

Abstract
Neutrophils must follow both endogenous and bacterial chemoattractant signals out of the vasculature and through the interstitium to arrive at a site of infection. By necessity, in the setting of multiple chemoattractants, the neutrophils must prioritize, favoring end target chemoattractants (e.g., fMLP and C5a) emanating from the site of infection over intermediary endogenous chemoattractants (e.g., IL-8 and LTB4) encountered en route to sites of infection. In this study, we propose a hierarchical model of two signaling pathways mediating the decision-making process of the neutrophils, which allows end target molecules to dominate over intermediary chemoattractants. In an under agarose assay, neutrophils predominantly migrated toward end target chemoattractants via p38 MAPK, whereas intermediary chemoattractant-induced migration was phosphoinositide 3-kinase (PI3K)/Akt dependent. When faced with competing gradients of end target and intermediary chemoattractants, Akt activation was significantly reduced within neutrophils, and the cells migrated preferentially toward end target chemoattractants even at 1/1,000th that of intermediary chemoattractants. End target molecules did not require chemotactic properties, since the p38 MAPK activator, LPS, also inhibited Akt and prevented migration to intermediary chemoattractants. p38 MAPK inhibitors not only reversed this hierarchy, such that neutrophils migrated preferentially toward intermediary chemoattractants, but also allowed neutrophils to be drawn out of a local end target chemoattractant environment and toward intermediary chemoattractants unexpectedly in an exaggerated (two- to fivefold) fashion. This was entirely related to significantly increased magnitude and duration of Akt activation. Finally, end target chemoattractant responses were predominantly Mac-1 dependent, whereas nondominant chemoattractants used primarily LFA-1. These data provide support for a two pathway signaling model wherein the end target chemoattractants activate p38 MAPK, which inhibits intermediary chemoattractant-induced PI3K/Akt pathway, establishing an intracellular signaling hierarchy.
Keywords: p38 MAPK; PI3K gamma; chemotaxis; neutrophils; signal transduction
Introduction
Neutrophils constitute the first line of defense against infectious agents and therefore play a central role in the inflammatory response. Upon infection and/or tissue damage, the neutrophils must first adhere to endothelium in response to chemokines presented at the endothelial interface and then migrate out of the microvasculature by following other chemokines secreted from nearby macrophages, mast cells, and other serosal cells (for review see Luster, 1998). These chemokines allow neutrophils to arrive in the general vicinity of the infection and would by definition have an intermediary role. Once in the vicinity, the neutrophils would then follow a series of end target chemoattractants including bacterial products and complement fragments to the final site of infection. Clearly, there are many chemoattractants that neutrophils encounter en route, and mechanisms must be in place for the cell to integrate and prioritize all of the multiple directional cues. Therefore, it would seem reasonable that a hierarchy of chemokines would exist, with some molecules being clearly dominant over others. Indeed, numerous investigators have demonstrated preferential migration toward end target chemoattractants, including fMLP and C5a, despite the presence of high concentrations of host-derived (intermediary) chemoattractants such as IL-8 or LTB4 (Campbell et al., 1997; Foxman et al., 1997, 1999; Shen et al., 2000). These data have led us to hypothesize that an intracellular signaling hierarchy also exists in the neutrophil decision-making process.
Despite the profound structural differences between the many chemoattractants, the receptors for these molecules are similar in that they belong to the seven-transmembrane helix receptor family (Damaj et al., 1996; Prossnitz et al., 1999). Once activated, these receptors transmit their signals to heterotrimeric G proteins, which activate downstream pathways leading to rapid cytoskeletal rearrangements and chemotaxis (Amatruda et al., 1995; Schraufstatter et al., 2001). Phosphoinositide 3-kinase (PI3K)* appears to be a key molecule in one of the intracellular pathways of neutrophil chemotaxis (Hirsch et al., 2000). Although multiple PI3K isoforms exist, chemotactic receptors utilize the p110γ catalytic isoform, which is responsible for downstream events including the conversion of membrane phospholipid phosphatidylinositol 4,5-biphosphate into phosphatidylinositol 3,4,5-triphosphate. This leads to the phosphorylation and activation of protein kinase B (also known as PKB or Akt). This is best evidenced by the fact that activation of chemotactic receptors in mice lacking p110γ manifests as no phosphorylation or no activation of PKB/Akt, leading to defects in chemotaxis (Hirsch et al., 2000).
However, P110γ-deficient neutrophils can still chemotax in significant numbers (Hirsch et al., 2000). Indeed, the p38 MAPK pathway has also been demonstrated to play a very important role in neutrophil chemotaxis in humans (Nick et al., 1997) and mice (Cara et al., 2001), and this pathway is still effectively activated in p110γ-deficient cells, perhaps explaining neutrophil recruitment to sites of inflammation in these mice (Sasaki et al., 2000). Whether the p38 MAPK and PI3K/Akt pathways are complementary, overlapping, or perhaps even opposing and whether they function in a hierarchical fashion remains unclear. Furthermore, whether there is any physiologic cross-talk between these pathways has never been addressed.
In this study, our data would support the proposed model that end target chemoattractants (fMLP and C5a) function primarily by stimulating p38 MAPK, whereas intermediary chemoattractants (IL-8 and LTB4) primarily function via the PI3K/Akt pathway. These pathways underlie the hierarchical dominance of end target chemoattractants over intermediary chemoattractants inasmuch as activation of the p38 MAPK pathway in neutrophils inhibits the PI3K/Akt pathway. Tonic inhibition of PI3K/Akt is apparent, since inhibition of p38 MAPK causes a marked increase in PI3K/Akt activity and a causally related exaggerated chemotactic response to intermediary chemokines.
Results
Dose response for IL-8 and fMLP
Fig. 1 a summarizes how chemotaxis was determined, and Fig. 1 b shows the under agarose design for the dose response experiments. Neutrophils were placed in two wells equidistant from a middle well that was the source of the chemoattractant. Neutrophils moved equally toward the central well from the left and the right well, suggesting that the chemoattractants diffused equally in both directions. We found that neutrophils migrate at concentrations between 0.1 and 10.0 pmol, with higher concentrations resulting in stasis and lower concentrations having no effect (Fig. 1 c). The optimal concentration of fMLP for neutrophil chemotaxis was found to be 1.0 pmol. At every concentration used, directional migration was observed in that 99% of neutrophils migrated toward (rather than away from) the source of chemoattractant and only one or two cells were seen moving in the opposite direction. The distance of neutrophil chemotaxis to fMLP followed an identical trend and was optimal at 1.0 pmol (unpublished data). At this concentration, neutrophils migrated 2,000 μm in 2 h or ∼15–20 μm/min. IL-8 produced a similar bell-shaped response curve for both the number of migrating cells and the distance traveled, with the optimal IL-8 dose seen at 10.0 pmol (unpublished data). In all of the following experiments, inhibitors caused a very similar decrease in the number of migrating cells, the distance traveled, and the migratory rate; therefore, only the number of cells is presented.
Figure 1.

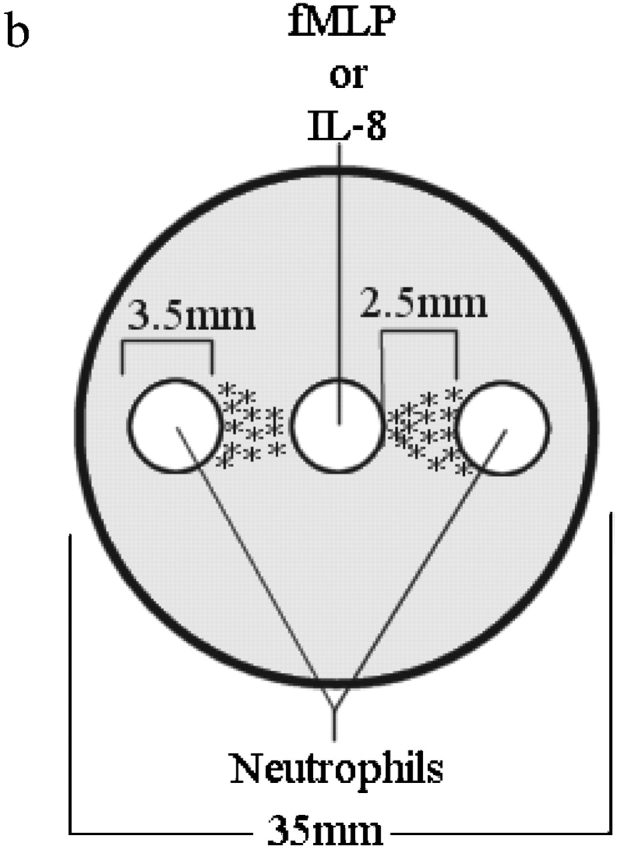

Gel setup and analysis. (a) Target areas used to measure chemotaxis. Randomly migrating neutrophils will appear in equal numbers in both target areas. Directionally migrating neutrophils will only be found in target area A. The number of chemotaxis cells can be determined by subtracting the number of cells in area B from the number of cells in area A. (b) Under agarose setup used for dose response experiments. (c) fMLP dose response. Data is shown as mean ± SEM (n = 8). *P < 0.05 compared with 0.0 pmol fMLP.
Effects of p38 MAPK and PI3K Inhibition on migration toward end target and intermediary chemoattractants
Two p38 MAPK inhibitors (SKF86002 and SB203858) and two PI3K inhibitors (wortmannin and LY294002) were used to determine which chemoattractants induce chemotaxis via the p38 MAPK and PI3K pathways. All inhibitors were used at optimal concentrations based on very extensive characterization by other groups (Zu et al., 1998) and by confirmation of these results in our own preliminary experiments. Fig. 2 a illustrates that neutrophil migration to 1.0 pmol of the end target chemoattractant fMLP was inhibited by >90% by both p38 MAPK inhibitors. Similarly, C5a-induced neutrophil migration was also inhibited completed by SB203580. In contrast, the PI3K inhibitors decreased migration to a much lesser degree in response to fMLP (35%) and by even less in response to C5a (Fig. 2 b). Treating the neutrophils with both a p38 MAPK inhibitor (SB203850) and a PI3K inhibitor (LY294002) inhibited chemotaxis as effectively as a p38 MAPK inhibitor alone (unpublished data). Clearly, the end target chemoattractants signal through a p38 MAPK–dominated pathway in which PI3K plays only a very minor role.
Figure 2.


Effect of p38 MAPK and PI3K in neutrophil chemotaxis from the left well to the right well in response to fMLP, IL-8, C5a, and LTB4. Neutrophils were pretreated for 60 min with either 30 μM SKF86002, 10 μM SB203850, 100 nM wortmannin, or 50 μM LY294002. UT, untreated neutrophils. (a) Effect of the p38 MAPK inhibitors on neutrophil chemotaxis. (b) Effect of the PI3K inhibitors on chemotaxis. Data is shown as mean ± SEM with a minimum of n = 8 for each experiment. *P < 0.05 compared with UT.
Fig. 2 a also shows the effects of the p38 MAPK inhibitors on neutrophil migration to 10.0 pmol of the intermediary chemoattractant, IL-8. The p38 MAPK inhibitor SKF86002 had no effect on the ability of neutrophils to chemotax to IL-8. The p38 MAPK inhibitor SB203850 had a minor effect on chemotaxis to IL-8; a 35% decrease was noted. A similar 30–40% decrease with SB203850 in chemotaxis was noted in response to LTB4. In complete contrast, both of the PI3K inhibitors dramatically decreased migration to IL-8, with wortmannin blocking 88% of chemotaxis and LY294002 blocking 78% of chemotaxis to IL-8 (Fig. 2 b). Similarly, LY294002 inhibited >90% of the chemotaxis in response to the intermediary chemoattractant LTB4. Treating neutrophils with both a p38 MAPK inhibitor (SB203850) and a PI3K inhibitor (LY294002) decreased chemotaxis as effectively as a PI3K inhibitor alone (unpublished data). These data suggest that the intermediary chemoattractants signal through a PI3K-dominated pathway in which p38 MAPK may play only a very minor role.
Competing gradients of end target and intermediary chemoattractants
In competing gradients consisting of optimal concentrations of fMLP (1 pmol; in right well) and 1/100th optimal concentration of IL-8 (0.1 pmol; in left well), neutrophils preferentially migrated toward fMLP (Fig. 3 a). When optimal concentrations of both fMLP and IL-8 were added, neutrophils still migrated toward fMLP, unexpectedly in much greater numbers. This pattern was repeated with competing gradients consisting of 1/100th optimal concentration of fMLP (0.01 pmol) and optimal concentrations of IL-8 (10.0 pmol). Under these conditions, neutrophils still preferentially migrated toward fMLP, although the effect was less pronounced (Fig. 3 a). It is noteworthy that this lower concentration of fMLP (without IL-8 present) does not induce any migration (Fig. 1 c), suggesting that not only does fMLP overcome any effects of IL-8, but the cells migrate more effectively in the presence of the opposing IL-8 gradient. Neutrophils also migrated toward fMLP in an opposing LTB4 gradient (Fig. 3 a). The other end target chemoattractant, C5a, induced neutrophil chemotaxis in a dominant fashion over optimal doses of LTB4 or IL-8 (Fig. 3 a). When opposing gradients of two end target chemoattractants (fMLP versus C5a) were used, there was no dominance for either chemoattractant, since neutrophils migrated equally well in both directions (unpublished data), consistent with the same rather than different signaling pathways being involved. An identical response was observed when the opposing gradients of the two intermediary chemoattractants (LTB4 versus IL-8) were used (unpublished data). For the remainder of the experiments, fMLP and IL-8 are used to further explore the mechanisms of action. Nevertheless, in numerous instances similar results are observed for C5a and LTB4.
Figure 3.


Neutrophils respond in a hierarchal manner to competing gradients of chemoattractants. (a) Migration of neutrophils in competing chemotactic gradients. Six gradients were tested: the first consisted of optimal fMLP (1.0 pmol) and 1/100th optimal IL-8 (0.1 pmol), the second consisted of 1/100th optimal fMLP (0.01 pmol) and optimal IL-8 (10.0 pmol), and the third consisted of optimal gradients of fMLP and IL-8. The fourth gradient consisted of optimal IL-8 and optimal C5a (10.0 pmol). The fifth gradient consisted of optimal LTB4 (10.0 pmol) and optimal fMLP. The final gradient consisted of optimal LTB4 and optimal C5a. (b) Migration of p38 MAPK inhibited neutrophils from a well containing 10.0 pmol IL-8 or 1.0 pmol fMLP to wells containing various concentrations of fMLP or IL-8. Results are shown as mean ± SE (n = 10). *P < 0.05 compared with migration toward fMLP.
fMLP is a smaller molecule then IL-8 (3 amino acids versus 88 amino acids), and therefore, the hierarchy may have been a result of faster diffusion of fMLP than IL-8. To show that this hierarchy was not a result of different diffusion rates, we used gels containing two wells; neutrophils and IL-8 were loaded into one well, and fMLP was loaded into a second distal well. Despite this very high local concentration of IL-8, neutrophils migrated to fMLP (Fig. 3 b). Once again, fMLP (10.0 pmol) induced twice the number of neutrophils to migrate out of the local IL-8 gradient then is observed without IL-8 (Fig. 1 c). fMLP receptor numbers were measured in the presence and absence of IL-8, and a 60% increase in fMLP receptor number was consistently noted (Table I).
Table I. Up-regulation of formyl-peptide receptor in response to IL-8 stimulation.
P < 0.05 compared to 0 nM IL-8.
Results are shown as the percentage of increase in fluorescence ± SEM.
We then reversed the positions of the chemoattractants (neutrophils and fMLP in one well and IL-8 into the second well). Under these conditions, neutrophils remained in the fMLP-containing well and did not migrate to IL-8 (Fig. 3 b). These results suggest that regardless of the concentrations of fMLP and IL-8 or the diffusion distance, neutrophils always prefer the end target chemokine fMLP.
Changes in downstream events of the IL-8 receptors mediates the chemotactic hierarchy
To determine why end target molecules dominated over intermediary chemoattractants, we next examined whether fMLP directly affects IL-8 receptor number and/or downstream events. Fig. 4 a illustrates that fMLP had absolutely no effect upon either CXCR1 or CXCR2 (the two known IL-8 receptors) at a wide range of concentrations. As a positive control, we show that CXCR1 and CXCR2 were both internalized in response to IL-8 (Fig. 4 a). Clearly, fMLP does not affect IL-8 receptor numbers.
Figure 4.


Internalization and signaling of CXCR1 and CXCR2. (a) Internalization of CXCR1 and CXCR2 in response to 100 nM fMLP and/or 100 nM IL-8 (n = 5). (b) Akt phosphorylation induced by 100 nM IL-8 (left) or 100 nM fMLP + 100 nM IL-8 (right). Results are shown as mean ± SEM (n = 4).
Since PI3K inhibition reduced IL-8 chemotaxis, we examined whether this pathway was affected by fMLP. To do this, we measured the phosphorylation of the downstream molecule Akt as an index of PI3K activity. Akt has been shown to be involved in the IL-8-induced PI3K activation as much as mice deficient in the P110γ catalytic isoform have no Akt phosphorylation. To confirm these findings in our human system, we determined that the PI3K inhibitor LY294002 prevented Akt phosphorylation in response to IL-8 (unpublished data). Fig. 4 b summarizes that Akt phosphorylation in response to IL-8 can be detected within 30 s, peaks within the first minute, and largely dissipates by 30 min. When neutrophils were stimulated with both fMLP and IL-8, the Akt phosphorylation at peak levels is less than half that seen with IL-8 alone and returns to control values by 5 min (Fig. 4 b). Together, these data suggest that fMLP has a direct inhibitory effect upon the PI3K/Akt pathway (Fig. 4 b) that is independent of IL-8 receptor number (Fig. 4 a).
Effect of PI3K or p38 MAPK inhibition on the fMLP/IL-8 hierarchy
We looked at the effect of PI3K inhibition on the fMLP/IL-8 hierarchy. Two gradients were tested: the first consisting of optimal fMLP (1.0 pmol) and 1/100th optimal IL-8 (0.1 pmol), and the second consisting of 1/100th optimal fMLP (0.01 pmol) and optimal IL-8 (10.0 pmol). In both gradients, neutrophils treated with the PI3K inhibitor LY294002 still preferentially migrated toward fMLP (Fig. 5 a).
Figure 5.



Migration of inhibitor-treated neutrophils in competing chemoattractant gradients. Five gradients were tested: the first consisted of optimal fMLP (1.0 pmol) and 1/100th optimal IL-8 (0.1 pmol) and the second consisted of 1/100th optimal fMLP (0.01 pmol) and optimal IL-8 (10.0 pmol). (a) Migration of PI3K inhibited neutrophils. (b) Migration of p38 MAPK inhibited neutrophils. (c) Migration of PI3K and p38 MAPK inhibited neutrophils. Results are shown as mean ± SEM (n = 10) except for LTB4 and C5a containing experiments where n = 8. *P < 0.05 compared with migration to IL-8 or LTB4.
By contrast, p38 MAPK inhibition not only blocked the preferential migration of neutrophils toward fMLP but also reversed and greatly enhanced migration of neutrophils toward IL-8. Indeed Fig. 5 b shows that when p38 MAPK–treated neutrophils were placed in competing gradients containing 1/100th optimal concentrations of fMLP (0.01 pmol) and optimal IL-8 concentrations (10.0 pmol), neutrophils preferentially migrated toward IL-8. Even more striking was the fact that even when the conditions were greatly biased toward fMLP, i.e., when optimal concentrations of fMLP (1.0 pmol) and 1/100th optimal concentrations of IL-8 (0.1 pmol) were used, p38 MAPK–treated neutrophils preferentially migrated toward IL-8. Similarly, when optimal concentrations of fMLP and LTB4 were tested, p38 MAPK–inhibited neutrophils migrated toward LTB4. Also shown in Fig. 5 b is the fact that the number of neutrophils that migrated toward IL-8 were approximately five times that normally seen in the absence of the opposing fMLP gradient. The enhanced migration to IL-8 occurs only in the presence of the p38 MAPK inhibitor and fMLP and not in the presence of the p38 MAPK inhibitor alone. This was not particular to this set of chemoattractants, since p38-inhibited neutrophils migrated in greater numbers to LTB4 in the presence of an opposing fMLP gradient. Clearly, when p38 MAPK is inhibited fMLP either activates additional pathways or increases PI3K/Akt to enhance migration toward IL-8.
Fig. 5 c demonstrates that alteration in PI3K/Akt was the mechanism by which fMLP induces p38 MAPK–inhibited neutrophils to migrate in greater numbers toward the intermediary chemoattractants. First, inhibition of PI3K with LY294002 eliminated p38-inhibited neutrophils from migrating toward IL-8 (Fig. 5 c). To examine whether inhibition of p38 MAPK increased PI3K activity, we examined Akt in the presence and absence of the p38 MAPK inhibitors. Fig. 6 shows that phosphorylation of Akt in response to fMLP and IL-8 peaks at ∼1 min and returns to control by 5 min. However, when p38 MAPK inhibited neutrophils are stimulated with both fMLP and IL-8, Akt phosphorylation is high and remains high for at least 60 min (Fig. 6). In additional experiments, p38 MAPK was activated by the nonchemotactic end target molecule, LPS, a potent activator of p38 MAPK (Haddad et al., 2001). LPS (10 ng/ml) pretreatment of neutrophils blocked migration to IL-8 (Fig. 7 a) in an identical fashion to fMLP. Inhibiting p38 MAPK with SB203850 before LPS treatment reversed the LPS-induced inhibition of migration to IL-8. However, the number of migrating neutrophils was not enhanced. Fig. 7 b shows that AKT phosphorylation induced by IL-8 was abolished in LPS-treated cells. p38 MAPK inhibition not only restored phosphorylation of AKT in IL-8– and LPS-stimulated cells, but once again the Akt phosphorylation was stronger and more prolonged then the phosphorylation induced by IL-8 alone (see Fig. 4 b for comparison). Clearly, this is a common feature of numerous end target molecules.
Figure 6.

Akt phosphorylation induced by simultaneous stimulation with 100 nM IL-8 and 100 nM fMLP in untreated cells and cells treated with 10 μM SB203580. Data is shown as mean ± SEM (n = 4).
Figure 7.


Effect of LPS on neutrophil chemotaxis and Akt phosphorylation induced by IL-8. (a) Neutrophils were pretreated for 1 h with 10 ng/ml LPS and then placed in wells adjacent to optimal concentrations of IL-8 (10.0 pmol). (b) Phosphorylation of Akt in LPS-treated neutrophils stimulated with IL-8 ± the p38 MAPK inhibitor SB203580. Results are shown as mean ± SEM with a minimum n = 9 for chemotaxis assay and n = 3 for Akt phosphorylation. *P < 0.05 compared with IL-8 alone.
Finally, to test how potent the reversal of the fMLP/IL-8 hierarchy with p38 MAPK inhibitors could be, p38-inhibited neutrophils were placed into a well with fMLP, and IL-8 was placed into a distant well. Under these conditions, p38 MAPK–treated neutrophils migrated out of the well containing fMLP and toward the IL-8 gradient (Fig. 8). It should be emphasized that IL-8 does not induce neutrophils to migrate away from a local fMLP environment in the absence of p38 MAPK inhibition (Fig. 3 b). By contrast, when p38 MAPK inhibited neutrophils were placed into an IL-8–containing well, they failed to migrate efficiently toward fMLP (Fig. 8).
Figure 8.

Migration of p38-MAPK inhibited neutrophils from a well containing 10.0 pmol IL-8 or 1.0 pmol fMLP to wells containing various concentrations of fMLP or IL-8. Results are shown as mean ± SEM (n = 10). *P < 0.05 compared with migration to IL-8.
IL-8 receptor number and function is not altered by p38 MAPK inhibition in opposing gradients
We examined several possibilities at the IL-8 receptor level to explain the reversal and enhancement of leukocyte chemotaxis away from end target chemoattractants and toward intermediary chemoattractants when p38 MAPK is inhibited. The first possibility was that fMLP and the p38 MAPK inhibitor together altered IL-8 receptor number. FACS® analysis was used to determine if receptor internalization was affected by p38 MAPK inhibition. Whereas IL-8 caused 78 and 72% of CXCR1 and CXCR2 internalization, respectively, fMLP had minimal effects on CXCR1 or CXCR2 (<15%). When p38 MAPK–treated neutrophils were stimulated with fMLP and IL-8, there was no alteration in the pattern of CXCR1 and CXCR2 internalization compared with fMLP and IL-8 stimulation of uninhibited cells (Table II). Both perturbations induced 80–90% internalization of CXCR1 and 70–75% of CXCR2 internalization.
Table II. CXCR1 and CXCR2 internalization in the presence of various stimuli.
| IL-8 receptor
|
IL-8 alone
|
fMLP alone
|
IL-8 + fMLP
|
IL-8 + fMLP + SB203580
|
|---|---|---|---|---|
| % | ||||
| CXCR1 | 78.3 ± 1.7 | 14.9 ± 13.8 | 82.8 ± 5.7 | 90.8 ± 1.5 |
| CXCR2 | 72.0 ± 9.6 | 12.5 ± 4.8 | 74.3 ± 1.2 | 69.6 ± 1.7 |
Results are shown as the percentage of internalization ± SEM. n = 2 for IL-8 + fMLP and IL-8 + fMLP + SB203580. n = 3 for IL-8 and fMLP.
Calcium flux was also unaltered by p38 MAPK inhibition. Fig. 9 a shows that activation of CXCR1 and CXCR2 was unaffected by p38 MAPK inhibition. fMLP stimulation induced Ca2+ influx of 60%. If IL-8 was then added to the cells, only a 20–25% Ca2+ influx was induced regardless of whether p38 MAPK was inhibited. Similarly, Fig. 9 b shows that IL-8 added first to neutrophils induced a 50–60% increase in Ca2+ influx. Subsequent addition of fMLP induced a 50–60% Ca2+ influx regardless of the presence of p38 MAPK inhibition. Clearly, Ca2+ influx was not related to the changes in chemotaxis.
Figure 9.
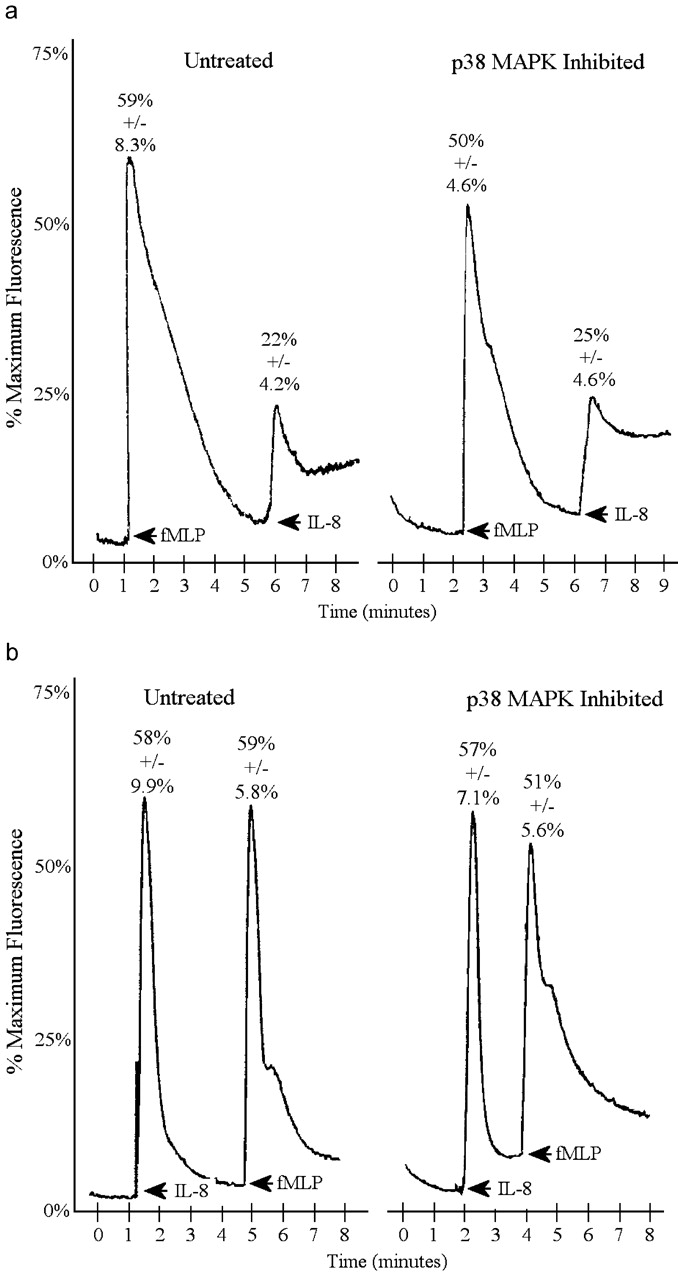
Effect of p38 MAPK inhibition on receptor activation measured by calcium flux. Effect of p38 MAPK inhibition on calcium flux induced by 100 nM fMLP followed by 100 nM IL-8 (a) or 100 nM IL-8 followed by 100 nM fMLP (b).
Previous research has shown that IL-8 induces chemotaxis primarily through CXCR1 and cell activation through CXCR2 (Jones et al., 1996; Godaly et al., 2000). We used blocking antibodies to both IL-8 receptors (CXCR1 and CXCR2) to see if receptor function changed when cells were treated with p38 MAPK inhibitors. IL-8–induced chemotaxis was entirely mediated by CXCR1 in our under agarose assay (unpublished data). Next, a competing gradient consisting of 1/100th optimal concentrations of fMLP (0.001 μM) and optimal concentrations of IL-8 (1.0 μM) was produced. Adding anti-CXCR1 antibodies to p38 MAPK–treated neutrophils reduced migration to IL-8 by 90% (Fig. 10). Adding anti-CXCR2 antibodies to p38 MAPK–treated cells had no effect on migration to IL-8 (Fig. 10). These results suggest that chemotaxis to IL-8 was CXCR1 mediated in p38 MAPK–treated neutrophils and that the pathways activated by CXCR1 and CXCR2 are unchanged by p38 MAPK inhibition.
Figure 10.

Effect of anti-CXCR1 and anti-CXCR2 antibodies on chemotaxis of p38 MAPK inhibited neutrophils in competing fMLP (0.01 pmol) and IL-8 (10.0 pmol) gradients. Results are shown as mean ± SEM (n = 5). *P < 0.05 compared with no antibody.
Downstream effectors
p38 MAPK and PI3K/Akt could activate the same adhesion molecules for chemotaxis or could activate specific subunits of the CD18 glycoprotein complex. Antibodies to LFA-1 (CD11a), Mac-1 (CD11b), and CD18 (the common unit to LFA-1 and Mac-1) were used to block these adhesion molecules. Anti-CD18 antibodies blocked migration to both IL-8 and fMLP by ∼75% (Fig. 11 a). Anti–LFA-1 antibodies had no impact on chemotaxis to fMLP, whereas anti–Mac-1 antibodies blocked 60% of chemotaxis to fMLP. Anti–LFA-1 antibodies blocked chemotaxis to IL-8 by 50%, but antibodies to Mac-1 had no significant impact on chemotaxis to IL-8. In preliminary work, the p38 MAPK inhibitors reduced Mac-1 up-regulation by 50–60% (unpublished data), suggesting downstream effects of these inhibitors on Mac-1 function.
Figure 11.

Role of LFA-1 and Mac-1 in chemotactic responses. (a) Migration of neutrophils to fMLP and IL-8 when treated with blocking antibodies to CD11a (LFA-1), CD11b (Mac-1), and CD18 (common subunit of CD11a and CD11b). (b) Migration of neutrophils treated with blocking antibodies to CD11a and CD11b in competing gradients of fMLP and IL-8. Results are shown as mean ± SEM (n = 12). *P < 0.05 compared with UT (isotype control).
We also looked at the impact of LFA-1 and Mac-1 inhibition in the fMLP/IL-8 hierarchy. We found that anti–LFA-1 had no significant impact on chemotaxis in this system, consistent with the PI3K/Akt pathway being turned off by fMLP upstream of LFA-1. Anti–Mac-1 again decreased migration by ∼50% (Fig. 11 b). Interestingly, anti–Mac-1 and anti–LFA-1 in tandem completely blocked migration in the p38 MAPK–inhibited fMLP/IL-8 hierarchy (unpublished data), suggesting that both adhesion molecules become important for IL-8–induced migration when p38 MAPK is inhibited in opposing gradients of fMLP.
Discussion
During an infection, many chemoattractants are released from various locations including the vascular endothelium, interstitial cells (macrophages and mast cells), and the infectious agent itself (Murdoch and Finn, 2000). These multiple sources of chemoattractants result in a very complex milieu of conflicting chemoattractant gradients that neutrophils must navigate through in order to reach the site of infection. One can envision numerous situations where neutrophils would be faced with having to make decisions between multiple gradients of different chemokines. In fact, one could argue that this would occur anytime a neutrophil encounters intermediary chemokines on the surface of endothelium, adheres, and now must migrate away from this site and toward a tissue source of end target chemoattractant (bacterial product or complement fragment). Therefore, we hypothesized that (a) endogenous-derived chemokines and end target bacterial chemoattractants would have different intracellular signaling pathways, (b) that these pathways would function in a hierarchical manner with end terminal pathways dominating, and (c) that an active inhibition by the end target signaling pathway of the intermediary chemoattractant-induced intracellular signaling is an important mechanism in the neutrophil decision-making process.
To study the decision-making (signaling) processes of neutrophils in complex chemotactic fields, we used the under agarose neutrophil migration assay first used by Nelson et al. (1975) and more recently by Campbell et al. (1997) and Foxman et al. (1997)(1999). This system allows for establishment of multiple simultaneous chemotactic fields. There are advantages of this assay over the more widely used Boyden chamber assay. Whereas the latter is used very effectively for large scale screening of compounds, it is a unidirectional fluid phase assay, which does not allow for the effective study of bidirectional decision making by the neutrophils in response to multiple chemotactic agents. Although the concentrations we used may appear larger than what has been used in Boyden chambers, the concentrations cannot be directly compared due to the more limited movement of the chemokines in the agarose versus fluid in a Boyden chamber. Previously, investigators have used this assay to report directional migration of neutrophils (Nelson et al., 1975; Foxman et al., 1997, 1999). We have modified the assay so that we could quantify the number of cells that migrated toward a particular chemotactic cue. This became extremely useful to report enhanced or decreased number of migrating neutrophils in response to various manipulations but required that we design the system in such a manner as to be able to limit and count the number of migrating cells. In this study, we report that end terminal chemoattractants at various concentrations and in various scenarios dominated over the chemotactic potential of intermediary chemoattractants, consistent with the work of Campbell et al., 1997, Foxman et al. (1997)(1999), and others (Knall et al., 1997) using the same under agarose assay system. We extend the work to show that several of the end target molecules induce chemotaxis via p38 MAPK, whereas in stark contrast the intermediary chemoattractants use the PI3K/Akt pathway in this system. More importantly, a hierarchy between the two pathways exists in that the former has inhibitory impact upon the latter. This may be the underlying problem in serious clinical situations like sepsis in which increased levels of LPS, fMLP, or C5a in the circulation may inhibit chemotaxis toward intermediary chemoattractants, thereby preventing neutrophil infiltrations to sites of infections. Indeed, the inability of neutrophils to emigrate is a well-described (Wagner and Roth, 1999) but unexplained phenomenon in septic patients. In addition, neutrophils migrated toward end target chemoattractants better when intermediary chemoattractants were present in an opposing gradient, suggesting an amplifying loop, which, in the case of fMLP, was at least in part due to increased fMLP receptor number. Along the same lines of cross-talk, we report that inhibition of the p38 MAPK pathway leads to an amplification of the PI3K/Akt pathway in both magnitude and duration. The p38 MAPK inhibition results in neutrophil migration away from end-terminal chemoattractants and toward intermediary chemoattractants in two- to fivefold greater numbers. Finally, and most importantly, the enhanced PI3K/Akt activation induced by p38 MAPK inhibitors is causally related to the increase in neutrophil chemotaxis, since inhibition of PI3K completely inhibited the enhanced chemotaxis in response to intermediary chemokines.
There is some supportive data for individual end terminal chemoattractants using p38 MAPK, whereas intermediary chemoattractants use PI3K/Akt. Previous work has clearly demonstrated that IL-8 activates PI3K/Akt, ERK, and p38 MAPK in primary human neutrophils (Knall et al., 1995; Capodici et al., 1998). Inhibition of PI3K with wortmannin inhibited ERK but not p38 MAPK activation, suggesting that the activation of PI3K/Akt and ERK was distinct from p38 MAPK activation (Knall et al., 1997). Moreover, p38 MAPK activation had no demonstrable role in the induction of neutrophil chemotaxis in response to IL-8, whereas PI3K/Akt was essential for IL-8–induced neutrophil chemotaxis (Knall et al., 1997). The bacterial compound fMLP also appears to activate multiple intracellular signaling proteins including PI3K/Akt, ERK, and p38 MAPK (Zu et al., 1998; Yasui and Komiyama, 2001). However, our data suggest that activation of p38 MAPK but not PI3K/Akt blocked fMLP-induced chemotaxis but had little impact on IL-8–induced neutrophil chemotaxis. We observed for the first time that when both pathways are activated simultaneously p38 MAPK dominates by depressing PI3K/Akt activity. Furthermore, p38 MAPK could interrupt and turn off previously activated PI3K/Akt in that when neutrophils were placed in a well with IL-8 and fMLP was placed in a distal well, the neutrophils would subsequently respond to fMLP, despite having IL-8/PI3K/Akt activated first. This latter observation is likely critical to neutrophils terminating all activity in response to intermediary chemoattractants and responding to end target chemoattractants.
Our data reveals that cross-talk between p38 MAPK and PI3K/Akt pathways is the inhibitory mechanism of action of end target chemoattractants on intermediary chemoattractant responses. Cross-talk among different signaling pathways is not without precedent. In fact, a significant amount of work has demonstrated that p38 MAPK and PI3K have opposing effects on apoptosis in neutrophils, endothelium, and other cells and that the PI3K/Akt pathway can inhibit p38 MAPK pathway (Berra et al., 1998; Alvarado-Kristensson et al., 2002). Although to date no one to our knowledge has shown that p38 MAPK negatively regulates PI3K/Akt, one study has shown that p38 MAPK can associate with Akt (Rane et al., 2001). In this study, we show that for chemotactic stimuli in neutrophils, p38 MAPK appears to inhibit PI3K. Moreover, when p38 MAPK is inhibited the effects of PI3K/Akt are greatly unmasked as evidenced by both a very substantial increase in Akt activity and a fivefold increase in PI3K-dependent chemotaxis.
A second site of inhibition by fMLP/p38 MAPK on IL-8/PI3K/Akt may be at the IL-8 receptor. Activation of the fMLP receptor has been shown to cross-desensitize G protein activation stimulated by IL-8 (Richardson et al., 1995; Ali et al., 1999). This process is mediated by receptor phosphorylation in which sites on the cytoplasmic end of the IL-8 receptors are phosphorylated by PKC. However, based on data presented herein, we feel that neither phosphorylation of the IL-8 receptor nor internalization is likely to be the reason for neutrophils not responding to IL-8 in the presence of fMLP. First, fMLP had absolutely no effect on the surface expression of either CXCR1 or CXCR2 in our system. Second, placing neutrophils into a local environment of IL-8 (Fig. 3 b) activated the IL-8 receptors and caused rapid homologous desensitization and internalization (Fig. 4). Nevertheless, even after the IL-8 receptors were internalized and phosphorylated by local IL-8, neutrophils still migrated toward fMLP placed in a distal well (in fact in greater numbers than without IL-8 present). Clearly, inhibition of the IL-8 pathway occurred down stream of the receptors and as our study suggests via p38 MAPK. Moreover, mutations at the phosphorylation sites on the IL-8 receptors do not inhibit responses to IL-8 stimulation (Richardson et al., 1998). As such, these authors predicted that another mechanism downstream of the G proteins was involved in maintaining the fMLP/IL-8 hierarchy. Our studies suggest that this mechanism is an inhibition of the PI3K pathway by p38 MAPK. Mechanisms by which p38 MAPK and PI3K/Akt differentially effect migration could include relocalization of CD11/CD18 toward the leading edge of neutrophils (preferentially toward end target chemoattractants), redistribution of CD11/CD18 into local microenvironments (e.g., lipid rafts) leading to improved chemotaxis and/or increased affinity, and/or avidity in response to end target chemoattractants but not intermediary chemokines. Our preliminary work suggests that the p38 MAPK and PI3K/Akt pathways may recruit different α subunits of the CD18 glycoprotein complex and that in opposing gradients Mac-1 seems to dominate over LFA-1. This would suggest that the two α subunits of CD18 oppose rather than cooperate with each other. There is much controversy regarding the relative importance of LFA-1 versus Mac-1 with studies indicating a dominant role for either the former or the latter (Gao and Issekutz, 1996; Ding et al., 1999; Hogg and Leitinger, 2001; Seo et al., 2001). Our observation that different chemoattractants may preferentially activate LFA-1 or Mac-1 separately would explain some of this controversy. Although the prevailing view is that LFA-1 and Mac-1 function in a cooperative additive manner, this theory does not explain the curious finding that for a particular stimulus, LFA-1 deficiency in mice reduces neutrophil recruitment, whereas Mac-1 deficiency actually increased neutrophil recruitment (Ding et al., 1999), raising the possibility of some opposing effects of LFA-1 and Mac-1.
Materials and methods
Reagents
The p38 MAPK inhibitors SKF86002 and SB203580, the PI3K inhibitor LY294002, and the chemoattractants C5a and LTB4 were purchased from Calbiochem. Ficoll-Histopaque 1077, anti-CXCR1 antibodies, anti-CXCR2 antibodies, mouse IgG1κ, Triton X-100, BSA, Tris HCl, Hepes, EGTA, fMLP, IL-8, β-mercaptoethanol, bromophenol blue, orthovanadate, leupeptin, aprotinin, and the PI3K inhibitor wortmannin were purchased from Sigma-Aldrich. HBSS, RPMI-1640 culture medium, and ultra pure agarose were purchased from Life Technologies. FBS was purchased from HyClone Labs. Dextran (mol wt = 250,000) was purchased from Spectrum. 35 mm × 10 mm tissue culture dishes and FITC-conjugated goat anti–mouse IgG were purchased from Becton Dickinson. Optilyse-B was purchased from Immunotech. SDS was purchased from Bio-Rad Laboratories. Highbond p+ membrane, EPO developing kit, and Hyperfilm MP were purchased from Amersham Biosciences. Rabbit anti–phospho-Akt (Ser 473) antibodies and HRP-conjugated anti–rabbit IgG antibodies were purchased from Cell Signaling. Quantity One software was purchased from Bio-Rad Laboratories.
Neutrophil isolation and preparation
Blood was collected from healthy human donors. Erythrocytes were removed using dextran sedimentation (6% dextran/0.9% NaCl) followed by two rounds of hypotonic lysis using ddH2O. Neutrophils were isolated from the resulting cell suspension using Ficol-Histopaque density centrifugation. The entire isolation was done at 4°C. Purified neutrophils were suspended in HBSS at a concentration of 1.0 × 107 cells/ml and were kept on ice until needed. Neutrophils were >95% viable and >97.5% pure as determined by FACS®.
Under agarose assay procedure
The under agarose assay was used as described previously (Nelson et al., 1975; Foxman et al., 1999) with minor modification. Falcon 35 mm × 10 mm culture dishes were filled with 3 ml of a 1.2% agarose solution containing 50% H2CO3-buffered HBSS and 50% RPMI-1640 culture medium with 20% heat-inactivated FCS. After the agarose solidified, three wells, 3.5 mm in diameter and 2.4 mm apart, were cut into a straight line in the gel. The gels were allowed to equilibrate for 1 h in a 37°C/5% CO2 incubator. Unless otherwise noted, the center well was loaded with purified neutrophils, and the outer wells were loaded with varying concentrations of chemoattractant. Once loaded, the gels were incubated for 2 h in a 37°C/5% CO2 incubator, which allowed sufficient time for neutrophils to migrate the entire distance between the wells. Results were recorded using a video camera attached to a ZEISS Axiovert 135 microscope.
For the dose response experiments, the two outer wells were loaded with neutrophils, and the inner well was loaded with chemoattractant. The maximum IL-8 concentration tested was 1.25 μM, which was the concentration of our stock solution.
p38 MAPK inhibitors were added to isolated neutrophils such that the final inhibitor concentration was 30 μM for SKF86002 and 10 μM for SB203580, which have been shown to be the optimal inhibitory concentration without nonspecific effects (Zu et al., 1998). The concentrations of PI3K inhibitors were 100 nM for wortmannin and 50 μM for LY294002 (Niggli and Keller, 1997). All inhibitors were incubated with the neutrophils for 10 min. All neutrophils were stored on ice until needed.
To determine the effects of competing fMLP and IL-8 gradients, one outer well was loaded with fMLP, the second outer well was loaded with IL-8, and the central well was loaded with neutrophils. We examined whether p38 MAPK was dominant over PI3K by adding p38 MAPK–inhibited, PI3K-inhibited, or PI3K- and p38 MAPK–inhibited neutrophils to the central well and IL-8 and fMLP into the two outer wells. To eliminate the possibility of diffusion rates accounting for preferential migration, p38 MAPK–inhibited or untreated neutrophils were loaded into a well containing IL-8, and fMLP was placed in an adjacent well (or vice versa).
To test the role of the two IL-8 receptors (CXCR1 and CXCR2) in the fMLP/IL-8 hierarchy, we treated neutrophils with either 2.5 μg/ml anti-CXCR1 or 10 μg/ml anti-CXCR2 antibodies and loaded these neutrophils into the central well of a gel containing IL-8 and fMLP in its outer wells.
Video analysis
The under agarose assay allows quantification of both random migration (chemokinesis) and directional migration (chemotaxis). To determine if the migration we observed was directional, two target areas were analyzed. Target A was a segment 500 μm × 2,300 μm between the neutrophil containing well and the chemoattractant containing well. Target B was of the same size as the first zone but extended from the neutrophil well to a point directly opposite the chemoattractant containing well (Fig. 1 a). The number of chemotaxing cells was determined by subtracting the number of migrating cells in target B from the number of migrating cells in area A.
IL-8 neutrophil receptor analysis using FACS®
Whole blood was diluted 1/10 with HBSS. Blood was stimulated using fMLP or IL-8 (1–100 nM) for 20 min at 37°C. Control tubes (cells alone) received no chemoattractant but were placed at 37°C for 20 min. Anti-CXCR1 or anti-CXCR2 primary antibody was added to experimental tubes at 2.5 and 10 μg/ml, respectively, and isotype IgG2a antibody was added to control tubes. All antibodies were incubated at 37°C for 15 min. Untreated and treated controls were used to ensure stimulation did not induce autofluorescence and nonspecific binding. After labeling, cells were washed and then lysed with Optilyze B solution for 10 min. Cells were washed again and incubated with FITC-labeled secondary antibody. After a 15 min, incubation cells were washed and resuspended in HBSS to a concentration of 1.0 × 106 cells/ml.
To test the effects of the p38 MAPK inhibitor, SB203580, on receptor internalization, neutrophils were treated with 10 μM SB203580 and then labeled using anti-CXCR1 or anti-CXCR2 antibodies as described previously. These cells were stimulated with 0.0001–0.1 μM fMLP and incubated for 20 min at 37°C. Fluorescent intensity was analyzed using flow cytometry gating on the neutrophil population.
Calcium flux
Isolated human neutrophils at a concentration of 10 million cells/ml were either left untreated or inhibited with the p38 MAPK inhibitor SB203580 for 1 h at 37°C. These cells were labeled with 4 μM fluo-3 dye at 37°C for 30 min with occasional agitation. Cells were washed twice with Hepes buffer with 1 mM calcium (145 mM NaCl, 5 mM KCl, 1 mM MgCl, 10 mM Hepes, and 10 mM glucose) and centrifuged at 1,300 rpm for 6 min. The neutrophils were diluted to one million cells per ml in Hepes buffer and pipetted into 4-mL cuvettes in PerkinElmer fluorescence spectrometer 650–10S with an excitation wavelength of 480 nm and emission recorded at 530 nm. Neutrophils were initially stimulated with 100 nM IL-8 or fMLP. Once the calcium levels stabilized, the cells were stimulated a second time with an additional 100 nM IL-8 or fMLP. Calcium flux was measured as the percentage of total calcium (fmax). fmax was determined by treating fluo-3–labeled cells with Triton X-100.
Akt blotting
p38 MAPK inhibited and uninhibited neutrophils at a concentration of 4.0 × 107 cells/ml were treated with fMLP and/or IL-8 such that the final concentration of either chemoattractant was 100 nM. Aliquots of these cell suspensions were removed at various time points and added to an equal volume of boiling 2× Laemmli buffer (125 mM Tris HCl, pH 6.8, 8% SDS, 10% β mercapto ethanol, 17% glycerol, 6.65% bromophenol blue, 5 mM orthovanadate, 10 μg/ml leupeptin, and 10 μg/ml aprotinin). Samples were then electrophoresed on an 8% SDS–polyacrylamide gel and transferred to a highbond p+ membrane. Blots were immunoblotted using anti–phospho-Akt antibody in TBS-tween with 2% BSA. The blots were then stripped and reprobed with anti-Akt antibodies to show that all lanes were loaded equally. Both blots were visualized on radiography film using a secondary antibody conjugated to HRP and an EPO chemiluminescence kit. Densitometry was performed using Quantity One software.
Statistics
All data are expressed as the arithmetic mean ± SEM. Data was compared using a single sample t test unless otherwise noted. Values of P < 0.05 were considered to be statistically significant.
Acknowledgments
All work was supported by a Canadian Institute for Health Research Group grant. P. Kubes is a Canada Research Chair and a Scientist of the Alberta Heritage Foundation for Medical Research.
Footnotes
Abbreviation used in this paper: PI3K, phosphoinositide 3-kinase.
References
- Ali, H., R.M. Richardson, B. Haribabu, and R. Snyderman. 1999. Chemoattractant receptor cross-desensitization. J. Biol. Chem. 274:6027–6030. [DOI] [PubMed] [Google Scholar]
- Alvarado-Kristensson, M., M.I. Porn-Ares, S. Grethe, D. Smith, L. Zheng, and T. Andersson. 2002. p38 Mitogen-activated protein kinase and phosphatidylinositol 3-kinase activities have opposite effects on human neutrophil apoptosis. FASEB J. 16:129–131. [DOI] [PubMed] [Google Scholar]
- Amatruda, T.T., III, S. Dragas-Graonic, R. Holmes, and H.D. Perez. 1995. Signal transduction by the formyl peptide receptor. Studies using chimeric receptors and site-directed mutagenesis define a novel domain for interaction with G-proteins. J. Biol. Chem. 270:28010–28013. [DOI] [PubMed] [Google Scholar]
- Berra, E., M.T. Diaz-Meco, and J. Moscat. 1998. The activation of p38 and apoptosis by the inhibition of Erk is antagonized by the phosphoinositide 3-kinase/Akt pathway. J. Biol. Chem. 273:10792–10797. [DOI] [PubMed] [Google Scholar]
- Campbell, J.J., E.F. Foxman, and E.C. Butcher. 1997. Chemoattractant receptor cross talk as a regulatory mechanism in leukocyte adhesion and migration. Eur. J. Immunol. 27:2571–2578. [DOI] [PubMed] [Google Scholar]
- Capodici, C., S. Hanft, M. Feoktistov, and M.H. Pillinger. 1998. Phosphatidylinositol 3-kinase mediates chemoattractant-stimulated, CD11b/CD18-dependent cell-cell adhesion of human neutrophils: evidence for an ERK-independent pathway. J. Immunol. 160:1901–1909. [PubMed] [Google Scholar]
- Cara, D.C., J. Kaur, M. Forster, D.M. McCafferty, and P. Kubes. 2001. Role of p38 mitogen-activated protein kinase in chemokine-induced emigration and chemotaxis in vivo. J. Immunol. 167:6552–6558. [DOI] [PubMed] [Google Scholar]
- Damaj, B.B., S.R. McColl, W. Mahana, M.F. Crouch, and P.H. Naccache. 1996. Physical association of Gi2alpha with interleukin-8 receptors. J. Biol. Chem. 271:12783–12789. [DOI] [PubMed] [Google Scholar]
- Ding, Z.M., J.E. Babensee, S.I. Simon, H. Lu, J.L. Perrard, D.C. Bullard, X.Y. Dai, S.K. Bromley, M.L. Dustin, M.L. Entman, et al. 1999. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 163:5029–5038. [PubMed] [Google Scholar]
- Foxman, E.F., J.J. Campbell, and E.C. Butcher. 1997. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J. Cell Biol. 139:1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxman, E.F., E.J. Kunkel, and E.C. Butcher. 1999. Integrating conflicting chemotactic signals. The role of memory in leukocyte navigation. J. Cell Biol. 147:577–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, J.X., and A.C. Issekutz. 1996. Mac-1 (CD11b/CD18) is the predominant beta 2 (CD18) integrin mediating human neutrophil migration through synovial and dermal fibroblast barriers. Immunology. 88:463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godaly, G., L. Hang, B. Frendeus, and C. Svanborg. 2000. Transepithelial neutrophil migration is CXCR1 dependent in vitro and is defective in IL-8 receptor knockout mice. J. Immunol. 165:5287–5294. [DOI] [PubMed] [Google Scholar]
- Haddad, E.B., M. Birrell, K. McCluskie, A. Ling, S.E. Webber, M.L. Foster, and M.G. Belvisi. 2001. Role of p38 MAP kinase in LPS-induced airway inflammation in the rat. Br. J. Pharmacol. 132:1715–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, E., V.L. Katanaev, C. Garlanda, O. Azzolino, L. Pirola, L. Silengo, S. Sozzani, A. Mantovani, F. Altruda, and M.P. Wymann. 2000. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 287:1049–1053. [DOI] [PubMed] [Google Scholar]
- Hogg, N., and B. Leitinger. 2001. Shape and shift changes related to the function of leukocyte integrins LFA-1 and Mac-1. J. Leukoc. Biol. 69:893–898. [PubMed] [Google Scholar]
- Jones, S.A., M. Wolf, S. Qin, C.R. Mackay, and M. Baggiolini. 1996. Different functions for the interleukin 8 receptors (IL-8R) of human neutrophil leukocytes: NADPH oxidase and phospholipase D are activated through IL-8R1 but not IL-8R2. Proc. Natl. Acad. Sci. USA. 93:6682–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knall, C., G.S. Worthen, A.M. Buhl, and G.L. Johnson. 1995. IL-8 signal transduction in human neutrophils. Ann. NY Acad. Sci. 766:288–291. [DOI] [PubMed] [Google Scholar]
- Knall, C., G.S. Worthen, and G.L. Johnson. 1997. Interleukin 8-stimulated phosphatidylinositol-3-kinase activity regulates the migration of human neutrophils independent of extracellular signal-regulated kinase and p38 mitogen-activated protein kinases. Proc. Natl. Acad. Sci. USA. 94:3052–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luster, A.D. 1998. Chemokines–chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 338:436–445. [DOI] [PubMed] [Google Scholar]
- Murdoch, C., and A. Finn. 2000. Chemokine receptors and their role in inflammation and infectious diseases. Blood. 95:3032–3043. [PubMed] [Google Scholar]
- Nelson, R.D., P.G. Quie, and R.L. Simmons. 1975. Chemotaxis under agarose: a new and simple method for measuring chemotaxis and spontaneous migration of human polymorphonuclear leukocytes and monocytes. J. Immunol. 115:1650–1656. [PubMed] [Google Scholar]
- Nick, J.A., N.J. Avdi, S.K. Young, C. Knall, P. Gerwins, G.L. Johnson, and G.S. Worthen. 1997. Common and distinct intracellular signaling pathways in human neutrophils utilized by platelet activating factor and FMLP. J. Clin. Invest. 99:975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niggli, V., and H. Keller. 1997. The phosphatidylinositol 3-kinase inhibitor wortmannin markedly reduces chemotactic peptide-induced locomotion and increases in cytoskeletal actin in human neutrophils. Eur. J. Pharmacol. 335:43–52. [DOI] [PubMed] [Google Scholar]
- Prossnitz, E.R., T.L. Gilbert, S. Chiang, J.J. Campbell, S. Qin, W. Newman, L.A. Sklar, and R.D. Ye. 1999. Multiple activation steps of the N-formyl peptide receptor. Biochemistry. 38:2240–2247. [DOI] [PubMed] [Google Scholar]
- Rane, M.J., P.Y. Coxon, D.W. Powell, R. Webster, J.B. Klein, W. Pierce, P. Ping, and K.R. McLeish. 2001. p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J. Biol. Chem. 276:3517–3523. [DOI] [PubMed] [Google Scholar]
- Richardson, R.M., H. Ali, E.D. Tomhave, B. Haribabu, and R. Snyderman. 1995. Cross-desensitization of chemoattractant receptors occurs at multiple levels. Evidence for a role for inhibition of phospholipase C activity. J. Biol. Chem. 270:27829–27833. [DOI] [PubMed] [Google Scholar]
- Richardson, R.M., H. Ali, B.C. Pridgen, B. Haribabu, and R. Snyderman. 1998. Multiple signaling pathways of human interleukin-8 receptor A. Independent regulation by phosphorylation. J. Biol. Chem. 273:10690–10695. [DOI] [PubMed] [Google Scholar]
- Sasaki, T., J. Irie-Sasaki, R.G. Jones, A.J. Oliveira-dos-Santos, W.L. Stanford, B. Bolon, A. Wakeham, A. Itie, D. Bouchard, I. Kozieradzki, et al. 2000. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 287:1040–1046. [DOI] [PubMed] [Google Scholar]
- Schraufstatter, I.U., J. Chung, and M. Burger. 2001. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 280:L1094–L1103. [DOI] [PubMed] [Google Scholar]
- Seo, S.M., L.V. McIntire, and C.W. Smith. 2001. Effects of IL-8, Gro-alpha, and LTB(4) on the adhesive kinetics of LFA-1 and Mac-1 on human neutrophils. Am. J. Physiol. Cell Physiol. 281:C1568–C1578. [DOI] [PubMed] [Google Scholar]
- Shen, W., B. Li, M.A. Wetzel, T.J. Rogers, E.E. Henderson, S.B. Su, W. Gong, Y. Le, R. Sargeant, D.S. Dimitrov, et al. 2000. Down-regulation of the chemokine receptor CCR5 by activation of chemotactic formyl peptide receptor in human monocytes. Blood. 96:2887–2894. [PubMed] [Google Scholar]
- Wagner, J.G., and R.A. Roth. 1999. Neutrophil migration during endotoxemia. J. Leukoc. Biol. 66:10–24. [DOI] [PubMed] [Google Scholar]
- Yasui, K., and A. Komiyama. 2001. Roles of phosphatidylinositol 3-kinase and phospholipase D in temporal activation of superoxide production in FMLP-stimulated human neutrophils. Cell Biochem. Funct. 19:43–50. [DOI] [PubMed] [Google Scholar]
- Zu, Y.L., J. Qi, A. Gilchrist, G.A. Fernandez, D. Vazquez-Abad, D.L. Kreutzer, C.K. Huang, and R.I. Sha'afi. 1998. p38 mitogen-activated protein kinase activation is required for human neutrophil function triggered by TNF-alpha or FMLP stimulation. J. Immunol. 160:1982–1989. [PubMed] [Google Scholar]