

Abstract
Listeria monocytogenes is a facultative intracellular bacterial pathogen that escapes from a phagosome and grows in the host cell cytosol. The pore-forming cholesterol-dependent cytolysin, listeriolysin O (LLO), mediates bacterial escape from vesicles and is ∼10-fold more active at an acidic than neutral pH. By swapping dissimilar residues from a pH-insensitive orthologue, perfringolysin O (PFO), we identified leucine 461 as unique to pathogenic Listeria and responsible for the acidic pH optimum of LLO. Conversion of leucine 461 to the threonine present in PFO increased the hemolytic activity of LLO almost 10-fold at a neutral pH. L. monocytogenes synthesizing LLO L461T, expressed from its endogenous site on the bacterial chromosome, resulted in a 100-fold virulence defect in the mouse listeriosis model. These bacteria escaped from acidic phagosomes and initially grew normally in cells and spread cell to cell, but prematurely permeabilized the host membrane and killed the cell. These data show that the acidic pH optimum of LLO results from an adaptive mutation that acts to limit cytolytic activity to acidic vesicles and prevent damage in the host cytosol, a strategy also used by host cells to compartmentalize lysosomal hydrolases.
Keywords: cytolysins; phagosome; virulence; molecular evolution; macrophages
Introduction
A common strategy among microbial pathogens is exploitation of the normal host process of endosomal/phagosomal acidification. For example, many enveloped viruses require acidification to trigger fusion of the viral envelope with an endosomal membrane (Hernandez et al., 1996). Similarly, many bacterial AB-toxins require acidification of the endosome to stimulate pore formation by the B-subunit and subsequent translocation of the enzymatic A-subunit into the host cytosol (Falnes and Sandvig, 2000). Lastly, some intracellular pathogens such as Trypanosoma cruzi and Listeria monocytogenes use acid-activatable pore-forming proteins to mediate escape from a phagosome into the host cell cytosol (Geoffroy et al., 1987; Tilney and Portnoy, 1989; Ley et al., 1990).
The capacity of L. monocytogenes to escape from an acidic phagosomal compartment is largely mediated by listeriolysin O (LLO),* an essential determinant of L. monocytogenes pathogenesis (Portnoy et al., 1992a). LLO is a member of a family of cholesterol-dependent cytolysins (CDCs) secreted by a large number of pathogenic gram-positive bacteria (Bayley, 1997). Other examples include streptolysin O, perfringolysin O (PFO), and pneumolysin. LLO is the only CDC that is made by an intracellular pathogen and has a pronounced acidic pH optimum. Although replacement of LLO with PFO complements for secreted cytolytic activity, it does not restore virulence (Jones and Portnoy, 1994a). L. monocytogenes secreting PFO are somewhat defective in escaping from a vacuole, but, strikingly, kill the host cell subsequent to entry into the cytosol. The cytotoxicity of PFO is due in part to the lack of a PEST-like sequence in PFO, which targets LLO for phosphorylation and/or degradation in the cytosol (Decatur and Portnoy, 2000). However, bacteria synthesizing PFO containing the PEST-like sequence are only partially restored in virulence. Unlike LLO, PFO does not have a pronounced acidic pH optimum, but rather is active at both acidic and neutral pHs (Portnoy et al., 1992b). We hypothesized that the acidic pH optimum of LLO is important for either efficient lysis of the phagosome and/or lack of toxicity in the cytosol.
The crystal structure of a soluble monomeric form of PFO has revealed a four domain protein with its COOH-terminal domain 4 mediating binding to cholesterol-rich membranes (Rossjohn et al., 1997). Subsequent to membrane binding, PFO oligomerizes into a large complex, containing up to 50 individual monomers, that forms large pores in the target membrane (Shepard et al., 2000). The structure of LLO awaits solution, but based on the overall similarity in sequence and function to PFO, it is reasonable to assume conservation in the relation of sequences to functions. However, because it has no direct correlate, the biochemical basis of LLO's acidic pH optimum cannot be inferred from the structure of PFO. Further, the role of LLO's acidic pH optimum in pathogenesis is not known. In this study, we identified a single residue in LLO that increased its activity at a neutral pH, making its activity profile similar to that of PFO. We then evaluated how this mutation affected L. monocytogenes pathogenesis.
Results
Identification of amino acid residues within LLO that confer an acidic pH optimum
We sought to isolate a mutation in LLO that increased its activity at a neutral pH and thus caused LLO to act like PFO. We began with the assumption that PFO contains a sequence that facilitates its activity over a broad pH range, and placing this sequence in LLO would alter LLO's pH profile. Because domain 4 of PFO was implicated in membrane binding and insertion (Nakamura et al., 1995), it was deemed a good candidate for regulating pH-dependent cytolysis. A chimeric protein consisting of the first three domains of LLO and the fourth domain of PFO was generated. The domain 4 chimera, LLO, and PFO were expressed as COOH-terminally his–tagged recombinant proteins in Escherichia coli and purified for analysis of hemolytic activity. The domain 4 chimera was less active than either PFO or LLO, yet its activity at both pH 5.5 and 7.4 was similar (Fig. 1 C). We interpreted these results to indicate that within the fourth domain of LLO were sequences that control the pH activity profile.
Figure 1.
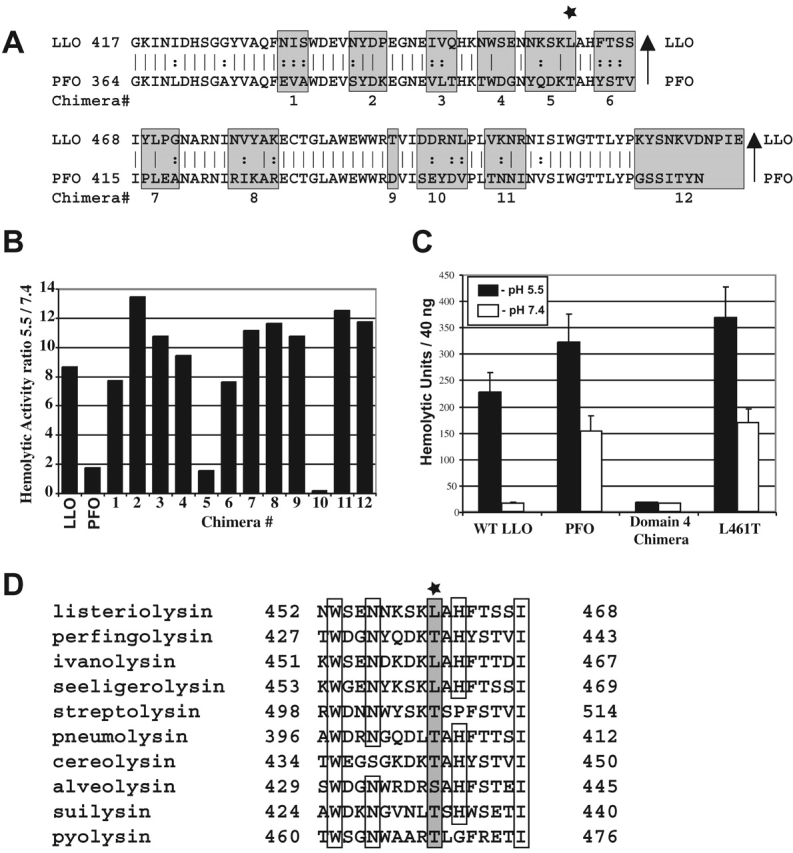
Identification of amino acid residues that control the pH dependence of LLO hemolytic activity. (A) Alignment of domain 4 of LLO and PFO. Amino acid sequences from PFO (bottom of the gray boxes) were exchanged with the amino acids within LLO (top of the gray boxes). Each chimera is numbered from 1 to 12 at the bottom of the gray boxes. The star indicates the L461T mutation. (B) Analysis of the hemolytic activity of the 12 chimeric proteins, expressed in E. coli, represented as the ratio of the activities at pH 5.5 and 7.4. (C) Hemolytic units at pH 5.5 and 7.4 of 40 ng of purified histidine-tagged wild-type LLO, PFO, the domain 4 chimera, and the L461T LLO mutant. Error bars represent the standard deviation. (D) Alignment of 10 CDCs in the 461 region. Star and the gray box indicate position 461 in LLO. Empty boxes indicate identity. The listed toxins are from the following organisms: listeriolysin, L. monocytogenes; perfringolysin, C. perfringens; ivanolysin, L. ivanovi; seeligeriolysin, L. seeligeri; streptolysin, Streptococcus pyogenes; pneumolysin, S. pneumoniae, cereolysin, Bacillus cereus; alveolysin, Paenibacillus alvei; suilysin, S. suis; pyolysin, Arcanobacterium pyogenes.
Next, we divided the fourth domain of LLO into 12 subdomains, each containing amino acids dissimilar to those of PFO, and swapped those regions of dissimilarity from PFO into LLO (Fig. 1 A). Two chimeras (5 and 10) showed a dramatic reduction in the ratio of activity at an acidic pH to that at a neutral pH (Fig. 1 B). However, chimera 10 was ∼10-fold less active than LLO (unpublished data) and was not studied further. The four amino acid changes in chimera 5 were then individually introduced into LLO. A single amino acid change, L461T, increased the hemolytic activity of LLO nearly 10-fold at a neutral pH without decreasing specific activity at pH 5.5. Thus, a single amino acid substitution is sufficient to confer the pH activity profile of PFO onto LLO (Fig. 1 C). Additionally, L461 is unique to the CDC of the pathogenic species of the Listeria genus (Fig. 1 D).
The L461T mutation in LLO reduces virulence
Having established that the L461T mutation conferred greater activity on purified LLO at a neutral pH, we introduced the mutation onto the chromosome of L. monocytogenes by allelic exchange. The resulting strain, DP-L4017, was used for further studies. The mutation had no effect on bacterial growth in vitro (unpublished data). Supernatant fluid derived from cultures of LLO L461T bacteria had a quantity of LLO similar to the wild type and had hemolytic activities at pH 5.5 and 7.4 similar to those of the purified proteins from E. coli (unpublished data).
The capacity of the LLO L461T mutant to grow in animals was evaluated by the lethal dose-50 (LD50) in the mouse listeriosis model. In BALB/c mice, the LD50 of the LLO L461T mutant was >3 × 106 as compared with an LD50 of 1–3 × 104 for wild-type bacteria (Table I). These data suggest that LLO pH dependence contributes to the in vivo growth of L. monocytogenes.
Table I.
L. monocytogenes lethal dose-50 and phagosomal escape measurements b
| Strain | Lethal dose-50 | Percent phagosomal escapec | Average pHc , d at perforation | |
|---|---|---|---|---|
| No treatment | 0.5 μM bafilomycin A1 | |||
| Wild type (10403S) | 1–3 × 104b | 72 ± 2 | 23 ± 6 | 5.7 ± 0.3 |
| LLO-minusa | 1 × 109 | 0 | 0 | ND |
| LLO L461T | >3 ×106 | 76 ± 2 | 25 ± 8 | 5.5 ± 0.2 |
The LLO-minus strain (2161) was published in Jones and Portnoy (1994a).
LD50 for 10403S was published in Portnoy et al. (1988).
Error indicates standard deviation.
Average represents 11 perforated phagosomes measured for wild type and 17 for LLO L461T.
The LLO L461T mutation does not affect the efficiency or pH of phagosomal escape
Based on the observation that LLO has an acidic pH optimum and the bacteria escape from phagosomes at an acidic pH (Beauregard et al., 1997), we hypothesized that a mutant LLO with greater activity at a neutral pH may act prematurely and not mediate escape efficiently. We used a fluorescence-based assay to monitor escape from the phagosome based on the observation that bacteria within the cytosol nucleate host actin filaments on their surface, whereas bacteria in vacuoles do not. We found that the LLO L461T mutant escaped from the phagosome of bone marrow–derived macrophages (BM∅) similarly to wild-type bacteria, 72 ± 2% versus 76 ± 2%, respectively, after 2 h (Table I).
Previous studies have indicated that preventing acidification of the phagosome with the vacuolar proton ATPase inhibitor bafilomycin A1 limits L. monocytogenes escape to the cytosol (Conte et al., 1996; Beauregard et al., 1997). We reasoned that escape of the LLO L461T mutant might not be affected by bafilomycin A1 treatment because its cytolysin was active at a neutral pH. However, when bafilomycin A1 was added to the macrophages, both the mutant and the wild-type bacteria escaped less efficiently (Table I). When bafilomycin A1 was present throughout the assay, both strains escaped with about a third of the efficiency of untreated controls.
Because treatment with bafilomycin A1 prevented escape of the LLO L461T mutant as well as the wild-type bacteria, it would appear that the additional activity of the mutant at a neutral pH does not eliminate the requirement for phagosome acidification. Therefore, we measured the pH of bacterium-containing phagosomes using a pH-sensitive fluid-phase fluorescent dye to determine if the LLO L461T mutant altered the phagosomal maturation process. We found that phagosomes containing the LLO L461T mutant reached an average minimum pH of 5.5 ± 0.3 before perforation, similar to that of the wild type, which reached a minimum mean pH of 5.7 ± 0.2 (Table I). We concluded that the LLO L461T mutation had no effect on phagosomal acidification or escape, and that phagosomal acidification was necessary for the escape of the LLO L461T mutant as well as for wild type. Therefore, it is unlikely that the LLO L461T mutant's virulence defect reflects a reduced ability to escape from phagosomes or an effect on phagosome maturation. The defect is likely due to the alteration of a different part of the pathogenic life cycle.
L. monocytogenes LLO L461T damages host cell membranes
Because the LLO L461T mutant had no defect in phagosomal escape, we next examined the capacity of the bacteria to grow in host cells using a quantitative tissue culture assay (Portnoy et al., 1988). In this assay, adding the antibiotic gentamicin to the culture medium kills extracellular bacteria but has no measurable effect on the growth of intracellular wild-type bacteria. Between 2 and 5 h after infection, the LLO L461T mutant grew well within J774 macrophages with an average apparent doubling time of 58 ± 8 min, slightly longer than the wild-type doubling time of 42 ± 4 min (Fig. 2 A). Strikingly, between 5 and 8 h after infection, the LLO L461T mutant grew with a nearly twofold longer average apparent doubling time (159 ± 30 min compared with the wild-type doubling time of 83 ± 8 min). Additionally, the LLO L461T mutant did not grow to as high a maximum number of bacteria.
Figure 2.
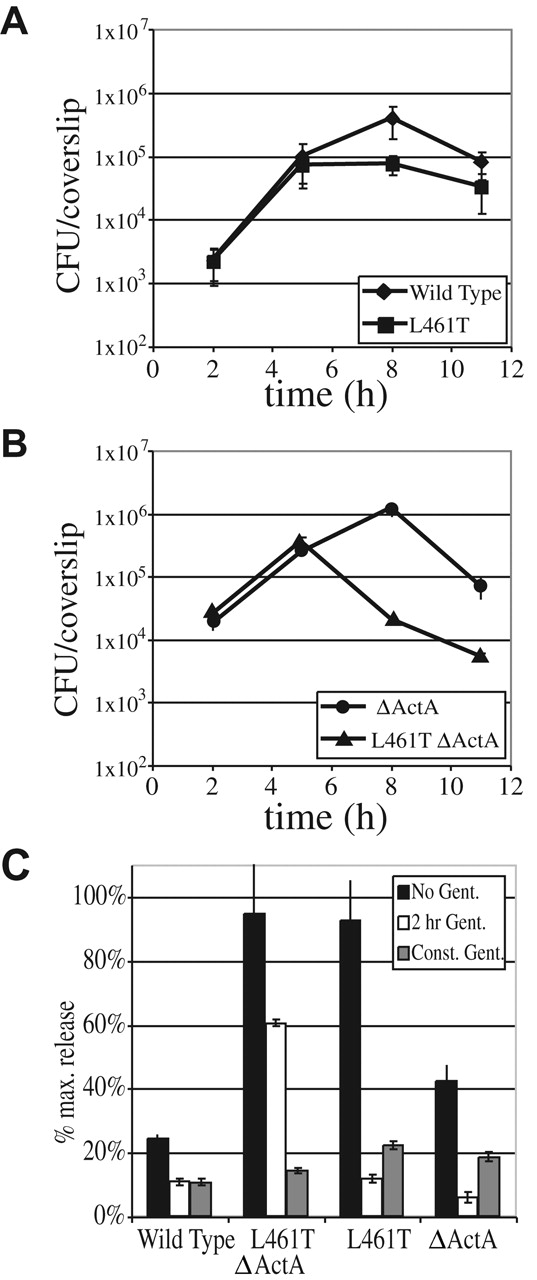
Growth of bacteria within J774 cells and release of LDH by infected cells. (A and B) Growth of bacteria within the J774 macrophage-like cell line in the presence of 50 mg/ml gentamicin. Gentamicin kills extracellular bacteria. Colony-forming units at each time point represent the average ± SD of results from three coverslips. (C) LDH release into tissue culture medium was used to monitor perforation of the host cell plasma membrane. Release of LDH after infection of J774 cells by each strain was compared with total LDH determined after lysis with detergent. LDH release was determined for different exposures of the cultures to gentamicin, which kills intracellular bacteria when host cells have been permeabilized. Error bars represent the SD of three infections.
We reasoned that the LLO L461T mutant's longer apparent doubling times and lower maximum bacterial numbers could reflect either a decrease in the overall growth rate or, more likely, an increase in the death of a subpopulation of intracellular bacteria. The analysis is complicated by the fact that after 5 h, L. monocytogenes spread from cell to cell (Sun et al., 1990; Gedde et al., 2000). To eliminate cell-to-cell spread from the analysis, an in-frame deletion was introduced within the actA gene. The resulting strain was fully capable of vacuolar escape and intracellular growth within the original host cell, but was unable to nucleate actin filaments and thus unable to enter the secondary cell's double-membraned vesicle or spread from cell to cell (unpublished data). As previously observed (Brundage et al., 1993), a ΔActA strain expressing wild-type LLO grew intracellularly for the first 8 h, after which bacterial numbers rapidly dropped due to death of the host cell and influx of gentamicin (Fig. 2 B). A corresponding drop in the number of LLO L461T ΔActA bacteria was observed, but the drop occurred at 5 h instead of at 8 h observed for the wild type. Treatment with the pharmacological inhibitor of actin polymerization, cytochalasin D, which prevents bacterial intracellular motility, led to similar growth defects as the deletion of ActA (unpublished data). Thus, the growth defect of LLO L461T bacteria was more pronounced when cell-to-cell spread was inhibited. (As shown in the next section, the LLO L461T mutant is not defective in the ability to spread from cell to cell.)
For both wild-type and LLO L461T bacteria, the drop in colony-forming units was only observed when gentamicin was present in the assay medium (unpublished data). When host membranes become permeabilized during a cell culture infection, gentamicin present in the culture medium enters the host cell and kills intracellular bacteria (Brundage et al., 1993; Decatur and Portnoy, 2000). Therefore, the gentamicin-dependent drop in numbers of intracellular bacteria suggested that host membranes had been permeabilized. Because the LLO L461T ΔActA mutant died earlier than the wild-type LLO ΔActA strain, and because this occurred in a gentamicin-dependent manner, we hypothesized that the greater activity of LLO L461T at a neutral pH led to earlier permeabilization of the host cell membrane. If this hypothesis were true, damage could be monitored by detecting release of the host cell enzyme lactate dehydrogenase (LDH) from the cytosol of the J774 cells into the culture medium. During a 7 h infection with wild-type bacteria, very little LDH was released either in the presence or absence of gentamicin (Fig. 2 C). In the absence of gentamicin, both the LLO L461T– and LLO L461T ΔActA–infected cells released nearly 100% of their LDH, indicating a major disruption of the host plasma membrane. Interestingly, when J774 cells were incubated in the constant presence of gentamicin, very little LDH was released during infection by any strain. Presumably, permeabilization of the cell allowed the influx of gentamicin, which then killed the intracellular bacteria (Fig. 2 B) and prevented further permeabilization and LDH release. When gentamicin was removed after 2 h, only the J774 cells infected with the LLO L461T ΔActA mutant released high quantities of LDH. When a monoclonal antibody that neutralizes LLO activity (Edelson and Unanue, 2001) was added extracellularly to the J774 cells, there was no effect on LDH release, indicating that toxicity is mediated by intracellular LLO (unpublished data).
A more sensitive method to test the integrity of the plasma membrane uses the membrane-impermeant dye propidium iodide. When membrane integrity is compromised, the dye enters the cell and increases its fluorescence upon binding cellular DNA. Staining can be measured by flow cytometry. After infection with the wild-type bacteria, most macrophage host cells still excluded the dye (Fig. 3 B). In contrast, infection with the LLO L461T mutant led to permeabilization of about half of the macrophages (Fig. 3 C), and infection with the LLO L461T ΔActA mutant permeabilized most of the macrophages (Fig. 3 E).
Figure 3.
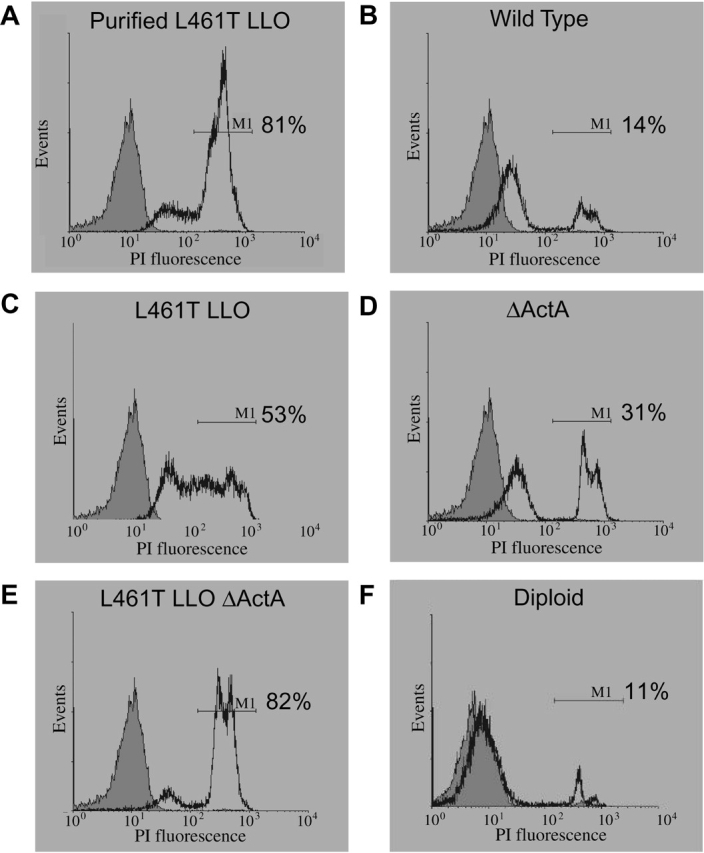
Assessment of host plasma membrane integrity using flow cytometry. (A–F) CD-1 murine BM∅s were stained with 2 μg/ml propidium iodide 7 h after infection with the indicated strains. The gray shaded histogram represents uninfected cells. The fluorescence range of cells scored as permeabilized, indicated by the marker M1, was defined by adding 106 hemolytic units of purified histidine-tagged LLO L461T to the macrophages. A total of 2.5 × 104 cells were scored in each assay; half are displayed in the histograms. The percentage of cells falling within the fluorescence range of marker M1 relative to the total number of cells counted is shown.
To address the possibility that the LLO L461T molecule had altered cytosolic stability, which could lead to increased cytotoxicity, we infected J774 cells and examined the steady-state quantity of cytosolic LLO. We found that there was approximately twofold more cytosolic LLO L461T than wild type (Fig. 4). However, when J774 cells were infected with a strain harboring two copies of the gene encoding LLO, so that they produce twice as many hemolytic units, a similar quantity of LLO to the LLO L461T mutant was observed (Fig. 4). However, despite the fact that infection with the merodiploid led to a concentration of LLO in the cytosol similar to the mutant, the merodiploid damaged the host cell's plasma membrane no more than the wild-type bacteria (Fig. 3 F). Together, these data suggested that the decreased growth of the LLO L461T mutant was associated with permeabilization of the host cells due to increased activity of the LLO L461T at a neutral pH, and was not due to an increased cytosolic concentration of LLO.
Figure 4.

LLO secreted by intracellular L. monocytogenes. [35S]Met-labeled LLO from J774 cells infected for 5 h was immunoprecipitated and examined using SDS-PAGE. Half of the protein was used to perform autoradiography, with a representative gel shown, and the other half was used for phosphorimaging. As published elsewhere (Moors et al., 1999; Decatur and Portnoy, 2000), two forms of LLO are observed, a full length 58-kD form and a truncated 55-kD form. Quantitation of the phosphorimaged samples is displayed on the bottom as pixel volume from full-length and the truncated form of LLO together. Quantitation of relative band intensity, versus wild type, is normalized for colony-forming units of each sample at the end of the 5-h experiment. The error measurement represents SD of the mean from three experiments.
L. monocytogenes strain LLO L461T is not defective in cell-to-cell spread
LLO plays an essential role in the escape of L. monocytogenes from both the primary phagosome and the secondary double membrane–bound vesicle formed during cell-to-cell spread (Tilney and Portnoy, 1989; Gedde et al., 2000). The results described above did not directly address whether the LLO L461T mutation affects bacterial cell-to-cell spread. We examined the ability of bacteria to spread from cell to cell by measuring the diameter of plaques formed in L2 monolayers after 3 d in the presence of different concentrations of gentamicin. Plaque diameter is a measure of a bacterium's ability to grow, move through the host cell cytosol, enter an adjacent cell, and escape from the secondary vesicle formed in the adjacent cell. At low gentamicin concentrations, the LLO L461T strain's plaque was equal in diameter to the wild type, whereas at high concentrations the mutant had a severe plaquing defect (Fig. 5). Thus, the capacity of an LLO L461T mutant to form plaques was gentamicin dependent.
Figure 5.

Cell-to-cell spread of L. monocytogenes in different concentrations of gentamicin. The diameter of bacterial plaques formed within L2 cell monolayers over 3 d was measured and expressed as a percentage of the diameter of wild-type plaques. ActA GGG is an L. monocytogenes strain with decreased actin-based motility. ΔplcA is an L. monocytogenes strain with an in-frame deletion in the gene encoding the bacterial phosphatidylinositol–specific phospholipase C, which leads to a reduced ability to escape from the phagosome. Diploid is a strain containing two copies of the gene coding for LLO. Error bars represent the SD of the mean from a minimum of three experiments. Representative wells are shown on the right.
Two L. monocytogenes mutants with slight defects in either actin-based motility or escape from the double-membraned vesicle were analyzed as controls. The corresponding reduction in the size of plaques formed by these mutants was independent of gentamicin concentration. Also, the merodiploid strain with two copies of LLO formed plaques identical to wild type at each gentamicin concentration. Therefore, we concluded that LLO L461T was fully capable of mediating cell-to-cell spread and escape from the double-membraned vesicle. Additionally, based on the data shown in Figs. 2, 3, and 5, we concluded that the plaque defect seen at high gentamicin concentrations reflected bacteriocidal activity of the antibiotic on intracellular bacteria that entered cells subsequent to LLO-mediated damage to the host cell membrane. Conversely, low concentrations of gentamicin did not allow the influx of sufficient quantities of the antibiotic to negatively affect the bacteria. These conclusions agree with the results observed for the merodiploid, which did not permeabilize host membranes to propidium iodide (Fig. 3) nor form gentamicin-sensitive plaques.
Discussion
The pore-forming protein LLO is a primary determinant of L. monocytogenes pathogenesis that has a pronounced acid pH optimum and acts in an acidic vacuolar compartment to mediate escape of the bacterium into the host cytosol. In this study, we have examined the role of the acidic pH optimum in the pathogenesis of L. monocytogenes infection. By swapping residues with the less pH-sensitive ortholog PFO, we identified an LLO mutant, L461T, that was 10-fold more active at a neutral pH than wild-type LLO. L. monocytogenes synthesizing LLO L461T were 100-fold less virulent in mice than bacteria secreting the wild-type LLO, indicating that the acid pH optimum of LLO plays an important role during infection. Tissue culture assays revealed that bacteria synthesizing LLO L461T were not defective in the extent or pH of phagosomal escape, cell-to-cell spread, or in escape from the double-membraned vesicle formed during cell-to-cell spread. However, bacteria synthesizing LLO L461T permeabilized host plasma membranes, as shown by (a) gentamicin sensitivity of intracellular bacteria, (b) release of LDH from host cells, and (c) staining of DNA within infected cells with a membrane-impermeant dye. Thus, the results of this study strongly suggest that the acidic pH optimum of LLO is necessary to avoid damage to infected host cells.
LLO is unique among the CDCs in that its activity is normally repressed at a neutral pH. The biochemical basis of the LLO pH optimum is not understood; it could reflect pH sensitivity in membrane binding, oligomerization, and/or pore formation. It was surprising that a single amino acid change, from leucine to threonine, could profoundly increase the hemolytic activity of LLO at a neutral pH. Neither leucine nor threonine alone contains properties that would suggest sensitivity to pH. The alteration from a nonpolar leucine to a polar threonine residue offers the potential of an additional hydrogen bond and a reduction in hydrophobicity, but in the absence of a detailed structure for LLO, it is difficult to determine the influence of these changes. However, if we assume a general structural similarity to PFO, because LLO and PFO are 42% identical (Rossjohn et al., 1997), then the L461T mutation is located on an outer loop of the fourth domain. Domain 4 has been implicated in both membrane binding and oligomerization (Nakamura et al., 1995; Bayley, 1997). Therefore, it is possible that the mutation may alter interactions with the target lipid bilayer or with other monomers in a pH-sensitive fashion. Also, there is evidence that domain 4 must undergo significant movement relative to the other domains when the monomer enters the lipid bilayer (Gilbert et al., 1999). In particular, the transmembrane hairpins of domain 3 have been proposed to pack with domain 4 when inserted in the membrane. The mutation of leucine 461 to threonine might affect intramolecular interactions in a manner that would make the cytolysin less sensitive to pH. The precise effect of the L461T mutation awaits additional biochemical analysis.
There are numerous examples of pathogens exploiting acidified compartments via pH-dependent proteins, including the viral hemagglutinins (Hernandez et al., 1996) and bacterial and protozoal toxins (Ley et al., 1990; Falnes and Sandvig, 2000). These precedents led us to hypothesize that the acidic optimum of LLO provides a mechanism to coordinate optimal pore-forming activity with maturation of the phagosome to allow efficient phagosomal escape, and any perturbation of this coordination would result in less efficient escape. Consistent with this hypothesis, a number of studies have shown that bafilomycin A1, a pharmacological inhibitor of phagosomal acidification, blocks the escape of L. monocytogenes from a phagosome (Conte et al., 1996; Beauregard et al., 1997). Therefore, it was surprising to find that the bacteria synthesizing a mutant LLO more active at a neutral pH escaped from the phagosome with a similar efficiency and at the same pH as wild-type bacteria. These data suggest that though acidification of the phagosome is important for bacterial escape, the pH sensitivity is not manifested through LLO activity. Given that bafilomycin A1 blocks maturation of endosomes (van Weert et al., 1995), these data suggest that a pH-dependent process other than LLO activity is involved in vacuolar perforation and escape.
Although the precise mechanism of vacuolar escape is not understood, there is recent evidence that CDCs, including LLO, may act as mediators of protein delivery to the host cytosol, similarly to the gram negative type III secretion system (Sibelius et al., 1996; Wadsworth and Goldfine, 1999; Madden et al., 2001). If indeed LLO acts to mediate cytosolic delivery from the phagosome, it is possible that the acid dependence is required for activation of some other pH-sensitive bacterial effector. At least one other L. monocytogenes virulence factor, a broad-range phospholipase C, is released upon vacuolar acidification (Marquis and Hager, 2000), so it is possible that LLO simply needs to form a pore to allow the transfer of the pH-sensitive effector to the cytosol to mediate vesicular escape. Another possibility is that a host factor involved in the escape process requires acidification to be activated or localized to the phagosome.
The most striking phenotype exhibited by L. monocytogenes expressing LLO L461T was the increased damage inflicted on the host cells' membranes. The simplest explanation for this observation is that LLO L461T is active within the neutral pH of the cytosol and passes a threshold at which the number of pores formed by the toxin overwhelms the host cell's membrane repair mechanisms (Walev et al., 2001). Alternately, it is possible that the mutation may lead to host cell damage through an unknown mechanism. However, our data clearly indicate that bacteria synthesizing LLO L461T permeabilize J774 host cells as early as 5 h after infection, whereas permeabilization by the wild type requires 8 h. It has been estimated that one bacterium secretes one LLO molecule per minute (Villanueva et al., 1995). Based on this rate and the doubling times for each strain, we calculated that at the time of permeabilization, the host cell is confronted with the production of ∼35 LLO L461T molecules per minute, enough monomers to assemble approximately one pore. In contrast, the wild-type bacteria are producing 420 LLO per minute when they permeabilize the host cell after 8 h of growth. These calculations indicate that bacteria synthesizing LLO L461T permeabilized the host cell with about one twelfth the production rate of wild type. This difference indicates that the unique acidic activity optimum of wild-type LLO enables L. monocytogenes to produce at least 10 times the number of progeny per infected host cell before significant host damage occurs.
Many factors contribute to the host cell's ability to deal with the presence of LLO within the cytosol. In light of studies suggesting that host cytosolic proteases cleave LLO (Villanueva et al., 1995; Decatur and Portnoy, 2000), an alternate explanation for the greater toxicity of LLO L461T is that it is less susceptible to proteolytic degradation. We observed an approximately twofold higher concentration of LLO L461T in the cytosol of J774 host cells, yet found that equal quantities of LLO were produced in vitro. Perhaps the greater concentration of LLO L461T reflected the fact that the cytolysin entered membranes more readily in the neutral pH of the cytosol where it was protected from proteolytic degradation, as has been shown for other CDCs (Nakamura et al., 1995). Regardless of the precise mechanism leading to the greater concentration of intracellular LLO L461T, we found that intracellular concentrations of LLO did not correlate with membrane permeabilization. A merodiploid LLO strain producing twice as much LLO was no more cytotoxic than the wild type, suggesting that a twofold increase in LLO concentration alone does not affect membrane damage under these conditions. However, a merodiploid strain synthesizing both the LLO L461T and the wild-type LLO acted similarly to the LLO L461T strain in the plaquing assay, producing gentamicin-sensitive plaques (unpublished data), suggesting that the mutant's cytotoxic phenotype is dominant over the wild type. These data suggest that the increased host membrane damage was due to the 10-fold greater activity of LLO L461T at a neutral pH and not simply intracellular LLO concentration.
The results of this study showed that L. monocytogenes expressing LLO L461T was ∼100-fold less virulent than the wild type. We hypothesize that the increased damage to the plasma membrane observed in tissue culture correlates with the defect in virulence in vivo. Additionally, we found that the LLO L461T grew less well in immunologically naive mice as early as 12 h after infection (unpublished data). Thus, the adaptive immune response probably plays little role in mediating the defect of the mutant bacteria. Instead, the defect is more likely to reflect the action of the innate immune response or a more basic defect in the bacterial life cycle in vivo. We can envision a number of scenarios that may lead to the mutant's virulence defect. One possibility is that a damaged cell no longer provides the advantages of an intracellular lifestyle, whether it is caused by the loss of access to nutrients, reduced actin-based motility, a loss of protection from humoral defenses, or another unknown mechanism. However, because the mutant grows and spreads normally at low gentamicin concentrations, a more likely possibility is that the mutant elicits a greater inflammatory response at the foci of infections due to greater cytotoxicity, thereby recruiting a greater number of leukocytes to the focus of infection. The hemolytic activity of LLO causes the production of inflammatory cytokines, leading to activation and chemotaxis of neutrophils and monocytes, which are largely responsible for containing the bacteria (Unanue, 1997; Kayal et al., 1999; Sibelius et al., 1999).
The results of this study support the concept that L. monocytogenes has evolved to minimize harm to its host cell. To achieve maximal virulence, the bacteria must maintain an equilibrium between producing a molecule that is cytolytic enough to mediate escape from the vesicle, yet not overly toxic to infected host cells. We hypothesize that L. monocytogenes has acquired a pathoadaptive mutation (Sokurenko et al., 1999) converting the primordial residue 461 to a leucine. This mutation is found solely in pathogenic Listeria and converts LLO from a toxin appropriate for an extracellular pathogen to that of an intracellular pathogen. Thus, LLO is highly active where it needs to be, in the acidic phagosome, yet relatively inactive in the neutral pH of the cytosol. Together with previously published data that LLO contains a PEST-like sequence that also reduces its toxicity (Decatur and Portnoy, 2000), our data imply that L. monocytogenes has evolved multiple failsafe mechanisms to regulate the activity of LLO, a molecule that has the potential to destroy its intracellular niche. Indeed, it has been hypothesized that the host cell uses these same acidic properties of the endocytic pathway to compartmentalize the activity of its own potentially hazardous lysosomal enzymes (Mellman et al., 1986).
Materials and methods
Bacterial strains, growth conditions, and reagents
The wild-type L. monocytogenes strain used for these studies was 10403S. L. monocytogenes strains with deletions of actA were constructed by allelic exchange as described previously (Skoble et al., 2000). The L. monocytogenes strain with an in-frame deletion of PI-PLC (ΔplcA, or DP-L1552) and strain ActA GGG (DP-L4032) were previously described (Camilli et al., 1993; Skoble et al., 2001). The merodiploid hly strain (DP-L4076) will be published in a subsequent manuscript (unpublished data). E. coli strains DH5α (GIBCO BRL) or XL-1 Blue (Stratagene) were used for cloning. E. coli strains BL21(DE3) or BL21(DE3)PlysS (Stratagene) were used for expression of proteins from pET vectors.
L. monocytogenes was grown in 3 ml brain heart infusion broth (BHI; Becton Dickinson) slanted without agitation in 15 ml conical tubes at 30°C overnight, unless otherwise noted. All E. coli strains were grown in Luria-Bertani broth (LB; Becton Dickinson) at 37°C shaking, unless otherwise noted. All tissue culture cells were grown in DME (GIBCO BRL), containing 7.5% heat-deactivated FBS (Hy-Clone) and 2 mM glutamine (DME; GIBCO BRL), at 37°C and 5% CO2, unless otherwise noted. All chemicals were purchased from Sigma-Aldrich, unless otherwise noted.
Sequences
The GenBank/EMBL/DDBJ accession nos. for the proteins examined in this study are the following: LLO, M29030; PFO, M36704; ivanolysin O, X60461; seeligeriolysin O, X60462; streptolysin O, M18638; pneumolysin, X52474; cereolysin, D21270; alveolysin, M62709; suilysin, Z36907; and pyolysin, U84782.
Cloning
Construction of the LLO expression vector.
DNA and protein analysis was performed using MacVector software (Genetics Computer Group). The region of hly coding for mature LLO was amplified by PCR, with the primers and templates described in Table II, using Vent polymerase (New England Biolabs, Inc.) to introduce a six histidine tag. The amplified fragment was then cut with restriction enzymes and ligated into pET29b (Novagen). This plasmid and all other plasmids were initially cloned in E. coli strain XL-1 Blue and then transferred into E. coli expression strain BL21(DE3), unless otherwise noted, to yield strain DP-3570.
Table II. Oligonucleotides and cloning strategies.
| Number | Sequence 5′→3′ (including enzyme site)a | Construct | Templateb | Cloningc method | Pair |
|---|---|---|---|---|---|
| 3140 | GGAATTCCATATGAAGGATGCATCTGCATTCAAT (Nde1) | His-LLO, p3570 | 10403S genomic DNA | B | 3232 |
| 3232 | CGGGATCCTTATTAGTGGTGGTGGTGGTGGTGTTCGATTG GATTATCTAC (BamH1) | His-LLO, p3570 | 10403S genomic DNA | B | 3140 |
| 3541 | GGAATTCCCATGGGAAAGGATATAACAGATAAAAATCA (Nco1) | His-PFO, p4167 | p1868 | B | 3542 |
| 3542 | CGGGATCCTTATTAGTGGTGGTGGTGGTGGTGATTGTAAG TAATACTAGATCCA (BamH1) | His-PFO, p4167 | p1868 | B | 3541 |
| 3543 | ACGCGTCGACTTATTAGTGGTGGTGGTGG (Sal1) | His-LLO(1-3)PFO4 | p1868 | S | 3578, 3575 |
| 3575 | GGAATTCCATATGAAGGATGCATCTGCA (Nde1) | His-LLO(1-3)PFO4 | p3570 | S | 3543, 3579 |
| 3578 | ACTATGATCTAAGTTTATTTTTCCATCTGTATAAGC | His-LLO(1-3)PFO4 | p1868 | S | 3543 |
| 3579 | GCTTATACAGATGGAAAAATAAACTTAGATCATAGT | His-LLO(1-3)PFO4 | p3570 | S | 3575 |
| 3740 | GGAGGATACGTTGCTCAATTCGAAGTAGCCTGGGATGAAG TAAATTATGAT | Chimera 1 | p3570 | Q | 3741 |
| 3741 | ATCATAATTTACTTCATCCCAGGCTACTTCGAATTGAGCAAC GTATCCTCC | Chimera 1 | p3570 | Q | 3740 |
| 3742 | AACATTTCTTGGGATGAAGTATCATATGACAAAGAAGGTAA CGAAATTGTTCAA | Chimera 2 | p3570 | Q | 3743 |
| 3743 | TTGAACAATTTCGTTACCTTCTTTGTCATATGATACTTCATCC CAAGAAATGTT | Chimera 2 | p3570 | Q | 3742 |
| 3744 | TATGATCCTGAAGGTAACGAAGTATTAACTCATAAAAACTG GAGCGAAAAC | Chimera 3 | p3570 | Q | 3745 |
| 3745 | GTTTTCGCTCCAGTTTTTATGAGTTAATACTTCGTTACCTTCA GGATCATA | Chimera 3 | p3570 | Q | 3744 |
| 3746 | AACGAAATTGTTCAACATAAAACATGGGATGGAAACAATAA AAGCAAGCTAGCT | Chimera 4 | p3570 | Q | 3747 |
| 3747 | AGCTAGCTTGCTTTTATTGTTTCCATCCCATGTTTTATGTTGA ACAATTTCGTT | Chimera 4 | p3570 | Q | 3746 |
| 3748 | CATAAAAACTGGAGCGAAAACTATCAAGATAAAACAGCTCA TTTCACATCGTCCATC | Chimera 5 | p3570 | Q | 3749 |
| 3749 | GATGGACGATGTGAAATGAGCTGTTTTATCTTGATAGTTTTC GCTCCAGTTTTTATG | Chimera 5 | p3570 | Q | 3748 |
| 3750 | AATAAAAGCAAGCTAGCTCATTATTCAACAGTAATCTATTTG CCTGGTAACGCG | Chimera 6 | p3570 | Q | 3751 |
| 3751 | CGCGTTACCAGGCAAATAGATTACTGTTGAATAATGAGCTA GCTTGCTTTTATT | Chimera 6 | p3570 | Q | 3750 |
| 3752 | GCTCATTTCACATCGTCCATCCCTCTTGAAGCTAACGCGAG AAATATTAATGTT | Chimera 7 | p3570 | Q | 3753 |
| 3753 | AACATTAATATTTCTCGCGTTAGCTTCAAGAGGGATGGACG ATGTGAAATGAGC | Chimera 7 | p3570 | Q | 3752 |
| 3754 | CCTGGTAACGCGAGAAATATTAGAATAAAAGCAAGAGAAT GCACTGGTTTAGCTTGG | Chimera 8 | p3570 | Q | 3755 |
| 3755 | CCAAGCTAAACCAGTGCATTCTCTTGCTTTTATTCTAATATTT CTCGCGTTACCAGG | Chimera 8 | p3570 | Q | 3754 |
| 3756 | TGGGAATGGTGGAGAGATGTAATTGATGACCGG | Chimera 9 | p3570 | Q | 3757 |
| 3757 | CCGGTCATCAATTACATCTCTCCACCATTCCCA | Chimera 9 | p3570 | Q | 3756 |
| 3758 | GGGAATGGTGGAGAACGGTAATTAGTGAATATGATGTTCC ACTTGTGAAAAATAGAAAT | Chimera 10 | p3570 | Q | 3759 |
| 3759 | ATTTCTATTTTTCACAAGTGGAACATCATATTCACTAATTACC GTTCTCCACCATTCCC | Chimera 10 | p3570 | Q | 3758 |
| 3760 | GACCGGAACTTACCACTTACAAATAATATAAATATCTCCATC TGGGGC | Chimera 11 | p3570 | Q | 3761 |
| 3761 | GCCCCAGATGGAGATATTTATATTATTTGTAAGTGGTAAGTT CCGGTC | Chimera 11 | p3570 | Q | 3760 |
| 3580 | AGATCCAGGGTATAAAGTGGTGCCCCAGATGGAGAT | Chimera 12 | p3570 | Q | 3581 |
| 3581 | ATCTCCATCTGGGGCACCACTTTATACCCTGGATCT | Chimera 12 | p3570 | Q | 3580 |
| 3799 | GAAAACAATAAAAGCAAGACAGCTCATTTCACATCGTCC | His-L461T and DP-L4017 | 10403S genomic DNA | QS | 3800 |
| 3800 | GGACGATGTGAAATGAGCTGTCTTGCTTTTATTGTTTTC | His-L461T and DP-L4017 | 10403S genomic DNA | QS | 3799 |
| 3931 | TTTCTGCAGAGAAACACGCGGATGAAATCGATA (Pst1) | DP-L4017 | 10403S genomic DNA | S | 3800, 3932 |
| 3932 | AAAAGAGCTCTCTGGAATTGAGGATGATTTCTTT (Sac1) | DP-L4017 | 10403S genomic DNA | S | 3799, 3931 |
Custom oligonucleotides were purchased from Operon Technologies and all restriction enzymes were from New England Biolabs, Inc.
Plasmid DNA was purified with a Qiagen plasmid midi kit and L. monocytogenes genomic DNA was isolated using a FastDNA® kit from Bio101.
B, basic PCR followed by plasmid ligation; S, splicing by overlap extension PCR and then plasmid ligation (Horton et al., 1990); Q, QuickchangeTM PCR (Stratagene).
Construction of the PFO expression vector.
Mature PFO was amplified by PCR from p1868 (Jones and Portnoy, 1994a), a plasmid carrying the coding sequence for mature PFO from Clostridium perfringens, using the primers, templates, and restriction sites described in Table II. This fragment was ligated into pET28a (Novagen) and later expressed with BL21(DE3)PlysS, strain DP-4167.
Construction of the domain chimeras, subdomain chimeras, and single amino acid mutation expression vectors.
The fourth domain of LLO was replaced by PFO domain 4 using splicing by overlap extension PCR (Horton et al., 1990) with the components and restriction sites described in Table II. The subdomain chimeras and the single amino acid mutations indicated in Fig. 1 were constructed by modifying p3570 with the protocol published in the Quickchange™ site-directed mutagenesis kit (Stratagene) and the primers listed in Table II.
Hemolytic activity screening of recombinant proteins
E. coli expression strains were grown overnight in LB containing 30 μg/ml kanamycin (LB-KAN). 400 μl of the overnight culture was added to 10 ml LB-KAN and grown for 1.5 h, and then 1 mM IPTG was added. This culture was incubated at 30°C, shaking for 3 h. Cultures were pelleted and then resuspended in 1 ml storage buffer (140 mM sodium chloride, 4 mM potassium chloride, 10 mM sodium phosphate, 0.5 mM DTT, pH 6.0) with 1 mM PMSF. The samples were sonicated on ice and cleared by centrifugation.
The quantitative assay was performed in a 96-well V-bottom styrene plate (Corning Inc.) with either neutral hemolysis buffer (35 mM sodium phosphate, 125 mM sodium chloride, 0.5 mg/ml BSA, pH 7.4, using acetic acid) or acidic hemolysis buffer (same as neutral hemolysis buffer but pH 5.5). Samples were serially diluted, and then 0.5% sheep red blood cells (HemoStat Laboratories) were added to each well. The plate was incubated, shaking at 37°C, and then pelleted in the V-bottom. Supernatant was transferred from the V-bottom plate into equivalent locations in a flexible polyvinyl chloride flat bottom 96-well plate (Becton Dickinson), and the absorbance at 450 nm for each well was measured (Spectramax340) and analyzed with SoftMax Pro v1.2 software (Molecular Devices Corp.). Hemolytic units were defined as the dilution of the sample at which 50% of the sheep red blood cells had been lysed.
Overexpression and purification of 6× his–tagged LLO proteins and 6× his–tagged PFO from E. coli
Recombinant strains were grown, shaking at 37°C, in LB-KAN to stationary phase. 20 ml of this culture was inoculated into 1 liter of LB-KAN and incubated, shaking at 30°C, for 90 min. Expression was induced by the addition of 1 mM IPTG, and the culture was incubated, shaking at 30°C, for 6 h. The bacterial pellet was harvested by centrifugation, resuspended in 40 ml cold lysis buffer (50 mM sodium phosphate, pH 8.0, 1 M sodium chloride, 20 mM imidazole, 10 mM 2-mercaptoethanol, 1 mM PMSF), and lysed in a French pressure cell at 12,000 psi. The lysate was centrifuged for 20 min at 17,000 g. The supernatant was collected and mixed into 5 ml Ni-NTA resin (QIAGEN) equilibrated in lysis buffer. The slurry was stirred at 4°C for 60 min to bind his-tagged protein to the resin. To remove unbound protein, the resin was packed into a column and washed with lysis buffer by dropwise gravity flow until UV absorbance of the eluate reached baseline, and then it was washed with wash buffer (lysis buffer, pH 6.0, 10% glycerol, 0.1% Tween 20). Washed resin was removed from the column, resuspended in elution buffer (lysis buffer, pH 6.0, and 800 mM imidazole), and incubated 10 min on ice, after which the supernatant was collected. This procedure was performed twice, yielding 6 ml eluate. Eluate was dialyzed in cassettes (Pierce Chemical Co.) within autoclaved storage buffer (lysis buffer, pH 6.0, with 1 mM EDTA). Both the Bradford method and UV280 absorbance determined protein concentrations. The procedure yielded ∼25 mg protein per liter starting culture. Aliquots not used immediately were stored in storage buffer with 50% glycerol at −80°C.
Allelic exchange of LLO L461T
To introduce the LLO L461T mutation onto the 10403S chromosome, a DNA fragment was produced by splicing by overlap extension PCR, using the primers, templates, and restriction enzymes in Table II, and then ligated into the temperature-sensitive plasmid vector pKSV7. Allelic exchange was performed as described previously (Camilli et al., 1993). Strains were verified initially by detecting the loss of an Nhe1 site in a chromosomal PCR product containing the mutation.
Animal studies
LD50 by intravenous infection was established as previously described using BALB/c mice (Portnoy et al., 1988).
Phagosomal escape assay
The percentage of bacteria that had escaped from the phagosome was determined by evaluating the presence of F-actin–coated bacteria within the macrophage, similar to an experiment previously described (Jones and Portnoy, 1994a). C57/BL6 BM∅s in DME, 10% FBS, with or without 0.5 μM bafilomycin A1 (Calbiochem), on a coverslip were infected for 15 min, resulting in a bacterium within 1/10 macrophages. Macrophages were washed with Ringer's buffer (5 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 10 mM Hepes, 10 mM glucose, pH 7.2) and 25 μg/ml gentamicin was added. At 120 min after infection, the macrophages were fixed for 15 min with cytoskeletal fixative (40 mM Hepes, 10 mM EGTA, 0.5 mM EDTA, 5 mM MgSO4, 33 mM potassium acetate, 0.02% sodium azide, 5% polyethylene glycol 400, 4% paraformaldehyde), washed, permeabilized with PBS, containing 2% goat serum and 0.3% Triton X-100, and stained with Texas red–phalloidin (Molecular Probes) and DAPI (Molecular Probes). A total of 50 macrophages harboring bacteria were examined for each bacterial strain in each of four experiments.
The determination of phagosomal pH was performed essentially as previously described (Beauregard et al., 1997) with the following modifications. In brief, fluid-phase fluorescein dextran, molecular weight 10,000 (Molecular Probes), was added to the bacteria-containing media used to infect macrophages. Phagosomes containing both 10-kD fluorescein dextran and bacteria were photographed every 30 s with a Quantix cooled charge-couple device camera (Photometrics) through fluorescent microscopy using a Nikon TE300 inverted microscope (Nikon), with phase-contrast and excitation wavelengths 485 and 440 nm and emission measurement at 520 nm. Images and the 485:440 ratio were collected until perforation was indicated by loss of dye from the vacuole. The 485:440 ratio measured just before perforation was compared with a standard curve to establish pH, as described in the published methods.
Cytotoxicity assays
Growth in J774 macrophage-like cells.
Intracellular growth of L. monocytogenes was performed as previously described (Jones and Portnoy, 1994b).
Flow cytometry.
Flow cytometry was performed on cultures of BMØs from CD-1 mice as previously described (Portnoy et al., 1988). BMØ were chosen for this assay because infected J774 are difficult to remove from tissue culture dishes without causing plasma membrane damage, whereas BMØ lift from the dish when incubated at 4°C. 106 macrophages were plated on 60-mm Lab-tek nontissue culture dishes (Fisher Scientific) overnight in bone marrow macrophage media (DME, 20% heat-deactivated FBS, 30% L cell supernatant containing CSF-1 in DME, 2 mM glutamine, 1 mM pyruvate, 0.1% 2-mercaptoethanol). Monolayers were infected with 107 washed bacteria for 30 min resulting in at least one bacterium per cell. 60 min after infection, 50 μg/ml gentamicin was added. 3 h after infection, the cell monolayer was washed to remove the gentamicin, and then fresh medium was added to the dish. 7 h after infection, medium from each culture was collected. 4°C PBS was then added and the dish was stored at 4°C for ∼30 min. Release of macrophages from the dish was monitored by microscopy. The macrophage-containing PBS was added to the previously removed media and centrifuged at 4°C. The pellet was washed with 4°C PBS, 10% FBS. Cell pellets were resuspended in PBS (10% FBS) and passed through a 70-μm cell strainer (Becton Dickinson). 1 μg propidium iodide (Molecular Probes) was added to each sample. Samples were analyzed with a flow cytometer (EPICS XL-MCL; Beckman Coulter).
LDH release assay.
LDH release assays were performed using the Cytotox 96® nonradioactive cytotoxicity assay (Promega), according to manufacturer's instructions and methods described previously (Decatur and Portnoy, 2000), with 2 × 104 J774 cells per well infected to achieve at least one bacterium per cell. Neutralizing anti-LLO monoclonal antibodies were supplied by Brian Edelson and Emil Unanue (Washington University School of Medicine, St. Louis, MO).
Intracellular LLO analysis
Intracellular levels of LLO were studied with previously established methods (Moors et al., 1999) and the following modifications. In brief, J774 cells were infected with L. monocytogenes strains for 30 min and then washed, and 50 μg/ml gentamicin was added at 60 min. 4 h after infection, methionine-starved cells were pulsed with [35S]methionine (NEN Life Science Products) for 1 h. At 5 h, macrophages were lysed, LLO was immunoprecipitated, and one half of the sample was subjected to SDS-PAGE for autoradiography and the other half run for analysis on a Phosphorimager 445 SI (Molecular Dynamics) and analyzed using Imagequant software (Molecular Dynamics). Monoclonal anti-LLO antibodies were supplied by Pascale Cossart (Institute Pasteur, Paris, France). The relative number of bacteria in each assay was established by lysing the infected J774 on coverslips in dishes processed in tandem with the radiolabeled dishes. Lysate was subsequently plated on LB-agar plates to determine colony-forming units.
Plaque assay
Plaquing assays within L2 cell monolayers were performed as described previously (Sun et al., 1990), with modifications to the methods of measurement (Skoble et al., 2000). In brief, L2 cells were grown to confluency in six-well tissue culture dishes and then infected with bacteria for 1 h. Subsequently, DME-agar containing gentamicin was added and plaques were grown for 3 d. Living cells were visualized by adding on day 3 an additional DME-agar overlay containing neutral red (GIBCO BRL) and incubating overnight.
Acknowledgments
We are grateful to Archie Bouwer for performing the LD50 assays, Darren Higgins for constructing the expression system for LLO, Pete Lauer and Rich Calendar for construction of the merodiploid bacteria, and Mary O'Riordan for guidance regarding the flow cytometer. Additional thanks are extended to Brian Edelson, Emil Unanue, and Pascale Cossart for supplying LLO monoclonal antibodies. We would also like to thank Norma Andrews (Yale University School of Medicine, New Haven, CT), Amy Decatur (University of Pennsylvania School of Medicine, Philadelphia, PA), Laurel Lenz (University of California, Berkeley, CA), Helene Marquis (Cornell University, Ithaca, NY), and Pamela Schnupf (University of California) for critical review of this manuscript.
This work was supported by National Institutes of Health grant AI27655 (D.A. Portnoy) and AI35950 (J.A. Swanson). M. Gedde was a Howard Hughes Medical Institute postdoctoral physician fellow.
I.J. Glomski and M.M. Gedde contributed equally to this work.
M.M. Gedde is currently at IntraBiotics Pharmaceuticals, 1245 Terra Bella Ave., Mountain View, CA 94043.
Footnotes
Abbreviations used in this paper: BMØ(s), bone marrow–derived macrophage(s); CDC(s), cholesterol-dependent cytolysin(s); LB, Luria-Bertani broth; LB-KAN, Luria-Bertani broth with 30 μg/ml kanamycin; LD50, lethal dose-50; LDH, lactate dehydrogenase; LLO, listeriolysin O; PFO, perfringolysin O.
References
- Bayley, H. 1997. Toxin structure: part of a hole? Curr. Biol. 7:R763–R767. [DOI] [PubMed] [Google Scholar]
- Beauregard, K.E., K.D. Lee, R.J. Collier, and J.A. Swanson. 1997. pH-dependent perforation of macrophage phagosomes by listeriolysin O from Listeria monocytogenes. J. Exp. Med. 186:1159–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage, R.A., G.A. Smith, A. Camilli, J.A. Theriot, and D.A. Portnoy. 1993. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc. Natl. Acad. Sci. USA. 90:11890–11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli, A., L.G. Tilney, and D.A. Portnoy. 1993. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol. Microbiol. 8:143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte, M.P., G. Petrone, C. Longhi, P. Valenti, R. Morelli, F. Superti, and L. Seganti. 1996. The effects of inhibitors of vacuolar acidification on the release of Listeria monocytogenes from phagosomes of Caco-2 cells. J. Med. Microbiol. 44:418–424. [DOI] [PubMed] [Google Scholar]
- Decatur, A.L., and D.A. Portnoy. 2000. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science. 290:992–995. [DOI] [PubMed] [Google Scholar]
- Edelson, B.T., and E.R. Unanue. 2001. Intracellular antibody neutralizes Listeria growth. Immunity. 14:503–512. [DOI] [PubMed] [Google Scholar]
- Falnes, P.O., and K. Sandvig. 2000. Penetration of protein toxins into cells. Curr. Opin. Cell Biol. 12:407–413. [DOI] [PubMed] [Google Scholar]
- Gedde, M.M., D.E. Higgins, L.G. Tilney, and D.A. Portnoy. 2000. Role of listeriolysin O in cell-to-cell spread of Listeria monocytogenes. Infect. Immun. 68:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy, C., J.L. Gaillard, J.E. Alouf, and P. Berche. 1987. Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. Infect. Immun. 55:1641–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, R.J., J.L. Jimenez, S. Chen, I.J. Tickle, J. Rossjohn, M. Parker, P.W. Andrew, and H.R. Saibil. 1999. Two structural transitions in membrane pore formation by pneumolysin, the pore-forming toxin of Streptococcus pneumoniae. Cell. 97:647–655. [DOI] [PubMed] [Google Scholar]
- Hernandez, L.D., L.R. Hoffman, T.G. Wolfsberg, and J.M. White. 1996. Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol. 12:627–661. [DOI] [PubMed] [Google Scholar]
- Horton, R.M., Z.L. Cai, S.N. Ho, and L.R. Pease. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques. 8:528–535. [PubMed] [Google Scholar]
- Jones, S., and D.A. Portnoy. 1994. a. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect. Immun. 62:5608–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S., and D.A. Portnoy. 1994. b. Intracellular growth of bacteria. Methods Enzymol. 236:463–467. [DOI] [PubMed] [Google Scholar]
- Kayal, S., A. Lilienbaum, C. Poyart, S. Memet, A. Israel, and P. Berche. 1999. Listeriolysin O-dependent activation of endothelial cells during infection with Listeria monocytogenes: activation of NF-kappa B and upregulation of adhesion molecules and chemokines. Mol. Microbiol. 31:1709–1722. [DOI] [PubMed] [Google Scholar]
- Ley, V., E.S. Robbins, V. Nussenzweig, and N.W. Andrews. 1990. The exit of Trypanosoma cruzi from the phagosome is inhibited by raising the pH of acidic compartments. J. Exp. Med. 171:401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden, J.C., N. Ruiz, and M. Caparon. 2001. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in gram-positive bacteria. Cell. 104:143–152. [DOI] [PubMed] [Google Scholar]
- Marquis, H., and E.J. Hager. 2000. pH-regulated activation and release of a bacteria-associated phospholipase C during intracellular infection by Listeria monocytogenes. Mol. Microbiol. 35:289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman, I., R. Fuchs, and A. Helenius. 1986. Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 55:663–700. [DOI] [PubMed] [Google Scholar]
- Moors, M.A., B. Levitt, P. Youngman, and D.A. Portnoy. 1999. Expression of listeriolysin O and ActA by intracellular and extracellular Listeria monocytogenes. Infect. Immun. 67:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, M., N. Sekino, M. Iwamoto, and Y. Ohno-Iwashita. 1995. Interaction of theta-toxin (perfringolysin O), a cholesterol-binding cytolysin, with liposomal membranes: change in the aromatic side chains upon binding and insertion. Biochemistry. 34:6513–6520. [DOI] [PubMed] [Google Scholar]
- Portnoy, D.A., P.S. Jacks, and D.J. Hinrichs. 1988. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 167:1459–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy, D.A., T. Chakraborty, W. Goebel, and P. Cossart. 1992. a. Molecular determinants of Listeria monocytogenes pathogenesis. Infect. Immun. 60:1263–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy, D.A., R.K. Tweten, M. Kehoe, and J. Bielecki. 1992. b. Capacity of listeriolysin O, streptolysin O, and perfringolysin O to mediate growth of Bacillus subtilis within mammalian cells. Infect. Immun. 60:2710–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossjohn, J., S.C. Feil, W.J. McKinstry, R.K. Tweten, and M.W. Parker. 1997. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell. 89:685–692. [DOI] [PubMed] [Google Scholar]
- Shepard, L.A., O. Shatursky, A.E. Johnson, and R.K. Tweten. 2000. The mechanism of pore assembly for a cholesterol-dependent cytolysin: formation of a large prepore complex precedes the insertion of the transmembrane beta-hairpins. Biochemistry. 39:10284–10293. [DOI] [PubMed] [Google Scholar]
- Sibelius, U., T. Chakraborty, B. Krogel, J. Wolf, F. Rose, R. Schmidt, J. Wehland, W. Seeger, and F. Grimminger. 1996. The listerial exotoxins listeriolysin and phosphatidylinositol-specific phospholipase C synergize to elicit endothelial cell phosphoinositide metabolism. J. Immunol. 157:4055–4060. [PubMed] [Google Scholar]
- Sibelius, U., E.C. Schulz, F. Rose, K. Hattar, T. Jacobs, S. Weiss, T. Chakraborty, W. Seeger, and F. Grimminger. 1999. Role of Listeria monocytogenes exotoxins listeriolysin and phosphatidylinositol-specific phospholipase C in activation of human neutrophils. Infect. Immun. 67:1125–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoble, J., D.A. Portnoy, and M.D. Welch. 2000. Three regions within ActA promote Arp2/3 complex-mediated actin nucleation and Listeria monocytogenes motility. J. Cell Biol. 150:527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoble, J., V. Auerbuch, E.D. Goley, M.D. Welch, and D.A. Portnoy. 2001. Pivotal role of VASP in Arp2/3 complex-mediated actin nucleation, actin branch-formation, and Listeria monocytogenes motility. J. Cell Biol. 155:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokurenko, E.V., D.L. Hasty, and D.E. Dykhuizen. 1999. Pathoadaptive mutations: gene loss and variation in bacterial pathogens. Trends Microbiol. 7:191–195. [DOI] [PubMed] [Google Scholar]
- Sun, A., A. Camilli, and D.A. Portnoy. 1990. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect. Immun. 58:3770–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney, L.G., and D.A. Portnoy. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109:1597–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unanue, E.R. 1997. Studies in listeriosis show the strong symbiosis between the innate cellular system and the T-cell response. Immunol. Rev. 158:11–25. [DOI] [PubMed] [Google Scholar]
- van Weert, A.W.M., K.W. Dunn, H.J. Geuze, F.R. Maxfield, and W. Stoorvogel. 1995. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J. Cell Biol. 130:821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva, M.S., A.J. Sijts, and E.G. Pam. 1995. Listeriolysin is processed efficiently into an MHC class I-associated epitope in Listeria monocytogenes-infected cells. J. Immunol. 155:5227–5233. [PubMed] [Google Scholar]
- Wadsworth, S.J., and H. Goldfine. 1999. Listeria monocytogenes phospholipase C-dependent calcium signaling modulates bacterial entry into J774 macrophage-like cells. Infect. Immun. 67:1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walev, I., S.C. Bhakdi, F. Hofmann, N. Djonder, A. Valeva, K. Aktories, and S. Bhakdi. 2001. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. USA. 98:3185–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]