

Abstract
Tropomyosin binds to actin filaments and is implicated in stabilization of actin cytoskeleton. We examined biochemical and cell biological properties of Caenorhabditis elegans tropomyosin (CeTM) and obtained evidence that CeTM is antagonistic to ADF/cofilin-dependent actin filament dynamics. We purified CeTM, actin, and UNC-60B (a muscle-specific ADF/cofilin isoform), all of which are derived from C. elegans, and showed that CeTM and UNC-60B bound to F-actin in a mutually exclusive manner. CeTM inhibited UNC-60B–induced actin depolymerization and enhancement of actin polymerization. Within isolated native thin filaments, actin and CeTM were detected as major components, whereas UNC-60B was present at a trace amount. Purified UNC-60B was unable to interact with the native thin filaments unless CeTM and other associated proteins were removed by high-salt extraction. Purified CeTM was sufficient to restore the resistance of the salt-extracted filaments from UNC-60B. In muscle cells, CeTM and UNC-60B were localized in different patterns. Suppression of CeTM by RNA interference resulted in disorganized actin filaments and paralyzed worms in wild-type background. However, in an ADF/cofilin mutant background, suppression of CeTM did not worsen actin organization and worm motility. These results suggest that tropomyosin is a physiological inhibitor of ADF/cofilin-dependent actin dynamics.
Keywords: myofibrils; thin filaments; actin; ADF/cofilin; tropomyosin
Introduction
Actin cytoskeleton is dynamic, and there is a constant exchange of actin subunits in the filaments by polymerization and depolymerization. However, within a single cell, different cytoskeletal structures exhibit variable rates of actin filament turnover. In lamellipodia of motile cells, rapid polymerization and depolymerization of actin filaments allow protrusion of the leading edges (Wang, 1985). In contrast, in stress fibers, actin filaments are less dynamic and the rate of monomer exchange within the filaments is relatively slow (Glacy, 1983; Amato and Taylor, 1986; Okabe and Hirokawa, 1989). These differences are regulated by a number of actin-binding proteins that affect assembly and disassembly of actin filaments (Cooper and Schafer, 2000; Pollard et al., 2000).
Actin depolymerizing factor (ADF)*/cofilin is one of the key players in enhancing actin filament turnover (Bamburg, 1999; Bamburg et al., 1999; Carlier et al., 1999). ADF/cofilin increases the rate of depolymerization from pointed ends of actin filaments (Carlier et al., 1997; Maciver et al., 1998) and severs filaments, thereby increasing the number of free ends (Maciver et al., 1991; Ichetovkin et al., 2000). These two activities can be uncoupled by point mutations (Moriyama and Yahara, 1999; Pope et al., 2000; Ono et al., 2001). The severing activity of ADF/cofilin is correlated with its binding to the side of filaments (Ono et al., 2001). When ADF/cofilin binds to actin filaments, it changes the twist of the filaments (McGough et al., 1997) which distorts the helical structure and increases the chance of filament breakage (McGough and Chiu, 1999). Genetic studies have shown that mutations that abolish only severing or F-actin binding by ADF/cofilin cause abnormal actin assembly in Caenorhabditis elegans (Ono et al., 1999) or severe defects in actin turnover and viability in yeast (Lappalainen and Drubin, 1997; Lappalainen et al., 1997), whereas a mutation that only impairs the depolymerizing activity causes no apparent phenotype in yeast (Moriyama and Yahara, 1999). In motile cells, ADF/cofilin is accumulated in lamellipodia (Bamburg and Bray, 1987; Svitkina and Borisy, 1999) and involved in the increase in the number of free barbed ends at the leading edge upon growth factor stimulation (Chan et al., 2000). Thus, although biochemical studies suggest that the effects of ADF/cofilin at the filament ends are sufficient to enhance actin turnover to a physiological range (Carlier et al., 1997; Didry et al., 1998), accumulated evidence suggests that the F-actin–binding/–severing activity of ADF/cofilin is important for its cellular function.
The activity of ADF/cofilin can be inhibited by several mechanisms, including phosphorylation of ADF/cofilin at a conserved serine residue (Morgan et al., 1993; Agnew et al., 1995; Moriyama et al., 1996), binding of phosphoinositides with ADF/cofilin (Yonezawa et al., 1990; Van Troys et al., 2000), and competition with tropomyosin (TM) (Bernstein and Bamburg, 1982; Nishida et al., 1984). Regulation of ADF/cofilin by phosphorylation/dephosphorylation or phosphoinositides is likely to be involved in cytoskeletal reorganization by intracellular signaling. However, the role of the competition of ADF/cofilin with TM is not clear. In biochemical studies, TM has been shown to protect actin depolymerization by chicken ADF (Bernstein and Bamburg, 1982) or slow down the kinetics of depolymerization by starfish depactin (Mabuchi, 1982) or porcine destrin (Nishida et al., 1985). Cofilins from porcine brain (Nishida et al., 1984) and chicken muscle (Abe et al., 1989) strongly bind to F-actin and dissociate TM from the filaments. This competition is not due to their overlapping binding sites on actin, but rather to different filament structures when TM or ADF/cofilin is bound (McGough, 1998). Microinjection of chicken myotubes with a high concentration of cofilin results in dissociation of TM from myofibrils (Nagaoka et al., 1995). In the growth factor–stimulated lamellipodia, cofilin is enriched at the leading edge, but TM is not present in the same region (Ogniewski Des Marais, V.M., I. Ichetovkin, M. Bailly, A. Chan, J.S. Condeelis, and S.E. Hitchcock-DeGregorio. 2001. 418 American Society for Cell Biology Annual Meeting. 2330 [Abstr.].). In budding yeast, cofilin is localized to cortical actin patches (Moon et al., 1993), whereas TM is localized to actin cables (Liu and Bretscher, 1989). In TM-null cells, cofilin is associated with actin cables (Belmont and Drubin, 1998) which could explain why actin cables are rapidly disassembled in conditional TM mutant cells at restrictive temperature (Pruyne et al., 1998).
Previous studies have suggested that TM has a function to stabilize actin filaments (Pittenger et al., 1994; Lin et al., 1997). TM directly affects the dynamic properties of actin by inhibiting spontaneous actin polymerization (Lal and Korn, 1986; Hitchcock-DeGregori et al., 1988) and depolymerization from the pointed ends (Broschat et al., 1989; Broschat, 1990). In addition, TM inhibits Arp2/3-nucleated actin polymerization (Blanchoin et al., 2001). Thus, these observations suggest that TM and ADF/cofilin regulate actin dynamics in opposite ways. However, it is not known whether these antagonistic effects are important for morphogenetic processes or stability of certain cytoskeletal structures.
In striated muscles, TM is a major thin filament protein and, together with troponin, regulates actomyosin interaction (Gordon et al., 2000). In spite of extensive investigations of TM in muscle contraction, its role in assembly and maintenance of myofibrils is currently unknown. Homozygous α-TM–null mice are embryonic lethal, whereas heterozygous knockout mice show no obvious phenotype (Blanchard et al., 1997; Rethinasamy et al., 1998). In Drosophila, mutations of a muscle TM isoform cause peripheral disruption of myofibrils and alterations in their mechanical properties (Kreuz et al., 1996). However, the structural defects in the central region of myofibrils are relatively mild, suggesting that TM is not very important for initial assembly of myofibrils (Kreuz et al., 1996). In C. elegans, TM is encoded by a single gene, lev-11/tmy-1, that gives rise to at least four TM isoforms (Kagawa et al., 1995; Anyanful et al., 2001). Mutations of this gene cause embryonic arrest at the twofold stage, but the actin organization in body wall muscle is not significantly altered (Williams and Waterston, 1994). One of the severe mutations introduces a premature stop codon at the end of exon 1 (Anyanful et al., 2001). However, due to the presence of multiple promoters and complex splicing patterns, it is not known how this mutation changes the composition of TM isoforms in the mutant muscle.
We have determined that UNC-60B, a muscle-specific ADF/cofilin isoform, is required for proper assembly of actin into myofibrils in C. elegans (Ono et al., 1999). Biochemical studies suggest that UNC-60B enhances actin filament dynamics by depolymerizing and severing actin filaments (Ono and Benian, 1998; Ono et al., 1999). However, myofibrils are very stable cytoskeletal structures. Therefore, we hypothesized the presence of a factor that inhibits UNC-60B–dependent actin dynamics and stabilizes actin filaments in myofibrils. Here, we present evidence that TM is a strong candidate for a physiological inhibitor of UNC-60B.
Results
Purification and characterization of C. elegans tropomyosin
Previously, Harris et al. (1977) reported purification of TM from C. elegans and its ability to bind to rabbit muscle actin. Here, we purified TM from wild-type C. elegans using a modified method and further characterized its biochemical properties. Purified TM (CeTM) migrated as a protein with an apparent molecular mass of 40 kD in SDS-PAGE (Fig. 1 A), as reported previously (Harris et al., 1977). CeTM shared two other biochemical properties with other TMs (Smillie, 1982): (a) CeTM was stable after boiling for 15 min; (b) electrophoretic mobility of CeTM was greatly retarded in the presence of 6 M urea (unpublished data). CeTM bound to C. elegans F-actin (Ce-actin) (Fig. 1 B). Binding of CeTM to actin was saturated at a molar ratio of 1:7.1 with a dissociation constant of 97 nM. CeTM bound to rabbit muscle actin in a similar manner (unpublished data).
Figure 1.

Purification and actin-binding activity of CeTM. (A) CeTM was purified from the soluble proteins after boiling worm extracts by ammonium sulfate fractionation (a) and Mono-Q column chromatography (b). (B) Binding of CeTM to Ce-actin was analyzed by a cosedimentation assay. Varied concentrations of CeTM were incubated with 10 μM F-Ce-actin and fractionated into supernatant (free) and pellets (bound) by ultracentrifugation. They were analyzed by SDS-PAGE (10% gel) and quantified by densitometric analysis of the Coomassie-stained gel.
CeTM and UNC-60B bind to F-actin in a mutually exclusive manner
We used CeTM, Ce-actin, and C. elegans ADF/cofilin (UNC-60B) to characterize their biochemical interactions. CeTM and UNC-60B bound to F-actin in a mutually exclusive manner in vitro (Fig. 2). When UNC-60B was preincubated with actin at a saturating concentration, binding of CeTM to actin was strongly inhibited (Fig. 2, A and B). CeTM at a high concentration (10 μM) failed to replace UNC-60B. Similarly, when CeTM was preincubated with F-actin at a saturating concentration, binding of UNC-60B to F-actin was inhibited (Fig. 2, C and D). Nevertheless, increasing amounts of UNC-60B decreased actin-bound CeTM and, instead, increased actin-bound UNC-60B (Fig. 2 D), indicating that UNC-60B competed with CeTM for actin binding. CeTM also inhibited UNC-60B–induced depolymerization of F-actin (Fig. 2 E). However, partial depolymerization still occurred in the presence of CeTM, which is probably due to partial dissociation of CeTM from F-actin. This incomplete inhibition suggests that, although CeTM protects F-actin from UNC-60B, it still allows UNC-60B–mediated actin filament turnover to a limited extent. Taken together, these results suggest that CeTM and UNC-60B bind to F-actin in a mutually exclusive manner.
Figure 2.
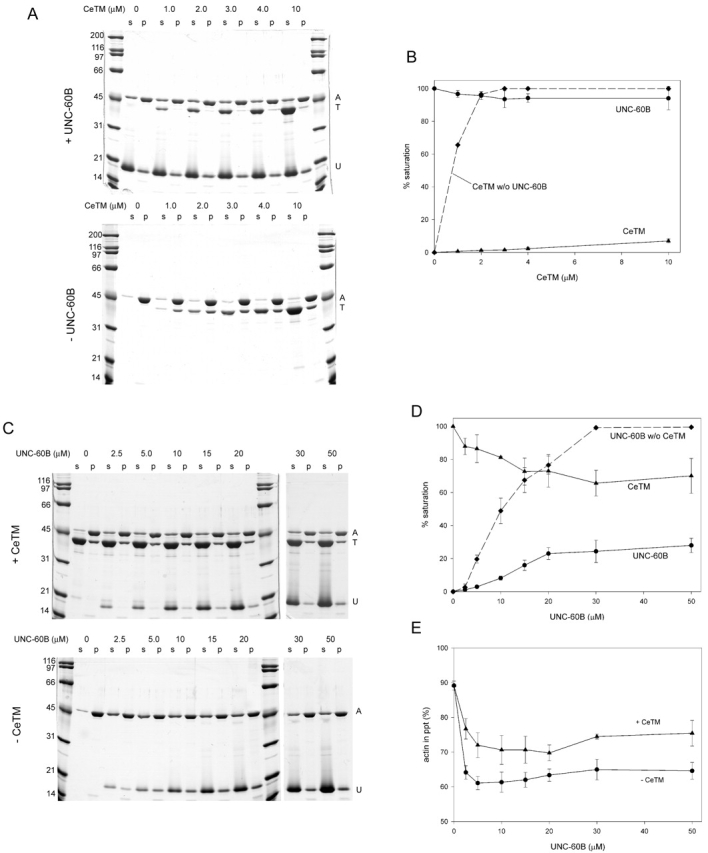
CeTM or UNC-60B interacts with F-Ce-actin in a mutually exclusive manner. (A and B) 10 μM F-actin was preincubated with or without 30 μM UNC-60B for 30 min, and, then, varied concentrations of CeTM were added to the mixtures. After 30 min, the reactions were analyzed by a cosedimentation assay. (A) Both supernatants (s) and pellets (p) were applied to Ca-SDS-PAGE (10% acrylamide gel) for a better separation of actin and CeTM. (B) Actin-bound CeTM or UNC-60B was quantified by densitometric analysis. Because F-actin was partially depolymerized in the presence of UNC-60B, the molar ratios of CeTM:actin or UNC-60B:actin in the pellets were calculated and presented as a percentage of saturation, where 100% was 1:7 for CeTM:actin or 1:1 for UNC-60B:actin. (C–E) 10 μM F-actin was preincubated with or without 10 μM CeTM for 30 min, and varied concentrations of UNC-60B were added to the mixtures. After 30 min of incubation, the reactions were analyzed by a cosedimentation assay. (C) Both supernatants (s) and pellets (p) were applied to Ca-SDS-PAGE. (D) Densitometric analysis of actin-bound CeTM or UNC-60B as described in (B). (E) Depolymerization of F-actin by UNC-60B in the absence (•) or presence (▴) of CeTM. Percentages of sedimented actin in the experiments shown in (C) are plotted on the vertical axis. Data shown in B, D, and E are means ± SD of three experiments.
CeTM inhibits UNC-60B–induced enhancement of actin polymerization
UNC-60B enhances the elongation rate of spontaneous actin polymerization by severing actin filaments and increasing the number of filament ends (Ono et al., 1999). At a molar ratio of 0.2:1 of UNC-60B to actin, the elongation phase (700–1,000 s) of polymerization was accelerated and the kinetics reached a plateau at 1,500 s (Fig. 3). However, CeTM strongly inhibited this activity (Fig. 3). At a molar ratio of 0.2:0.2:1 of CeTM:UNC-60B:actin, the kinetics of polymerization was indistinguishable from that of actin alone (Fig. 3). The inhibition was observed when UNC-60B was increased up to a ratio of 0.2: 1:1 of CeTM:UNC-60B:actin (unpublished data). In contrast, CeTM itself did not show an apparent effect on actin polymerization (Fig. 3). The slight increase in the turbidity in the presence of CeTM is probably due to binding of CeTM to F-actin as we confirmed by a pelleting assay (unpublished data). The results suggest that CeTM is a strong inhibitor of filament severing by UNC-60B.
Figure 3.

Effects of CeTM and UNC-60B on spontaneous polymerization of Ce-actin. 5 μM G-Ce-actin was incubated with or without 1 μM CeTM or 1 μM UNC-60B, and polymerized by adding salt (time 0). Time course of polymerization was measured as changes in turbidity (absorbance at 310 nm).
CeTM is a major component of thin filaments and excludes UNC-60B
To obtain evidence for in vivo competition between CeTM and UNC-60B for actin binding, native thin filaments were isolated from wild-type worms and their components examined. Actin and CeTM were detected as two major components of the thin filaments (Fig. 4 B, lane P3). UNC-60B was detected in the thin filament fraction by Western blot, but not visible by Coomassie staining of the gel (unpublished data). Quantification of actin, CeTM, and UNC-60B revealed that actin binding by CeTM is nearly saturated in the thin filaments (actin:CeTM = 1: 0.141 = 7.09: 1) and UNC-60B is a very minor component (actin:UNC-60B = 1: 9.44 × 10−7 = 1.06 × 106: 1) (Table 1).
Figure 4.

Competitive interactions of CeTM or UNC-60B with isolated thin filaments. (A) Schematic representation of fractionation of C. elegans thin filaments as described in Materials and methods. S1–6 and P1–6 were analyzed by SDS-PAGE (B). (C) The salt-extracted thin filaments (P2) were further examined for their interaction with CeTM and UNC-60B. The P2 fraction in A was resuspended with 0–2.0 μM CeTM and incubated for 30 min. Then, a final 10 μM of UNC-60B was added to some reactions (+), and after 30 min of incubation they were analyzed by a cosedimentation assay.
Table I. Quantification of actin, CeTM, and UNC-60B in the thin filament fraction.
| Amounts in 10 μg of isolated thin filaments | |||
|---|---|---|---|
| μg | nmol | mol/mol actin | |
| Actin | 7.21 ± 0.29* | 172 ± 7.2 | 1 |
| CeTM | 1.87 ± 0.064 | 24.3 ± 0.82 | 0.141 |
| UNC-60B | 2.75 × 10−3 ± 0.68 × 10−3 | 0.162 × 10−3 ± 0.040 × 10−3 | 9.44 × 10−7 |
Data shown are means ± SD of four separate experiments.
There are two possible explanations for the poor association of UNC-60B with the thin filaments: (a) interaction of UNC-60B with actin is prevented by some factors; or (b) UNC-60B was dissociated from F-actin during the fractionation. To test if UNC-60B is able to bind to the thin filaments, active recombinant UNC-60B was incubated with the isolated thin filaments and their possible interaction was examined by a cosedimentation assay, (Fig. 4). Recombinant UNC-60B cosedimented with thin filaments only at a trace amount (Fig. 4 B, lane P4) and caused partial actin depolymerization and dissociation of CeTM from the filaments (Fig. 4 B, compare S3 and S4). When the thin filaments were extracted with a buffer containing 0.6 M KCl, most of CeTM and several other proteins were dissociated from the filaments and released into the supernatant (Fig. 4 B, lane S2). After the high-salt extraction, UNC-60B was able to bind to the filaments (Fig. 4 B, lane P6). There was no detectable difference in these interactions in the presence or absence of Ca2+ (unpublished data). The results indicate that binding of UNC-60B to thin filaments is inhibited by a component(s) of the thin filaments that can be extracted by a high concentration of salt.
Because CeTM was the major protein that was extracted by high salt (Fig. 4 B, lane S2), it was a likely component that inhibited UNC-60B binding to the filaments. We experimentally confirmed the role of CeTM in this competition by an in vitro reconstitution (Fig. 4 C). After thin filaments had been extracted by 0.6 M KCl and resuspended in a buffer containing 0.1 M KCl, purified CeTM was added back to the filaments (Fig. 4 C, lanes e and f). The added CeTM bound to the filaments (Fig. 4 C, lane f). This addition of CeTM was sufficient to inhibit UNC-60B binding to the filaments (Fig. 4 C, compare lanes d and h). This inhibitory effect of CeTM was dose dependent, such that filament binding by UNC-60B was increased when added CeTM was decreased (Fig. 4 C, lanes g–l). These results indicate that CeTM is sufficient to inhibit binding of UNC-60B to muscle thin filaments.
CeTM and UNC-60B are localized in different patterns during muscle development
Relative localization of CeTM and UNC-60B during muscle development was examined in wild-type C. elegans embryos by immunofluorescence microscopy (Fig. 5). Both UNC-60B and CeTM became detectable in muscle cells at a very early stage of muscle development (∼290 min), nearly at the same time (Fig. 5, a and b). At the comma stage (290–350 min), both UNC-60B and CeTM are diffusely localized in the cytoplasm and positions of nuclei are devoid of staining (Fig. 5, a–d; DAPI staining of the nuclei is not shown). After the 1.5-fold stage (∼430 min), CeTM was gradually localized to a narrow region (Fig. 5 f) which represented myofibrils as determined by double staining with anti-actin antibody (unpublished data), whereas UNC-60B remained in the diffuse cytoplasm (Fig. 5 e). This differential localization of UNC-60B and CeTM was maintained throughout the later embryonic development (Fig. 5, g and h).
Figure 5.
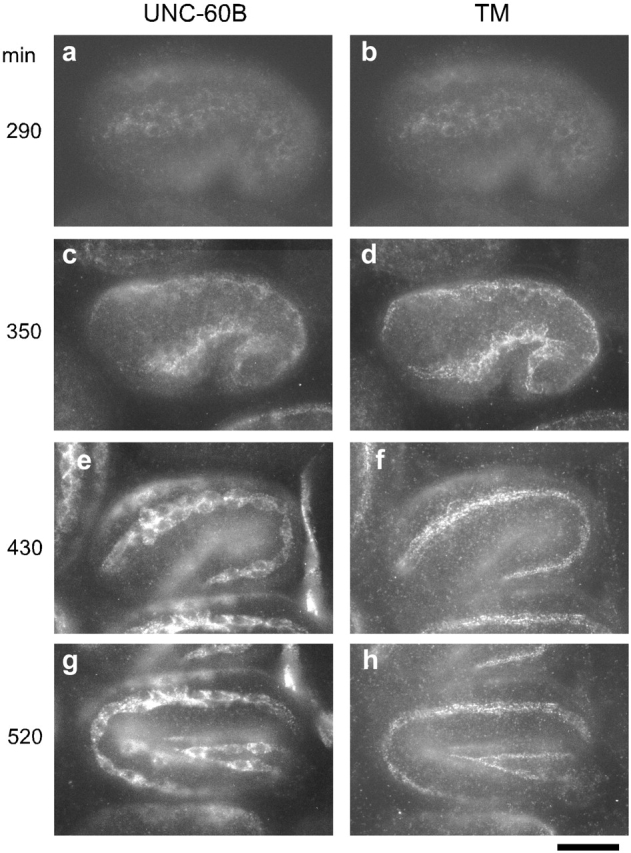
Expression and localization of UNC-60B and CeTM in C. elegans embryos. Embryos from wild-type C. elegans were double stained with anti-UNC-60B (a, c, e, and g) and anti-CeTM (b, d, f, and h) antibodies. Approximate timing of the embryos after the first cleavage is indicated on the left. Bar, 10 μm.
In adult body wall muscle, CeTM and UNC-60B were also localized to different regions of thin filaments (Fig. 6). Interestingly, the staining patterns of CeTM and actin were different (Fig. 6, A–C). CeTM was localized in linear patterns (Fig. 6 B), whereas actin was localized in ladder-like patterns (Fig. 6 A) in which the regions of dense bodies were devoid of staining (Francis and Waterston, 1985). UNC-60B was stained as dotted lines (Fig. 6 E) between two lines of CeTM with little overlapping zones (Fig. 6 D). Double staining of actin and UNC-60B showed that UNC-60B was localized to the central areas of the actin ladders between dense bodies (Fig. 6, G–I). Double staining of vinculin, as a marker for dense bodies, and UNC-60B revealed that UNC-60B filled the gaps between dense bodies but was not colocalized with them (Fig. 6, J–L). We also noted that the shape of the UNC-60B–positive dots was irregular (Fig. 6, E, H, and K), suggesting that association of UNC-60B with myofibrils is unstable and dynamic. As summarized in Fig. 6 M, actin filaments in the nematode body wall muscle are obliquely aligned with their barbed ends attached to the middle of the I-bands, where dense bodies are located instead of Z-lines in vertebrate striated muscles. Our results show that CeTM is associated with the outer parts of the I-bands, whereas UNC-60B is localized near the barbed ends of the actin filaments.
Figure 6.
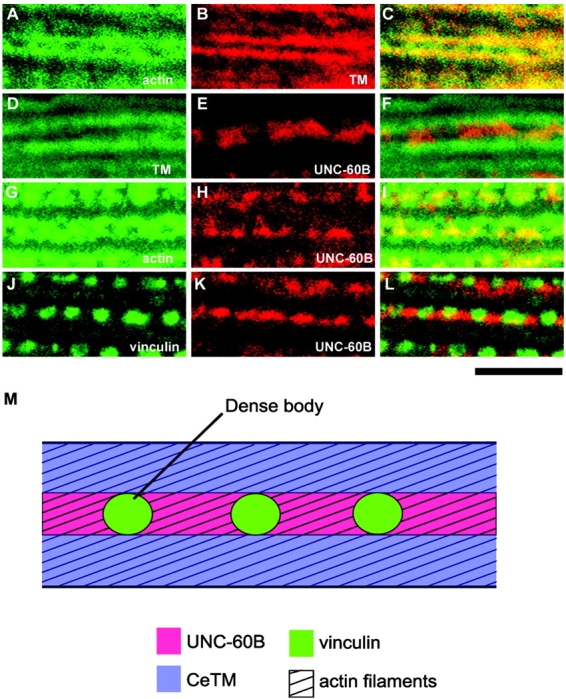
Localization of UNC-60B and CeTM in adult body wall muscle. Adult nematodes were double-stained with anti-actin (A) and anti-CeTM (B), or anti-CeTM (D), anti-actin (G), or anti-vinculin (J), and anti-UNC-60B (E, H, and K) antibodies. Merged images are shown in C, F, I, and L. Images are enlarged parts of the I-band regions. Bar, 5 μm. (M) Schematic representation of locations of UNC-60B, CeTM, vinculin, and actin in the I-bands.
Suppression of CeTM causes disorganization of actin filaments in wild-type, but not in unc-60 mutants
To address the role of CeTM in the actin filament organization in vivo we suppressed the expression of CeTM by RNA-mediated interference (RNAi) and examined its effect on the actin cytoskeleton. The C. elegans TM gene, tmy-1, gives rise to at least four isoforms using two different promoters and alternative splicing (Kagawa et al., 1995; Anyanful et al., 2001). The promoter analysis by Kagawa and colleagues (1995) has suggested that CeTMI and CeTMII are expressed in body wall muscle, whereas CeTMIII and CeTMIV are expressed in pharyngeal muscle. However, the actual tissue distribution of these proteins was not extensively studied. Therefore, we constructed two vectors for RNAi: (a) pTM1 contains a common sequence for CeTMI and CeTMII, but not for CeTMIII and CeTMIV; and (b) pTM2 contains a common sequence for all four isoforms. dsRNAs were expressed from these vectors in bacteria and delivered into worms by feeding with them. pTM1 and pTM2 successfully suppressed the expression of CeTM to different extents (Fig. 7 A). pTM1 and pTM2 reduced the amount of CeTM to ∼50% and 10%, respectively, as compared to wild-type worms grown under standard conditions. A control vector with no CeTM insert had no effect on the level of CeTM (Fig. 7 A). The levels of UNC-60B and actin were not significantly affected by CeTM RNAi (Fig. 7 A).
Figure 7.

Effects of suppression of CeTM on worm motility. (A) Protein levels of CeTM, UNC-60B, and actin were examined by Western blot. Total lysates (25 μg protein) from wild-type or unc-60 (r398) homozygous worms under standard conditions (St), or control-, TM1-, or TM2-fed worms were separated by SDS-PAGE, and subjected to Coomassie staining (CBB) or Western blots with anti-CeTM, anti–UNC-60B, or anti-actin antibodies. Molecular mass markers in kD are indicated on the left of the gel. (B) Motility of control and CeTM-suppressed worms. Data shown are means ± SD; n = 10. (C–H) Micrographs of control and CeTM-suppressed worms on agar plates. Wild-type (C–E) or unc-60 (r398). (F–H) worms were fed with control (C and F), TM1 (D and G), or TM2 (E and H). Bar, 1.0 mm.
CeTM RNAi affected motility of ∼100% of wild-type worms examined, which became slow or paralyzed (Fig. 7 B). pTM1 caused slightly mild effects: the worms could slowly move, but their tails tended to bend whereas the anterior halves of their bodies did not move well (Fig. 7, B and D). pTM2 produced much more severe paralysis, and the affected worms barely moved (Fig. 7, B and E). The motility defects in CeTM-suppressed worms were most severe when they were L4 larva to adults. In both cases, the affected worms were sterile and laid no eggs (Fig 7, D and E), but the reason for this is currently under investigation.
In contrast, CeTM RNAi did not worsen motility of an ADF/cofilin mutant strain (Fig. 7, B and F–H). unc-60 (r398) is a weak loss-of-function allele that has a premature stop codon in the unc-60B coding region, which truncates three amino acids from the C terminus. The r398 mutant UNC-60B protein binds to G-actin, but is unable to bind to and sever F-actin (Ono et al., 1999, 2001). unc-60 (r398) homozygotes move much slower than wild-type (Ono et al., 1999) and control RNAi with no CeTM inserts had no detectable effects on motility (Fig. 7 B). Interestingly, pTM1 slightly improved motility, whereas pTM2 did not have apparent effects on motility (Fig. 7, B and G). The protein levels of CeTM were decreased in the unc-60 animals to a similar extent to those in wild-type (Fig. 7 A). A t-test suggests that pTM1 produced significantly different effects from the control (P < 0.01), but pTM2 did not (P > 0.05). In addition, CeTM RNAi did not affect fecundity of unc-60 (r398) homozygotes. The CeTM-suppressed unc-60 mutants laid many fertilized eggs (Fig. 7, G and H) and produced equivalent number of progeny to the mutant worms under standard culture conditions.
Phalloidin staining of body wall muscle revealed that suppression of CeTM caused disorganization of actin filaments in wild-type but did not worsen the phenotype in unc-60 mutants (Fig. 8). In wild-type, striated organization of actin filaments were disrupted by CeTM RNAi and abnormal wavy bundles were formed in body wall muscle (Fig. 8, C and E). In the unc-60 (r398) mutants, although aggregates of actin were formed, some filaments were organized into striated patterns in the center of the cells under the control conditions (Fig. 8 B). However, CeTM RNAi did not cause major alterations in the actin filament organization in this unc-60 mutant (Fig. 8, D and F). These results suggest that in CeTM-suppressed cells, actin filaments are destabilized in wild-type, whereas they are stable when the activity of ADF/cofilin is decreased.
Figure 8.

Effects of suppression of CeTM on actin organization in body wall muscle. Actin filaments in body wall muscle were visualized by staining with rhodamine-phalloidin. Micrographs of wild-type (A, C, and E) and unc-60 (r398) (B, D, and F) that had been fed with control (A and B), TM1 (C and D), or TM2 (E and F). Bar, 10 μm.
In CeTM-suppressed cells, UNC-60B was unevenly distributed in the cells and associated with bundles where actin was also localized (Fig. 9, B, D, and F), whereas, in the control cells, UNC-60B was evenly distributed and associated with a part of myofibrils (Fig 9, A, C, and E). The UNC-60B–decorated bundles in CeTM-suppressed cells did not appear to be normal myofibril structures. These bundles were often thick and sometimes wavy (Fig. 9 B). This is strong evidence that UNC-60B heavily binds to a subset of actin filaments in CeTM-suppressed cells, which leads to destabilization of myofibril structures.
Figure 9.
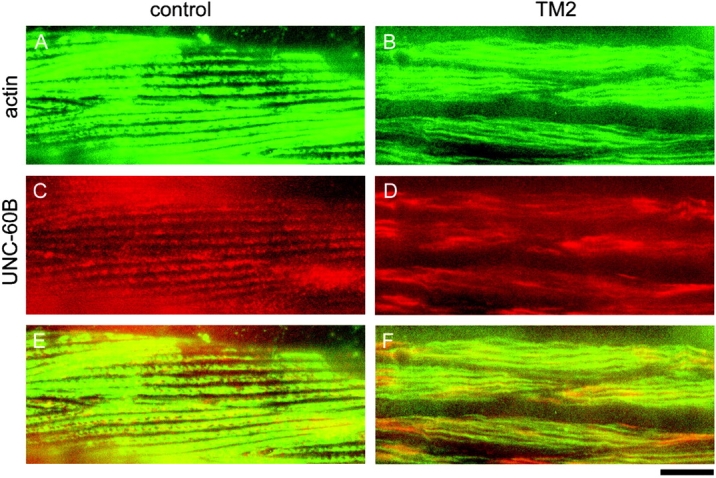
Effects of suppression of CeTM on localization of UNC-60B. Control (A, C, and E) or TM2-fed (B, D, and F) nematodes were double-stained with anti-actin (A and B) and anti–UNC-60B (C and D) antibodies. Merged images are shown in E and F. Bar, 10 μm.
CeTM is mislocalized in an unc-60 mutant
On the other hand, in the unc-60 (r398) mutant cells, considerably strong staining of CeTM was found in the aggregates, whereas some striated staining of CeTM in the myofibrils was present as well (Fig. 10 A). The r398 mutant UNC-60B protein was predominantly localized in the aggregates but barely detectable in the striated myofibrils (Fig. 10 B). Because the r398 mutant UNC-60B protein binds to G-actin but fails to bind to F-actin in vitro (Ono et al., 1999), the aggregates may contain both G- and F-actin. These results suggest that integration of CeTM into myofibrils depends on proper reorganization of actin filaments by UNC-60B.
Figure 10.
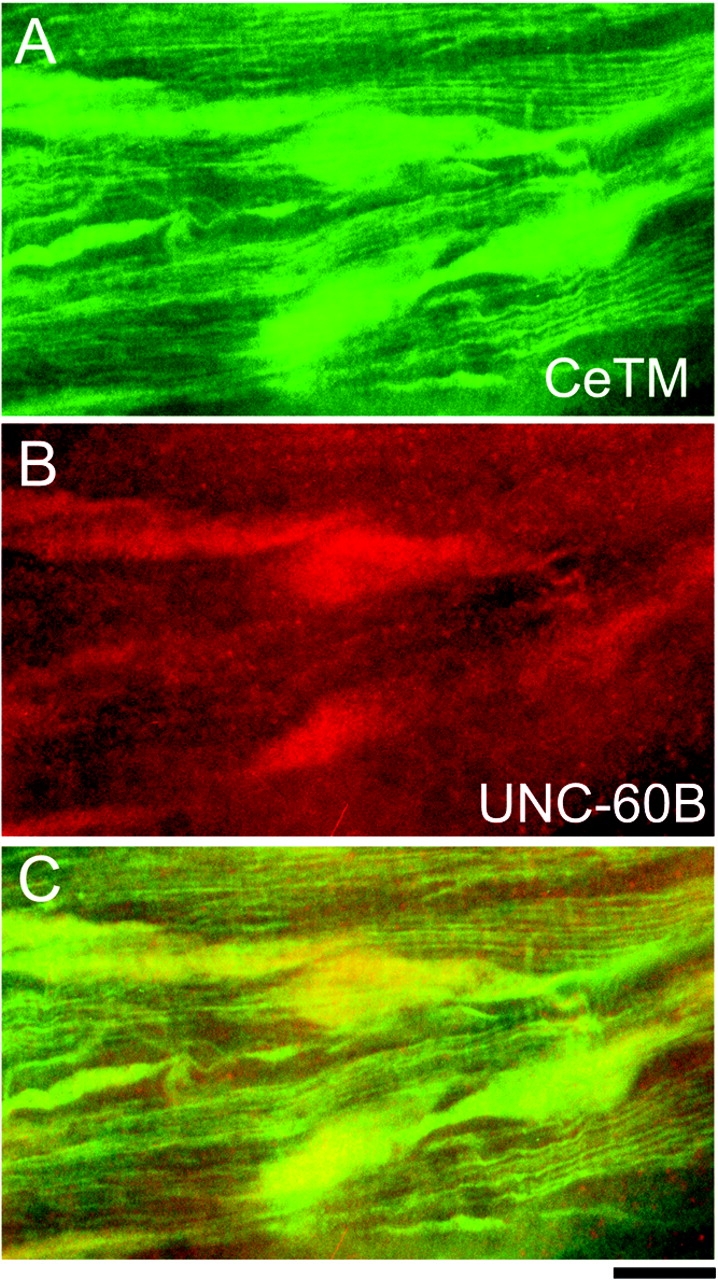
Localization of CeTM in the unc-60 (r398) mutant. Adult body wall muscle of unc-60 (r398) was double stained with anti-CeTM (A) and anti-UNC-60B (B) antibodies. Merged image is shown in C. Bar, 10 μm.
Discussion
In this study, we provide both in vitro and in vivo evidence that TM inhibits ADF/cofilin-mediated actin filament dynamics. Biochemical experiments using purified C. elegans proteins show that binding of TM or ADF/cofilin to F-actin is mutually exclusive. TM and ADF/cofilin are localized in different patterns in muscle cells. Suppression of TM disrupts actin filament organization in wild-type genetic background but not in an ADF/cofilin mutant background. These experimental results support a cellular mechanism that TM plays a role in regulating actin filament dynamics by competing with ADF/cofilin for F-actin binding.
Previous biochemical studies have shown that binding of TM to F-actin inhibits interaction of ADF/cofilin with actin. However, our study is the first to demonstrate biochemical interaction and competition of these three proteins from a single organism and confirms the competition between TM and ADF/cofilin. Binding of UNC-60B with F-actin was relatively stable in vitro, so that when UNC-60B and actin preformed a complex, CeTM did not readily replace UNC-60B. However, early muscle development is a rapid process that takes only 2 h from expression of muscle genes to forming contractile apparatuses (Epstein et al., 1993; Hresko et al., 1994). We observed expression of UNC-60B and CeTM as early as 290 min after the first cleavage, and localization of CeTM to the myofibrils at 420 min. This rapid reorganization suggests that there is a mechanism to enhance dynamic exchange of actin-bound UNC-60B with CeTM during muscle development. Candidates for such regulatory factors include a family of LIM-kinases that phosphorylate ADF/cofilins and inhibit their actin binding (Arber et al., 1998; Yang et al., 1998), and phosphoinositides that bind to ADF/cofilins and compete for actin binding (Yonezawa et al., 1990; Van Troys et al., 2000). The role of LIM-kinases in muscle cells has not been reported. In contrast, phosphatidylinositol 4,5-bisphosphate has been implicated in inhibition of cofilin-induced actin reorganization in chicken skeletal muscle cells (Nagaoka et al., 1995).
In the isolated nematode thin filaments, UNC-60B is a minor component. Our in vitro reconstitution experiments suggest that CeTM excludes UNC-60B from the thin filaments. However, we noted that UNC-60B bound to actin in the thin filaments at a lesser amount than to purified actin, even after CeTM was removed by the high-salt extraction. This observation suggests that thin filaments have other factors that prevent UNC-60B from binding to them. A strong candidate of such a factor is UNC-87, a calponin-like protein (Goetinck and Waterston, 1994a). UNC-87 is localized to thin filaments in C. elegans body wall muscle and essential for maintenance of myofibril structures (Goetinck and Waterston, 1994b). UNC-87 binds to TM-bound actin filaments in vitro (Kranewitter et al., 2001). When UNC-87 is expressed in mammalian cells, it is localized to stress fibers and remains associated with cytoskeletal fractions after extraction with 0.8 M KCl (Kranewitter et al., 2001). Therefore, the extraction of the nematode thin filaments with 0.6 M KCl to remove CeTM probably did not dissociate UNC-87 from the filaments. It will be interesting to examine the effect of UNC-87 and other calponin-like proteins on ADF/cofilin-mediated actin filament dynamics. Interestingly, in chicken muscle cells, when the myofibrils become mature, microinjected cofilin replaces TM on myofibrils without disrupting the myofibril structure (Nagaoka et al., 1995), suggesting the presence of an actin filament stabilizer in addition to TM.
By immunostaining, we found that UNC-60B was localized to the center of the I-bands where CeTM was not found. However, UNC-60B was a very minor component in our thin filament preparations. This discrepancy might be because UNC-60B–bound filaments were unstable, so that they were disassembled during the preparation. Alternatively, UNC-60B might be associated with structural components other than actin filaments. In the previous model of the C. elegans body wall muscle, thin filaments are proposed to be anchored to dense bodies which are 1.5–2.0 μm apart from each other (Waterston, 1988). If this model is correct, the staining of actin would become discontinuous and be absent in the center of the I-bands. However, actual staining for actin appears as continuous lines in the I-bands and is present in the center of the I-bands between dense bodies. Therefore, it would be more likely that the thin filaments are attached not only to dense bodies but also to regions between dense bodies where UNC-60B is localized. Indeed, in the ultrastructure of body wall muscle of the nematode Ascaris, dense bodies are linked together by strands of the “supporting fibrils,” which are located throughout the center of the I-bands (Rosenbluth, 1965, 1967). Therefore, the equivalent structure may be present in C. elegans and function as a scaffold for thin filaments and UNC-60B.
We demonstrated that suppression of CeTM expression by RNAi caused disorganization of muscle actin filaments in wild-type but not in an unc-60 mutant strain. In the CeTM-suppressed worms, the phenotypes were most severe when they were adults, suggesting that, after assembly of myofibrils, actin filaments are gradually deteriorated by the activity of UNC-60B. That also implies that UNC-60B is still active in mature muscle cells and has the ability to enhance actin dynamics. The competitiveness between TM and ADF/cofilin suggests that the balance of two kinds of factors, stabilizers and destabilizers of actin filaments, is important for maintaining the integrity of myofibrils. In C. elegans, enhancers of actin dynamics, UNC-60B (ADF/cofilin) (Ono et al., 1999) and UNC-78 (actin-interacting protein 1) (Ono, 2001), are required for assembly of actin into myofibrils. However, it is not clear whether these factors are important for maintenance of myofibrils because mutations of these genes cause formation of disorganized myofibrils from the assembly stage in embryos. Myofibrils are a form of very stable actin cytoskeleton, and yet myofibrils in cultured muscle cells are capable of incorporating microinjected actin without altering the length of the thin filaments (Imanaka-Yoshida et al., 1993; Komiyama et al., 1993; Littlefield et al., 2001). In mammals, a muscle-specific cofilin isoform is expressed in striated muscles (Ono et al., 1994; Mohri et al., 2000; Thirion et al., 2001) and is a strong candidate for an enhancer of actin dynamics. To understand this problem, a conditional allele of ADF/cofilin would allow us to investigate a role of ADF/cofilin and its functional relationship with stabilizers of actin filaments after functional myofibrils had been assembled.
Materials and methods
Nematode strains
Nematodes were grown at 20°C as described (Brenner, 1974). Wild-type strain N2 was obtained from the Caenorhabditis Genetics Center (St. Paul, MN). unc-60 (r398) was described previously (McKim et al., 1988).
Proteins
Actin was purified from wild-type C. elegans as described (Ono, 1999). Recombinant UNC-60B was expressed in Escherichia coli and purified as described (Ono and Benian, 1998).
Purification of tropomyosin from wild-type C. elegans
Frozen nematodes (30–40 ml as packed volume) were thawed in 2 vol (1 vol refers to an initial volume of packed worms) of a homogenizing buffer (50 mM NaCl, 1 mM EDTA, 20 mM Tris-HCl, 1 mM DTT, 1 mM PMSF, pH 8.0), and homogenized by passing twice through a French pressure cell at 5,000–8,000 lb/in2. The homogenate was centrifuged at 10,000 g for 10 min, and the pellet was washed twice by suspending in the homogenizing buffer and pelleting at 10,000 g for 10 min. The washed pellet was extracted with 1 vol of 0.6 M KCl, 5 mM ATP, 5 mM MgCl2, 0.2 mM EGTA, 20 mM Tris-HCl, 1 mM DTT, 1 mM PMSF, pH 8.0, and centrifuged at 10,000 g for 10 min. The extraction was repeated once more. The combined supernatant was mixed with 1/20 vol of 10% Triton X-100 and centrifuged at 100,000 g for 2 h. The supernatant was heated in a boiling water bath for 15 min, and then centrifuged at 10,000 g for 15 min. The supernatant was fractionated at 55–80% saturation of ammonium sulfate and dialyzed extensively against 0.1 M KCl, 20 mM Tris-HCl, pH 8.0. It was applied to a Mono-Q column that had been equilibrated with the same buffer and eluted with a linear gradient of KCl (0.1–0.6 M). CeTM was eluted at 0.44–0.48 M KCl. The fractions containing pure CeTM were dialyzed against 0.1 M KCl, 20 mM Hepes-NaOH, pH 7.5, and concentrated by Centricon (MWCO 30,000) (Millipore). The protein concentration was determined by a BCA Protein Assay Kit (Pierce Chemical Co.). The molar concentration of CeTM was calculated as a dimer using a M r of 66,000 that was deduced from the cDNA sequences of CeTMI and CeTMII (Kagawa et al., 1995).
Assays for actin-binding and actin polymerization
Cosedimentation assays of Ce-actin with UNC-60B and/or CeTM were performed as described previously (Ono and Benian, 1998) in a buffer containing 0.1 M KCl, 2 mM MgCl2, 20 mM Hepes-NaOH, 1 mM DTT, pH 7.5. Ultracentrifugation was performed with a Beckman Airfuge at 28 psi for 20 min. The supernatants and pellets were adjusted to the same volume and analyzed by SDS-PAGE. In standard SDS-PAGE, actin and CeTM migrated so closely that densitometric quantification of the two bands was very difficult. We found that inclusion of CaCl2 at 1 mM in a 10% polyacrylamide gel and at 0.1 mM in running buffer slightly retards the migration of actin, but not CeTM. Therefore, calcium-containing electrophoresis was used for densitometric analysis of CeTM and Ce-actin.
The kinetics of spontaneous actin polymerization was monitored as changes in turbidity at a wavelength of 310 nm (Carlier et al., 1997) under conditions as described previously (Ono et al., 1999).
Isolation of nematode thin filaments
Nematode thin filaments were isolated as described by Harris et al. (Harris et al., 1977) with slight modifications. Nematodes (6 ml as a pellet) were suspended in 12 ml of Buffer A (50 mM NaCl, 10 mM Imidazole, 1 mM EDTA, 5 mM DTT, 1 mM PMSF, pH 7.0) and homogenized by passing twice through a French pressure cell at 5,000–8,000 lb/in2. The homogenate was centrifuged at 8,000 g for 10 min, and the pellet was washed twice by suspending in Buffer A and pelleting at 8,000 g for 10 min. The pellet was extracted 3 min on ice in 4 ml of a buffer containing 50 mM KCl, 5 mM MgCl2, 0.2 mM EGTA, 5 mM ATP, 10 mM MES-KOH, 1 mM DTT, 1 mM PMSF, pH 6.0 and centrifuged at 10,000 g for 10 min. The supernatant, which is designated as “worm low-salt/ATP extract,” was ultracentrifuged at 140,000 g for 20 min. The resultant pellet contained actin and TM as major components and was considered as a thin filament fraction. The protein concentration was determined by a BCA Protein Assay Kit (Pierce Chemical Co.). Amounts of Ce-actin and CeTM in the thin filament fraction were quantified on Coomassie-stained gels followed by a densitometric analysis using purified Ce-actin and CeTM as standards. Amounts of UNC-60B in the thin filaments were quantified by Western blot with anti–UNC-60B antibody using purified UNC-60B as a standard. Quantification was performed in ranges where band intensity of the standard proteins was linearly correlated with their amounts.
Cosedimentation assays with thin filaments
The worm low-salt/ATP extract (50 μl at 1.5 mg/ml) was supplemented with the same volume of 50 mM or 3 M KCl solutions to adjust KCl to final 50 mM or 0.6 M and ultracentrifuged by a Beckman Airfuge at 28 psi for 20 min. The pellets were resuspended in 0.1 M KCl, 2 mM MgCl2, 1 mM DTT, 20 mM Hepes-NaOH, pH 7.5, by gentle pipetting, incubated with or without 10 μM UNC-60B in a total volume of 50 μl for 30 min at room temperature, and ultracentrifuged by a Beckman Airfuge at 28 psi for 20 min. Both supernatants and pellets were adjusted to 100 μl and analyzed by SDS-PAGE.
For reconstitution with CeTM, after the worm low-salt/ATP extract was treated with 0.6 M KCl and ultracentrifuged, the pellets were resuspended in 0.1 M KCl, 2 mM MgCl2, 1 mM DTT, 20 mM Hepes-NaOH, pH 7.5, with 0 to 2.0 μM of purified CeTM in a total volume of 50 μl, and incubated for 15 min at room temperature. They were then supplemented with UNC-60B at final volume of 10 μM or the same volume of the buffer, incubated for 30 min at room temperature, and ultracentrifuged by a Beckman Airfuge at 28 psi for 20 min.
Production of antibodies
Purified CeTM was used to immunize guinea pigs by a standard protocol at Spring Valley Laboratories (Woodvine, MD). The antiserum was specific for CeTM by Western blot without further purification and used at 1:500 to 1:1,000 dilutions for Western blot and immunofluorescent staining.
Fluorescent microscopy
Worm embryos were obtained by cutting adults on poly-lysine–coated slides, freeze cracked as described (Epstein et al., 1993), and fixed with methanol at –20°C for 5 min, and, subsequently, with 4% formaldehyde in cytoskeleton buffer (138 mM KCl, 3 mM MgCl2, 2 mM EGTA, 10 mM MES-KOH, pH 6.0) containing 0.32 M sucrose (Cramer and Mitchison, 1993) for 15 min at room temperature. They were washed with PBS containing 30 mM glycine for 10 min and stained with antibodies diluted in 1% BSA in PBS. Anti-CeTM and anti–UNC-60B antibodies were used as primary antibodies, which are visualized by Alexa488-labeled goat anti–guinea pig IgG (Molecular Probes) and Cy3-labeled goat anti–rabbit IgG (Jackson ImmunoResearch Laboratories). Immunofluorescent staining of adult nematodes was performed as described (Finney and Ruvkun, 1990) using anti-CeTM, anti-UNC-60B, anti-actin (C4; ICN Biomedicals), or anti-vinculin (MH24; Francis and Waterston, 1985), provided by Dr. M. Hresko (Washington University School of Medicine, Seattle, WA). Phalloidin staining of adult worms was performed as described previously (Ono, 2001). Samples were viewed by epifluorescence using a Zeiss Axioskop microscope with a Plan-Neofluar 100× objective and photographs were taken on Kodak TMAX 400 film. The negatives were scanned at 2,400 dot per inch and the images processed by Adobe Photoshop 6.0.
RNA interference experiments
Two fragments of cDNA for CeTM were amplified from a C. elegans cDNA library, provided by Dr. A. Fire (Carnegie Institution of Washington, Washington, D.C.) by PCR with REDTaq DNA polymerase (Sigma-Aldrich). TM1 is a 549-bp fragment corresponding to position 5–553 of the cDNA for CeTMI (GenBank/EMBL/DDBJ accession no. D38540), which is common with CeTMII but not with CeTMIII and IV. TM2 is a 383-bp fragment corresponding to position 839–1,221 of the cDNA for CeTMI, which is common for all four isoforms. The amplified DNA fragments were digested by Bgl II and Nhe I at the sites introduced by the PCR primers and cloned into L4440 at the cloning site between two oppositely oriented T7 promoters (Timmons and Fire, 1998). Forward and reverse PCR primers for TM1 are 5′-CATGAGATCTCCCCTCCAATTTGAATCGTCG and 5′-CATGGCTAGCTCGTATTTGCGGTCAGCCTCC. Forward and reverse PCR primers for TM2 are 5′-CATGAGATCTGAATTGGTGCACGAGAAGGAAC and 5′-CATGGCTAGCGCTGCCGGCTCTTGTAAACG.
E. coli HT115(DE3), an RNase III–deficient strain (Timmons et al., 2001), was transformed with the L4440 vector with the CeTM inserts and used to suppress the expression of CeTM by RNA-mediated interference (RNAi) by feeding worms (Timmons and Fire, 1998). RNAi was performed essentially as described (Fraser et al., 2000). Briefly, the transformed bacteria were cultured overnight at 37°C in LB containing 50 μg/ml ampicillin, applied onto 10-cm plates containing NGM agar with 1 mM IPTG and 25 μg/ml carbenicillin, and incubated overnight at room temperature. Ten worms at the L4 stage were transferred onto the plates and cultured 24 h at 20°C. The worms were transferred onto new plates and cultured for 3 d at 20°C, and phenotypes were analyzed in their F1 generation.
Western blot
Worms were harvested off NGM plates with M9 buffer (22 mM KH2PO4, 42 mM Na2HPO4, 85.5 mM NaCl, 1 mM MgSO4) and washed with M9 three times. After removing M9, the worms were suspended in SDS-lysis buffer (2% SDS, 80 mM Tris-HCl, 5% β-mercaptoethanol, 15% glycerol, 0.05% bromophenol blue, pH 6.8), heated at 97°C for 2 min, homogenized by brief sonication, and heated again at 97°C for 2 min. Protein concentrations of the SDS lysates were determined by a filter paper dye-binding assay (Minamide and Bamburg, 1990). 25 μg of each lysate was separated on a 12% SDS-polyacrylamide gel and transferred onto polyvinylidene difluoride membrane (Immobilon-P; Millipore). The membranes were blocked in 5% nonfat milk in PBS containing 0.1% Tween 20 and incubated for 1 h with anti-CeTM, anti-UNC-60B or anti-actin (C4) antibodies followed by treatment with peroxidase-labeled goat anti–guinea pig IgG (ICN Biomedicals), goat anti–rabbit IgG (Pierce Chemical Co.), or goat anti–mouse IgG (Pierce Chemical Co.). The reactivities were detected with a SuperSignal chemiluminescence reagent (Pierce Chemical Co.).
Motility assay
A motility assay was performed as described (Epstein and Thomson, 1974). Briefly, adult worms were placed in M9 buffer. Then, one beat was counted when a worm swung its head to either right or left. The total number of beats in 30 s was recorded.
Acknowledgments
We thank F. Matsumura for technical advice on electrophoresis, and H. Joshi and G. Benian for access to their equipment and comments on the manuscript. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources.
This work was supported by grants from the American Heart Association, the National Science Foundation (MCB-0110464), and the University Research Committee of Emory University to S. Ono.
Footnotes
Abbreviations used in this paper: ADF, actin-depolymerizing factor; CeTM, Caenorhabditis elegans tropomyosin; RNAi, RNA-mediated interference; TM, tropomyosin.
References
- Abe, H., S. Ohshima, and T. Obinata. 1989. A cofilin-like protein is involved in the regulation of actin assembly in developing skeletal muscle. J. Biochem. 106:696–702. [DOI] [PubMed] [Google Scholar]
- Agnew, B.J., L.S. Minamide, and J.R. Bamburg. 1995. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J. Biol. Chem. 270:17582–17587. [DOI] [PubMed] [Google Scholar]
- Amato, P.A., and D.L. Taylor. 1986. Probing the mechanism of incorporation of fluorescently labeled actin into stress fibers. J. Cell Biol. 102:1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anyanful, A., Y. Sakube, K. Takuwa, and H. Kagawa. 2001. The third and fourth tropomyosin isoforms of Caenorhabditis elegans are expressed in the pharynx and intestines and are essential for development and morphology. J. Mol. Biol. 313:525–537. [DOI] [PubMed] [Google Scholar]
- Arber, S., F.A. Barbayannis, H. Hanser, C. Schneider, C.A. Stanyon, O. Bernard, and P. Caroni. 1998. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 393:805–809. [DOI] [PubMed] [Google Scholar]
- Bamburg, J.R. 1999. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 15:185–230. [DOI] [PubMed] [Google Scholar]
- Bamburg, J.R., and D. Bray. 1987. Distribution and cellular localization of actin-depolymerizing factor. J. Cell Biol. 105:2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg, J.R., A. McGough, and S. Ono. 1999. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol. 9:364–370. [DOI] [PubMed] [Google Scholar]
- Belmont, L.D., and D.G. Drubin. 1998. The yeast V159N actin mutant reveals roles for actin dynamics in vivo. J. Cell Biol. 142:1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, B.W., and J.R. Bamburg. 1982. Tropomyosin binding to F-actin protects the F-actin from disassembly by brain actin-depolymerizing factor (ADF). Cell Motil. 2:1–8. [DOI] [PubMed] [Google Scholar]
- Blanchard, E.M., K. Iizuka, M. Christe. D.A. Conner, A. Geister-Lowrance, F.J. Schoen, D.W Maughan, C.E. Seideman, and J.G. Seideman. 1997. Targeted ablation of the murine α-tropomyosin gene. Circ. Res. 81:1005–1010. [DOI] [PubMed] [Google Scholar]
- Blanchoin, L., T.D. Pollard, and S.E. Hitchcock-DeGregori. 2001. Inhibition of the Arp2/3 complex-nucleated actin polymerization and branch formation by tropomyosin. Curr. Biol. 11:1300–1304. [DOI] [PubMed] [Google Scholar]
- Brenner, S. 1974. The genetics of Caenorhabditis elegans. Genetics. 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broschat, K.O. 1990. Tropomyosin prevents depolymerization of actin filaments from the pointed end. J. Biol. Chem. 265:21323–21329. [PubMed] [Google Scholar]
- Broschat, K.O., A. Weber, and D.R. Burgess. 1989. Tropomyosin stabilizes the pointed end of actin filaments by slowing depolymerization. Biochemistry. 28:8501–8506. [DOI] [PubMed] [Google Scholar]
- Carlier, M.F., V. Laurent, J. Santolini, R. Melki, D. Didry, G.X. Xia, Y. Hong, N.H. Chua, and D. Pantaloni. 1997. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J. Cell Biol. 136:1307–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier, M.F., F. Ressad, and D. Pantaloni. 1999. Control of actin dynamics in cell motility. Role of ADF/cofilin. J. Biol. Chem. 274:33827–33830. [DOI] [PubMed] [Google Scholar]
- Chan, A.Y., M. Bailly, N. Zebda, J.E. Segall, and J.S. Condeelis. 2000. role of cofilin in epidermal growth factor–stimulated actin polymerization and lamellipod protrusion. J. Cell Biol. 148:531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, J.A., and D.A. Schafer. 2000. Control of actin assembly and disassembly at filament ends. Curr. Opin. Cell Biol. 12:97–103. [DOI] [PubMed] [Google Scholar]
- Cramer, L., and T.J. Mitchison. 1993. Moving and stationary actin filaments are involved in spreading of postmitotic PtK2 cells. J. Cell Biol. 122:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didry, D., M.F. Carlier, and D. Pantaloni. 1998. Synergy between actin depolymerizing factor/cofilin and profilin in increasing actin filament turnover. J. Biol. Chem. 273:25602–25611. [DOI] [PubMed] [Google Scholar]
- Epstein, H.F., and J.N. Thomson. 1974. Temperature-sensitive mutation affecting myofilament assembly in Caenorhabditis elegans. Nature. 250:579–580. [DOI] [PubMed] [Google Scholar]
- Epstein, H.F., D.L. Casey, and I. Ortiz. 1993. Myosin and paramyosin of Caenorhabditis elegans embryos assemble into nascent structures distinct from thick filaments and multifilament assemblages. J. Cell Biol. 122:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney, M., and G. Ruvkun. 1990. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 63:895–905. [DOI] [PubMed] [Google Scholar]
- Francis, G.R., and R.H. Waterston. 1985. Muscle organization in Caenorhabditis elegans: localization of proteins implicated in thin filament attachment and I-band organization. J. Cell Biol. 101:1532–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, A.G., R.S. Kamath, P. Zipperlen, M. Martinez-Campos, M. Sohrmann, and J. Ahringer. 2000. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 408:325–330. [DOI] [PubMed] [Google Scholar]
- Glacy, S.D. 1983. Subcellular distribution of rhodamine-actin microinjected into living fibroblastic cells. J. Cell Biol. 97:1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetinck, S., and R.H. Waterston. 1994. a. The Caenorhabditis elegans muscle-affecting gene unc-87 encodes a novel thin filament-associated protein. J. Cell Biol. 127:79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetinck, S., and R.H. Waterston. 1994. b. The Caenorhabditis elegans UNC-87 protein is essential for maintenance, but not assembly, of bodywall muscle. J. Cell Biol. 127:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, A.M., E. Homsher, and M. Regnier. 2000. Regulation of contraction in striated muscle. Physiol. Rev. 80:853–924. [DOI] [PubMed] [Google Scholar]
- Harris, H.E., M.Y. Tso, and H.F. Epstein. 1977. Actin and myosin-linked calcium regulation in the nematode Caenorhabditis elegans. Biochemical and structural properties of native filaments and purified proteins. Biochemistry. 16:859–865. [DOI] [PubMed] [Google Scholar]
- Hitchcock-DeGregori, S.E., P. Sampath, and T.D. Pollard. 1988. Tropomyosin inhibits the rate of actin polymerization by stabilizing actin filaments. Biochemistry. 27:9182–9185. [DOI] [PubMed] [Google Scholar]
- Hresko, M.C., B.D. Williams, and R.H. Waterston. 1994. Assembly of body wall muscle and muscle cell attachment structures in Caenorhabditis elegans. J. Cell Biol. 124:491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichetovkin, I., J. Han, K.M. Pang, D.A. Knecht, and J.S. Condeelis. 2000. Actin filaments are severed by both native and recombinant Dictyostelium cofilin but to different extents. Cell Motil. Cytoskel. 45:293–306. [DOI] [PubMed] [Google Scholar]
- Imanaka-Yoshida, K., J.M. Sanger, and J.W. Sanger. 1993. Contractile protein dynamics of myofibrils in paired adult rat cardiomyocytes. Cell Motil. Cytoskeleton. 26:301–312. [DOI] [PubMed] [Google Scholar]
- Kagawa, H., K. Sugimoto, H. Matsumoto, T. Inoue, H. Imadzu, K. Takuwa, and Y. Sakube. 1995. Genome structure, mapping and expression of the tropomyosin gene tmy-1 of Caenorhabditis elegans. J. Mol. Biol. 251:603–613. [DOI] [PubMed] [Google Scholar]
- Komiyama, M., K. Kouchi, K. Maruyama, and Y. Shimada. 1993. Dynamics of actin and assembly of connectin (titin) during myofibrillogenesis in embryonic chick cardiac muscle cells in vitro. Dev. Dyn. 196:291–299. [DOI] [PubMed] [Google Scholar]
- Kranewitter, W.J., J. Ylanne, and M. Gimona. 2001. UNC-87 is an actin-bundling protein. J. Biol. Chem. 276:6306–6312. [DOI] [PubMed] [Google Scholar]
- Kreuz, A.J., A. Simcox, and D. Maughan. 1996. Alterations in flight muscle ultrastructure and function in Drosophila tropomyosin mutants. J. Cell Biol. 135:673–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal, A.A., and E.D. Korn. 1986. Effect of muscle tropomyosin on the kinetics of polymerization of muscle actin. Biochemistry. 25:1154–1158. [DOI] [PubMed] [Google Scholar]
- Lappalainen, P., and D.G. Drubin. 1997. Cofilin promotes rapid actin filament turnover in vivo. Nature. 388:78–82. [DOI] [PubMed] [Google Scholar]
- Lappalainen, P., E.V. Fedorov, A.A. Fedorov, S.C. Almo, and D.G. Drubin. 1997. Essential functions and actin-binding surfaces of yeast cofilin revealed by systematic mutagenesis. EMBO J. 16:5520–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J.J., K.S. Warren, D.D. Wamboldt, T. Wang, and J.L. Lin. 1997. Tropomyosin isoforms in nonmuscle cells. Int. Rev. Cytol. 170:1–38. [DOI] [PubMed] [Google Scholar]
- Littlefield, R., A. Almenar-Queralt, and V.M. Fowler. 2001. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat. Cell Biol. 3:544–551. [DOI] [PubMed] [Google Scholar]
- Liu, H.P., and A. Bretscher. 1989. Disruption of the single tropomyosin gene in yeast results in the disappearance of actin cables from the cytoskeleton. Cell. 57:233–242. [DOI] [PubMed] [Google Scholar]
- Mabuchi, I. 1982. Effects of muscle proteins on the interaction between actin and an actin-depolymerizing protein from starfish oocytes. J. Biochem. 92:1439–1447. [DOI] [PubMed] [Google Scholar]
- Maciver, S.K., H.G. Zot, and T.D. Pollard. 1991. Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J. Cell Biol. 115:1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciver, S.K., B.J. Pope, S. Whytock, and A.G. Weeds. 1998. The effect of two actin-depolymerizing factors (ADF/cofilins) on actin filament turnover: pH sensitivity of F-actin binding by human ADF, but not of Acanthamoeba actophorin. Eur. J. Biochem. 256:388–397. [DOI] [PubMed] [Google Scholar]
- McGough, A. 1998. F-actin-binding proteins. Curr. Opin. Struct. Biol. 8:166–176. [DOI] [PubMed] [Google Scholar]
- McGough, A., and W. Chiu. 1999. ADF/cofilin weakens lateral contacts in the actin filament. J. Mol. Biol. 291:513–519. [DOI] [PubMed] [Google Scholar]
- McGough, A., B. Pope, W. Chiu, and A. Weeds. 1997. Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J. Cell Biol. 138:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim, K.S., M.F. Heschl, R.E. Rosenbluth, and D.L. Baillie. 1988. Genetic organization of the unc-60 region in Caenorhabditis elegans. Genetics. 118:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamide, L.S., and J.R. Bamburg. 1990. A filter paper dye-binding assay for quantitative determination of protein without interference from reducing agents or detergents. Anal. Biochem. 190:66–70. [DOI] [PubMed] [Google Scholar]
- Mohri, K., H. Takano-Ohmuro, H. Nakashima, K. Hayakawa, T. Endo, K. Hanaoka, and T. Obinata. 2000. Expression of cofilin isoforms during development of mouse striated muscles. J. Muscle Res. Cell Motil. 21:49–57. [DOI] [PubMed] [Google Scholar]
- Moon, A.L., P.A. Janmey, K.A. Louie, and D.G. Drubin. 1993. Cofilin is an essential component of the yeast cortical cytoskeleton. J. Cell Biol. 120:421–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, T.E., R.O. Lockerbie, L.S. Minamide, M.D. Browning, and J.R. Bamburg. 1993. Isolation and characterization of a regulated form of actin-depolymerizing factor. J. Cell Biol. 122:623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama, K., and I. Yahara. 1999. Two activities of cofilin, severing and accelerating directional depolymerization of actin filaments, are affected differentially by mutations around the actin-binding helix. EMBO J. 18:6752–6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama, K., K. Iida, and I. Yahara. 1996. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells. 1:73–86. [DOI] [PubMed] [Google Scholar]
- Nagaoka, R., K. Kusano, H. Abe, and T. Obinata. 1995. Effects of cofilin on actin filamentous structures in cultured muscle cells. Intracellular regulation of cofilin action. J. Cell Sci. 108:581–593. [DOI] [PubMed] [Google Scholar]
- Nishida, E., S. Maekawa, and H. Sakai. 1984. Cofilin, a protein in porcine brain that binds to actin filaments and inhibits their interactions with myosin and tropomyosin. Biochemistry. 23:5307–5313. [DOI] [PubMed] [Google Scholar]
- Nishida, E., E. Muneyuki, S. Maekawa, Y. Ohta, and H. Sakai. 1985. An actin-depolymerizing protein (destrin) from porcine kidney. Its action on F-actin containing or lacking tropomyosin. Biochemistry. 24:6624–6630. [DOI] [PubMed] [Google Scholar]
- Okabe, S., and N. Hirokawa. 1989. Incorporation and turnover of biotin-labeled actin microinjected into fibroblastic cells: an immunoelectron microscopic study. J. Cell Biol. 109:1581–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, S. 1999. Purification and biochemical characterization of actin from Caenorhabditis elegans: its difference from rabbit muscle actin in the interaction with nematode ADF/cofilin. Cell Motil. Cytoskel. 43:128–136. [DOI] [PubMed] [Google Scholar]
- Ono, S. 2001. The Caenorhabditis elegans unc-78 gene encodes a homologue of actin-interacting protein 1 required for organized assembly of muscle actin filaments. J. Cell Biol. 152:1313–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, S., and G.M. Benian. 1998. Two Caenorhabditis elegans actin depolymerizing factor/cofilin proteins, encoded by the unc-60 gene, differentially regulate actin filament dynamics. J. Biol. Chem. 273:3778–3783. [DOI] [PubMed] [Google Scholar]
- Ono, S., N. Minami, H. Abe, and T. Obinata. 1994. Characterization of a novel cofilin isoform that is predominantly expressed in mammalian skeletal muscle. J. Biol. Chem. 269:15280–15286. [PubMed] [Google Scholar]
- Ono, S., D.L. Baillie, and G.M. Benian. 1999. UNC-60B, an ADF/cofilin family protein, is required for proper assembly of actin into myofibrils in Caenorhabditis elegans body wall muscle. J. Cell Biol. 145:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, S., A. McGough, B.J. Pope, V.T. Tolbert, A. Bui, J. Pohl, G.M. Benian, K.M. Gernert, and A.G. Weeds. 2001. The C-terminal tail of UNC-60B (ADF/cofilin) is critical for maintaining its stable association with F-actin and is implicated in the second actin-binding site. J. Biol. Chem. 276:5952–5958. [DOI] [PubMed] [Google Scholar]
- Pittenger, M.F., J.A. Kazzaz, and D.M. Helfman. 1994. Functional properties of non-muscle tropomyosin isoforms. Curr. Opin. Cell Biol. 6:96–104. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., L. Blanchoin, and R.D. Mullins. 2000. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29:545–576. [DOI] [PubMed] [Google Scholar]
- Pope, B.J., S.M. Gonsior, S. Yeoh, A. McGough, and A.G. Weeds. 2000. Uncoupling actin filament fragmentation by cofilin from increased subunit turnover. J. Mol. Biol. 298:649–661. [DOI] [PubMed] [Google Scholar]
- Pruyne, D.W., D.H. Schott, and A. Bretscher. 1998. Tropomyosin-containing actin cables direct the Myo2p-dependent polarized delivery of secretory vesicles in budding yeast. J. Cell Biol. 143:1931–1945. [DOI] [PubMed] [Google Scholar]
- Rethinasamy, P., M. Muthuchamy, T. Hewett, G. Boivin, B.M. Wolska, C. Evans, R.J. Solaro, and D.F. Wieczorek. 1998. Molecular and physiological effects of α-tropomyosin ablation in the mouse. Circ. Res. 82:116–123. [DOI] [PubMed] [Google Scholar]
- Rosenbluth, J. 1965. Ultrastructural organization of obliquely striated muscle fibers in Ascaris lumbricoides. J. Cell Biol. 25:495–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth, J. 1967. Obliquely striated muscle III. Contraction mechanism of Ascaris body muscle. J. Cell Biol. 34:15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie, L.B. 1982. Preparation and identification of alpha- and beta-tropomyosins. Methods Enzymol. 85:234–241. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1999. Arp2/3 complex and actin-depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirion, C., R. Stucka, B. Mendel, A. Gruhler, M. Jaksch, K.J. Nowak, N. Binz, N.G. Laing, and H. Lochmuller. 2001. Characterization of human muscle type cofilin (CFL2) in normal and regenerating muscle. Eur. J. Biochem. 268:3473–3482. [DOI] [PubMed] [Google Scholar]
- Timmons, L., and A. Fire. 1998. Specific interference by ingested dsRNA. Nature. 395:854. [DOI] [PubMed] [Google Scholar]
- Timmons, L., D.L. Court, and A. Fire. 2001. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 263:103–112. [DOI] [PubMed] [Google Scholar]
- Van Troys, M., D. Dewitte, J.L. Verschelde, M. Goethals, J. Vandekerckhove, and C. Ampe. 2000. The competitive interaction of actin and PIP2 with actophorin is based on overlapping target sites: design of a gain-of-function mutant. Biochemistry. 39:12181–12189. [DOI] [PubMed] [Google Scholar]
- Wang, Y.L. 1985. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J. Cell Biol. 101:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston, R.H. 1988. Muscle. The Nematode C. elegans. W.B. Wood, editor. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 281–335.
- Williams, B.D., and R.H. Waterston. 1994. Genes critical for muscle development and function in Caenorhabditis elegans identified through lethal mutations. J. Cell Biol. 124:475–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N., O. Higuchi, K. Ohashi, K. Nagata, A. Wada, K. Kangawa, E. Nishida, and K. Mizuno. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 393:809–812. [DOI] [PubMed] [Google Scholar]
- Yonezawa, N., E. Nishida, K. Iida, I. Yahara, and H. Sakai. 1990. Inhibition of the interactions of cofilin, destrin, and deoxyribonuclease I with actin by phosphoinositides. J. Biol. Chem. 265:8382–8386. [PubMed] [Google Scholar]