., and K.P. Campbell. 1999. Dystroglycan inside and out. Curr. Opin. Cell Biol. 11:602–607. [DOI] [PubMed] [Google Scholar]
- Huh, K.H., and C. Fuhrer. 2002. Clustering of nicotinic acetylcholine receptors: from the neuromuscular junction to interneuronal synapses. Mol. Neurobiol. 25:79–112. [DOI] [PubMed] [Google Scholar]
- Jacobson, C., F. Montanaro, M. Lindenbaum, S. Carbonetto, and M. Ferns. 1998. alpha-dystroglycan functions in acetylcholine receptor aggregation but is not a coreceptor for agrin-MuSK signaling. J. Neurosci. 18:6340–6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, C., P. Cote, S. Rossi, R. Rotundo, and S. Carbonetto. 2001. The dystroglycan complex is necessary for stabilization of acetylcholine receptor clusters at neuromuscular junctions and formation of the synaptic basement membrane. J. Cell Biol. 152:435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, K.B., J.R. Swedlow, D.O. Morgan, and H.E. Varmus. 1995. c-Src enhances the spreading of src−/− fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 9:1505–1517. [DOI] [PubMed] [Google Scholar]
- Lin, W., R.W. Burgess, B. Dominguez, S.L. Pfaff, J.R. Sanes, and K.F. Lee. 2001. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 410:1057–1064. [DOI] [PubMed] [Google Scholar]
- Marangi, P.A., J.R. Forsayeth, P. Mittaud, S. Erb-Vogtli, D.J. Blake, M. Moransard, A. Sander, and C. Fuhrer. 2001. Acetylcholine receptors are required for agrin-induced clustering of postsynaptic proteins. EMBO J. 20:7060–7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, P.T., and J.R. Sanes. 1997. Integrins mediate adhesion to agrin and modulate agrin signaling. Development. 124:3909–3917. [DOI] [PubMed] [Google Scholar]
- Meier, T., D.M. Hauser, M. Chiquet, L. Landmann, M.A. Ruegg, and H.R. Brenner. 1997. Neural agrin induces ectopic postsynaptic specializations in innervated muscle fibers. J. Neurosci. 17:6534–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittaud, P., P.A. Marangi, S. Erb-Vogtli, and C. Fuhrer. 2001. Agrin-induced activation of acetylcholine receptor-bound src family kinases requires rapsyn and correlates with acetylcholine receptor clustering. J. Biol. Chem. 276:14505–14513. [DOI] [PubMed] [Google Scholar]
- Mohamed, A.S., K.A. Rivas-Plata, J.R. Kraas, S.M. Saleh, and S.L. Swope. 2001. Src-class kinases act within the agrin/MuSK pathway to regulate acetylcholine receptor phosphorylation, cytoskeletal anchoring, and clustering. J. Neurosci. 21:3806–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanaro, F., S.H. Gee, C. Jacobson, M.H. Lindenbaum, S.C. Froehner, and S. Carbonetto. 1998. Laminin and alpha-dystroglycan mediate acetylcholine receptor aggregation via a MuSK-independent pathway. J. Neurosci. 18:1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanaro, F., M. Lindenbaum, and S. Carbonetto. 1999. α-Dystroglycan is a laminin receptor involved in extracellular matrix assembly on myotubes and muscle cell viability. J. Cell Biol. 145:1325–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noakes, P.G., M. Gautam, J. Mudd, J.R. Sanes, and J.P. Merlie. 1995. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature. 374:258–262. [DOI] [PubMed] [Google Scholar]
- Patton, B.L., J.H. Miner, A.Y. Chiu, and J.R. Sanes. 1997. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J. Cell Biol. 139:1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton, B.L., J.M. Cunningham, J. Thyboll, J. Kortesmaa, H. Westerblad, L. Edstrom, K. Tryggvason, and J.R. Sanes. 2001. Properly formed but improperly localized synaptic specializations in the absence of laminin alpha4. Nat. Neurosci. 4:597–604. [DOI] [PubMed] [Google Scholar]
- Sander, A., B.A. Hesser, and V. Witzemann. 2001. MuSK induces in vivo acetylcholine receptor clusters in a ligand-independent manner. J. Cell Biol. 155:1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes, J.R., and J.W. Lichtman. 2001. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2:791–805. [DOI] [PubMed] [Google Scholar]
- Smith, C.L., P. Mittaud, E.D. Prescott, C. Fuhrer, and S.J. Burden. 2001. Src, Fyn, and Yes are not required for neuromuscular synapse formation but are necessary for stabilization of agrin-induced clusters of acetylcholine receptors. J. Neurosci. 21:3151–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama, J.E., D.J. Glass, G.D. Yancopoulos, and Z.W. Hall. 1997. Laminin-induced acetylcholine receptor clustering: an alternative pathway. J. Cell Biol. 139:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, S.M., and J.S. Brugge. 1997. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13:513–609. [DOI] [PubMed] [Google Scholar]
- Thomas, J.W., B. Ellis, R.J. Boerner, W.B. Knight, G.C. White II, and M.D. Schaller. 1998. SH2- and SH3-mediated interactions between focal adhesion kinase and Src. J. Biol. Chem. 273:577–583. [DOI] [PubMed] [Google Scholar]
- Wagner, K.R., J.B. Cohen, and R.L. Huganir. 1993. The 87K postsynaptic membrane protein from Torpedo is a protein-tyrosine kinase substrate homologous to dystrophin. Neuron. 10:511–522. [DOI] [PubMed] [Google Scholar]
- Wallace, B.G. 1989. Agrin-induced specializations contain cytoplasmic, membrane, and extracellular matrix-associated components of the postsynaptic apparatus. J. Neurosci. 9:1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, B.G. 1994. Staurosporine inhibits agrin-induced acetylcholine receptor phosphorylation and aggregation. J. Cell Biol. 125:661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, B.G. 1995. Regulation of the interaction of nicotinic acetylcholine receptors with the cytoskeleton by agrin-activated protein tyrosine kinase. J. Cell Biol. 128:1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X., W. Li, E.D. Prescott, S.J. Burden, and J.C. Wang. 2000. DNA topoisomerase IIbeta and neural development. Science. 287:131–134. [DOI] [PubMed] [Google Scholar]
- Yang, X., S. Arber, C. William, L. Li, Y. Tanabe, T.M. Jessell, C. Birchmeier, and S.J. Burden. 2001. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 30:399–410. [DOI] [PubMed] [Google Scholar]