

Abstract
The small GTPase RhoA controls activity of serum response factor (SRF) by inducing changes in actin dynamics. We show that in PC12 cells, activation of SRF after serum stimulation is RhoA dependent, requiring both actin polymerization and the Rho kinase (ROCK)–LIM kinase (LIMK)–cofilin signaling pathway, previously shown to control F-actin turnover. Activation of SRF by overexpression of wild-type LIMK or ROCK-insensitive LIMK mutants also requires functional RhoA, indicating that a second RhoA-dependent signal is involved. This is provided by the RhoA effector mDia: dominant interfering mDia1 derivatives inhibit both serum- and LIMK-induced SRF activation and reduce the ability of LIMK to induce F-actin accumulation. These results demonstrate a role for LIMK in SRF activation, and functional cooperation between RhoA-controlled LIMK and mDia effector pathways.
Keywords: LIM kinase; mDia; ROCK; actin; SRF
Introduction
The Rho family of Ras-related GTPases are involved in the control of many cellular processes, including cytoskeletal organization, cell cycle control, and transformation, and transcriptional regulation (for reviews see Van Aelst and D'Souza-Schorey, 1997; Bishop and Hall, 2000). The Rho family GTPase RhoA controls the formation of actin structures by interacting with a set of effector proteins that includes the Rho kinases (ROCKs; also known as ROKs), myosin light chain (MLC)* phosphatase, PI4-P 5-kinase, and the Diaphanous group of formin proteins (mDia proteins; for reviews see Kaibuchi et al., 1999; Amano et al., 2000). The ROCK kinases play several roles in RhoA-induced actin reorganization. They control actin filament bundling by phosphorylating MLC and inhibiting MLC phosphatase (Amano et al., 1996; Kimura et al., 1996); regulate synthesis of PI-4,5P, an important regulator of cytoskeletal remodelling (Sechi and Wehland, 2000), by controlling the activity of PI-4P 5-kinase (Oude Weernink et al., 2000); and promote F-actin accumulation by activating LIM kinases (LIMKs), which phosphorylate and inactivate the actin filament depolymerizing/severing factor cofilin (Arber et al., 1998; Yang et al., 1998; Maekawa et al., 1999; Sumi et al., 1999; Ohashi et al., 2000). The mDia proteins also induce F-actin assembly. In this case, binding of Rho appears to relieve an autoinhibitory interaction between the mDia NH2- and COOH-terminal sequences; this allows their conserved formin-homologous domains to promote F-actin assembly via an unknown mechanism (Watanabe et al., 1997, 1999; Tominaga et al., 2000; Alberts, 2001). The mDia proteins cooperate with ROCK kinases to promote actin stress fiber formation (Nakano et al., 1999; Watanabe et al., 1999; Tominaga et al., 2000), but the involvement of ROCK-induced LIMK activation in this process has not been addressed directly.
Recent studies have identified a direct link between the ability of Rho GTPases to control actin polymerization and the ability to regulate transcriptional activation by serum response factor (SRF). SRF, a MADS-box transcription factor, controls the activities of many growth factor–inducible and muscle-specific genes (Shore and Sharrocks, 1995; Arsenian et al., 1998). Serum- and mitogen-activated SRF activity is RhoA dependent, and activated forms of Rho family GTPases can activate SRF in the absence of extracellular stimuli (Hill et al., 1995). Signal-induced SRF activation can be inhibited both by blockade of actin polymerization and by overexpression of nonpolymerizable derivatives of cytoskeletal actin (Sotiropoulos et al., 1999; Tominaga et al., 2000; unpublished data). In addition, certain actin-binding drugs and constitutively active forms of regulators of actin polymerization, such as LIMK, mDia, the Wiskott-Aldrich Syndrome protein (WASP) family, and vasodilator-stimulated phosphoprotein (VASP), can potentiate SRF activity in the absence of extracellular signals (Sotiropoulos et al., 1999; Tominaga et al., 2000; unpublished data). These results have led to the proposal that SRF is somehow activated in response to depletion of the cellular G-actin pool (Sotiropoulos et al., 1999).
The RhoA–actin signaling pathway regulates a subset of SRF target genes including vinculin, β-actin, SRF itself, SM22, and sm-α actin (Sotiropoulos et al., 1999; Gineitis and Treisman, 2001; Mack et al., 2001). However, the signaling pathways linking RhoA to actin polymerization and SRF activity appear to vary between cell types. In NIH3T3 cells, SRF activation requires the RhoA effector mDia (Tominaga et al., 2000; unpublished data) but not ROCK or LIMK (Sahai et al., 1998, 1999; Sotiropoulos et al., 1999). In rat aortic smooth muscle cells, by contrast, ROCK does contribute to SRF activation, although it remains unclear whether LIMK and mDia are also involved (Mack et al., 2001). In this work, we have investigated the role of LIMK signaling to SRF using the rat PC12 phaeochromocytoma cell line, prompted by previous reports that LIMK expression is pronounced in cells of neural origin (Cheng and Robertson, 1995; Proschel et al., 1995). We show that in PC12 cells, the ROCK–LIMK–cofilin–actin pathway plays an important role in signaling to SRF. In addition, both serum- and LIMK-induced SRF activity is dependent on mDia, which is also required for LIMK1-induced F-actin accumulation in PC12 cells. Our results reveal functional cooperation between mDia and LIMK, consistent with a model in which Rho-dependent LIMK activation regulates the stability of mDia-dependent F-actin.
Results
Activation of SRF via the ROCK–LIMK–cofilin pathway in neuronal cell lines
LIMK has been reported to be expressed at high level in the CNS (Cheng and Robertson, 1995; Proschel et al., 1995). Moreover, stimulation of N1E-115 neuroblastoma cells with lysophosphatidic acid induces the phosphorylation and activation of LIMKs via the RhoA effector ROCK (Maekawa et al., 1999). We therefore investigated the involvement of the ROCK–LIMK effector pathway in SRF activation in neuronal cell lines. N1E-115, PC12, SKNMC, and Neuro2A cells were transfected with the SRF-controlled reporter gene 3D.Aluc, maintained in 0.5% serum for 40 h and then restimulated with 15% serum for 8 h before analysis of reporter gene activity (see Materials and methods). Serum induction of the reporter gene in N1E-115 cells was inefficient and not pursued further (unpublished data). In PC12, SKNMC, and Neuro2A cells, however, serum stimulation resulted in robust reporter gene activation; this was dependent on RhoA, as it was abolished by coexpression of C3 transferase, which ADP ribosylates and thereby inactivates RhoA (Fig. 1 A).
Figure 1.

Serum-induced activation of SRF reporter requires the ROCK–LIMK–cofilin pathway in PC12, SKNMC, and Neuro2A cells. (A) The different cell types were transfected with the SRF reporter together with expression plasmids encoding C3 transferase (50 ng), nonphosphorylatable cofilin S3A (0.5 or 2.0 μg), kinase-inactive LIMK1 kinase domain (K1C.D460A) (0.5 or 1.0 μg), or kinase-inactive ROCKΔ3 K105A (0.5 or 2.0 μg) as indicated, or treated with latrunculin B (LB; 0.5 μM) or Y27632 (10 μM). Serum induction of reporter activity was measured and quantitated as described in the Materials and methods; error bars show SEM (n = 3) for PC12 and Neuro2A, and half range (n = 2) for SKNMC. (B) SRF activation by serum stimulation does not require the ROCK–LIMK–cofilin pathway in NIH3T3 cells. NIH3T3 cells were transfected with the SRF reporter and expression plasmids as in A. Reporter activity was measured and quantitated taking serum-induced level as 100%. Error bars show half range (n = 2). (C) Activation of SRF by Rho GTPases and actin-binding drugs in PC12 cells. Cells were transfected with the SRF reporter and either cotransfected with expression plasmids encoding RhoA.V14 (1.0 μg), Rac1.V12 (0.3 μg), Cdc42.V12 (0.3 μg), or N-WASP minimal F-actin–nucleating VCA domain (0.3 μg), or treated with 2 μM cytochalasin D or 0.5 μM jasplakinolide before reporter activity was measured. (D) Serum-induced activation of the SRF target gene vinculin requires functional ROCK in PC12 cells. Serum-starved PC12 cells were serum stimulated with or without Y27632 pretreatment as indicated. Total RNA was analyzed for vinculin and GAPDH transcripts by RNA'ase protection.
We next evaluated the potential contribution of the ROCK–LIMK–cofilin pathway to serum-induced SRF activation. Overexpression of the nonphosphorylatable cofilin mutant cofilin S3A can block activation of SRF by overexpressed LIMK1 (Sotiropoulos et al., 1999). To inhibit ROCK activity, we exploited the kinase-inactive ROCK mutant ROCKΔ3-K105A (Ishizaki et al., 1997) and the small molecule ROCK inhibitor Y27632 (Uehata et al., 1997; Ishizaki et al., 2000). In PC12, SKNMC, and Neuro2A cells, serum induction of the SRF reporter was substantially blocked by expression of cofilin S3A or ROCKΔ3-K105A (Fig. 1 A). Because PC12 exhibited both a robust induction of the reporter and a high absolute level of activity, we focused our attention on these cells for further experiments. In PC12 cells, SRF reporter activity was also effectively blocked by expression of a kinase-inactive LIMK1 derivative D460A (Edwards and Gill, 1999), and upon inhibition of ROCK activity by Y27632 (Fig. 1 A). Together these data suggest that in all three neuronal cell lines, the ROCK–LIMK–cofilin pathway is essential for SRF activation. In contrast, neither cofilin nor LIMK appeared required for SRF activation in NIH3T3 cells (Fig. 1 B), in agreement with our previous finding that ROCK activity is not essential for SRF activation in these cells (Sahai et al., 1999).
We used PC12 cells to characterize the involvement of Rho signaling and actin dynamics in signaling to SRF in more detail. SRF reporter activation was blocked by treatment of the cells with latrunculin B, which inhibits actin polymerization by sequestration of G-actin (Fig. 1 A, left). Expression of the GTPase-defective RhoA mutant RhoA.V14 activated SRF as efficiently as serum stimulation (Fig. 1 C), as previously observed in NIH3T3 fibroblasts (Hill et al., 1995). In addition, the actin-binding drugs jasplakinolide and cytochalasin D strongly activate SRF in PC12 as well as in NIH3T3 cells (Fig. 1 C) (Sotiropoulos et al., 1999). Activated forms of the Rho-related GTPases Rac1 and Cdc42 also activated SRF in PC12 cells, this was more efficient than RhoA, as did the minimal actin filament–nucleating domain of neural (N)-WASP, VCA (reviewed by Higgs and Pollard, 2001), which functions only weakly in NIH3T3 cells (Sotiropoulos et al., 1999) (Fig. 1 C).
We used Y27632 treatment to test the involvement of ROCK signaling in activation in PC12 cells of a bona fide SRF target gene, vinculin, which in fibroblasts is dependent on RhoA–actin signaling (Sotiropoulos et al., 1999; Gineitis and Treisman, 2001). Vinculin transcripts increased approximately fourfold over a 2-h period after serum stimulation, and this was substantially blocked by Y27632 pretreatment (Fig. 1 D). Thus, in PC12 cells, SRF regulation is dependent on a RhoA–ROCK–LIMK–cofilin signaling cascade.
Cofilin phosphorylation in PC12 cells is Rho dependent
To obtain more direct evidence for changes in LIMK activity upon serum stimulation of PC12 cells, we used two-dimensional (2D) gel electrophoresis and immunoblotting to examine cofilin phosphorylation. In both cycling or serum-starved PC12 cell extracts, cofilin migrated as two predominant species of 19 kD, presumably representing cofilin and actin depolymerizing factor, which should cross react with the antiserum used (Fig. 2 A, top, arrows). Serum stimulation induced the appearance of an additional spot, which was sensitive to treatment of the cells with Y27632 (Fig. 2 A, middle, arrow). This species was also sensitive to phosphatase treatment, indicating that it is a phosphorylated derivative of cofilin (Fig. 2 A, bottom, arrow).
Figure 2.

Cofilin phosphorylation via the Rho pathway in PC12 cells. The 2D gels are shown with acidic isoelectric point to the right, SDS-PAGE top to bottom. (A) Serum-induced phosphorylation of endogenous cofilin requires ROCK activity. PC12 cells were maintained in 0.5% serum before stimulation with 15% serum with or without Y27632 (Y) pretreatment. Cell extracts were prepared and fractionated by 2D gel electrophoresis; cofilin was detected by immunoblotting. Where indicated, extracts were treated with alkaline phosphatase (P'tase) before analysis. (B) FLAG–cofilin reporter assay. PC12 cells transfected with the indicated FLAG–cofilin expression plasmids (0.2 μg) were analyzed by 2D gel electrophoresis and immunoblotting with anti-FLAG antibody; phosphatase treatment was performed on anti-FLAG immunoprecipitates. (C) Phosphorylation status of FLAG–cofilin under different growth conditions. Cells transfected with wild-type FLAG–cofilin expression plasmids were maintained as indicated and analyzed as in part B. Only the cofilin species are shown; the positions of unphosphorylated and phosphorylated forms are indicated above the figure. (D) Serum-induced phosphorylation of FLAG–cofilin requires RhoA and ROCK activity. Cells transfected with expression plasmids encoding wild-type FLAG–cofilin and C3 transferase (50 ng), as indicated, were serum starved and restimulated with 15% serum for 5 min; Y27632 treatment (Y) was as indicated.
To allow investigation of the role of RhoA and its effectors in signaling to SRF via LIMK, we set up a simple cofilin reporter assay. Extracts from transfected cells expressing FLAG epitope–tagged cofilin were subjected to 2D gel electrophoresis followed by immunoblotting with FLAG antibody. Wild-type FLAG–cofilin generated two species in this assay. The more acidic of these represents the phosphorylated form, because it is not generated by the nonphosphorylatable mutant cofilin S3A, and is sensitive to phosphatase treatment (Fig. 2 B, arrows). The ratio of phosphorylated to nonphosphorylated FLAG–cofilin observed in serum-starved cells varied between experiments, increasing as cofilin expression increased (Fig. 2 D, middle and bottom), perhaps reflecting homeostatic adjustment of the treadmilling machinery to maintain the F-actin level. We therefore only compared FLAG–cofilin phosphorylation levels within a given experiment. Serum starvation caused a reduction in the amount of phosphorylated FLAG–cofilin relative to the unphosphorylated form (Fig. 2 C, top and middle). Upon serum stimulation, increased phosphorylation of FLAG–cofilin was detectable within 5 min and persisted for at least 30 min (Fig. 2 C, bottom). Serum-induced FLAG–cofilin phosphorylation did not occur in cells expressing C3 transferase or treated with Y27632, indicating that it depends on RhoA–ROCK signaling (Maekawa et al., 1999; Ohashi et al., 2000) (Fig. 2 D, top and middle); C3 or Y27632 treatment also slightly but consistently reduced the proportion of phosphorylated FLAG–cofilin in serum-starved cells, suggesting that the basal level of LIMK activity is also significantly dependent on RhoA (Fig. 2 D, bottom).
SRF activation via ROCK and LIMK requires functional RhoA
The data presented above show that ROCK–LIMK signaling is required for serum-induced SRF activation in PC12 cells. We used C3 transferase expression to test whether this is the only RhoA-dependent event required for signaling to SRF in these cells. In parallel, we examined the effects of C3 transferase upon cofilin phosphorylation using the FLAG–cofilin transfection assay. As expected, C3 transferase expression abolished SRF activation by RhoA.V14 (Fig. 3 A). Surprisingly, C3 expression also abolished SRF activation by overexpressed LIMK1 (Fig. 3 A), although it did not inhibit induction of FLAG–cofilin phosphorylation by LIMK1 (Fig. 3 B, top). This result suggests that LIMK activation of SRF requires an additional RhoA-dependent event, even in starved cells with only basal levels of RhoA activity.
Figure 3.
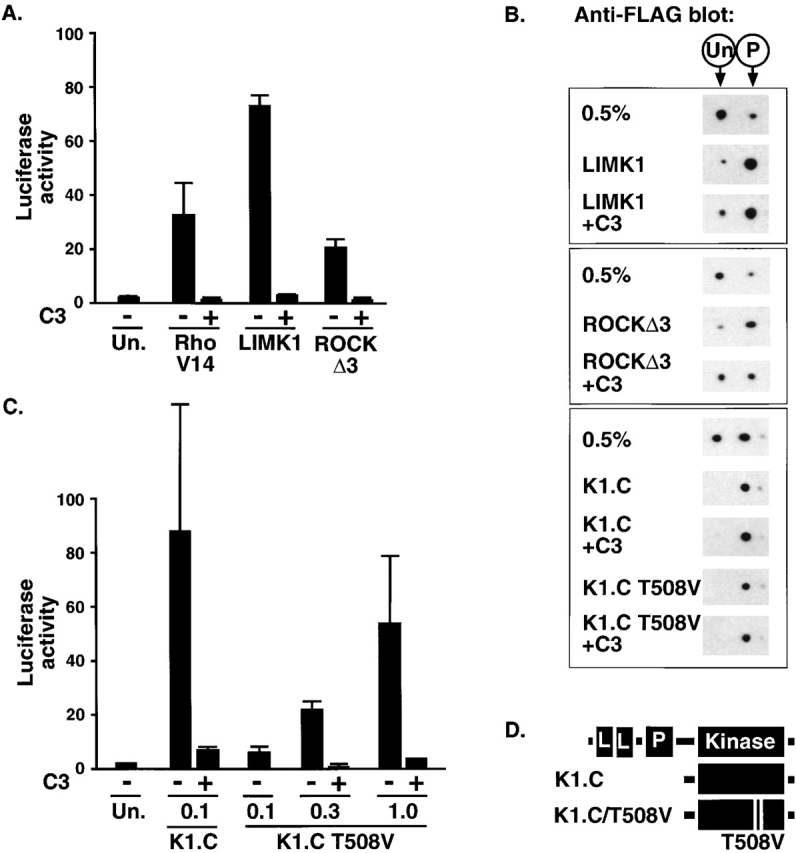
LIMK1 requires an additional RhoA-dependent event to activate SRF. (A) Sensitivity of SRF activation to C3 transferase. PC12 cells were transfected with the SRF reporter and expression plasmids encoding RhoA.V14 (1.0 μg), LIMK1 (1.0 μg), or ROCKΔ3 (60 ng), and C3 transferase (50 ng) as indicated, and reporter activity was measured. (B) FLAG–cofilin phosphorylation does not require RhoA. Cells were transfected with expression plasmids encoding wild-type FLAG–cofilin (0.2 μg) alone or with LIMK1 (1.0 μg), ROCKΔ3 (30 ng), LIMK1 kinase domain K1.C (0.1 μg), or ROCK-insensitive LIMK1 kinase domain mutant K1.C/T508V (1.0 μg), and C3 transferase (50 ng) as indicated, before processing by 2D gel electrophoresis followed by anti-FLAG immunoblot. Only the FLAG–cofilin species are shown; the positions of unphosphorylated and phosphorylated forms are indicated above the figure. (C) SRF activation by LIMK1 T508V requires RhoA. Cells were transfected with the SRF reporter and expression plasmids encoding LIMK1 kinase domain K1.C (0.1 μg) or ROCK-insensitive LIMK1 kinase domain mutant K1.C/T508V (0.1, 0.3, or 1.0 μg), with C3 transferase (50 ng) as indicated, and reporter activity was measured. (D) LIMK1 derivatives. The LIMK1 coding region is indicated at the top with LIM, PDZ, and kinase domains shown as boxes; the K1.C kinase domain (residues 293–646) is shown below and its ROCK-insensitive mutant T508V is also indicated.
The RhoA effector kinase ROCK activates LIMK1 by phosphorylating it at a single site, T508 (Maekawa et al., 1999; Ohashi et al., 2000), and we therefore tested whether the sensitivity of LIMK1 to C3 transferase reflected the involvement of RhoA–ROCK signaling in LIMK activation. First, we examined whether functional RhoA is required for SRF activation induced by the activated ROCK derivative, ROCKΔ3, which should activate LIMK1 independently of RhoA (Maekawa et al., 1999; Ohashi et al., 2000). ROCKΔ3 activated SRF with an efficiency comparable to that of RhoA.V14, but as observed with LIMK1, this was sensitive to C3 transferase (Fig. 3 A). Expression of ROCKΔ3 also induced phosphorylation of FLAG–cofilin, which was not blocked by C3 expression although it was somewhat reduced (Fig. 3 B, middle). Next, we examined SRF activation by LIMK T508V, in which the target site for ROCK phosphorylation is replaced with a nonphosphorylatable valine residue (Fig. 3 D); such mutants retain some catalytic activity both in vitro and in vivo (Edwards and Gill, 1999; Ohashi et al., 2000). Expression of the wild-type LIMK1 kinase domain efficiently activated SRF, and this was again substantially RhoA dependent (Fig. 3 C), although at high expression levels some C3-resistant SRF activation was observed (unpublished data; see Discussion). Overexpression of the LIMK1-T508V kinase domain also activated SRF, although substantially less efficiently than wild type; however, activation was still RhoA dependent (Fig. 3 C). Under transfection conditions in which expression levels of wild-type and T508V LIMK were adjusted to give similar levels of SRF activation, both proteins induced a similar degree of FLAG–cofilin phosphorylation, which was not impaired by coexpression of C3 transferase (Fig. 3 B, bottom). Taken together, these results show that to activate SRF in PC12 cells, LIMK requires a second Rho-dependent signal, which must regulate a process distinct from cofilin phosphorylation.
mDia is required for SRF activation in PC12 cells
The RhoA effector mDia promotes F-actin accumulation and is required for serum-induced SRF activation in NIH3T3 cells (Tominaga et al., 2000; unpublished data). We therefore investigated its role in signaling to SRF in PC12 cells. An activated form of mDia1, mDia1*, which lacks the RhoA-binding domain (Watanabe et al., 1999), efficiently activated SRF in PC12 cells (Fig. 4 A). In contrast to SRF activation by LIMK, activation of SRF by mDia1* was substantially unaffected by coexpression of C3 transferase, indicating that it did not require functional Rho (Fig. 4 B). Expression of activated mDia1 did not alter the proportion of phosphorylated FLAG–cofilin generated in the cofilin reporter assay (Fig. 4 C, top), indicating that it does not activate SRF by regulating LIMK activity. Thus, mDia provides an alternative route to the ROCK–LIMK pathway for signaling to SRF downstream of RhoA.
Figure 4.

mDia activity is required for SRF activation in PC12 cells. (A) mDia1 derivatives. The mDia1 coding region is indicated at the top with RhoA-binding domain as a circle and formin homology regions 1–3 as boxes; below are shown the activated mDia1 derivative Dia1* (residues 256–1255), the inactive interfering derivative dnDia1 (residues 571–1181, lacking 750–771), and the control inactive noninterfering derivative ΔDia1 (residues 256–567). (B) SRF activation by mDia1 in PC12 cells. Cells were transfected with the SRF reporter and expression plasmids encoding Dia1* (0.3 μg) with C3 transferase (50 ng), as indicated, and reporter activity was measured. The effect of C3 transferase on reporter activation by the synthetic activator SRF-VP16 (50 ng) is shown for comparison. Error bars show half range (n = 2). (C) mDia1 does not promote cofilin phosphorylation. Cells were transfected with expression plasmids encoding wild-type FLAG–cofilin (0.2 μg) alone or with Dia1* (0.3 μg), dnDia1 (2.0 μg), or LIMK1 (1.0 μg). After maintenance in 0.5% serum with serum stimulation as indicated, cells were analyzed for FLAG–cofilin phosphorylation by 2D gel electrophoresis followed by anti-FLAG immunoblot. Only the FLAG–cofilin species are shown; the positions of unphosphorylated and phosphorylated forms are indicated above the figure. (D) Serum-induced SRF activation requires functional mDia. Left, cells were transfected with the SRF reporter and an expression plasmid encoding dnDia1 (2.0 μg), as indicated, and either maintained in 0.5% serum or serum stimulated before measurement of reporter activity. Serum-stimulated reporter activity is taken as 100%. Error bars show SEM (n = 4). (E) Cofilin S3A potentiates the effect of interfering mDia. Cells were transfected with the SRF reporter together with expression plasmids encoding dnDia1 (2.0 μg) and/or cofilin S3A (0.5 or 2.0 μg), as indicated, and either was maintained in 0.5% serum or was serum stimulated before quantitation of reporter activity. Serum-induced reporter activity is taken as 100%. Error bars show half range (n = 2).
To evaluate the contribution of mDia proteins to serum-induced SRF activation, we used an inactivated mDia1 derivative that can interfere with SRF activation when overexpressed in NIH3T3 cells. This mutant, dnDia1, lacks the NH2-terminal RhoA-binding domain of mDia1 and sequences within the COOH-terminal domain (Fig. 4 A; unpublished data). In PC12 cells, dnDia1 expression did not activate SRF, but instead substantially inhibited SRF activation after serum stimulation (Figs. 4 D and 5 A). Importantly, expression of dnDia1 did not prevent the increase in FLAG–cofilin phosphorylation that occurred upon serum stimulation (Fig. 4 C, middle), indicating that its effect on SRF is specific and does not result from a nonspecific impairment of Rho signaling. Expression of dnDia1 was not toxic, as it affected neither the number of GFP-positive cells nor the thymidine kinase–renilla control reporter (unpublished data). Expression of cofilin S3A and dnDia1 inhibited reporter induction more effectively than dnDia1 alone (Fig. 4 E). Taken together, these data show that in addition to the ROCK–LIMK pathway, functional mDia is required for serum-induced SRF activation in these cells, and activation of SRF by mDia occurs independently of cofilin phosphorylation.
Functional mDia is required for SRF activation by LIMK
The data in the preceding sections show that RhoA-mediated activation of SRF in PC12 cells requires both LIMK and mDia signaling, and establish mDia as a candidate for the Rho-dependent function required by LIMK for SRF activation. We therefore tested whether dnDia1 can interfere with LIMK-induced SRF activity. Expression of dnDia1 substantially inhibited SRF activation by overexpressed LIMK1; in contrast, ΔDia1, an inactive mDia1 derivative that does not inhibit serum-induced SRF activation, did not inhibit LIMK1-induced SRF activation (Fig. 5 A). Expression of dnDia1 did not affect the ability of overexpressed LIMK1 to induce cofilin phosphorylation (Fig. 5 B). Thus, mDia is likely to represent the additional RhoA-dependent signal required for SRF activation by LIMK.
Figure 5.

mDia is required for LIMK1-induced SRF activation and F-actin assembly. (A) LIMK1-induced SRF activation requires mDia. PC12 cells were transfected with expression plasmids encoding LIMK1 (1.0 μg), inactive mDia derivative ΔDia1 (Dia1 256–567; 3.0 μg), or interfering mDia1 derivative dnDia1 (Figure 4 A; 3.0 μg), as indicated, together with the SRF reporter plasmid and maintained in 0.5% serum before quantitation of reporter activity, taking LIMK1+ΔDia1 as 100%. Error bars show SEM (n = 3). (B) dnDia1 does not affect LIMK1- induced cofilin phosphorylation. Cells were transfected with expression plasmids encoding wild-type FLAG–cofilin (0.2 μg) alone or with LIMK1 (1.0 μg) and dnDia1 (2.0 μg) as indicated. After maintenance in 0.5% serum, cells were analyzed for FLAG–cofilin phosphorylation by 2D gel electrophoresis and anti-FLAG immunoblot. Only the FLAG–cofilin species are shown; the positions of unphosphorylated and phosphorylated forms are indicated above the figure. (C) LIMK-induced F-actin accumulation requires mDia activity. PC12 cells were transfected with expression plasmids encoding LIMK1 (1.0 μg), inactive mDia derivative ΔDia1 (Dia1 256–567; 3.0 μg), or interfering mDia1 derivative dnDia1 (Figure 4 A; 3.0 μg). The mean F-actin content in transfected cells was determined using FACS® analysis and expressed relative to the mean F-actin content of untransfected cells from the same population, indicated by the dotted line (Materials and methods). The chart summarizes three independent experiments of this type, presenting the average of the mean F-actin content in each population ± SEM; statistical significance was estimated using the unpaired t test.
Because SRF activation is dependent on alterations in actin dynamics, these results suggest that the ability of LIMK to induce increases in F-actin will also be dependent on mDia activity. We therefore investigated whether dnDia1 can inhibit the ability of LIMK1 to increase cellular F-actin content. PC12 cells were transfected with expression plasmids encoding epitope-tagged LIMK1 and mDia1 derivatives, and FACS® analysis of FITC-phalloidin–stained cells was then used to quantify and compare the mean F-actin content in transfected and untransfected cell populations (Howard and Meyer, 1984; Bleul et al., 1996; Burger et al., 1999). We first assessed the ability of dnDia1 to affect the F-actin level. Expression of dnDia1 had no significant effect on mean F-actin content (Fig. 5 C). A similar result was obtained with a control mDia1 derivative, ΔDia1, which neither activates SRF nor interferes with serum-induced SRF activity in PC12 cells (Fig. 5 C; unpublished data). We then assessed the ability of these two mDia derivatives to affect LIMK1-induced F-actin assembly. Expression of LIMK1 led to an ∼60% increase in the F-actin level in the presence or absence of ΔDia1; (Fig. 5 C; unpublished data). In contrast, expression of dnDia1 together with LIMK1 led to a significant reduction in the ability of LIMK to increase F-actin content, which was increased to only 25% above untransfected cells (Fig. 5 C). Thus, functional mDia is required for LIMK1 both to promote F-actin assembly and activate SRF.
Discussion
ROCK-mediated activation of LIMK and phosphorylation of cofilin are required for serum-induced signaling to SRF in the PC12 neuronal cell line. In contrast, LIMK1 apparently does not play a significant role in SRF activation in NIH3T3 cells, even though SRF is regulated by actin dynamics in these cells (Sotiropoulos et al., 1999). However, both serum- and LIMK-induced SRF activation in PC12 cells require a second RhoA-controlled pathway involving the mDia proteins, which forms the main regulatory pathway to SRF in NIH3T3 cells (Tominaga et al., 2000; unpublished data). These results, summarized in Fig. 6, demonstrate functional cooperation between LIMK and mDia proteins, and strongly support our model in which SRF activation is brought about by changes in actin dynamics (Sotiropoulos et al., 1999). Actin assembly and turnover can also be regulated by other Rho family GTPases (Edwards et al., 1999; Higgs and Pollard, 2001), and our finding that Cdc42- and Rac1-induced signaling activates SRF efficiently in PC12 cells suggests that inputs from these GTPases into the actin treadmilling cycle may also play a role in SRF regulation in these cells (Fig. 6). Indeed, there is evidence that RhoA may signal to cofilin exclusively through LIMK2 and Rac through LIMK1, though there is still some uncertainty regarding this point (Ohashi et al., 2000; Sumi et al., 2001). However, our experiments do not address whether it is LIMK1 or LIMK2 that participates in Rho-dependent signaling to SRF in PC12 cells.
Figure 6.

Regulatory pathways to SRF via actin dynamics. The actin treadmilling cycle is shown schematically with actin monomer as a wedge, profilin as a rectangle, and cofilin as a circle. The treadmilling cycle is regulated by two Rho effector pathways. The Dia proteins promote F-actin accumulation via an uncharacterized mechanism, perhaps involving recruitment of profilin–actin to the Dia FH1 domain. The ROCK–LIMK–cofilin cascade negatively regulates cofilin activity, thereby increasing the F-actin level by inhibiting monomer dissociation and F-actin severing. Serum-induced SRF activation in PC12 cells is dependent on Rho signaling and actin polymerization, consistent with previous findings that SRF activity responds to depletion of the cellular G-actin pool or subpopulation of it (Sotiropoulos et al., 1999; unpublished data). Maximal serum-induced activity in PC12 cells requires operation of both ROCK–LIMK–cofilin and Dia pathways. The ability of LIMK both to activate SRF and to increase the F-actin level in PC12 cells is dependent on Dia proteins, suggesting that the activity of LIMK is targeted to a pool of Dia-dependent F-actin.
Previous studies have shown that Rho family GTPases use multiple pathways to control the accumulation of F-actin. For example, RhoA both promotes F-actin assembly via the mDia proteins (Watanabe et al., 1997, 1999; unpublished data) and inhibits its turnover via ROCK–LIMK signaling (Maekawa et al., 1999; Sumi et al., 1999). In contrast, Rac1 and Cdc42 induce F-actin nucleation via WASP/Scar family proteins and inhibit F-actin turnover through PAK–LIMK signaling (Edwards et al., 1999). Although it would appear reasonable to suppose that in both cases these pathways are somehow constrained to act on the same pool of F-actin, no direct evidence for such functional cooperation has been presented. Our finding that functional mDia is required for LIMK to induce accumulation of F-actin provides direct evidence for functional cooperation between the F-actin assembly and turnover pathways downstream of RhoA. A simple model consistent with this observation is that the primary target for LIMK action is a pool of F-actin whose accumulation is dependent on a basal level of RhoA signaling through mDia (Fig. 6). At high expression levels, however, both LIMK and especially its isolated kinase domain can activate SRF largely independently of RhoA (Sotiropoulos et al., 1999; unpublished data), perhaps because under such conditions LIMK becomes mislocalized and acts on RhoA-independent F-actin. Our results suggest that the previously observed cooperation between mDia and ROCK in stress fiber formation reflects both the F-actin accumulation and actin filament bundling pathways downstream of ROCK (Nakano et al., 1999; Watanabe et al., 1999; Tominaga et al., 2000).
Our studies indicate that the relative importance of different RhoA effector pathways for SRF activation differs between cell types. Although mDia function is required both in NIH3T3 fibroblasts and PC12 cells, ROCK and LIMK function plays a significant role only in PC12 cells. We speculate that ROCK–LIMK signaling to SRF may be a general feature of neuronal cell types. Functional ROCK is also required for maximal SRF activation in rat aortic smooth muscle cells, although in this case the roles of LIMK and mDia have not been addressed (Mack et al., 2001). At present, the molecular basis for the differential involvement of LIMK signaling in SRF activation remains unclear. We think it unlikely that it reflects cell type–specific expression of LIMK in the nervous system because LIMKs are detectable in many cell types. It is unlikely that LIMK is not effectively coupled to RhoA signaling in NIH3T3 cells, because serum-induced alterations in cofilin phosphorylation are readily detectable using the FLAG–cofilin assay (unpublished data). Similarly, the requirement for mDia activity in PC12 cells indicates both that this RhoA effector pathway must be functional in these cells and that it is likely providing a substrate upon which LIMK signaling can act. It may be that the different F-actin structures in PC12 cells are organized such that RhoA-dependent regulation is mediated predominantly by regulation of cofilin rather than mDia activity. Alternatively, it is possible that Diaphanous and LIMK signaling in NIH3T3 and PC12 cells reflects a difference between the treadmilling rates of RhoA-dependent F-actin in the two cell types: in cells with a low basal mDia activity and treadmilling rate, F-actin levels would be highly responsive to increased mDia activity, whereas in cells with relatively high basal mDia activity, F-actin might be effectively regulated via LIMK; this might apply in PC12 cells. Direct measurements of treadmilling rates in the two cell types and the development of reagents specific for the activated states of LIMK and mDia should also allow direct insights into these issues.
Materials and methods
Plasmids
LIMK1, N-WASP, and cofilin constructs are described elsewhere (Ishizaki et al., 1997; Sahai et al., 1998; Sotiropoulos et al., 1999). Diaphanous constructs encode mDia1 sequences as follows: Dia1*, residues 256–1255 (ΔN1+C; Watanabe et al., 1999); ΔDia1, 256–567; and dnDia1, 571–1181 (lacking 750–771) (unpublished data). The 3D.Aluc reporter plasmid contains the promoter sequences from 3D.ACAT (Sotiropoulos et al., 1999) inserted into pGL3 (Promega).
Cell culture, transfection, and gene expression assays
PC12 cells were maintained on collagen-coated plates in DME with 6% FCS and 6% horse serum and seed at 6 × 105 cells per 6-cm dish before transfection using FuGene reagent (Roche) according to the manufacturer's instructions. Each transfection mix (4 μg total plasmid DNA) included SRF reporter plasmid 3D.ALuc (firefly luciferase; 0.1 μg), reference standard pRLTK (thymidine kinase promoter controlling renilla luciferase; 0.4 μg), EF-eGFP (0.2 μg) to monitor transfection efficiency, and EFplink expression vector or derivatives encoding activator proteins or FLAG–cofilin (3.3 μg). For phosphorylation studies, cofilin expression plasmids were used at 0.2 μg per transfection. From control immunoblotting experiments with transfection efficiency estimated using GFP, we estimate that FLAG–cofilin was overexpressed by between two- and fivefold (unpublished data). Transfected cells were maintained in 0.5% serum for 18 h before stimulation with 15% serum; lysates were prepared 8 h later. For reporter experiments, activities were normalized to the activity of a parallel control transfection in which reporter activation was achieved using the constitutively active SRF derivative SRFVP16 (50 ng; Dalton and Treisman, 1992), taken as 100. Each reporter figure shows mean ± SEM for at least three independent transfection experiments. RNA analysis and transfection of mouse NIH3T3 cells were performed as previously described (Sotiropoulos et al., 1999); total RNA (10 μg) was analyzed using mouse GAPDH and vinculin probes with RNase T1 digestion (8 U per hybridization). Drug treatments were as follows: cytochalasin D (2 μM; Calbiochem); jasplakinolide (0.5 μM; Molecular Probes); pretreatments for 45 min with Y27632 (10 μM; gift from Yoshitomi Corp, Osaka, Japan) or latrunculin B (0.5 μM; Calbiochem).
2D. gel electrophoresis assay for cofilin phosphorylation
Cells were washed with ice cold phosphate-buffered saline and fixed with 10% TCA for 15 min on ice before scraping and collection. The acetone-washed cell pellet was solubilized with sonication in 2D sample buffer (8 M urea, 4% CHAPS, 40 mM Tris base, 0.5% IPG buffers, pH 3–10; Amersham Pharmacia Biotech), and cleared by centrifugation before loading Immobilon strips with a linear pH gradient from 3 to 10 (Amersham Pharmacia Biotech). First dimension electrophoresis was performed using the IPGphor isoelectric focusing system (Amersham Pharmacia Biotech); second dimension was by 18% SDS-PAGE. Immunoblotting was performed by standard procedures with detection by anti-FLAG antibody (M2; Sigma-Aldrich) for transfected cells expressing FLAG–cofilin or the anti-cofilin antibody (Cytoskeleton).
Phosphatase treatment
Cells were washed with ice cold PBS and lysed directly on the plate (6-cm dish) with 100 μl of SDS lysis buffer (1% SDS, 10 mM EDTA, 5 mM NaF, 10 mM KH2PO4, 50 mM Tris, pH 8.1, with protease inhibitors) for 10 min on ice and transferred to a test tube. The lysate was then diluted with 900 μl of IP dilution buffer (0.01% SDS, 1.1% triton, 1.2 mM EDTA, 167 mM NaCl, 5 mM NaF, 10 mM KH2PO4, 16.7 mM Tris, pH 8.0, with protease inhibitors) and clarified by centrifugation before immunoprecipitation using anti-FLAG beads (25 μl) for 2 h at 4°C. Beads were then washed twice using IP buffer and dispersed in 100 μl of phosphatase buffer (50 mM Tris-HCl, pH 8.5, 1 mM EDTA) containing 20 U of alkaline phosphatase (Boehringer); control beads were dispersed in 100 μl phosphatase buffer containing 5 mM NaF and 10 mM KH2PO4. Incubation was at 37°C for 1 h; beads were then collected and cofilin was eluted using 2D sample buffer. For phosphatase treatment of endogenous cofilin, lysates were prepared as above in IP buffer lacking NaF and KH2PO4 and treated directly with 40 U alkaline phosphatase for 1 h at 37°C. Proteins were recovered by TCA precipitation and processed for 2D gel analysis as above.
FACS® quantitation of F-actin
For analysis of F-actin levels, FACS® analysis was used to quantitate FITC-phalloidin staining in fixed cell populations (Howard and Meyer, 1984; Bleul et al., 1996; Burger et al., 1999). Five 6-cm plates of cells were transfected as above using 1 μg LIMK1 and 3 μg mDia construct; one plate was retained for a parallel luciferase assay. Trypsinized cells were washed sequentially with DME/1% horse serum and PBS, and then resuspended in 0.5 ml PBS. Cells were fixed by the addition of 0.5 ml 8% para-formaldehyde in PBS for 10 min at room temperature, washed in PBS, permeabilized in 1 mL of 0.3% Triton X-100 in PBS, and washed again before incubation with anti-myc 9E10 antibody in 5% FCS in PBS for 45 min at 37°C. After removal of the primary antibody, the cells were stained with a 1/500 dilution of Cy3-conjugated anti–mouse antibody (Jackson ImmunoResearch Laboratories) to identify transfected cells and 33 nM FITC-phalloidin to measure F-actin content in 5% FCS in PBS for 45 min at 37°C. Stained cells were sorted on a FACScan® machine (Becton Dickinson) and the phalloidin staining was quantified for transfected (Cy3 positive) and nontransfected (Cy3 negative) cells in each sample. Gates for Cy3 were established using a mock transfected control cell population. Mean F-actin content was evaluated and expressed as a percentage of that of untransfected cells from the same experiment.
Acknowledgments
We thank the following colleagues from the London Research Institute of Cancer Research UK for advice, technical assistance, and helpful comments on the manuscript: David Frith (2D gel facility); Cathy Simpson and Derek Davies (FACS® facility); and Caroline Hill, Gipi Schiavo, Nassia Sotiropoulos, Sharon Tooze, and Michael Way.
O. Geneste was supported by postdoctoral fellowships from the Human Frontier Science Program and the ICRF. J. Copeland was supported by a postdoctoral fellowship from the Hitchings-Elion Program of the Burroughs Wellcome Fund.
O. Geneste's present address is Division de Cancerologie, Institut de Recherches Servier, 125 Chemin de Ronde, 78290 Croissy sur Seine, France.
Cancer Research UK London Research Institute comprises the Lincoln's Inn Fields and Clare Hall Laboratories of the former Imperial Cancer Research Fund (ICRF) after the merger of the ICRF with the Cancer Research Campaign in February, 2002.
Footnotes
Abbreviations used in this paper: 2D, two dimensional; LIMK, LIM kinase; MLC, myosin light chain; N-WASP, neural Wiskott-Aldrich Syndrome protein; ROCK, Rho kinase; SRF, serum response factor.
References
- Alberts, A.S. 2001. Identification of a carboxy-terminal diaphanous-related formin homology protein autoregulatory domain. J. Biol. Chem. 276:2824–2830. [DOI] [PubMed] [Google Scholar]
- Amano, M., M. Ito, K. Kimura, Y. Fukata, K. Chihara, T. Nakano, Y. Matsuura, and K. Kaibuchi. 1996. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271:20246–20249. [DOI] [PubMed] [Google Scholar]
- Amano, M., Y. Fukata, and K. Kaibuchi. 2000. Regulation and functions of Rho-associated kinase. Exp. Cell Res. 261:44–51. [DOI] [PubMed] [Google Scholar]
- Arber, S., F.A. Barbayannis, H. Hanser, C. Schneider, C.A. Stanyon, O. Bernard, and P. Caroni. 1998. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 393:805–809. [DOI] [PubMed] [Google Scholar]
- Arsenian, S., B. Weinhold, M. Oelgeschlager, U. Ruther, and A. Nordheim. 1998. Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J. 17:6289–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, A.L., and A. Hall. 2000. Rho GTPases and their effector proteins. Biochem. J. 348:241–255. [PMC free article] [PubMed] [Google Scholar]
- Bleul, C.C., R.C. Fuhlbrigge, J.M. Casasnovas, A. Aiuti, and T.A. Springer. 1996. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J. Exp. Med. 184:1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger, J.A., M. Burger, and T.J. Kipps. 1999. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 94:3658–3667. [PubMed] [Google Scholar]
- Cheng, A.K., and E.J. Robertson. 1995. The murine LIM-kinase gene (limk) encodes a novel serine threonine kinase expressed predominantly in trophoblast giant cells and the developing nervous system. Mech. Dev. 52:187–197. [DOI] [PubMed] [Google Scholar]
- Dalton, S., and R. Treisman. 1992. Characterization of SAP-1, a protein recruited by serum response factor to the c-fos serum response element. Cell. 68:597–612. [DOI] [PubMed] [Google Scholar]
- Edwards, D.C., and G.N. Gill. 1999. Structural features of LIM kinase that control effects on the actin cytoskeleton. J. Biol. Chem. 274:11352–11361. [DOI] [PubMed] [Google Scholar]
- Edwards, D.C., L.C. Sanders, G.M. Bokoch, and G.N. Gill. 1999. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1:253–259. [DOI] [PubMed] [Google Scholar]
- Gineitis, D., and R. Treisman. 2001. Differential usage of signal transduction pathways defines two types of SRF target gene. J. Biol. Chem. 276:24531–24539. [DOI] [PubMed] [Google Scholar]
- Higgs, H.N., and T.D. Pollard. 2001. Regulation of actin filament network formation through the ARP2/3 complex: activation by a diverse array of proteins. Annu. Rev. Biochem. 70:649–676. [DOI] [PubMed] [Google Scholar]
- Hill, C.S., J. Wynne, and R. Treisman. 1995. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 81:1159–1170. [DOI] [PubMed] [Google Scholar]
- Howard, T.H., and W.H. Meyer. 1984. Chemotactic peptide modulation of actin assembly and locomotion in neutrophils. J. Cell Biol. 98:1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki, T., M. Naito, K. Fujisawa, M. Maekawa, N. Watanabe, Y. Saito, and S. Narumiya. 1997. p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett. 404:118–124. [DOI] [PubMed] [Google Scholar]
- Ishizaki, T., M. Uehata, I. Tamechika, J. Keel, K. Nonomura, M. Maekawa, and S. Narumiya. 2000. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol. Pharmacol. 57:976–983. [PubMed] [Google Scholar]
- Kaibuchi, K., S. Kuroda, and M. Amano. 1999. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu. Rev. Biochem. 68:459–486. [DOI] [PubMed] [Google Scholar]
- Kimura, K., M. Ito, M. Amano, K. Chihara, Y. Fukata, M. Nakafuku, B. Yamamori, J. Feng, T. Nakano, K. Okawa, et al. 1996. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 273:245–248. [DOI] [PubMed] [Google Scholar]
- Mack, C.P., A.V. Somlyo, M. Hautmann, A.P. Somlyo, and G.K. Owens. 2001. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J. Biol. Chem. 276:341–347. [DOI] [PubMed] [Google Scholar]
- Maekawa, M., T. Ishizaki, S. Boku, N. Watanabe, A. Fujita, A. Iwamatsu, T. Obinata, K. Ohashi, K. Mizuno, and S. Narumiya. 1999. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 285:895–898. [DOI] [PubMed] [Google Scholar]
- Nakano, K., K. Takaishi, A. Kodama, A. Mammoto, H. Shiozaki, M. Monden, and Y. Takai. 1999. Distinct actions and cooperative roles of ROCK and mDia in Rho small G protein-induced reorganization of the actin cytoskeleton in Madin-Darby canine kidney cells. Mol. Biol. Cell. 10:2481–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi, K., K. Nagata, M. Maekawa, T. Ishizaki, S. Narumiya, and K. Mizuno. 2000. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J. Biol. Chem. 275:3577–3582. [DOI] [PubMed] [Google Scholar]
- Oude Weernink, P.A., P. Schulte, Y. Guo, J. Wetzel, M. Amano, K. Kaibuchi, S. Haverland, M. Voss, M. Schmidt, G.W. Mayr, and K.H. Jakobs. 2000. Stimulation of phosphatidylinositol-4-phosphate 5-kinase by Rho-kinase. J. Biol. Chem. 275:10168–10174. [DOI] [PubMed] [Google Scholar]
- Proschel, C., M.J. Blouin, N.J. Gutowski, R. Ludwig, and M. Noble. 1995. Limk1 is predominantly expressed in neural tissues and phosphorylates serine, threonine and tyrosine residues in vitro. Oncogene. 11:1271–1281. [PubMed] [Google Scholar]
- Sahai, E., A.S. Alberts, and R. Treisman. 1998. RhoA effector mutants reveal distinct effector pathways for cytoskeletal reorganization, SRF activation, and transformation. EMBO J. 17:1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai, E., T. Ishizaki, S. Narumiya, and R. Treisman. 1999. Transformation mediated by RhoA requires activity of ROCK kinases. Curr. Biol. 9:136–145. [DOI] [PubMed] [Google Scholar]
- Sechi, A.S., and J. Wehland. 2000. The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P(2) influences cytoskeletal protein activity at the plasma membrane. J. Cell Sci. 113:3685–3695. [DOI] [PubMed] [Google Scholar]
- Shore, P., and A.D. Sharrocks. 1995. The MADS-box family of transcription factors. Eur. J. Biochem. 229:1–13. [DOI] [PubMed] [Google Scholar]
- Sotiropoulos, A., D. Gineitis, J. Copeland, and R. Treisman. 1999. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell. 98:159–169. [DOI] [PubMed] [Google Scholar]
- Sumi, T., K. Matsumoto, Y. Takai, and T. Nakamura. 1999. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 147:1519–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi, T., K. Matsumoto, and T. Nakamura. 2001. Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J. Biol. Chem. 276:670–676. [DOI] [PubMed] [Google Scholar]
- Tominaga, T., E. Sahai, P. Chardin, F. McCormick, S.A. Courtneidge, and A.S. Alberts. 2000. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell. 5:13–25. [DOI] [PubMed] [Google Scholar]
- Uehata, M., T. Ishizaki, H. Satoh, T. Ono, T. Kawahara, T. Morishita, H. Tamakawa, K. Yamagami, J. Inui, M. Maekawa, and S. Narumiya. 1997. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 389:990–994. [DOI] [PubMed] [Google Scholar]
- Van Aelst, L., and C. D'Souza-Schorey. 1997. Rho GTPases and signaling networks. Genes Dev. 11:2295–2322. [DOI] [PubMed] [Google Scholar]
- Watanabe, N., P. Madaule, T. Reid, T. Ishizaki, G. Watanabe, A. Kakizuka, Y. Saito, K. Nakao, B.M. Jockusch, and S. Narumiya. 1997. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 16:3044–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, N., T. Kato, A. Fujita, T. Ishizaki, and S. Narumiya. 1999. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat. Cell Biol. 1:136–143. [DOI] [PubMed] [Google Scholar]
- Yang, N., O. Higuchi, K. Ohashi, K. Nagata, A. Wada, K. Kangawa, E. Nishida, and K. Mizuno. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 393:809–812. [DOI] [PubMed] [Google Scholar]