

Abstract
T he organization of transcriptionally active ribosomal genes in animal cell nucleoli is investigated in this study in order to address the long-standing controversy with regard to the intranucleolar localization of these genes. Detailed analyses of HeLa cell nucleoli include direct localization of ribosomal genes by in situ hybridization and their indirect localization via nascent ribosomal transcript mappings. On the light microscopy (LM) level, ribosomal genes map in 10–40 fluorescence foci per nucleus, and transcription activity is associated with most foci. We demonstrate that each nucleolar focus observed by LM corresponds, on the EM level, to an individual fibrillar center (FC) and surrounding dense fibrillar components (DFCs). The EM data identify the DFC as the nucleolar subcompartment in which rRNA synthesis takes place, consistent with detection of rDNA within the DFC. The highly sensitive method for mapping nascent transcripts in permeabilized cells on ultrastructural level provides intense and unambiguous clustered immunogold signal over the DFC, whereas very little to no label is detected over the FC. This signal is strongly indicative of nascent “Christmas trees” of rRNA associated with individual rDNA genes, sampled on the surface of thin sections. Stereological analysis of the clustered transcription signal further suggests that these Christmas trees may be contorted in space and exhibit a DNA compaction ratio on the order of 4–5.5.
Keywords: cell nucleolus; rRNA genes; ribosomal RNA; immunohistochemistry; in situ hybridization
Introduction
Approximately 400 copies of ribosomal genes are present in the human diploid genome; they are organized in the form of head-to-tail tandem repeats at well-described positions within five pairs of chromosomes (30–50 repeats per chromosome; Hadjiolov, 1985). Transcriptionally active ribosomal genes of eukaryotic organisms are accommodated in a special nuclear organelle termed the nucleolus, which also represents the site at which the assembly and maturation of preribosomal particles occur (e.g., Scheer and Benavente, 1990). Three basic structural compartments are commonly distinguished within the nucleolar architecture: fibrillar centers (FCs),* dense fibrillar components (DFCs), and granular components (Hadjiolov, 1985).
The molecular organization of ribosomal transcripts in the form of so-called “Christmas trees” was described on spreads of nuclear contents from amphibian oocytes more than 30 years ago (Miller and Beatty, 1969). Similar structures were also reported later in other species (e.g., Trendelenburg et al., 1974; Puvion-Dutilleul et al., 1977). Surprisingly, a description of the morphological equivalent of Christmas trees in in situ systems only recently appeared. Christmas trees in a condensed form were observed in somatic cells of Pisum sativum (Gonzalez-Melendi et al., 2001). In animal cells, similar structures have only been observed in specialized nucleoli of insect cell oocytes (Scheer et al., 1997).
In addition, a schism exists in the nucleolar literature regarding in which nucleolar compartment the rRNA synthesis takes place in somatic animal cells. On one hand, rRNA transcription sites were reported to be localized to the interior of the FC, whereas other authors found them in the DFC, together with the DFC/FC border zone (e.g., Granboulan and Granboulan, 1965; Scheer and Benavente, 1990; Raska et al., 1995; Cmarko et al., 2000; Thiry et al., 2000). The discrepancies in published results can, at least to a large extent, be explained by a highly compact structure of the nucleolus, particularly of DFC (Stanek et al., 2001). Consequently, in situ hybridization (ISH) and immunocytochemical approaches may fail, and different kinds of cell treatment that provide a more accessible structure (Koberna et al., 1999) may provide clearer signal and less equivocal results.
Keeping the compact nucleolar structure in mind, the problem of in situ organization of active ribosomal genes in human HeLa cells has been addressed using improved methods to label newly synthesized RNA within nucleolar structure at the ultrastructural level, and to compare this with light microscopic analysis. Using a combination of light microscopy (LM) and EM approaches, we show that the pattern of the transcription signal seen in ultrathin nucleolar sections corresponds to marked labeling of the DFC, and further provides evidence of individual compacted Christmas trees detected at high-resolution within the DFC.
Results and discussion
The rDNA coding sequences are localized within multiple fluorescence foci
The efficiency and pattern of the fluorescence ISH rDNA signal strongly depended on the fixation/permeabilization treatment used (unpublished data). In this respect, the ISH efficiency was highest after methanol/acetone treatment. Although this treatment resulted in apparent ultrastructural changes, these structure alterations were below the resolution limit of LM (Stanek et al., 2001).
At the LM resolution, the ISH signal consisted of 10–40 fluorescence foci per cell nucleus (Fig. 1, A–C). Most of this signal was confined to the nucleoli. Few rDNA foci were found next to the nucleolus, probably in the perinucleolar condensed chromatin. Rare isolated rDNA foci, which were distant from the nucleoli, were also visualized. We identified these foci with so-called micronucleoli (Smetana and Busch, 1974).
Figure 1.
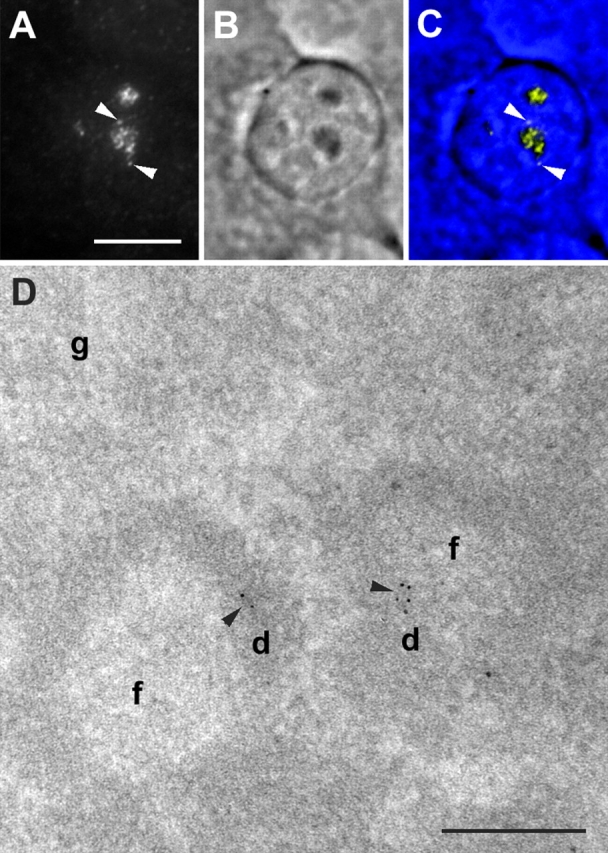
Localization of ribosomal genes in HeLa cells. (A–C) Wide-field LM mapping of ribosomal genes. Fluorescence mapping of ribosomal genes (A and C, yellow), phase contrast image (B and C, blue), and the merge image (C) are shown. Ribosomal genes are located in a number of fluorescent foci. Most of the foci are situated in the nucleolus. Two foci are situated in the perinucleolar chromatin (arrowheads). (D) EM mapping of ribosomal genes. The signal (arrowheads) is located in DFC. (f, d and g) FCs, DFCs, and granular components, respectively, and the same designation is used in other EM images. Bars: (A–C) 10 μm; (D) 200 nm.
Next, we mapped the ribosomal genes at the EM level, by a postembedding labeling procedure. In contrast to preembedding, it enabled the convenient preservation of the ultrastructure. The sensitivity of the postembedding EM ISH was much lower than the preembedding labeling, as most target sequences were hidden in a volume of the section and the ribosomal genes sampled to the surface of thin sections could be revealed at best. However, the sectioning procedure itself could help in exposing a long enough rDNA sequence at the section surface (Raska et al., 1995). In agreement with this previous study in which a different ISH approach with the sense probes to 18-S rDNA was used, the ISH signal in aldehyde-fixed and thin-sectioned nucleoli of HeLa cells consisted only of rare clusters of gold particles. In 25 images tested, 42 gold clusters were detected; only 10 of them were located outside the DFC region, and consequently, ∼75% of ISH signal mapped into DFC (Fig. 1 D).
ISH approach was not able to differentiate between transcriptionally active and inactive genes. Thus, the bromouridine (BrU) labeling of growing transcripts, reporting the active genes position exclusively, was performed next.
Most nucleolar rDNA foci exhibit transcriptional activity
We mapped the nucleolar transcription by means of BrU, which was incorporated during 5 min into newly synthesized transcripts both in hypotonically treated cells that incorporated BrU in vivo and in permeabilized cells (Fig. 2, A and B). After this time interval, the majority of the signal corresponded in living cells to nascent precursor rRNA (Stanek et al., 2001). The lower rate of the transcription in permeabilized cells incorporating BrU was reported (Jackson et al., 1993), such that in permeabilized cells, the large majority of labeled transcripts had to correspond to nascent transcripts. In both cases, the LM mapping of the nucleolar RNA synthesis provided a pattern consisting of 10–40 fluorescence foci per nucleus. However, some spatially close foci appeared to fuse due to the limited resolution of LM (see below). This phenomenon was also observed in the case of rDNA foci mappings. Both in cells that underwent the hypotonic shift and in permeabilized cells, the nucleolar, but not extranucleolar transcription, as distinguished by phase-contrast microscopy, was abolished by the low concentration of actinomycin D. At the same time, the nucleolar signal was not sensitive to α-amanitin treatment in concentrations sufficient to inhibit RNA polymerases II and III, but not RNA polymerase I (Stanek et al., 2001; unpublished data). Thus, we concluded that the BrU incorporated into the nucleolar RNA corresponded to the synthesis of the rRNA precursors.
Figure 2.

LM mapping of nucleolar transcription in HeLa cells. (A and B) Wide-field images of nucleolar transcription pattern seen in both hypotonically treated (A) and permeabilized (B) cells. The pattern consists of numerous fluorescent foci. Note that the foci in A are more blurred with respect to the foci in B. (C–E) Light microscopic image of ribosomal genes (C and E, red) with respect to the position of nucleolar transcription sites (D and E, green). The merged image is in E. One confocal section after image restoration is shown. Most of rDNA foci exhibit the transcription activity. An rDNA focus without transcription signal is indicated by the arrow. Bars: (B) 10 μm; (E) 5 μm.
The nucleolar signal in permeabilized cells was more prominent with regard to the extranucleolar signal if compared with cells, which underwent the hypotonic shift. We ascribed this phenomenon in permeabilized cells to the fact that, in contrast to the nucleoplasm, the nucleolar structure was loosened/extracted only to a limited extent. In addition, the fluorescence foci in living cells were more blurred than in permeabilized cells (Fig. 2, A and B). This difference was still more pronounced after the prolongation of incorporation time (unpublished data). Results observed in the living cells were compatible with previously reported rapid changes in localization of BrU labeled RNA (Stanek et al., 2001). In permeabilized cells, a prolongation of the incubation time in the transcription buffer led to an increase of signal intensity without influencing the focal pattern of the fluorescence.
Next, we performed the simultaneous mapping of nucleolar rDNA sequences and transcription (Fig. 2, C and D). The observed fluorescence patterns were compatible with the above single labeling experiments. The whole transcription signal within the nucleolar body was colocalized with the rDNA foci, and only a minor part of the nucleolar rDNA foci did not colocalize with the transcription signal. Therefore, in the frame of the resolution of LM, most nucleolar rDNA foci were identified with transcriptionally active foci. We considered the minor cases of nucleolar rDNA foci not exhibiting the transcription activity as pointing to the detection of (transcriptionally) silent rDNA sequences, probably replicating rDNA (unpublished data). The rDNA foci observed outside the nucleolar body did not contain a transcription signal.
Intense ultrastructural transcription signal in the form of clustered gold particles in nucleoli of permeabilized cells points to the presence of Christmas trees in DFC
Next, we performed EM mapping of transcription. In cells, which underwent the hypotonic shift, the ultrastructural details of all three nucleolar compartments were preserved in an excellent way (Fig. 3 A). The signal was observed in DFC and at the FC/DFC border after 5 min of incorporation of BrU, whereas FC was devoid of signal. The clustering of gold particles was recognized frequently.
Figure 3.
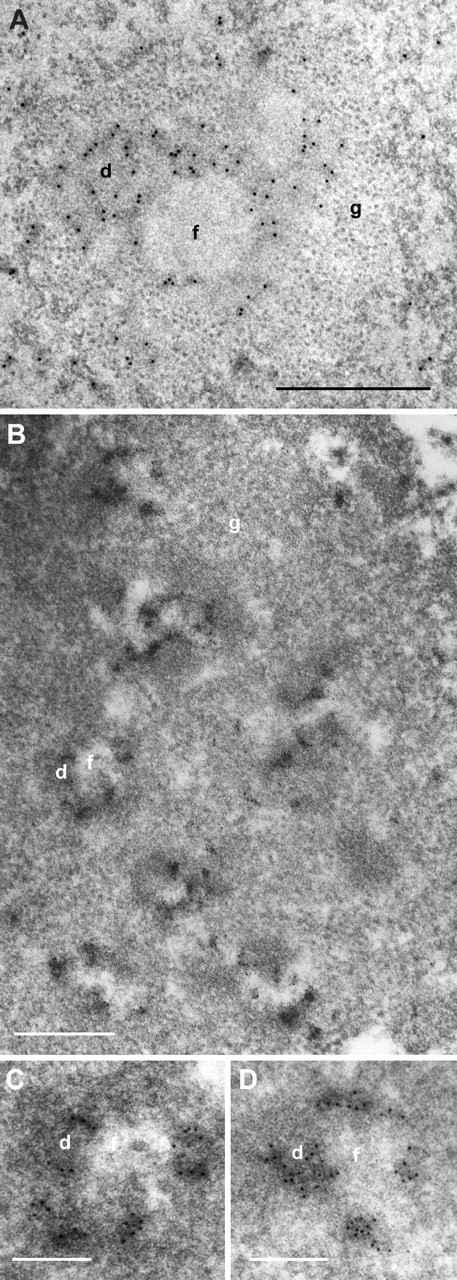
EM mapping of nucleolar transcription in HeLa cells. (A) Nucleolar EM transcription signal observed in hypotonically treated cells is confined to the DFC. (B–D) EM localization of transcription in thin-sectioned permeabilized cell. (C and D) Detailed parts of B. Nucleolar transcription signal consists of numerous clusters of gold particles accumulated in the DFC. (A) Bars: 0.2 μm; (B–D) 0.5 μm.
In permeabilized cells, the ultrastructure of nucleoli was, in contrast to highly extracted nucleoplasm, well preserved, and all the nucleolar compartments could be readily identified. The transcription signal was also confined to DFC and the FC/DFC border after 5 min of BrU incorporation. However, the signal consisted entirely of clusters of up to several tens of gold particles confined to subdomains of DFC (Fig. 3, B–D). The clusters were frequently elongated with a maximum length of 200 nm and a mean width 45 nm. Multiple clusters of gold particles were regularly detected in DFC surrounding individual FC, and even tens of such clusters could be observed in a large nucleolar section encompassing several FC profiles. The pattern of the transcription signal was, to a large extent, insensitive to the prolongation of BrUTP incorporation (unpublished data). If incorporation time was shortened to 1 min, the signal was lowered progressively. Individual gold particles or clusters of a few gold particles were observed in only in some EM profiles in the DFC and DFC/FC border (unpublished data).
The clear-cut and intense EM signal of newly synthesized rRNA sampled to the surface of individual thin sections (Fig. 3) was much higher than the signal revealed by ribosomal genes ISH. Long enough sequences had to be available for successful ISH, and only a very small fraction of them was exposed to the section surface during the postembedding detection. In contrast, there had to be ∼100 nascent rRNA chains present in each rRNA gene (Jackson et al., 1998), with many incorporated BrUs in each transcript, so that thousands of brome epitopes had to be present in nascent rRNA synthesized from one gene. This allowed us to suggest that the very high transcription signal in the form of clustered gold particles in nucleoli of permeabilized cells reflected the presence of Christmas trees in the DFC. Our conclusion is in harmony with results of Gonzalez-Melendi et al. (2001) that identified the clustered bromouridine signal with the condensed Christmas trees in plant cells.
Nucleolar BrU focus observed by LM corresponds to individual FC and surrounding DFC on the electron microscopic level
A simultaneous LM and EM mapping of rDNA transcription was performed next in order to correlate the rDNA transcription pattern revealed by fluorescence and EM. The number of FCs was identical to the number of fluorescence foci or was, due to the resolution limit of LM, slightly higher. The superposition of concomitant LM and EM images of the same nucleolar section identified each fluorescence focus with the individual FC (or two or more closely spaced FCs) together with the surrounding DFC labeled by gold particles at the EM level (Fig. 4). The concomitant LM and EM mapping of transcription unambiguously showed that LM alone did not resolve FC from DFC. Within the resolution limit of the LM, individual FC, together with the surrounding rim of DFC, was in most cases visualized as a single focus.
Figure 4.

Concomitant LM and EM mapping of transcription signal in a thin section of permeabilized cell. (B) Detailed part of A. The colored insert in A represents the wide-field LM picture of the same area depicting newly synthesized RNA (red) and DNA counterstained with YOYO (green). (C) Merge of an inverse EM image from A in cyan, and a transcription LM signal from the upper insert in red. Note the overlap of the LM and EM transcription signals. Interestingly, because of the consecutive EM and LM labeling performed, an electron-dense stain due to antibodies accumulated at the surface of thin section can be recognized. Bars: (A) 0.5 μm; (B) 0.2 μm.
Based on a serial sectioning, a three-dimensional view of the arrangement of focal transcription sites in the nucleolus is shown in Fig. 5. Regarding the arrangement of active ribosomal genes, the pattern of foci testified to the presence of structural nucleolar units, consisting of the individual FC together with the surrounding DFC.
Figure 5.
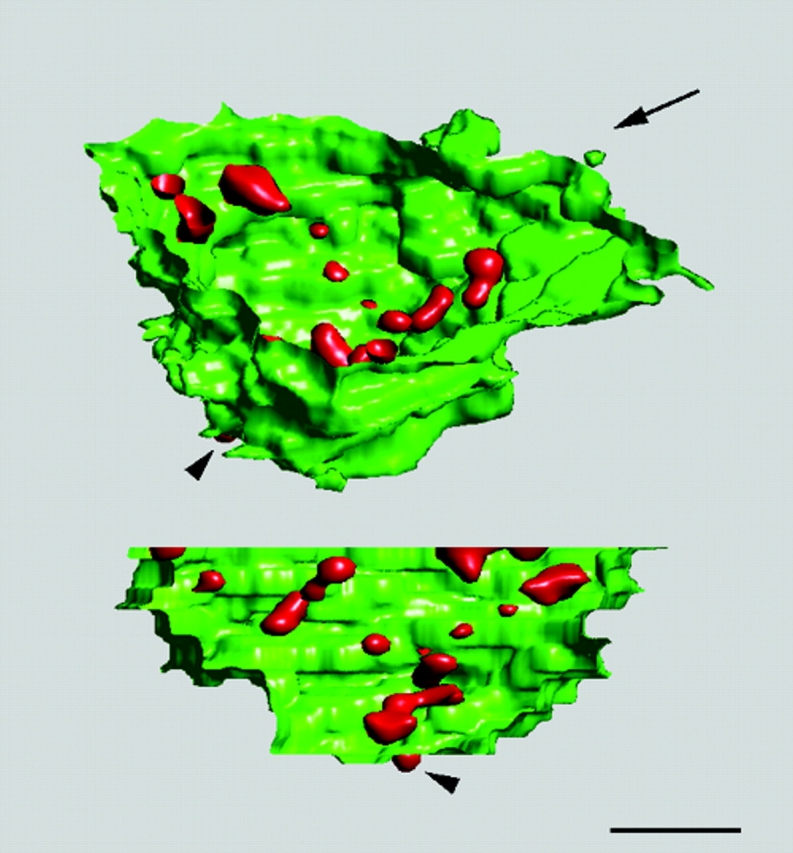
Three-dimensional view of transcription sites within nucleolus of a HeLa cell. 26 thin sections of a serially sectioned permeabilized cell were used for the three-dimensional reconstruction of the confocal fluorescence transcription signal (red) within the border of nucleolus together with perinucleolar condensed chromatin (YOYO-1 staining, green). Two different views of the reconstructed image are presented. Direction of the view presented in the lower part of the image is indicated by the arrow. Two-dimensional image restoration was applied on the transcription signal. Bar, 1 μm.
Ribosomal transcription units are compacted and contorted within DFC
The clear visualization of clusters of transcription signals allows us to make some approximations as to the compaction of individual rDNA transcription units. The in situ length of the ribosomal transcribed unit was shown to be ∼300 nm in a pea (Pisum sativum; Gonzalez-Melendi et al., 2001) and >400 nm in a grasshopper (Locusta migratoria; Scheer et al., 1997). These lengths corresponded to 6.6- and 7-kb sequences of DNA, respectively. The transcription unit size in human cells is 13 kb (Hadjiolov, 1985). As the length of clusters of gold particles observed in nucleoli of permeabilized HeLa cells did not exceed 200 nm, the transcription units may be highly contorted in space. In order to estimate the length of the transcription unit, a stereological approach was used. 549 clusters were detected in 200 random nuclear sections. The total volume occupied by newly synthesized rRNAs was 0.035% of the nuclear volume. The mean nuclear volume as assessed by LM was 730 ± 80 μm3. Keeping in mind that there were 150 transcriptionally active ribosomal genes in the HeLa cell nucleus (Jackson et al., 1993), we computed a volume of BrU labeled parts of the Christmas tree. Approximating the shape of the labeled part of the Christmas tree as a cone, we could roughly estimate its length to be ∼800 nm. In other words, the packing ratio of transcribing ribosomal genes was ∼5.5. This number was ∼4 when the cylindrical shape of the observed clusters was assumed instead. These numbers should be understood as lower limits of the legitimate value, as we cannot exclude that some transcriptional markers were not detected, particularly at the 5′ end of the transcription unit.
The presence of a transcription signal in the DFC both in living cells and in permeabilized cells, and the identification of a transcription signal in permeabilized cells with Christmas trees enabled us to contribute to the debate about the position of the active ribosomal genes within the nucleolus of somatic animal cells. Our results are not in agreement with findings of Thiry et al. (2000) who in cultured HeLa cells situated the bulk of the rDNA transcription into the interior of FC. Our results are compatible with the model presented by Cook (1999), in which the transcriptionally active rDNAs are aligned along the border between FC and DFC. However, in contrast to the model proposed (Cook, 1999), we consider the process of transcription to generate such an environment for the rDNA that the transcriptionally active part of the gene becomes, in morphological terms, engulfed in the DFC. Despite of the differences in the organization of the animal and plant nucleolus (Gonzalez-Melendi et al., 2001), the scheme of the DFC as an environment of the active ribosomal genes seems to be common both in plant and animal somatic cells.
The number of transcriptionally active ribosomal genes in the HeLa cell nucleus several times exceeded the number of FCs or transcription foci, respectively. Therefore, there had to be several active rDNAs present in each focus. From our data, the average number fell between 4 and 15 per focus. On the basis of our results, identifying each fluorescent focus of transcriptionally active ribosomal genes with an individual fibrillar center together with its surrounding the DFC (Fig. 4), the focus was considered to serve as the structural unit in the organization of active ribosomal genes. This concept is supported by numerous observations that testified to the presence in FC and DFC of various factors involved in the transcription of ribosomal genes, particularly of RNA polymerase I (Scheer and Benavente, 1990; Raska et al., 1995). We emphasize in this respect that the discrimination between actively transcribing polymerases from the whole pool of RNA polymerase I molecules could not be established by the mapping experiments up to now.
Materials and methods
Cell culture and drugs treatment
HeLa cells were cultured as described previously (Koberna et al., 1999). In all experiments, we limited ourselves to cells showing in phase contrast regular nucleoli, i.e., to truly interphase cells. We also avoided multinuclear cells as well as cells with large nuclei possessing apparently highly elevated genome copies, which were occasionally seen in the culture.
The inhibition of RNA polymerase I transcription or extranucleolar transcription was achieved by the addition of actinomycin D or α-amanitin, respectively (Raska et al., 1995; Koberna et al., 1999).
Incorporation of BrU into nascent RNA
Two different approaches for the labeling of rDNA transcripts with BrU were used: the hypotonic shift enabling the incorporation of BrU in vivo was performed as described previously (Koberna et al., 1999); for permeabilized cells, the protocol of Raska et al. (1995) was followed with these modifications: 0.03% Triton X-100 was used for cell permeabilization and the BrU incorporation was performed in a buffer containing 15% glycerol at 35°C.
In situ hybridization of rDNA
The DNA probe was prepared using an Enzo DNA labeling kit from a pA plasmid construct (Erickson et al., 1981), provided by Jo Sylvester (Nemours Children's Clinic Research, Orlando, FL). This construct involved a spanning sequence for downstream ∼200 nucleotides of 18 S rRNA, internal transcribed spacer 1, sequence for 5.8 S rRNA, internal transcribed spacer 2 and upstream ∼4,500 nucleotides of 28 S rRNA. The probe was labeled with digoxygenin-11-dUTP by nick translation.
The cells growing on coverslips were washed in cold PBS and fixed/permeabilized in methanol at −20°C for 30 min, rinsed in acetone for 20 s, and air dried. Before their denaturation, the cells were incubated with 100 μg/ml RNAse A (Roche) for 2 h at 37°C, gradually dehydrated in ice-cold 30, 50, 70, 96, and 100% ethanol, and air-dried. The denaturation of the cells was performed in 70% deionized formamide/2 × SSC, pH 7.0, at 75°C for 2–3 min. The cells were then placed in ice-cold 70% ethanol, dehydrated, and air dried. The probe was denatured in deionized formamide at 75°C for 8 min and placed on ice. 6 μl of a hybridization mix containing 25 ng of probe, 0.5 mg/ml sonicated salmon sperm DNA, 2 mg/ml Escherichia coli tRNA, 50% deionized formamide, 2 × SSC, 0.2% BSA, and 10% dextran sulfate, was used per each coverslip. The ISH was performed overnight at 37°C in a humidified chamber. After the posthybridization washes, coverslips were incubated with 200 U/ml of RNAase H (Sigma-Aldrich) at 37°C for 2 h for the elimination of possible RNA–DNA duplexes. The combined detection of BrU labeled transcripts and ISH was performed as described previously (Haaf and Ward, 1996). After immunolabeling, the cells were postfixed overnight with 3:1 mixture of methanol:acetic acid at −20°C, and then proceeded to hybridization as described above.
LM
Permeabilized cells were fixed either in 2% formaldehyde in PBS for 10 min after transcription, or fixed/permeabilized in methanol at −20°C for 30 min, rinsed in acetone for 20 s, and air dried. Before immunolabeling, the cells were washed in PBS and incubated for 10 min with 0.5% BSA in order to block nonspecific binding. Subsequently, the cells were incubated with rat anti-BrU antibody (Harlan SeraLab) and donkey anti–rat antibody conjugated with Cy3 fluorochrome (Jackson ImmunoResearch Laboratories) for 60 min at room temperature. The DNA probe was visualized by a combination of mouse anti-digoxygenin (Roche) and goat anti–mouse FITC-conjugated antibodies (Jackson ImmunoResearch Laboratories).
Specimens were embedded in a ProLong antifade kit (Molecular Probes) and viewed using an AX70 Provis (Olympus) microscope equipped with a charge-coupled device camera (PXL with KAF 1400 chip; Photometrics) or TCS NT (Leica) confocal microscope. In the case of confocal imaging, three-dimensional sets of optical sections 120 nm apart were collected and restored using the maximum likelihood estimation algorithm (Huygens 2; Scientific Volume Imaging BV). In order to estimate the nuclear volume, the cells were counterstained with YOYO-1 and a series of confocal sections were collected and analyzed using AnalySIS software (Soft Imaging Systems).
EM and LM on ultrathin sections
The postembedding rDNA ISH was performed as described previously (Wachtler et al., 1992) on lowicryl sections. BrU-labeled RNA was detected on lowicryl sections according to Raska et al. (1995).
6 or 12 nm gold anti–mouse adduct and goat anti–mouse antibody conjugated with Cy3 fluorochrome were used as secondary antibodies for EM and LM respectively (Jackson ImmunoResearch Laboratories). Sections for LM were counterstained with YOYO-1 (Molecular Probes). It should be mentioned here that we were unable to standardize the relevant EM double labeling mapping with sufficient intensities of both ISH and transcription signals.
For the simultaneous visualization of BrU at the LM and EM levels, the sections on the EM grid were incubated with mouse anti-BrU antibody, and followed by incubation with gold adduct. Then antibody with fluorochrome was applied. Sections were stained with YOYO-1 and observed by AX70 Provis (Olympus) microscope. After observation, the sections were stained with uranyl acetate and viewed using an EM 900 (Zeiss) electron microscope equipped with a KeenView CCD camera (Soft Imaging Systems). Fluorescence BrU incorporation pattern was restored using the maximum likelihood estimation algorithm (Huygens 2, Scientific Volume Imaging BV) constrained to two-dimensional images. In the fluorescence imaging, thin sections represented the confocal sections themselves. This method allowed us to reach the axial resolution of ∼70 nm (an average thickness of sections).
The quantitative evaluation of the ultrastructural rRNA signal was performed using the stereological approach (Gundersen et al., 1988). Each Christmas tree was estimated to occupy a conical volume of the base radius equal to the main width of detected clusters of gold particles. In the case of cylindrical approximation, the main width represented a diameter of the cylinder.
Acknowledgments
The authors would like to thank Professor J. Sylvester for the rDNA plasmids, and Jackson ImmunoResearch Laboratories for the donation of most of their reagents. Due to limited space, they apologize to all those whose work was not directly cited.
Footnotes
Abbreviations used in this paper: BrU, bromouridine; DFC, dense fibrillar component; FC, fibrillar center; ISH, in situ hybridization; LM, light microscopy.
References
- Cmarko, D., P.J. Verschure, L.I. Rothblum, D. Hernandez-Verdun, F. Amalric, R. van Driel, and S. Fakan. 2000. Ultrastructural analysis of nucleolar transcription in cells microinjected with 5-bromo-UTP. Histochem. Cell Biol. 113:181–187. [DOI] [PubMed] [Google Scholar]
- Cook, P.R. 1999. The organization of replication and transcription. Science. 284:1790–1795. [DOI] [PubMed] [Google Scholar]
- Erickson, J.M., C.L. Rushford, D.J. Dorney, G.N. Wilson, and R.D. Schmickel. 1981. Structure and variation of human ribosomal DNA: molecular analysis of cloned fragments. Gene. 16:1–9. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Melendi, P., B. Wells, A.F. Beven, and P.J. Shaw. 2001. Single ribosomal transcription units are linear, compacted Christmas trees in plant nucleoli. Plant J. 27:223–233. [DOI] [PubMed] [Google Scholar]
- Granboulan, N., and P. Granboulan. 1965. Cytochimie ultrastructurale du nucléole II. Etude des sites de synthese du RNA dans le nucléole et le noyau. Exp. Cell Res. 38:604–619. [DOI] [PubMed] [Google Scholar]
- Gundersen, H.J., P. Bagger, T.F. Bendtsen, S.M. Evans, L. Korbo, N. Marcussen, A. Moller, K. Nielsen, J.R. Nyengaard, B. Pakkenberg, et al. 1988. The new stereological tools: dissector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis. APMIS. 96:857–881. [DOI] [PubMed] [Google Scholar]
- Haaf, T., and D.C. Ward. 1996. Inhibition of RNA polymerase II transcription causes chromatin decondensation, loss of nucleolar structure, and dispersion of chromosomal domains. Exp. Cell Res. 224:163–173. [DOI] [PubMed] [Google Scholar]
- Hadjiolov, A.A. 1985. The nucleolus and ribosome biogenesis. Cell Biology Monographs. A.M. Beermann, L. Goldstein, K.R. Portrer, and P. Sitte, editors. New York. 1–263.
- Jackson, D.A., A.B. Hassan, R.J. Errington, and P.R. Cook. 1993. Visualization of focal sites of transcription within human nuclei. EMBO J. 12:1059–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, D.A., F.J. Iborra, E.M. Mers, and P.R. Cook. 1998. Numbers and organization of RNA polymerases, nascent transcripts, transcription units in HeLa nuclei. Mol. Biol. Cell. 9:1523–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koberna, K., D. Stanek, J. Malínskya, M. Eltsov, A. Pliss, V. Ctrnacta, S. Cermanová, and I. Raska. 1999. Nuclear organization studied with the help of a hypotonic shift: its use permits hydrophilic molecules to enter into living cells. Chromosoma. 108:325–335. [DOI] [PubMed] [Google Scholar]
- Miller, O.L., and B.R. Beatty. 1969. Visualization of nucleolar genes. Science. 164:955–957. [DOI] [PubMed] [Google Scholar]
- Puvion-Dutilleul, F., J.P. Bachellerie, A. Bernadac, and J.P. Zalta. 1977. Transcription complexes in subnuclear fractions isolated from mammalian cells: ultrastructural study. C.R. Acad. Sci. Hebd. Seances Acad. Sci. D. 284:663–666. [PubMed] [Google Scholar]
- Raska, I., M. Dundr, K. Koberna, I. Melcák, M.C. Risueno, and I. Török. 1995. Does the synthesis of ribosomal RNA take place within nucleolar fibrillar centers or dense fibrillar components? J. Struct. Biol. 114:1–22. [DOI] [PubMed] [Google Scholar]
- Scheer, U., and R. Benavente. 1990. Functional and dynamic aspects of the mammalian nucleolus. Bioessays. 12:14–21. [DOI] [PubMed] [Google Scholar]
- Scheer, U., B. Xia, H. Merkert, and D. Weisenberger. 1997. Looking at Christmas trees in the nucleolus. Chromosoma. 105:470–480. [DOI] [PubMed] [Google Scholar]
- Smetana, K., and H. Busch. 1974. The nucleolus and nucleolar DNA. The Cell Nucleus. H. Busch, editor. Academic Press, New York. 173–147.
- Stanek, D., K. Koberna, A. Pliss, J. Malínsky, M. Masata, J. Vecerová, M.C. Risueño, and I. Raska. 2001. Non-isotopic mapping of ribosomal RNA synthesis and processing in the nucleolus. Chromosoma. 110:460–470. [DOI] [PubMed] [Google Scholar]
- Thiry, M., T. Cheutin, M.F. O'Donohue, H. Kaplan, and D. Ploton. 2000. Dynamics and three-dimensional localization of ribosomal RNA within the nucleolus. RNA. 6:1750–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg, M.F., H. Spring, U. Scheer, and W.W. Franke. 1974. Morphology of nucleolar cistrons in a plant cell, Acetabularia mediterranea. Proc. Natl. Acad. Sci. USA. 71:3626–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachtler, F., C. Schöfer, W. Mosgöller, K. Weipoltshammer, H.G. Schwarzacher, M. Guichaoua, M. Hartung, A. Stahl, J.L. Berge-Lefranc, I. Gonzalez, et al. 1992. Human ribosomal RNA gene repeats are localized in the dense fibrillar component of nucleoli: light and electron microscopic in situ hybridization in human Sertoli cells. Exp. Cell Res. 198:135–143. [DOI] [PubMed] [Google Scholar]