

Abstract
Toxoplasma gondii is an obligatory intracellular parasite, an important human pathogen, and a convenient laboratory model for many other human and veterinary pathogens in the phylum Apicomplexa, such as Plasmodium, Eimeria, and Cryptosporidia. 22 subpellicular microtubules form a scaffold that defines the cell shape of T. gondii. Its cytoskeleton also includes an intricate apical structure consisting of the conoid, two intraconoid microtubules, and two polar rings. The conoid is a 380-nm diameter motile organelle, consisting of fibers wound into a spiral like a compressed spring. FRAP analysis of transgenic T. gondii expressing YFP-α-tubulin reveals that the conoid fibers are assembled by rapid incorporation of tubulin subunits during early, but not late, stages of cell division. Electron microscopic analysis shows that in the mature conoid, tubulin is arranged into a novel polymer form that is quite different from typical microtubules.
Keywords: Apicomplexan parasites; microtubules; motility; cytoskeleton; cryoelectron microscopy
Introduction
Toxoplasma gondii is an obligate intracellular parasite and an important human pathogen, causing a chronic infection affecting ∼30% of the U.S. population. It is a common opportunistic pathogen in AIDS patients, where reactivation of encysted latent parasites leads to potentially fatal uncontrolled proliferation (Luft et al., 1993). T. gondii is also a convenient laboratory model for many other human and veterinary pathogens in the phylum Apicomplexa, including Plasmodium, Eimeria, and Cryptosporidia (Roos et al., 1999, 2000). The cytoskeleton of T. gondii includes 22 subpellicular microtubules (Nichols and Chiappino, 1987) that, together with a set of flattened vesicles underlying the plasma membrane (the inner membrane complex) (Cintra and de Souza, 1985) and a network of filamentous proteins (IMC1 and IMC2; Morrissette et al., 1997; Mann and Beckers, 2001), form a scaffold that defines the cell shape. T. gondii also displays an intricate apical structure from which the phylum Apicomplexa takes its name, consisting of the conoid, two intraconoid microtubules, and two polar rings. The conoid is a truncated cone, 280 nm in length and 380 nm in diameter. Whereas parasites are inside a host cell, the conoid remains enclosed within the shell formed by the subpellicular microtubules. However, when the parasites are swimming extracellularly, the conoid intermittently protrudes beyond the apical end of the microtubules. Protrusion of the conoid is sensitive to parasite cytoplasmic calcium concentration, and can be induced by calcium ionophore treatment (Mondragon and Frixione, 1996; Pezzella et al., 1997; Stommel et al., 1997). EM studies have shown that the conoid consists of fibers wound into a spiral like a compressed spring (de Souza, 1974; Nichols and Chiappino, 1987; Morrissette et al., 1997). Based on their width, it has been thought that the conoid fibers might be microtubules, but several observations argue against this view: tubulin antibodies normally fail to label the conoid (Schwartzman et al., 1985); electron microscopic studies of the spiral elements revealed neither tubulin protofilaments nor a circular cross-section (Nichols and Chiappino, 1987); and normal microtubules are believed to be too rigid to form a stable structure with a radius of curvature <200 nm (Amos and Amos, 1991b; Gittes et al., 1993).
Many different polymeric arrangements of tubulin occur in vivo, and an even larger number can be induced to form in vitro (Dustin, 1984; Murray, 1991). All the various arrangements presumably share a relatively small set of bonding patterns between subunits, thus accounting for the shared symmetry elements apparent in their architecture. These bonding preferences lead to straight chains of tubulin dimers associated end to end (protofilaments), which in turn are associated side by side into curved sheets of parallel columns. In cross-section, the profile of the various structures built from these curved sheets is a circle or an arc of a circle, as expected if all the protofilament–protofilament interactions are identical. Profiles lacking circular symmetry have not been described.
Here we report that the conoid fibers of T. gondii are assembled by a rapid incorporation of tubulin subunits during early but not late stages of cell division, and that in the mature conoid, tubulin is arranged into a novel polymer form that is quite different from typical microtubules.
Results
YFP-α-tubulin labels all the tubulin-containing structures in T. gondii
We have previously described transgenic T. gondii expressing the fluorescent protein YFP fused to the N terminus of α-tubulin, as shown in Fig. 1 (Striepen et al., 2000). All of the known tubulin containing structures are labeled in these parasites, including subpellicular microtubules, centrioles, and spindles. To our surprise, YFP-α-tubulin also brightly labels the apical end of the parasite in the region of the conoid. This region contains several structures believed to be important for host cell invasion, including rhoptries and micronemes (membrane-bound secretory organelles) (Nichols and Chiappino, 1987; Dubremetz et al., 1989; Carruthers and Sibley, 1997), two polar rings and two short microtubules, and the conoid, a motile organelle of intriguing structure but unknown function (Scholtyseck et al., 1970; de Souza, 1974; Russell and Burns, 1984; Nichols and Chiappino, 1987; Dubremetz et al., 1989; Carruthers and Sibley, 1997) (Fig. 1, B–E). Using deconvolution microscopy and quantitative fluorescence measurements, we determined precisely the amount of YFP fluorescence per unit microtubule length (Swedlow et al., 2002), and applying these calculations to the intensely labeled apical spot indicated a tubulin content equivalent to 5.5 μm of microtubule. The two intraconoid microtubules are only 0.35 μm long (see below), much too small to account for the apical YFP-α-tubulin by themselves.
Figure 1.
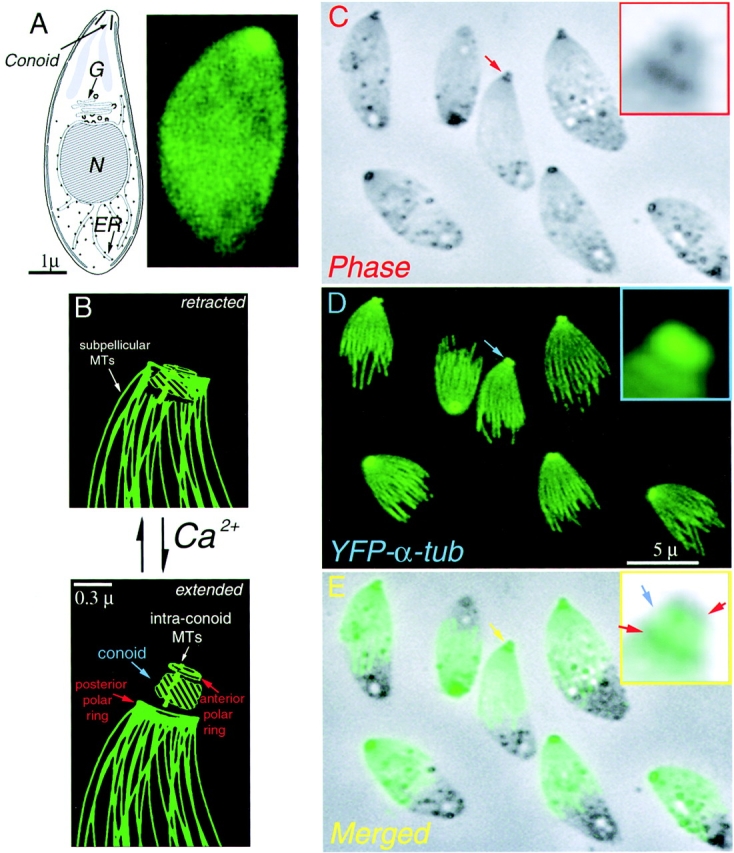
Drawing and LM images of Toxoplasma gondii. (A) (left) Drawing of T. gondii. showing the nucleus (N), Golgi (G), endoplasm reticulum (ER), and conoid. (right) Fluorescence image of living YFP-α-tubulin transgenic T. gondii. Intact cells appear uniformly green due to cytoplasmic YFP-α-tubulin. In extracted parasites (C–E), individual microtubules are visible. (B) Cartoon of conoid movement, modified from (Morrissette, 1995). In extracellular parasites, the conoid alternates between the retracted and extended states. Increase in cytoplasmic [Ca2+] induces extension. (C) Phase contrast image of A23187 treated YFP-α-tubulin transgenic T. gondii, some having extended conoids (red arrow). Two phase dark regions in the extended conoid (inset) correspond to the upper and lower polar rings (see B). (D) The brightly fluorescent apical end of YFP-α-tubulin transgenic parasites (blue arrow) protrudes together with the conoid (C, red arrow). (E) Merged phase contrast and fluorescence images. The apical spot of YFP fluorescence colocalizes with the phase light region (inset, blue arrow).
The normal location of the conoid is at the apical end of the parasite, surrounded by the lower polar ring (Fig. 1 B). During host cell invasion, or after treatments that raise the internal [Ca2+], the upper polar ring and conoid are extended until the base of the conoid protrudes beyond the lower polar ring (Mondragon and Frixione, 1996) (Fig 1, C–E). In extracellular parasites, the apical complex appears as a small phase dark spot (Fig. 1 C), and this dark spot coincides with the apical fluorescent spot observed in YFP-α-tubulin transgenic parasites. When parasites are treated with the calcium ionophore A23187, the apical fluorescent spot protrudes together with the phase dark apical complex (Fig. 1, C–E). Some substructure can be observed in the apical complex after detergent extraction of ionophore treated parasites: the upper and lower polar rings remain phase dark, whereas the conoid region (located between the two polar rings) emerges as a phase-bright region. In YFP-α-tubulin transgenics, the bright apical spot of YFP fluorescence lies between the phase dark polar rings (Fig. 1 E). These observations strongly suggest that the bright apical spot of fluorescence arises from the conoid itself, rather than from other structures in the apical complex, consistent with the conclusion drawn from the quantitative measurements of YFP fluorescence cited above (Swedlow et al., 2002).
The presence of tubulin in the apical complex was confirmed by indirect immunofluorescence with monoclonal antibodies against α- and β-tubulin, as shown in Fig. 2. Anti-tubulin antibodies label only the subpellicular microtubules in cells treated as for normal immunocytochemical labeling, as previously reported (Schwartzman et al., 1985). However, more vigorous detergent treatment, or treatment with detergent plus high salt extraction, permitted labeling of the conoid. Anti-tubulin strongly labels both the 22 subpellicular microtubules and the apical end of the parasites, colocalizing with the structures containing YFP (Fig. 2, A–C). Western blot analysis of whole parasite extracts demonstrates YFP antibody reactivity with full-length YFP-α-tubulin only. No other form of YFP is present in detectable amount (Fig. 2 D).
Figure 2.
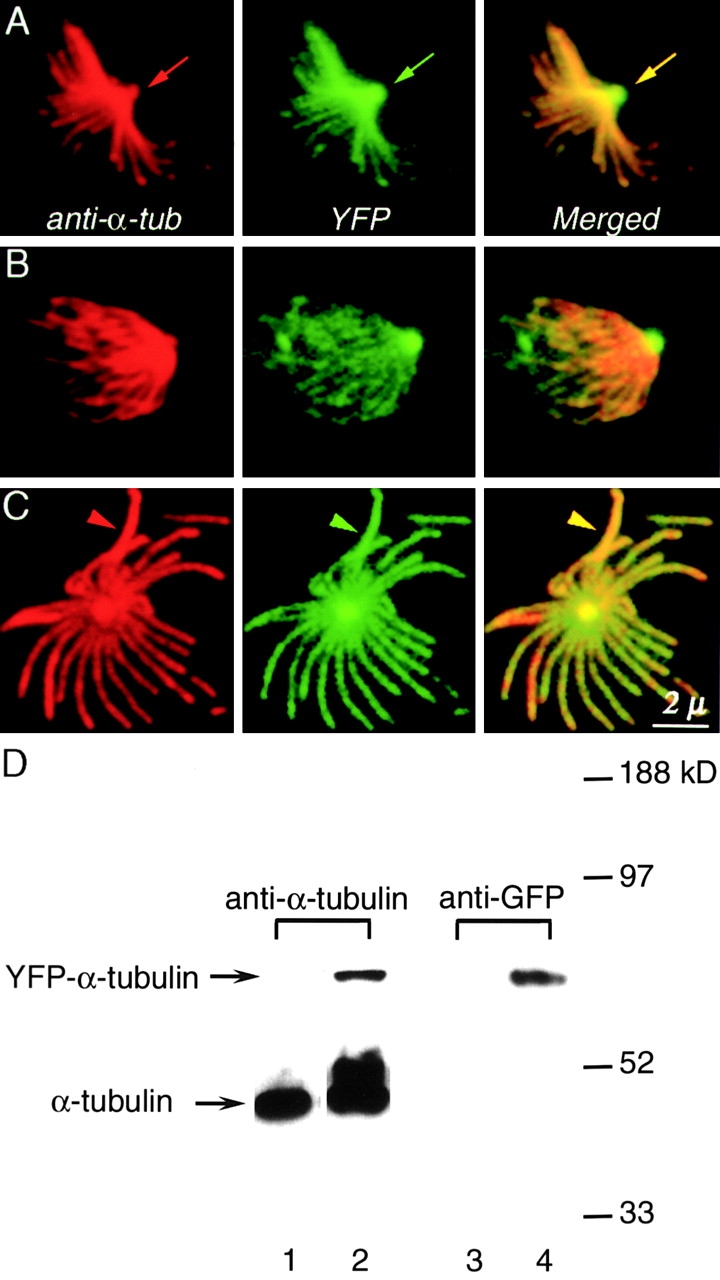
Colocalization of YFP and anti-tubulin antibody in transgenic T. gondii. (A–C) (left column, red) Anti–α-tubulin staining of deoxycholate extracted YFP-α-tubulin transgenic T. gondii. (middle column, green) YFP fluorescence. (right column) Merged YFP and immunofluorescence images. YFP-α-tubulin fluorescence colocalizes with antibody labeling both in the apical region (A, arrows) and in the subpellicular microtubules (C, arrows). (D) Western blot analysis of whole cell extracts of wild type RH parasites (lane 1 and 3) and YFP-α-tubulin transgenic parasites (lanes 2 and 4). Lanes 1 and 2 were probed with anti–α-tubulin antibody. Lanes 3 and 4 were probed with anti-GFP antibody. Lanes 2 and 4 were loaded with approximately twice as many parasites as lane 1 in order to show the YFP-α-tubulin band clearly.
Tubulin is rapidly incorporated into the conoid early in the cell cycle, but not later
FRAP experiments revealed several different patterns of dynamic behavior in the various tubulin-containing structures of T. gondii. Bleached subpellicular microtubules of mature parasites do not recover, as shown in Fig. 3 A. The bleached regions of subpellicular microtubules in daughter cells also remain dark, but are extended to produce fluorescent “tails ” (Fig. 3 A). We conclude that fluorescent YFP-α-tubulin subunits are continuously added to the growing ends of the subpellicular microtubules during daughter cell assembly. During later stages of cell division (Fig. 3 B), when the subpellicular microtubules are no longer growing, the bleached subpellicular microtubules remain dark even after fluorescent tubulin has reappeared in the cytoplasm.
Figure 3.
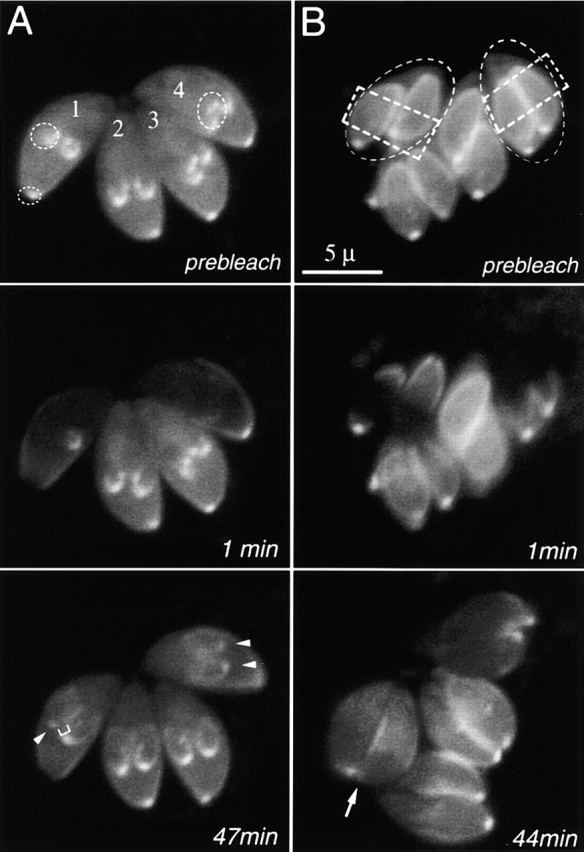
FRAP analysis of YFP-α-tubulin flux in living T. gondii. (A) (prebleach) A vacuole with four parasites at an early stage of cell division. The microtubule scaffolds of daughter cells have begun forming but are still small. Dotted circles, targets for photobleaching. (1 min postbleach) One daughter and the apical region of parasite #1, and both daughters of parasite #4 have been photobleached. (47 min postbleach) The maternal apical region shows no recovery, but the apical spots of the bleached daughters have reappeared (white arrow heads). The bleached region of the daughter's scaffold has not recovered, marked by the darker region between the recovered conoid and the elongating subpellicular microtubules (white bracket). (B) (prebleach) A vacuole with four parasites (two of which are outlined by the dashed ellipses), each containing two daughters nearing completion of cell division. Dotted rectangles, targets for photobleaching. (44 min postbleach) There is very little recovery in the daughter apical spot (white arrow). The bright spot next to the arrow is the apical spot of the adjacent daughter, which was not bleached. The bleached subpellicular microtubules remain dark, and no new fluorescent tail is observed, indicating that elongation of the subpellicular microtubules had ceased before photobleaching occurred. Individual microtubules are not resolvable in these images, due to the poor signal/noise ratio in images from this Zeiss LSM510 confocal (Swedlow et al., 2002).
The apical fluorescent spots within daughter cells recover after photobleaching, but at different rates, depending on the cell cycle. Daughter apices bleached during the early stages of cell division show obvious recovery, averaging 16% of the fluorescence of unbleached controls at 20 min postphotobleaching, and reaching a maximum of 34% (SEM 8%) at 40–45 min. (Fig. 3 A, arrowheads). In contrast, daughter apical complexes bleached during the late stages of cell division showed barely detectable recovery (average 6% after 45 min; Fig 3 B, arrow). No recovery of the bleached mother cell apical complex was observed. These observations imply that the apical tubulin-containing structure is assembled only during the very early stages of cell division, but is relatively static (very low exchange or incorporation rate) after it becomes mature at late stages of cell division.
The conoid fibers are composed of tubulin
The apical elements under study are ∼0.3 μm long by 0.4 μm wide, too small to be well resolved by light microscopy. Electron microscopic images of thin sections of the apical region (Fig. 4) reveal several distinctive organelles, including the conoid, polar rings, rhoptries, and intraconoid microtubules (Scholtyseck et al., 1970; de Souza, 1974; Russell and Burns, 1984; Nichols and Chiappino, 1987; Dubremetz et al., 1989; Carruthers and Sibley, 1997). The conoid is a truncated cone ∼280 nm long (Fig. 4 A). Transverse thin sections through the apical end of parasites treated with tannic acid show the conoid as a 380-nm diameter ring of electron dense material (Fig. 4 C). The conoid fibers give rise to a set of spiral markings modulating the density of this ring. Outside the conoid, subpellicular microtubules emanate from the lower polar ring (postulated to be a microtubule-organizing center; Russell and Burns, 1984) spiraling outward and towards the posterior end of the parasite (Fig. 4 C, arrows). Inside the conoid, the two intraconoid microtubules are clearly visible, exhibiting canonical microtubule structure (a circle of 13 protofilaments). In the image shown, these two microtubules are sandwiched between two rhoptries, membrane-bounded secretory organelles believed to be involved in host-cell invasion. Glancing sections sometimes include a single wall of the conoid, in which the highly curved conoid fibers are visible over a short distance (Fig. 4 B). Faint longitudinal striations can be seen running along the fibers.
Figure 4.
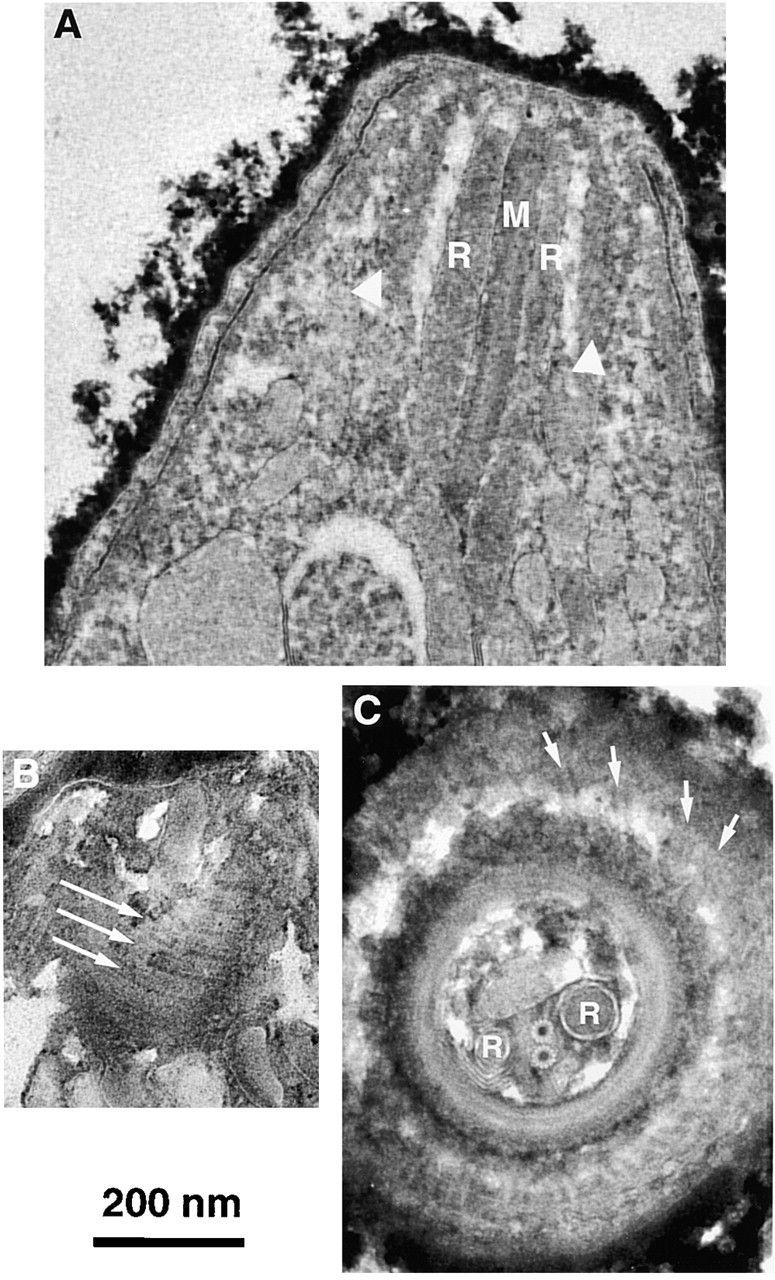
EM images of sections through the apical end of T. gondii with conoid retracted. (A) Sagittal section showing intraconoid microtubules (M), rhoptries (R) and walls of the conoid (white arrowheads). (B) A parasagittal section that grazed the wall of the conoid, revealing several of the spirally wound conoid fibers (long arrows). Faint longitudinal striations along each fiber can be seen. (C) Cross section of the apical end. The conoid appears as a circle of electron dense material with spiral striations. Just outside the conoid is a portion of the lower polar ring, to which the subpellicular microtubule (arrows) attach. Inside the conoid lie two rhoptries (R) positioned on either side of the two intraconoid microtubules.
The conoid is seen most clearly in its extended state, induced by treating extracellular parasites with the calcium ionophore A23187 (Mondragon and Frixione, 1996). Detergent-extracted parasite cytoskeletons were deposited on a holey carbon film and negatively stained, as shown in Fig 5. The conoid is marked by a prominent basket-weave pattern, created by superimposition of the conoid fibers on the front and back sides of the organelle (Fig. 5 A). Quantitative analysis suggests 14 fibers ∼430 nm in length, but the difficulty of counting the superimposed fibers introduces an uncertainty of ±1. Each fiber completes ∼1/2 turn around the conoid as it spirals from apex to base. Longitudinal striations with a spacing of 5 nm can sometimes be seen along the length of each fiber. Interestingly, the pitch angle of the fibers changes with the state of extension of the conoid. In extended conoids, the fibers intersect the polar ring at angles of 35–50°, whereas in retracted conoids the pitch is usually shallower (25–35°).
Figure 5.
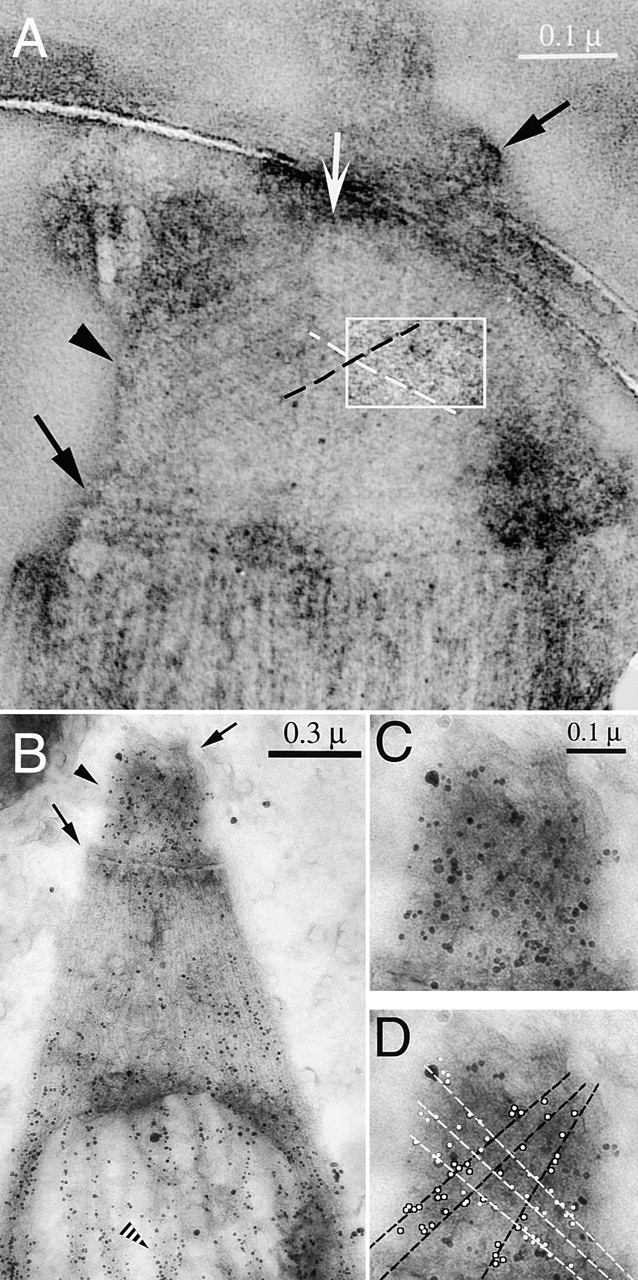
Immunogold labeling of T. gondii using anti-tubulin antibodies. (A) EM image of a negatively stained conoid of T. gondii, suspended over a hole in a carbon film. White arrow, intra-conoid microtubules; black arrows, upper and lower polar rings; black arrowhead, conoid region. Each conoid fiber is outlined by a dark line of electron-dense stain deposited in the groove between the fibers. The region within the white rectangle has been filtered and displayed with enhanced contrast to increase the visibility of the fibers and their longitudinal striations. (B) Immunogold labeling of T. gondii using a mixture of anti–α-tubulin and anti–β-tubulin antibodies. Black arrows and arrowheads as in A; striped arrowhead, a subpellicular microtubule, heavily decorated with gold particles. (C and D). Enlarged view of the conoid region from B. Most gold particles lie along conoid fibers, as indicated by the black and white lines in D.
ImmunoEM was carried out to determine which structures within the apical region contain tubulin. Although secondary antibodies labeled with fluorophores intensely stain anti-tubulin in both the conoid region and on subpellicular microtubules (as shown in Fig. 2), secondary antibodies coupled to conventional gold particles penetrated the conoid poorly and labeled only the subpellicular microtubules. However, secondary antibody coupled to very small (1.5 nm) gold particles gave modest labeling of the conoid in parasites extracted with deoxycholate, and this labeling was dramatically improved by using secondary antibodies conjugated with ultrasmall gold particles (< = 0.8 nm) followed by silver enhancement (Lah et al., 1990) to make the gold particles visible (Fig. 5, B and C). Many of the silver-enhanced gold particles align with the diagonal stripes of the conoid fibers (Fig. 5 D), suggesting that these fibers are assembled from tubulin.
Tubulin in the conoid fibers is assembled into protofilaments
To further characterize these tubulin containing fibers, Toxoplasma was examined by electron microscopy using specimens prepared in several different ways, including negative staining of detergent extracted cytoskeletons with phosphotungstic acid, cryoelectron microscopy of unfixed, unstained, frozen-hydrated cytoskeletons, and longitudinal and cross-sections of fixed, embedded parasites (both intact and detergent extracted).
In thin sagittal sections, the walls of the conoid appear as two vertical bands of electron-dense material ∼280 nm long and 35 nm wide (Figs. 4 A and 6, A and B). The conoid fibers are embedded in this dense matrix, and in appropriately oriented regions they can be seen in cross section. If the plane of section is parallel to the axis of the conoid, the conoid fibers are cut obliquely as in Fig. 4 A. Tilting the section in the microscope brings the conoid fibers on one side of the conoid into an end-on view, whereas on the other side they are brought into a side view (Fig. 6, A and B). If the direction of tilt is such that the top of the conoid moves away from the viewer, then the conoid fibers on the left-hand side of the conoid become parallel to the direction of view (Fig. 6 A), whereas the right hand fibers become perpendicular to the view direction. Tilting in the opposite direction (Fig. 6 B, top towards viewer) reverses the appearance of the left and right sides. These observations establish that the conoid fibers follow a left-handed helical path.
Figure 6.
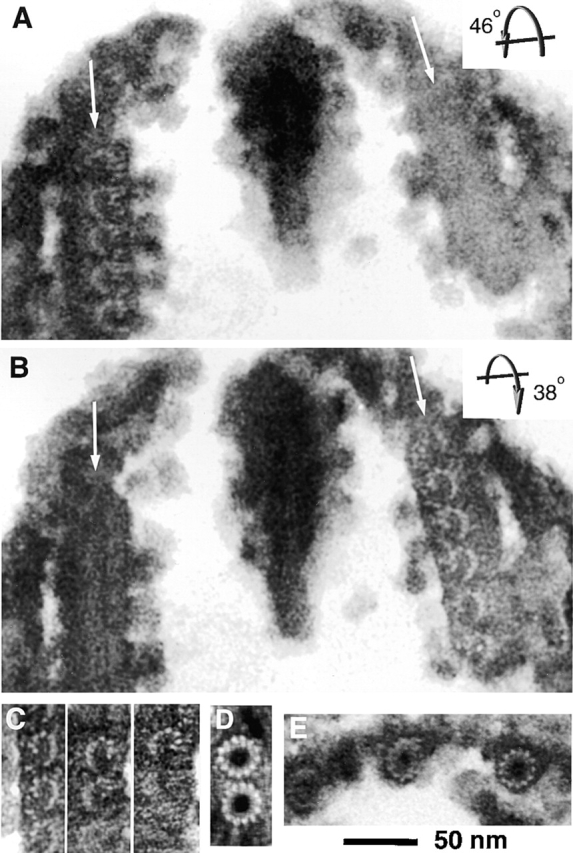
Cross sections of conoid fibers, intraconoid microtubules, and subpellicular microtubules. (A) EM image of a thin sagittal section of the conoid in detergent extracted T. gondii treated with tannic acid. Arrows indicate walls of the conoid. The section was tilted 46° (top away from observer), which oriented the conoid fibers on the left-hand side parallel to the direction of view. The comma shape and protofilament substructure of the fibers are revealed. (B) The same section as in A, titled 38° in the opposite direction (top toward the observer). The fibers on the right are now seen end on, revealing the same arrangement of protofilaments as in A. These two tilted views establish that the conoid fibers follow a left-handed helical path. (C) Montage of images showing the protofilament arrangement in tilted views of sections from three other conoids. (D) End-on view of intraconoid microtubules, showing the canonical circular profile with 13 protofilaments. (E) Subpellicular microtubules, viewed end on, again revealing a circular profile and 13 protofilaments.
We used the tannic acid staining protocol (Mizuhira and Futaesaku, 1971; Futaesaku et al., 1972; Tilney et al., 1973) to make the protofilaments in the conoid fibers visible. The end-on view of the conoid fibers shows clear protofilament structure, indicating that tubulin subunits are arranged in columns parallel to the fiber axis, just as in microtubules (Fig. 6, A–C). However, unlike microtubules, the protofilaments are not arranged as a closed tube, and instead form ribbons of nine protofilaments folded into a comma shape. The concave side of the comma always faces the interior of the conoid, and the tail of the comma always points toward the conoid base. This profile is reliably observed (on both sides of the conoid) in any fiber when the angle and axis of tilt are set appropriately, and this is the only cross-sectional appearance that we have seen for these fibers (Fig. 6 C), in both extended and retracted conoids. Even when slight misorientation of the fibers blurs the individual protofilaments (as is often the case in the more highly curved fibers of retracted conoids), the comma-shaped profile is still apparent. The profile of these fibers differs radically from the profiles of intraconoid and subpellicular microtubules in the same sections (Fig. 6, D and E, respectively).
Rotationally and translationally aligned cross-sectional profiles of a number of conoid fibers were superimposed to obtain an average (Fig 7). In the best individual cross-sections, and in the average, several features can be seen that distinguish these fibers from typical singlet or doublet microtubules. Most obviously, the protofilaments of conoid fibers are not wrapped into a closed tube with circular cross-section. Nor do these protofilaments lie along an arc of a circle, i.e., these protofilaments assemble into a nonsymmetric structure, they are not simply incomplete microtubules missing a small sector. In addition to the nine protofilaments that are clearly outlined in negative contrast by the tannic acid staining protocol (Fig. 7 B, black ellipses), additional small structures with lower contrast are consistently seen (Fig. 7 B, gray “arms”). These are likely to represent microtubule-associated proteins (MAPs)* (or motor molecules) attached to the tubulin protofilaments, as suggested by Fourier analysis of images of frozen hydrated specimens (discussed below). We note that the sum of 14 fibers, 430 nm long, with nine protofilaments, plus the two 350-nm intraconoid microtubules closely matches the estimated tubulin content of the apical region (Swedlow et al., 2002).
Figure 7.

Comparison of conoid fibers, singlet, and doublet microtubules. (A) Three copies of an average formed by superimposing the rotationally and translationally aligned images of multiple conoid fibers. The three copies are positioned as adjacent fibers in the wall of the conoid. (B) Interpretive drawing to identify probable protofilaments (dark ellipses) and associated proteins (elongated gray blobs) in A. (C) EM images of cross sections of singlet (upper) and doublet (lower) microtubules, for comparison with B.
Longitudinal periodicities in T. gondii microtubules and in conoid fibers
Having tentatively established the radial and azimuthal distribution of tubulin in the conoid fibers, we sought to determine the longitudinal packing arrangement of tubulin subunits, using cryoelectron microscopy of frozen hydrated specimens (native microtubule structure is more reliably preserved by this method (Taylor and Glaeser, 1976; Mandelkow and Mandelkow, 1985, 1986; Murray, 1986b; Mandelkow et al., 1991). Fig. 8 A shows an unfixed, unstained T. gondii parasite, embedded in amorphous solid water at −170°C suspended over a hole in a holey carbon film. This conoid (Fig 8 A, brackets) is in the retracted position, posterior to the lower polar ring (p2), and therefore partially shielded from view by the curtain of subpellicular microtubules (m). Fig. 8 B shows the apical region of a detergent- extracted parasite in which the conoid (brackets) is extended beyond the lower polar ring (p2). The conoid fibers are now clearly seen, along with both the intraconoid and subpellicular microtubules (i and m, respectively). Longitudinal striations are faintly visible in some of the conoid fibers (best seen by viewing Fig. 8 B at a glancing angle, looking from bottom right to top left). Note that this appearance is distinctly different from the intraconoid microtubules, in which the striations arising from overlapping protofilaments are clearly visible, and diagnostic of classical 13 protofilament microtubules (Mandelkow et al., 1986; Wade et al., 1990; Wade and Chretien, 1993).
Figure 8.
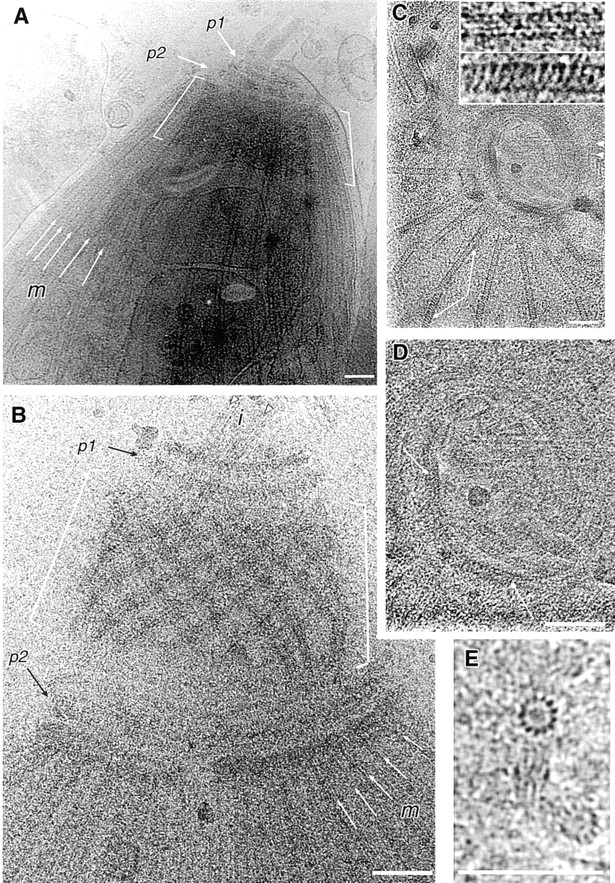
Cryoelectron micrographs of frozen hydrated T. gondii. The specimens are unfixed, unstained, embedded in a thin layer of amorphous solid water at –170°C, and suspended over holes in a carbon film. (A) Apical region of a nearly intact T. gondii. Both polar rings (p1, p2) are in front of the retracted conoid (between the white brackets). The conoid is seen through the curtain of subpellicular microtubules (m), some of which are indicated by white arrows. (B) Apical cytoskeleton from a detergent-extracted T. gondii, with extended conoid. The conoid (brackets) now lies between the two polar rings (p1, p2). The 14 individual conoid fibers, 430 nm long, are clearly seen. The two intraconoid microtubules, 350 nm long, (i, arrowheads) project through the upper polar ring. Subpellicular microtubules (m, white arrows) attach to the lower polar ring. (C) End-on view of conoid, polar rings, and attached subpellicular microtubules splayed out radially from the lower polar ring. Two segments of a broken subpellicular microtubule that have rotated with respect to one another (arrows) display dramatically different striation patterns, shown at higher magnification in the insets. Arrowheads, intra-conoid microtubules. (D) Enlarged view of the conoid from C, to show the periodic beading along the spirally arranged conoid fibers (white arrows). (E) Rare end-on views of two unfixed, unstained frozen-hydrated microtubules. In one of the microtubules, the orientation is nearly perfect, allowing the 13 individual protofilaments to be counted. Bars (A–E), 100 nm.
An end-on view of the conoid is sometimes obtained from frozen hydrated cytoskeletons of detergent-extracted parasites deposited onto holey carbon films (Fig. 8 C). Again, the conoid fibers give rise to spiral markings around the circumference of the conoid. Careful examination of these images reveals a periodic density modulation along some regions of these fibers (Fig. 8 D, arrows).
Longitudinal periodicities in individual conoid fibers (Fig. 8 B) are difficult to see by eye, but Fourier analysis of images from frozen hydrated samples reveals a rich set of periodic features. Power spectra were computed from individual conoid fibers that were digitally cut out of the image and digitally straightened. Summing the power spectra from several conoid fibers, clear 8- and 4-nm layer lines are visible (Fig. 9, top). This periodicity is characteristic of MAP-decorated microtubules (Amos and Klug, 1974; Amos, 1977; Kim et al., 1979; Murray, 1986a; Amos and Amos, 1991a). In a few cases, these two-layer lines could be seen in power spectra from individual conoid fibers. Remarkably, power spectra from conoid fiber images in glancing sections such as Fig. 4 B, whereas somewhat noisy, nevertheless showed the 8-nm longitudinal periodicity. The distribution of intensities along the equator of the power spectra (corresponding to the Fourier transform of the radial mass distribution of the object) is similar for subpellicular and intraconoid microtubules, but the outermost strong equatorial peak occurs at slightly smaller reciprocal space position for the conoid fibers than for the other two structures (5.4 nm vs. 5.0 nm). Weaker layer lines are also visible in the power spectra from some conoid fibers, suggesting that their true repeat length is 96 nm (Amos and Klug, 1974; Amos, 1977), as it is for the intraconoid and the subpellicular microtubules (Fig. 9, middle two spectra).
Figure 9.
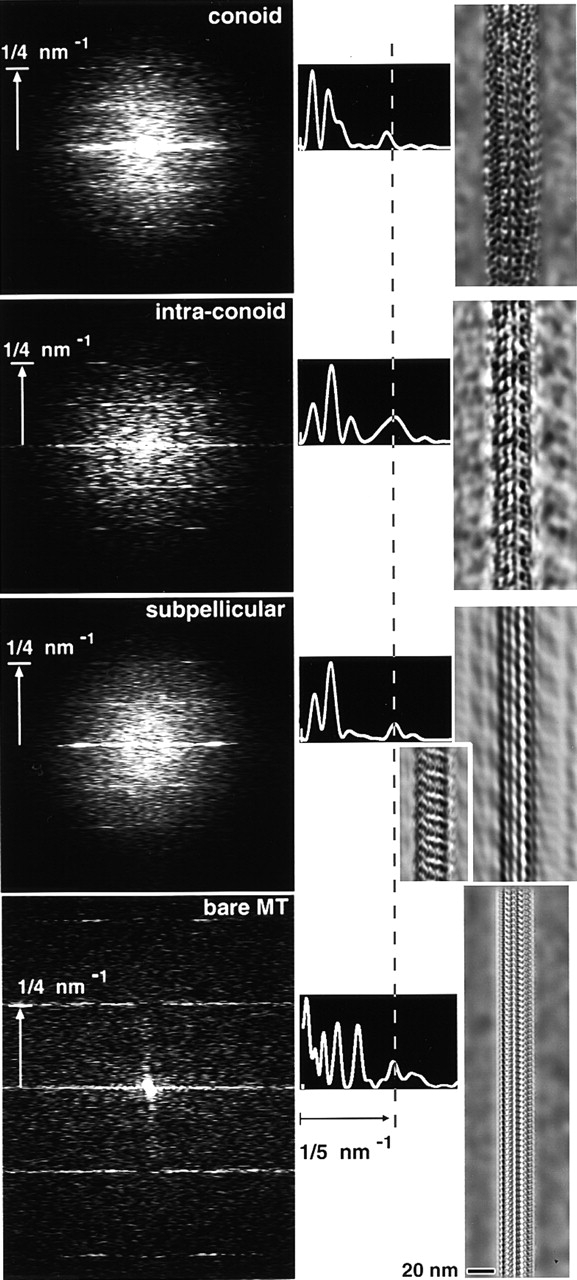
Fourier analysis of images of frozen hydrated T. gondii conoid fibers, intraconoid microtubules, subpellicular microtubules, and a bare typical cellular microtubule. (left column) Power spectra from the four structures, oriented with fiber axes vertical. The sum of three particularly good power spectra from conoid fibers (“unbent” to straighten the fiber axis before Fourier transformation) is shown at the top. The lower three power spectra are from single images of an intraconoid microtubule, subpellicular microtubule, and an undecorated microtubule purified from fertilized sea urchin egg, representative of a typical bare microtubule. In the three T. gondii spectra, higher intensities along layer lines indicate strong periodic density fluctuations at intervals of 4 nm, 8 nm, and larger spacings, most of which index as higher orders of a fundamental 96 nm repeat. The bare microtubule shows only a 2- and 4-nm repeat. (middle column) Intensity along the right half of the equator of each power spectrum is displayed as a graph. The outermost peak (∼1/5 nm−1, vertical dotted line) occurs at slightly smaller radius for the conoid fibers. (right column) Filtered images calculated by setting intensities to zero outside of the equator and layer lines (4 nm, 8 nm, and brightest of the remaining layer lines for T. gondii; 2 and 4 nm for the bare microtubule) in the Fourier transform, then inverse transforming. For subpellicular microtubules, two different images were filtered to show the two different patterns of periodic density fluctuations commonly observed. Sighting along the filtered images at a glancing angle makes the protofilament overlap pattern more obvious.
In all three types of tubulin polymers from T. gondii, the distribution of intensity along the layer lines is quite different from that typically observed for in vitro polymerized microtubules or undecorated microtubules purified from cells (Mandelkow and Mandelkow, 1985; Murray, 1991; Amos, 1995). An undecorated cellular microtubule, purified from fertilized sea urchin egg, is shown in Fig. 9 (bottom spectrum) for comparison. Note the presence of the 2-nm layer line (indicative of exceptionally good structural preservation) and the absence of layer lines for periodicities longer than 4 nm for this bare microtubule. In particular, the 8-nm dimer periodicity is undetectable, indicating that the difference between α- and β-tubulin is indiscernible in a bare microtubule by this technique. Filtered images of the various structures were generated by inverse transforming the Fourier transforms after masking to include only the strongest layer lines. This lengthwise averaging greatly increases the contrast of the longitudinal periodic repeats for the intraconoid, subpellicular, and bare cellular microtubules. Although this approach is less successful for the conoid fibers than for the microtubules (due to the very short lengths of straight fiber that are available), additional structure attributable to as-yet-unidentified proteins is evident in all three T. gondii structures.
Several different patterns of periodic longitudinal density modulation are seen in subpellicular microtubules (Fig. 8 C, arrows), depending on direction of view and extent of defocus of the objective lens. Some fragments show a faint beading along the straight longitudinal protofilament striations (Fig. 8 C, upper inset) as is typical of microtubules in other types of cells (Amos and Amos, 1991a; Wade and Chretien, 1993). Other fragments show strong 8-nm striations running across the microtubule, almost perpendicular to the long axis (Fig. 8 C, lower inset). In a few cases, we have found microtubules standing on end in these preparations (Fig. 8 E), a view that has not previously been reported for native microtubules. These end-on views may arise from intraconoid microtubules, which, in contrast to conoid fibers and subpellicular microtubules, are always perfectly straight.
Discussion
The nature of conoid fibers in Apicomplexan parasites has been the subject of much debate. Combining improved extraction/fixation and staining with a variety of microscopic imaging techniques, we have unequivocally demonstrated that the conoid fibers of T. gondii are composed of tubulin. Under suitable conditions, the conoid incorporates YFP-α-tubulin (Figs. 1–3), is recognized by anti-tubulin antibodies (Figs. 2 and 5), and exhibits tubulin protofilament structure in both cross-section (Figs. 6 and 7) and longitudinally (Figs. 4 B, 5 A, 8 B, and 9).
T. gondii has one copy each of genes for α- and β-tubulin (Nagel and Boothroyd, 1988), which are utilized for several different macromolecular assemblies, including spindle, intraconoid and subpellicular microtubules, and conoid fibers. Surprisingly, although the conoid fibers are composed of the same tubulin isoforms and have several structural features in common with microtubules, they are not microtubules at all. In microtubules, columns of dimers (protofilaments) can be seen in cross sections if care is take to orient the microtubule exactly parallel to the direction of view. In this orientation, the microtubule looks perfectly circular, with distinct, countable protofilaments around its entire circumference (Figs. 6, D and E, and 8 E). Very slight misorientation (∼5° in a typical EM section) blurs the images so that individual protofilaments can no longer be counted on two sides, but the profile remains circular; misorientation of ∼20° causes the profile to become elliptical.
Side views of conoid fibers (Figs. 4 B, 5 A, and 8 B) show longitudinal striations, suggesting that their subunits are arranged in straight columns similar to microtubule protofilaments. We confirmed this arrangement by showing that careful orientation does reveal distinct protofilaments in cross sections of the conoid (Figs. 6 and 7). However, unlike microtubules, the conoid fibers are not tubular and contain only ∼9 protofilaments. Protofilaments are more difficult to count in conoid fibers than in microtubules because perfect orientation is much harder to achieve (due to the curvature of the fibers themselves as they spiral around the conoid wall), but the cross-sectional profile of tubulin protofilaments is the same on both sides of the conoid shown in Fig. 6 (for example), confirming the proper orientation of this sample. Thus, it is clear that the observed deviation from circular symmetry inherent in the comma shape of conoid fibers cannot be an artifact of misorientation.
The lack of circular symmetry also indicates that the conoid fibers are not derived from ordinary microtubules simply through loss of a few protofilaments. Selective protofilament loss has been observed with heat or other treatments that partially depolymerize microtubules in other types of cells (Behnke and Forer, 1967; Witman et al., 1972; Tilney et al., 1973), but the remnant structures are always C- or S-shaped in cross-section; i.e., arcs of circles. Sheets of protofilaments that fail to form a closed tube can also be observed under certain polymerizing conditions in vitro, but these also consist of arcs of circles when viewed in cross section (Burton and Himes, 1978; Mandelkow and Mandelkow, 1979; Pierson et al., 1979; McIntosh and Euteneuer, 1984; Amos and Amos, 1991a; Mandelkow et al., 1991; Chretien et al., 1995). A transition between a closed tube form and a helical ribbon has been reported in Allogromia (Welnhofer and Travis, 1998), but that structure does not resemble the comma shape observed in conoid fibers. We conclude that the conoid fibers represent a previously unknown arrangement of tubulin.
Along the length of the conoid fibers, tubulin molecules are spaced 4 nm apart, just as in microtubules. It seems probable that conoid protofilaments exhibit parallel, as opposed to antiparallel, orientation (implying that the fiber as a whole is a polar structure) as has been observed in all naturally occurring tubulin polymers, but the signal-to-noise ratio in images of conoid fibers obtained to date is too low to confirm this supposition. However, the lack of circular symmetry in cross-section means that the lateral association of protofilaments in conoid fibers must be quite different from microtubules. The noncircular cross-sectional profile implies that the bonds between adjacent protofilaments are very different at one end of the comma compared to the other: adjacent protofilaments at the curved end must be rotated by ∼30° with respect to each other (similar to the 360/13 = 27.7 degrees in a canonical 13 protofilament microtubule), whereas adjacent protofilaments at the tail of the comma are only reoriented by ∼15°. Could this asymmetric arrangement be an accommodation to the extreme curvature of the fibers as they spiral around the 380-nm diameter conoid? At this point one can only speculate that the energy required to maintain this nonequivalence (Amos and Amos, 1991b) is provided by the binding energy of other proteins associated with the conoid fibers (CAPS). Weak layer lines in the power spectra computed from conoid fiber images hint that a periodic super-lattice with a long repeat is present, indexing on 96 nm (as is typical for MAPS; Amos and Klug, 1974; Amos, 1977; Kim et al., 1979; Murray, 1986a; Amos and Amos, 1991a). Monoclonal antibodies against unknown peptides associated with the conoid have been reported (Morrissette and Roos, 1998; Sasai et al., 1998), and it will be interesting to see if these proteins associate directly with the conoid fibers, and/or T. gondii microtubules, and whether they exhibit significant similarity to known microtubule binding proteins (Lewis et al., 1988; Noble et al., 1989; Hemphill et al., 1992; Doll et al., 1993; Perez et al., 1997).
FRAP measurements reveal rapid incorporation cytoplasmic tubulin into developing conoids during the early stages of daughter parasite assembly, but not into mature conoids. Similarly, the subpellicular microtubules of daughter parasites incorporate tubulin at their growing ends, but are not in equilibrium with the cytoplasmic pool after elongation has ceased. This change in dynamic properties is consistent with results from previous studies using drugs that affect microtubule stability (Stokkermans et al., 1996). Formation of subpellicular microtubules is blocked upon treatment with oryzalin (Shaw et al., 2000; Morrissette and Sibley, 2002), but preformed microtubules are stable in the presence of the drug. Neither taxol nor dinitroanilines affect preformed conoids (Shaw et al., 2000). The lower polar ring has been postulated to be the microtubule-organizing center for the subpellicular microtubules (Russell and Burns, 1984). Daughter conoids are apparently formed de novo in each cell cycle at some distance from the maternal conoid, implying that no template is needed for initiation of conoid fiber assembly.
The conoid undergoes a Ca2+-dependent translation of ∼0.5 μm relative to the lower polar ring (Mondragon and Frixione, 1996), causing it to extend at the apical end of the parasite. We have now observed a second form of motion correlated with extension, an increase in pitch of the conoid fibers. It is not clear whether extension and increased pitch result from independent motors responding to the same activation signal, or whether one motion is a simple mechanical consequence of the other. The nature of the motor(s) is unknown. The two intraconoid microtubules are in the right location to serve as tracks for a dynein/kinesin powered movement, but none has yet been reported in this location. Cytochalasin D, which blocks the Ca2+-dependent motile activity required for parasite exit from the host cell (Dobrowolski and Sibley, 1996, 1997), has been reported to partially block conoid protrusion in response to Ca2+ ionophore treatment (Mondragon and Frixione, 1996), suggesting involvement of a myosin. However, the maximum inhibition observed was only ∼50%. Another important unanswered question is how the matrix surrounding the conoid fibers participates in these movements. The new polymer expands our knowledge of tubulin assemblies, but also lengthens the list of questions to be answered.
Materials and methods
Parasite culture
T. gondii tachyzoites (strain RH) were cultivated in human foreskin fibroblast cells as previously described (Roos et al., 1994). The YFP-α-tubulin transgenics were generously provided by Dr. B. Striepen (University of Georgia, Athens, GA) (Striepen et al., 2000). Parasites were harvested soon after emerging from host cells, passed through a 3-μm filter (Nucleopore), centrifuged at 1,000 g for 12 min, and then washed once with PBS.
A23187 treatment and detergent extraction
YFP-α-tubulin parasites (1 T25 flask) were harvested and washed, then incubated in 10 μl 20 μ M A23187 in PBS at room temperature for 1 h. For extraction, parasites were centrifuged and resuspended in 20 μl 10 mM deoxycholate in distilled water (pH 7.0). 2–3 μl extracted parasite suspension was put on a slide, covered with a coverslip, sealed with nail polish, and imaged immediately.
Immunofluorescence labeling
A 10-μl drop of parasites was allowed to adhere to a coverslip for 1 h in a hydrated chamber, fixed 10 min with 4% formaldehyde in PBS, extracted 40 min with 10 mM deoxycholate in Mg2+PBS, blocked with 3% BSA in PBS, and labeled with monoclonal anti–α-tubulin antibody (B5-1-2, diluted 1:1,000 in PBS + 3%BSA; Sigma-Aldrich) followed by Alexa594 goat anti–mouse IgG (1:1,000 diluted in PBS + 3% BSA; Molecular Probes).
Immunogold labeling
RH parasites were resuspended in 30 μl 10 mM deoxycholate and rotated at 20°C for 30 min. 3 μl of extracted parasite suspension was allowed to settle on a carbon coated nickel grid for 7 min in a humid chamber. Grids were washed 5 min with water, fixed 5 min with 4% formaldehyde in PBS, and then washed 3 × 5 min with PBS. Free aldehyde groups were blocked by incubation with 50 mM glycine in PBS for 20 min (pH 7.5) followed by 15 min incubation with 0.1% NaBH4 in PBS. Grids were washed twice with PBS, blocked with 5% BSA + 0.1% fish gelatin in PBS for 30 min, washed 2 × 5 min with incubation buffer (0.8% BSA and 0.1% fish gelatin in PBS, 15 mM NaN3), incubated with primary antibodies (a mixture of mouse anti-α [B5-1–2; Sigma-Aldrich] and anti-β tubulin [E7; Sigma-Aldrich] diluted 1:50 in incubation buffer with 15 mM NaN3) for 3 h, washed 6 × 5 min in incubation buffer, inverted on 10-μl drops of secondary antibody (goat anti–mouse IgG-conjugated with ultra-small gold (EMS Inc.) diluted 1:20 in incubation buffer), incubated overnight at 4°C, and then washed 6 × 5 min with incubation buffer and 2 × 5 min with PBS. The samples were postfixed 5 min with 1% glutaraldehyde in PBS, followed by 3 × 5 min wash in distilled water. Silver enhancement was carried out using the HQ silver enhancement kit (Nanoprobes Inc.) by floating grids on mixtures of the initiator, activator, and modulator for 45 s to 1 min in a light-tight chamber, and then washing 2 × 5 min with water. Grids were negatively stained using 2% phosphotungstic acid (pH 7.0).
Western blotting
Parasites were harvested, washed with PBS, lysed with 0.5% NP40, 2 mM EDTA in PBS, immediately put in SDS gel loading buffer, and heated at 70°C for 20 min. Samples were run on a 4–12% Bis-Tris SDS gradient gel (Invitrogen) at 200 V for 1 h and electroblotted onto nitrocellulose membranes. After incubating in blocking buffer (5% fat-free dry milk in PBS with 0.02% Tween-20), blots were labeled with monoclonal mouse anti-GFP (Clontech Laboratories, Inc.) or monoclonal mouse anti–α-tubulin (Sigma-Aldrich; B5-1–2) diluted 1:1,000 in blocking buffer. Bound antibody was detected with goat anti–mouse IgG conjugated to horseradish peroxidase (1:3,000 in blocking buffer; Bio-Rad Laboratories), using the Supersignal West Pico Kit (Pierce Chemical Co.) according to the manufacturer's protocol.
FRAP
Host cell monolayers were cultivated in ΔT chambers (Bioptechs, Inc.) and infected with parasites. 10 mM Hepes (pH 7.0) was added to the medium immediately prior to imaging at 37°C, using a Bioptechs heated microscope stage and objective lens heater. FRAP analysis was performed on a Zeiss LSM510 confocal microscope using the 488-nm line of an Ar/Kr laser.
Cryoelectron microscopy
Parasites were isolated and treated with A23187 to induce conoid protrusion, followed by deoxycholate extraction. 1–2 μl extracted parasite suspension was placed on a grid covered with holey carbon film and plunged into rapidly stirred cryogen (liquid propane + 5% isopentane, maintained at liquid nitrogen temperature) as previously described (Murray and Ward, 1987a, 1987b; Ward and Murray, 1987). Frozen grids were searched with a Philips 400 EM at low illumination with a defocus of ∼0.8 mm to find appropriate regions for imaging. The EM was focused off axis at ×220,000, and micrographs were acquired using the Philips low dose unit at ×22,000, ×28,000 or ×36,000 magnification, with an electron dose of ∼500 e−/nm2, at 120 kev and defocus of 1.6 or 2.0 μm, on Kodak SO-163 film developed for maximum contrast.
Conventional EM of thin sections
2 T25 flasks of RH parasites were harvested, treated with A23187, and suspended in 2 ml 0.1 M NaMES, 2 mM MgCl2, 100 μM CaCl2, 1 mM CaEGTA, pH 6.6 (Buffer 1). Parasites were lysed by adding TX-100 to 0.5%, incubated 15 min, washed with Buffer 1, fixed in Buffer 1 plus 1% glutaraldehyde and 1% tannic acid for 6 h at 37°C, centrifuged, and resuspended in 1 ml 15 mM NaH2PO4 (pH 6.0) plus 1.3% OsO4 at 4°C for 1 h. After centrifugation, the pellet was rinsed with H2O, stained with 2% UrAc for 10 h, dehydrated, and embedded in Spurr resin. Dark grey sections (estimated to be 25–30 nm thick) were cut and stained with saturated UrAc:MeOH (1:1) for 1.5 h, and then with lead citrate for 4 min. Sections were imaged at 120 kev in a Philips 400 electron microscope.
Image analysis
Micrographs were scanned on a Perkin-Elmer 1010G microdensitometer at a pixel spacing of 5, 10, or 25 μm on the film. Data was transferred to an Indigo Iris work station for analysis using the Semper image processing software (Synoptics Ltd.).
Acknowledgments
We thank Drs. Boris Striepen for YFP-α-tubulin transgenic T. gondii, Naomi Morrissette for help with earlier EM studies of T. gondii, and Con Beckers for helpful discussion.
This research was supported by National Institutes of Health grants R01 A149301 (J.M. Murray and D.S. Roos) and R21 GM60216 (J.M. Murray). D.S. Roos is a Burroughs-Wellcome Scholar in Molecular Parasitology.
Footnotes
Abbreviation used in this paper: MAP, microtubule-associated protein.
References
- Amos, L.A. 1995. The microtubule lattice: 20 years on. Trends Cell Biol. 5:48–51. [DOI] [PubMed] [Google Scholar]
- Amos, L.A. 1977. Arrangement of high molecular weight associated proteins on purified mammalian brain microtubules. J. Cell Biol. 72:642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos, L.A., and A. Klug. 1974. Arrangement of subunits in flagellar microtubules. J. Cell Sci. 14:523–549. [DOI] [PubMed] [Google Scholar]
- Amos, L.A., and W.B. Amos. 1991a. Molecules of the Cytoskeleton. The Guilford Press, New York. 253 pp.
- Amos, L.A., and W.B. Amos. 1991. b. The bending of sliding microtubules imaged by confocal light microscopy and negative stain electron microscopy. J. Cell Sci. 14:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke, O., and A. Forer. 1967. Evidence for four classes of microtubules in individual cells. J. Cell Sci. 2:169–192. [DOI] [PubMed] [Google Scholar]
- Burton, P.R., and R.H. Himes. 1978. Electron microscope studies of pH effects on assembly of tubulin free of associated proteins. J. Cell Biol. 77:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers, V.B., and L.D. Sibley. 1997. Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur. J. Cell Biol. 73:114–123. [PubMed] [Google Scholar]
- Chretien, D., S.D. Fuller, and E. Karsenti. 1995. Structure of growing microtubule ends: two-dimensional sheets close into tubes at variable rates. J. Cell Biol. 129:1311–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cintra, W.M., and W. de Souza. 1985. Immunochemical localization of cytoskeletal proteins and electron microscopy of detergent extracted tachyzoites of T. gondii. J. Submicrosc. Cytol. 17:503–508. [PubMed] [Google Scholar]
- de Souza, W. 1974. Fine structure of the conoid of T. gondii Rev. Instit. Medicina Tropical Sao Paulo. 16:32–38. [PubMed] [Google Scholar]
- Dobrowolski, J.M., and L.D. Sibley. 1996. Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell. 84:933–939. [DOI] [PubMed] [Google Scholar]
- Dobrowolski, J., and L.D. Sibley. 1997. The role of the cytoskeleton in host cell invasion by Toxoplasma gondii Behring Inst Mitt: 90–96. [PubMed]
- Doll, T., M. Meichsner, B.M. Riederer, P. Honegger, and A. Matus. 1993. An isoform of microtubule-associated protein 2 (MAP2) containing four repeats of the tubulin-binding motif. J. Cell Sci. 106:633–639. [DOI] [PubMed] [Google Scholar]
- Dubremetz, J., E. Ferreira, and C. Dissous. 1989. Isolation and partial characterization of rhoptries and micronemes from Eimeria nieschulzi zoites. Parasitol. Res. 75:449–454. [DOI] [PubMed] [Google Scholar]
- Dustin, P. 1984. Microtubules. Springer-Verlag, Berlin. 481 pp.
- Futaesaku, Y., V. Mizuhira, and H. Nakamura. 1972. In Histochemistry and Cytochemistry. Proc. 4th Int. Congr. Histochem. Cytochem. T. Takeuchi et al., editors. Nakanishi Printing Co., Kyoto, Japan. 155 pp.
- Gittes, F., B. Mickey, J. Nettleton, and J. Howard. 1993. Flexural rigidity of microtubules and actin-filaments measured from thermal fluctuations in shape. J. Cell Biol. 120:923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemphill, A., M. Affolter, and T. Seebeck. 1992. A novel microtubule-binding motif identified in a high molecular weight microtubule-associated protein from Trypanosoma brucei. J. Cell Biol. 117:95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H., L.I. Binder, and J.L. Rosenbaum. 1979. The periodic association of MAP2 with brain microtubules in vitro. J. Cell Biol. 80:266–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lah, J.J., D.M. Hayes, and R.W. Burry. 1990. A neutral pH silver development method for the visualization of 1-nm gold particles in pre-embedding EM immunocytochemistry. J. Histochem. Cytochem. 38:503–508. [DOI] [PubMed] [Google Scholar]
- Lewis, S.A., D.H. Wang, and N.J. Cowan. 1988. Microtubule-associated protein MAP2 shares a microtubule binding motif with tau protein. Science. 242:936–939. [DOI] [PubMed] [Google Scholar]
- Luft, B.J., R. Hafner, A.H. Korzun, C. Leport, D. Antoniskis, E.M. Bosler, D.D.d. Bourland, R. Uttamchandani, J. Fuhrer, and J. Jacobson. 1993. Toxoplasmic encephalitis in patients with the acquired immunodeficiency syndrome. New Eng. J. Med. 329:995–1000. [DOI] [PubMed] [Google Scholar]
- Mandelkow, E.M., and E. Mandelkow. 1979. Junctions between microtubule walls. J. Mol. Biol. 129:135–148. [DOI] [PubMed] [Google Scholar]
- Mandelkow, E.M., and E. Mandelkow. 1985. Unstained microtubules studied by cryo-electron microscopy. Substructure, supertwist and disassembly. J. Mol. Biol. 181:123–135. [DOI] [PubMed] [Google Scholar]
- Mandelkow, E., and E.M. Mandelkow. 1986. Quick-frozen microtubules studied by cryoelectron microscopy and image processing. Ann. N.Y. Acad. Sci. 483:13–23. [DOI] [PubMed] [Google Scholar]
- Mandelkow, E.M., R. Schultheiss, R. Rapp, M. Muller, and E. Mandelkow. 1986. On the surface lattice of microtubules: helix starts, protofilament number, seam, and handedness. J. Cell Biol. 102:1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow, E.M., E. Mandelkow, and R.A. Milligan. 1991. Microtubule dynamics and microtubule caps: a time-resolved cryo-electron microscopy study. J. Cell Biol. 114:977–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, T., and C. Beckers. 2001. Characterization of the subpellicular network, a filamentous membrane skeletal component in the parasite T. gondii. Mol. Biochem. Parasitol. 115:257–268. [DOI] [PubMed] [Google Scholar]
- McIntosh, J.R., and U. Euteneuer. 1984. Tubulin hooks as probes for microtubule polarity: an analysis of the method and an evaluation of data on microtubule polarity in the mitotic spindle. J. Cell Biol. 98:525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuhira, V., and Y. Futaesaku. 1971. Ann. Proc. EMSA, 29th, Boston, MA. 494 pp.
- Mondragon, R., and E. Frixione. 1996. Ca(2+)-dependence of conoid extrusion in T. gondii tachyzoites. J. Eukaryot. Microbiol. 43:120–127. [DOI] [PubMed] [Google Scholar]
- Morrissette, N.S. 1995. The Apical Cytoskeleton of T. gondii: Ph.D. thesis. University of Pennsylvania, Philadelphia, PA. 191 pp.
- Morrissette, N.S., J.M. Murray, and D.S. Roos. 1997. Subpellicular microtubules associate with an intramembranous particle lattice in the protozoan parasite T. gondii. J. Cell Sci. 110:35–42. [DOI] [PubMed] [Google Scholar]
- Morrissette, N.S., and D.S. Roos. 1998. T. gondii: a family of apical antigens associated with the cytoskeleton. Exp. Parasitol. 89:296–303. [DOI] [PubMed] [Google Scholar]
- Morrissette, N.S., and L.D. Sibley. 2002. Disruption of microtubules uncouples budding and nuclear division in T. gondii. J. Cell Sci. In press. [DOI] [PubMed]
- Murray, J.M. 1986. a. Electron microscopy of frozen hydrated eukaryotic flagella. J. Ultrastruct. Molec. Struct. Res. 95:196–209. [DOI] [PubMed] [Google Scholar]
- Murray, J.M. 1986. b. Studies of eukaryotic flagella by cryoelectron microscopy. Ann. N.Y. Acad. Sci. 483:24–30. [DOI] [PubMed] [Google Scholar]
- Murray, J.M. 1991. Structure of flagellar microtubules. Int. Rev. Cytol. 125:47–93. [DOI] [PubMed] [Google Scholar]
- Murray, J.M., and R. Ward. 1987. a. Preparation of holey carbon-films suitable for cryoelectron microscopy. J. Electron Microsc. Tech. 5:285–290. [Google Scholar]
- Murray, J.M., and R. Ward. 1987. b. Principles for the construction and operation of a device for rapidly freezing suspensions for cryoelectron microscopy. J. Electron Microsc. Tech. 5:279–284. [Google Scholar]
- Nagel, S.D., and J.C. Boothroyd. 1988. The α- and β-tubulins of T. gondii are encoded by single copy genes containing multiple introns. Molec. Biochem. Parasitol. 29:261–273. [DOI] [PubMed] [Google Scholar]
- Nichols, B.A., and M.L. Chiappino. 1987. Cytoskeleton of T. gondii. J. Protozool. 34:217–226. [DOI] [PubMed] [Google Scholar]
- Noble, M., S.A. Lewis, and N.J. Cowan. 1989. The microtubule binding domain of microtubule-associated protein MAP1B contains a repeated sequence motif unrelated to that of MAP2 and tau. J. Cell Biol. 109:3367–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, M., K. Aloria, J.C. Zabala, and J. Avila. 1997. A putative beta-tubulin phosphate-binding motif is involved in lateral microtubule protofilament interactions. Eur. J. Biochem. 248:840–847. [DOI] [PubMed] [Google Scholar]
- Pezzella, N., A. Bouchot, A. Bonhomme, L. Pingret, C. Klein, H. Burlet, G. Balossier, P. Bonhomme, and J.M. Pinon. 1997. Involvement of calcium and calmodulin in T. gondii tachyzoite invasion. Eur. J. Cell Biol. 74:92–101. [PubMed] [Google Scholar]
- Pierson, G.B., P.R. Burton, and R.H. Himes. 1979. Wall substructure of microtubules polymerized in vitro from tubulin of crayfish nerve cord and fixed with tannic acid. J. Cell Sci. 39:89–99. [DOI] [PubMed] [Google Scholar]
- Roos, D.S., R.G. Donald, N.S. Morrissette, and A.L. Moulton. 1994. Molecular tools for genetic dissection of the protozoan parasite T. gondii. Methods Cell Biol. 45:27–78. [DOI] [PubMed] [Google Scholar]
- Roos, D.S., M.J. Crawford, R.G. Donald, L.M. Fohl, K.M. Hager, J.C. Kissinger, M.G. Reynolds, B. Striepen, and W.J. Sullivan, Jr. 1999. Transport and trafficking: Toxoplasma as a model for Plasmodium. Novartis Found. Symp. 226:176–195. [DOI] [PubMed] [Google Scholar]
- Roos, D.S., J.A. Darling, M.G. Reynolds, K.M. Hager, B. Striepen, and J.C. Kissinger. 2000. Toxoplasma as a model parasite: apicomplexan biochemistry, cell biology, molecular genetics, genomics, and beyond. Biology of Parasitism. C. Tschud and E.J. Pearce, editors. Kluwer Academic Publishers, Boston. 143–167.
- Russell, D.G., and R.G. Burns. 1984. The polar ring of coccidian sporozoites: a unique microtubule-organizing centre. J. Cell Sci. 65:193–207. [DOI] [PubMed] [Google Scholar]
- Sasai, K., H.S. Lillehoj, A. Hemphill, H. Matsuda, Y. Hanioka, T. Fukata, E. Baba, and A. Arakawa. 1998. A chicken anti-conoid monoclonal antibody identifies a common epitope which is present on motile stages of Eimeria, Neospora, and Toxoplasma. J. Parasitol. 84:654–656. [PubMed] [Google Scholar]
- Scholtyseck, E., H. Melhorn, and K. Friedhoff. 1970. The fine structure of the conoid of sporozoa and related organisms. Z. Parasitenkd. 34:68–94. [Google Scholar]
- Schwartzman, J.D., E.C. Krug, L.I. Binder, and M.R. Payne. 1985. Detection of the microtubule cytoskeleton of the coccidian T. gondii and the hemoflagellate Leishmania donovani by monoclonal antibodies specific for beta-tubulin. J. Protozool. 32:747–749. [DOI] [PubMed] [Google Scholar]
- Shaw, M.K., H.L. Compton, D.S. Roos, and L.G. Tilney. 2000. Microtubules, but not actin filaments, drive daughter cell budding and cell division in T. gondii. J. Cell Sci. 113:1241–1254. [DOI] [PubMed] [Google Scholar]
- Stokkermans, T.J., J.D. Schwartzman, K. Keenan, N.S. Morrissette, L.G. Tilney, and D.S. Roos. 1996. Inhibition of T. gondii replication by dinitroaniline herbicides. Exp. Parasitol. 84:355–370. [DOI] [PubMed] [Google Scholar]
- Stommel, E.W., K.H. Ely, J.D. Schwartzman, and L.H. Kasper. 1997. T. gondii: dithiol-induced Ca2+ flux causes egress of parasites from the parasitophorous vacuole. Exp. Parasitol. 87:88–97. [DOI] [PubMed] [Google Scholar]
- Striepen, B., M.J. Crawford, M.K. Shaw, L.G. Tilney, F. Seeber, and D.S. Roos. 2000. The plastid of T. gondii is divided by association with the centrosomes. J. Cell Biol. 151:1423–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedlow, J.R., K. Hu, P.D. Andrews, D.S. Roos, and J.M. Murray. 2002. Measurement of tubulin content in the conoid and spindle pole of the parasite T. gondii Proc. Nat'l. Acad. Sci. In press. [Google Scholar]
- Taylor, K.A., and R.M. Glaeser. 1976. Electron microscopy of frozen hydrated biological specimens. J. Ultrastruct. Res. 55:448–456. [DOI] [PubMed] [Google Scholar]
- Tilney, L.G., J. Bryan, D.J. Bush, K. Fujiwara, M.S. Mooseker, D.B. Murphy, and D.H. Snyder. 1973. Microtubules: evidence for 13 protofilaments. J. Cell Biol. 59:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade, R.H., and D. Chretien. 1993. Cryoelectron microscopy of microtubules. J. Struct. Biol. 110:1–27. [DOI] [PubMed] [Google Scholar]
- Wade, R.H., D. Chretien, and D. Job. 1990. Characterization of microtubule protofilament numbers. How does the surface lattice accommodate? J. Mol. Biol. 212:775–786. [DOI] [PubMed] [Google Scholar]
- Ward, R., and J.M. Murray. 1987. Natural propane cryogen for frozen hydrated biological specimens. J. Elect. Micro. Tech. 5:275–277. [Google Scholar]
- Welnhofer, E.A., and J.L. Travis. 1998. Evidence for a direct conversion between two tubulin polymers—microtubules and helical filaments—in the foraminiferan, Allogromia laticollaris. Cell Motil. Cytoskeleton. 41:107–116. [DOI] [PubMed] [Google Scholar]
- Witman, G.B., K. Carlson, J. Berliner, and J.L. Rosenbaum. 1972. Chlamydomonas flagella. I. Isolation and electrophoretic analysis of microtubules, matrix, membranes, and mastigonemes. J. Cell Biol. 54:507–539. [DOI] [PMC free article] [PubMed] [Google Scholar]