

Abstract
The ability to heal wounds is vital to all organisms. In mammalian tissues, alterations in intermediate filament (IF) gene expression represent an early reaction of cells surviving injury. We investigated the role of keratin IFs during the epithelialization of skin wounds using a keratin 6α and 6β (K6α/K6β)-null mouse model. In skin explant culture, null keratinocytes exhibit an enhanced epithelialization potential due to increased migration. The extent of the phenotype is strain dependent, and is accompanied by alterations in keratin IF and F-actin organization. However, in wounded skin in vivo, null keratinocytes rupture as they attempt to migrate under the blood clot. Fragility of the K6α/K6β-null epidermis is confirmed when applying trauma to chemically treated skin. We propose that the alterations in IF gene expression after tissue injury foster a compromise between the need to display the cellular pliability necessary for timely migration and the requirement for resilience sufficient to withstand the rigors of a wound site.
Keywords: mouse skin grafting; keratin; skin; injury; wound healing; migration
Introduction
Tissue injury triggers an immediate and elaborate response designed to reestablish those functions essential to survival. In metazoans, a universal aspect of the response consists of the mobilization of surviving cells proximal to the wound site toward restoration of the damaged tissue. This activation is reflected through changes in gene transcription and protein regulation at a posttranslational level (Martin, 1997). In vertebrates, intermediate filament (IF) proteins are among the cellular constituents whose regulation is altered within hours after injury. A large family of conserved genes (n > 67) encode a diverse group of IF proteins (mol. wt range: 40–280 kD) capable of self-assembly into 10–12-nm-wide filaments (Fuchs and Weber, 1994; Quinlan et al., 1994). The properties of IF polymers differ from those of F-actin and microtubules, the other mainstream constituents of the cytoskeleton. A primary role of IFs is to act as a flexible scaffold enabling cells to resist physical stress (Fuchs and Cleveland, 1998; Coulombe et al., 2000). Accordingly, defects in IFs engender cell fragility and underlie a wide array of inherited diseases (Fuchs and Cleveland, 1998; Irvine and McLean, 1999). Additional roles, including protection against chemical stress and proapoptotic signals, are fulfilled by specific IFs in explicit cell types (Coulombe and Omary, 2002; Oshima, 2002). A fundamental question in cytoskeletal research concerns the functional significance of the diversity encountered among IF proteins. Given this, it is of interest to define the contribution of IF proteins in the response of cells and tissues to injury.
With its ∼50 members partitioned into two sequence types (Hesse et al., 2001), the epithelial-restricted keratins provide a unique handle to explore the significance of the multiplicity and differential regulation of IF genes. Type I (K9-K23; Ha1-Ha9) and type II (K1-K8; Hb1-Hb6) keratin genes are regulated in a pairwise fashion, reflecting a heteropolymerization requirement (Fuchs and Weber, 1994). Most pairs of keratin genes are regulated in a differentiation-specific fashion in epithelia (Moll et al., 1982). For instance, in interfollicular epidermis, progenitor basal cells express K5/K14 as their main pair of type II/type I keratins, whereas early differentiating keratinocytes express the K1/K10 pair. This blueprint varies depending on regional differences in epidermis, on disease, or environmental challenges (McGowan and Coulombe, 1998a). This is the case after epidermal injury, which triggers the induction of K6 isoforms and type I K16 and K17 in keratinocytes undergoing activation at the wound edge. This transcriptional event occurs at the expense of K1 and K10, and correlates with striking alterations in the morphology and other properties of keratinocytes. Expression of K6, K16, and K17 persists as wound-activated keratinocytes migrate into the site of injury, but is reversible upon wound closure (Mansbridge and Knapp, 1987; Paladini et al., 1996; Takahashi et al., 1998).
Along with their partners K16 and K17, K6 paralogues, of which there are many in mammalian genomes (Tyner and Fuchs, 1986; Ramirez et al., 1995; Takahashi et al., 1995, 1998), exhibit a complex regulation that includes constitutive expression in specific compartments within all epithelial appendages (McGowan and Coulombe, 1998a). This has complicated the assessment of their role during adult wound repair; for instance, mice null for the two functional K6 genes, keratin 6α and 6β (K6α/K6β), die rapidly after birth owing to the fragility of stress-bearing epithelia within the oral mucosa (Wong et al., 2000; Wojcik et al., 2001). Here, we exploit an ex vivo skin explant culture assay along with in vivo skin tissue grafting to analyze the response of K6α/K6β null to various forms of challenges including injury. We show that K6α/K6β and K16 profoundly impact the potential of keratinocytes for wound epithelialization, and are essential to the maintenance of keratinocyte integrity in activated epidermis. We propose a model in which these keratins function to provide a resilient cytoskeletal scaffold able to maintain adequate structural integrity while providing sufficient pliability for effective migration into the wound site. This hypothesis may also explain in part the diversity and differentiation-related distribution of IF proteins.
Results
K6α/K6β-null keratinocytes exhibit an enhanced epithelialization potential
To assess the wound epithelialization potential in K6α/K6β-null mice, we used a skin explant culture assay that mimics the behavior of keratinocytes at the edge of skin wounds in vivo. This assay offers a qualitative and quantitative assessment of keratinocytes' potential for epithelialization, while avoiding effects from dermis-mediated contraction of the wound bed (Mazzalupo et al., 2002) or limitations from severe postnatal phenotypes (K6α/K6β-null mice die shortly after birth; Wong et al., 2000). Full thickness skin punches from 2–4-d-old wild-type, hemizygous, and K6α/K6β-null backskins from the original mixed genetic strain background (129/Sv-C57Bl/6-DBA2) were cultured for 8 d. K6α/K6β-null explants exhibited a statistically significant 1.8-fold enhancement of epithelial outgrowth as compared with wild-type and hemizygous explants (Fig. 1, A and B). The availability of a K14-null mouse model (Lloyd et al., 1995) provided an opportunity to test for keratin specificity and the potential impact of the NeoR cassette found within the targeting vector. There was only a modest enhancement in keratinocyte outgrowth in K14-null explants (1.25-fold; Fig. 1 C). Thus, the epithelialization potential of keratinocytes is more sensitive to the loss of K6α/K6β proteins than loss of K14, another major keratin present in this setting.
Figure 1.
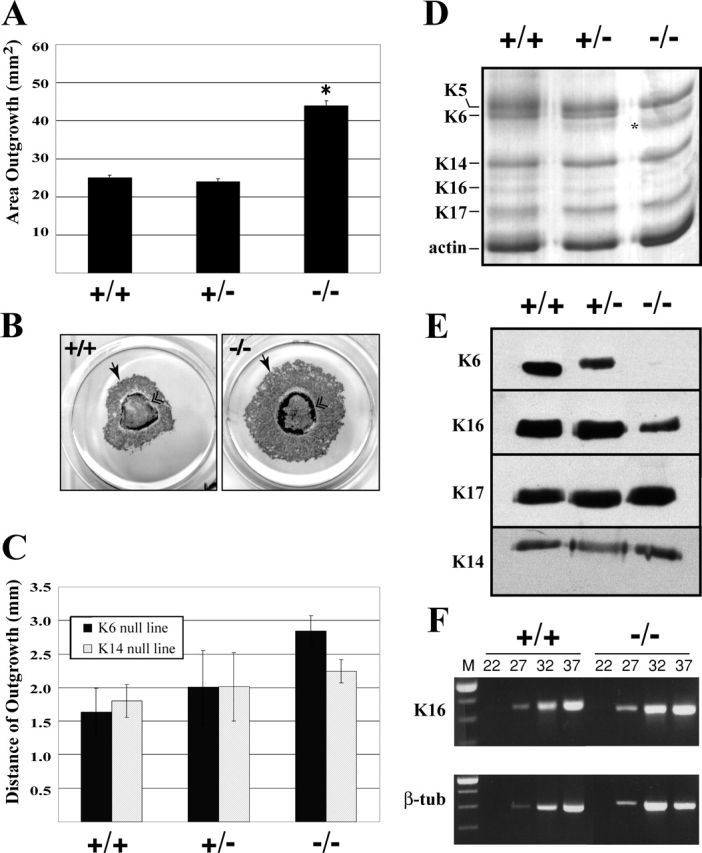
K6α/K6β-null keratinocytes exhibit enhanced epithelialization potential in skin explant culture. (A) Quantitation of keratinocyte outgrowth from explants. Skin punches were cultured for 8 d, and keratinocytes were identified by K17 immunostaining. Eight skin punches per mouse with a minimum of 13 mice per genotype were analyzed. Mean ± SEM values are shown. Asterisks depict statistically significant difference from wild type (P < 0.0001). (B) Examples of wild-type and K6α/K6β-null skin explants processed for K17 immunostaining after 8 d in culture. Arrow indicates leading edge of keratinocyte outgrowth and double arrowheads denote edge of explant biopsy. (C) The performance of K6α/K6β- and K14-null skin explants was compared by assessing the distance between keratinocytes located at the distal edge of the outgrowth and the explant edge. The error bars represent SD. (D and E) Cellular proteins were prepared from pooled explants in culture for 6 d (n > 60 explants per genotype; see Materials and methods), resolved using SDS-PAGE, and stained with Coomassie blue or Western blotting to determine keratin content. In D, asterisk denotes a band that is present in variable amounts between preparations and which likely represents a degradation product from a larger protein. (F) Semi-quantitative RT-PCR was performed on RNA collected from 6-d-old outgrowths. K16 specific primers were used quantify the amount of K16 mRNA transcripts. β-Tubulin primers were used as an internal control. Samples were collected after 22, 27, 32, and 37 PCR cycles.
Total protein extracts were prepared from cellular outgrowths and analyzed to determine whether loss of K6α/K6β had an impact on the levels of other keratins. At least 20 explants from each of three mice were pooled for each genotype for this purpose. Wild-type and hemizygous null keratinocyte outgrowths feature K5, K6, K14, K17, and to a lesser extent, K16, as their main keratins (Fig. 1 D). In addition to a complete loss of K6α/K6β, null samples showed a partial loss of K16 but no change in K5, K14, or K17 (Fig. 1, D and E). In contrast to K16 protein, K16 mRNA levels were similar in K6α/K6β-null and wild-type samples (Fig. 1 F). These data are consistent with the notion that unlike K14 and K17, K16 forms unstable heterotypic complexes with either K5 or K6 (Wawersik et al., 1997). This property likely accounts for the selective loss of K16 in a competitive environment in which type II keratin binding partners are limiting. Enhanced epithelial outgrowth in skin explant culture, thus, correlates with a partial loss of K16 protein in addition to the complete loss of K6α/K6β.
Enhanced outgrowth of K6α/K6β-null keratinocytes results from migration and is accompanied by changes in keratin and F-actin organization, and in p120ctn
Re-epithelialization of skin wounds in vivo results from increases in mitotic activity and migration of keratinocytes located at the wound margins (Martin, 1997). Likewise, over a period of 8 d in culture, migration and mitosis equally contribute to the outgrowth of keratinocytes from skin explants (Wawersik et al., 2001). We addressed the role of mitotis and migration in null explants using a three-prong approach. First, explants were treated with the nucleotide analogue BrdU 2 h before harvest to visualize cells engaged in DNA replication. The density of BrdU-positive keratinocytes at the explant tissue edge, where they are most abundant, was similar in wild-type and null explants (Fig. 2, A and B). Second, analysis of proteins from explant outgrowths revealed no change in levels of proliferating cell nuclear antigen or myc relative to actin (Fig. 2 C). Third, the outgrowth of keratinocytes was assessed in two complementary ways after treatment with the irreversible mitosis inhibitor mitomycin C (Wawersik et al., 2001). In the first experiment, explants were treated with mitomycin C at 24 h after seeding, cultured for 8 d, and processed for analysis. Relative to their wild-type controls, treated and untreated K6α/K6β-null explants show a comparable enhancement in outgrowth area (Fig. 1 D). In the second experiment, treatment with mitomycin C was performed at 48 h after seeding and the outgrowth distance calculated after treatment, and at days 4, 6, and 8 thereafter, for the same set of live explants. Under such conditions, K6α/K6β-null keratinocytes migrated further than wild-type or hemizygous keratinocytes (Fig. 2 E). Collectively, these analyses suggest that the increased epithelialization potential manifested by K6α/K6β-null samples in skin explant culture results mainly from enhanced migration rather than increased mitosis.
Figure 2.
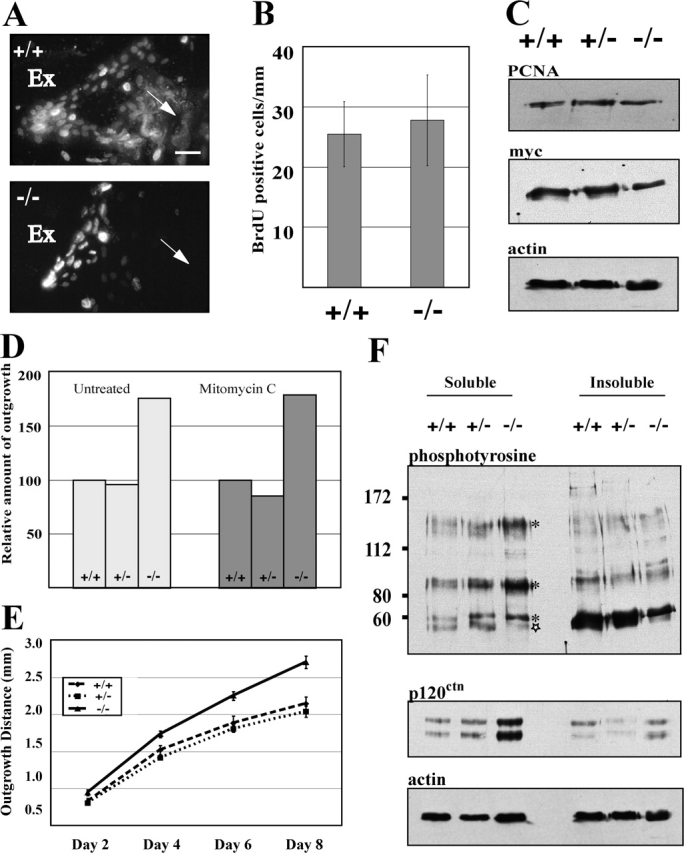
Enhanced K6α/K6β-null epithelialization likely results from increased keratinocyte migration. (A) Immunofluorescence staining for BrdU in cellular outgrowths from 6-d-old skin explants. The region shown is located next to the explant edge (Ex). Arrows depict the direction of outgrowth. Bar, 50 μm. (B) Density of BrdU-positive keratinocytes in wild-type and K6α/K6β-null samples (expressed per millimeter of explant tissue perimeter). The error bars represent SD. (C) Western blot analysis for myc and proliferating cell nuclear antigen in protein extracts prepared from 6-d-old explants. Actin is the loading control. (D and E) Keratinocyte migration in skin explants after mitomycin C treatment. (D) Total area of keratinocyte outgrowth after 8 d, after treatment or not at 24 h, is shown for hemizygous and homozygous K6α/K6β-null explants relative to wild type (n = 62 wt; 132 hemizygous; 35 null explants). (E) Keratinocyte outgrowth in wild-type, hemizygous and homozygous K6α/K6β-null explants as a function of time in live skin explant cultures after treatment at 48 h after seeding. Distance measurements were made at days 2, 4, 6, and 8 d in culture (n = 6 wt; 13 hemizygous; 12 null explants). The error bars represent SEM. (F) Pooled cellular outgrowths from skin explants cultured for 6 d (n > 60 per genotype) were solubilized with lysis buffer, and insoluble proteins were pelleted, solubilized, and electrophoresed (2 μg protein) before Western blot analysis using α-p120ctn, α-phosphotyrosine, and α-actin antibodies. Phosphotyrosine epitopes that are increased in K6α/K6β-null samples compared with controls are identified with an asterisk, whereas those present in lower amounts are identified with a star. The two bands in the p120ctn blot likely represents distinct isoforms of p120ctn (Anastasiadis and Reynolds, 2001).
We subjected wild-type and K6α/K6β-null explants to morphological and biochemical analyses. As is the case at wound margins in vivo, keratinocytes migrate as a stratified sheet out of skin explants in ex vivo culture, and the number of cell layers progressively decreases with distance from the skin biopsy (Mazzalupo et al., 2002). An increase in cell size does not account for the enhanced outgrowth in K6α/K6β-null explants. Indirect immunofluorescence using antibodies to K6α/K6β revealed a pan-cytoplasmic keratin filament network in both wild-type and hemizygous null samples, whereas null cells are negative (unpublished data). Antibodies to either K16 or K17 revealed the presence of abnormal filament bundles in a subset of suprabasal cells located in the stratified portion of the outgrowth, near the explant edge (Fig. 3 A, arrowheads). Such anomalies were not seen in controls (Fig. 3 A, arrows). We examined F-actin organization given that K6α/K6β-null keratinocytes show enhanced migration. Relative to wild-type, actin filaments in K6α/K6β-null keratinocytes stained with increased intensity. In suprabasal cells located near the explant edge, prominent F-actin staining occurred in the cortex of K6α/K6β-null keratinocytes (Fig. 3 B, double arrowheads). In matrix-attached keratinocytes located at the leading edge of the outgrowth, the most impressive change in K6α/K6β-null cells consisted of an increase in stress fibers (Fig. 3 B, double arrowheads).
Figure 3.
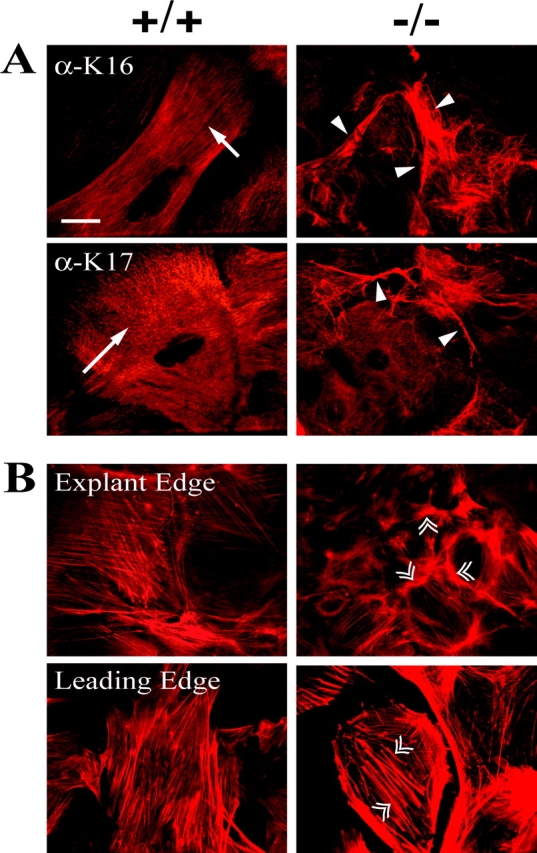
Immunofluorescence staining of keratinocyte outgrowths reveals alterations in the keratin and actin cytoskeletons. Wild-type and K6α/K6β-null explants were immunostained for (A) K16, K17, and (B) actin. (A) Single arrows identify pan-cytoplasmic keratin filaments in wild-type samples, whereas arrowheads point to partially collapsed K16- and K17-containing filament networks in K6α/K6β-null keratinocytes at the explant edge. (B) Double arrowheads identify the increased intensity and number of actin filaments in the K6α/K6β-null keratinocytes. Bar, 50 μm.
The changes in migration potential and F-actin organization observed in K6α/K6β-null explants likely reflect alterations in key protein regulators via tyrosine phosphorylation. To examine this issue, protein samples from 6-d-old cellular outgrowths were fractionated into detergent-soluble and -insoluble pools, and phosphotyrosine epitopes assessed by Western blotting. Relative to wild-type and hemizygous null controls, three bands, with apparent masses of ∼120, ∼90, and ∼60 kD, were increased in the soluble protein pool prepared from the K6α/K6β-null sample (Fig. 2 F, bands marked with an asterisk). Conversely, an ∼55–56-kD antigen was reduced in both the soluble and insoluble fractions of the K6α/K6β-null sample (Fig. 2 F, star). Immunoprecipitation showed that the ∼120-kD phosphoepitope is likely to be p120ctn (unpublished data), a catenin known to associate with E-cadherin (Anastasiadis and Reynolds, 2001). The levels of p120ctn protein are increased as well in the soluble fraction of the K6α/K6β (Fig. 2 F). No obvious change could be detected in the subcellular localization of either p120ctn or E-cadherin when using indirect immunofluorescence (unpublished data). Likewise, no change could be detected in the steady-state levels or tyrosine phosphorylation of the EGF receptor. Additional analyses will be required to ascertain the identity of the ∼90-, ∼60-, and ∼55–56-kD phosphotyrosine antigens. Meanwhile, our studies identified p120ctn as a potential player in the keratinocyte migration phenotype observed.
Influence of strain background affects the behavior of skin explant cultures
The genetic background can influence the consequences of keratin-null mutations in mice. Depending on the strain background, K8-null mice may exhibit colorectal hyperplasia or embryonic lethality (Baribault et al., 1993, 1994), whereas K17-null mice may exhibit striking alopecia or not (McGowan et al., 2002). K6α/K6β-null mice are able to survive their severe oral lesions in a specific strain background (compare Wong et al., 2000, with Wojcik et al., 2001). The latter finding is relevant here, considering that Wojcik et al. (2001) also did not find evidence of altered wound repair in the skin of their K6α/K6β-null mice.
The K6α/K6β-null allele was backcrossed into the C57Bl/6 and 129/SvJ inbred strains for at least seven generations, and the epithelialization potential was evaluated in skin explant culture. Wild-type explants derived from inbred and mixed background strains showed the same amount of outgrowth suggesting equal epithelialization potential (Fig. 4). In the C57Bl/6 strain, hemizygous and homozygous null K6α/K6β-null keratinocytes exhibit an epithelialization potential similar to the mixed genetic background (Fig. 4). However, in the 129/SvJ strain, K6α/K6β-null keratinocytes exhibit a substantially larger (3.1-fold) increase in epithelialization potential compared with wild type (Fig. 4). In this instance, hemizygous null explants display a statistically significant increase in outgrowth compared with wild type. Therefore, genetic background influences the extent to which the epithelialization potential is enhanced in K6α/K6β-null keratinocytes. A gene dosage effect occurs in the 129/SvJ strain, indicating that the number of K6-encoding alleles is important in at least some settings, as predicted by Takahashi et al. (1998).
Figure 4.

Enhanced epithelialization potential in skin explant culture is affected by genetic strain background. Mixed K6α/K6β-null mice (129/Sv × C57Bl/6 × DBA2) were backcrossed into two different inbred strains, 129/SvJ and C57Bl/6, for a minimum of seven generations. The extent of keratinocyte outgrowth after 8 d of skin explant culture was quantitated and compared with the mixed genetic background. Mean ± SEM values are displayed. Genotypes that are statistically distinct from wild-type explants are identified with an asterisk (P ≤ 0.0002).
K6α/K6β-null keratinocytes are fragile in skin tissue subjected to challenge
To study the functional importance of K6α/K6β in the natural context of mature skin tissue, we grafted backskins derived from newborn C57Bl/6 wild-type, hemizygous, and K6α/K6β-null mice onto immunocompromised recipient mice. Grafted backskins developed normally irrespective of genotype, as evidenced by the growth of a thick fur (Fig. 5, A and B). Likewise, the regrowth of hair taking place after chemical depilation and anagen induction (Stenn and Paus, 2001) was similar in all genotypes (unpublished data). These results corroborate those of Wojcik et al. (2001) showing that K6α/K6β, whose expression is restricted to the companion layer in anagen stage hair follicles (Takahashi et al., 1998), is not essential to hair formation and cycling.
Figure 5.


Grafted K6α/K6β-null skin tissue exhibit epithelial fragility after incisional wounding. (A and B) Dorsal view of wild-type and K6α/K6β-null backskins grafted onto immunocompromised mice. (C and E) Micrographs of H&E stained sections of unwounded grafted backskin from wild-type and K6α/K6β-null animals. (D and F) Micrographs of similar sections at 3 d after full skin thickness incisional wounding of the grafted tissue. Arrowheads depict migrating keratinocytes at the wound edge. (G and H) Higher magnification of activated epithelium in (G) wild-type and (H) K6α/K6β-null skin grafts at 3 d after injury. Arrows point to the occurrence of degenerative changes in the activated epithelium at the wound edge and to intracellular lysis in migrating keratinocytes. Open arrowheads point to floating nuclei within the suprabasal layers. (I) Immunostaining for K17 in wound edge tissue from a K6α/K6β-null skin graft, confirming that cleavage, as indicated by arrows, occurs within the cytoplasm. epi, epidermis; HF, hair follicle; AK, activated keratinocytes at the wound edge; and Sc, scab. Bars: (C–F) 100 μm; (G–I) 50 μm.
Grafted skins were subjected to full thickness wounding and histologically assessed 3 d later to examine the ability of K6α/K6β-null epithelia to become activated and mount a re-epithelialization response (Fig. 5, C–F). Wild-type, hemizygous, and K6α/K6β-null skin grafts all exhibited the hallmarks of keratinocyte activation at the wound edge, as manifested by cellular hypertrophy and production of a band of migrating keratinocytes under the fibrin clot (Fig. 5 F, arrowheads). BrdU incorporation showed the hyperproliferative zone was present and comparable in all genotypes (unpublished data). Strikingly, however, swelling and lysis of keratinocytes was seen in K6α/K6β-null tissue at locations normally showing prominent K6 expression, such as the upper suprabasal layer of wound edge epidermis and in the migrating epithelium (Fig. 5, D and F). The occurrence of intracellular keratinocyte lysis in such areas was confirmed by examination at a higher magnification (Fig. 5, G and H) along with immunostaining for an intracellular antigen such as K17 (Fig. 5 I). Such lysis was never seen in wild-type and hemizygous null samples.
We examined wild-type and K6α/K6β-null grafted skin in an alternative setting in which the expression of K6α/K6β is induced. Grafted backskins were treated with PMA, a substance known to induce hyperproliferation and K6α/K6β expression (Takahashi et al., 1998) without abrogating terminal differentiation (Bernot et al., 2002). Significant thickening of the epidermis occurred in the suprabasal compartment of PMA-treated wild-type and K6α/K6β-null grafted backskin (Fig. 6, A and D), providing further evidence that loss of K6α/K6β does not alter the mitotic potential of keratinocytes (Fig. 2). The resilience of the thickened epidermis was tested via gentle rubbing of PMA-treated skin with a pencil eraser (10 measured strokes) followed by histological analysis. In the specific case of K6α/K6β-null skin grafts, the stress test had to be interrupted because of severe loss of integrity. Upon histological examination of these samples, cytolysis and intra-epidermal cleavage had occurred in the suprabasal layers of epidermis (Fig. 6, E and F). Wild-type (Fig. 6, B and C) and hemizygous skin grafts (not depicted) did not show any such fragility or cytolysis under these conditions. These studies demonstrate the role of K6α/K6β as a crucial element responsible for maintaining keratinocyte integrity in challenged skin tissue, and in migrating keratinocytes found at sites of tissue injury.
Figure 6.
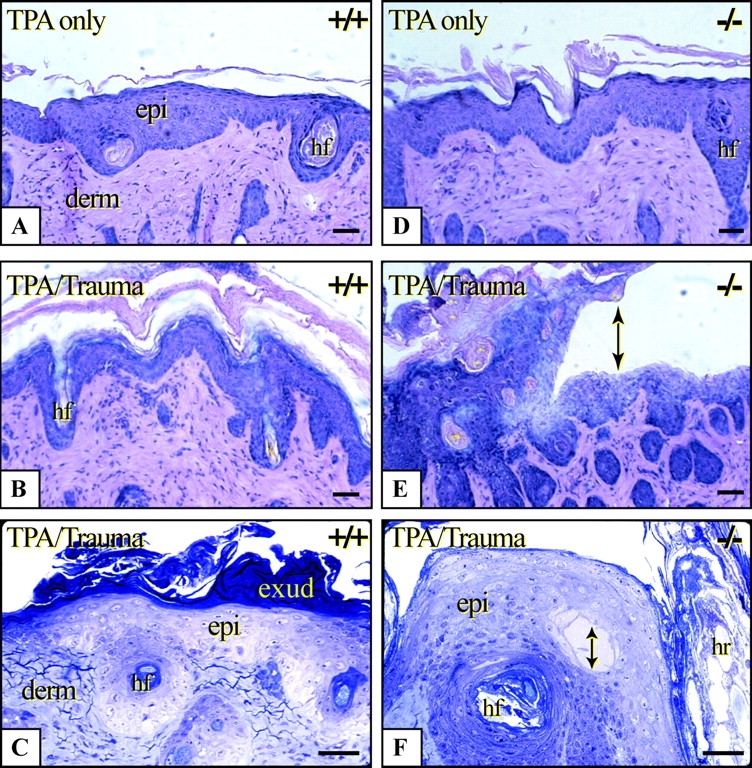
Trauma-induced lysis of K6α/K6β-null keratinocytes in grafted backskin subjected to chemical induction. PMA was applied on days 1, 3, and 5, and the grafted backskins harvested on day 7. (A and D) Micrographs of H&E stained sections of wild-type and K6α/K6β-null PMA treated grafts without mechanical trauma. (B and E) Gentle friction was applied to treated grafts immediately before harvest. Arrows depict intracellular lysis in the suprabasal layers of K6α/K6β-null epidermis. (C and F) Wild-type and K6α/K6β-null grafted tissue were processed for plastic embedding, sectioned (0.5-μm thick), and stained with toluidine blue. epi, epidermis; derm, dermis; exud, exudate; hr, hair; and hf, hair follicle. Bars, 50 μm.
Discussion
We examined the role of the wound-inducible keratins by subjecting K6α/K6β-null mice to complementary ex vivo and in vivo analyses. In skin explant culture, which reproduces key epithelial events occurring at the wound edge in vivo, K6α/K6β-null keratinocytes show a markedly enhanced epithelialization potential owing to an improved ability to migrate. Loss of K6α/K6β is accompanied by a selective decrease in K16 protein. The phenotype correlates with a perinuclear collapse of K16- and K17-containing filaments, altered F-actin content, and increased levels of p120ctn. However, when assessed in grafted skin tissue, K6α/K6β-null keratinocytes located at the edge of acute wounds exhibit lysis typical of keratin deficiency, presumably in response to stress. Fragility of K6α/K6β-null keratinocytes was confirmed when trauma was applied to grafted backskin treated with a classical inducer of epidermal thickening and K6α/K6β expression. The enhanced migratory properties shown by K6α/K6β-null keratinocytes in the idealized setting of skin explant culture, thus, appears negated by their fragility once in the harsher environment of a wound in vivo. These results have direct implications for the significance of the changes in keratin expression at the edge of skin wounds, and for the involvement of IFs in the response to injury.
Significance of altered keratin expression during wound epithelialization in skin
The type II K6α/K6β and type I K16 and K17 keratin proteins are related, respectively, in primary structure to K5 and K14, which are constitutively expressed in basal keratinocytes of the skin (Lloyd et al., 1995). Accumulation of K6α/K6β, K16, and K17 at the wound edge runs concurrently with the down-regulation of K1 and K10 (Mansbridge and Knapp, 1987; Paladini et al., 1996), whose expression normally parallels differentiation in the suprabasal layers of epidermis. This inductive response is evolutionary conserved (Estrada et al., 1993) and occurs in a number of other complex epithelia, including the oral mucosa and cornea (Schermer et al., 1989; Takahashi and Coulombe, 1997). The enhanced migratory potential exhibited by K6α/K6β-null skin keratinocytes, thus, appears counterintuitive considering that evolution has selected for the involvement of K6 proteins after injury. The relevance of our findings is supported by studies conducted in transgenic mice overexpressing K16 protein. In this instance, a delay is observed in the keratinocyte outgrowth produced during skin explant culture. Such mice also exhibit a delay in the closure of skin wounds in vivo (Wawersik et al., 2001).
Mixed results have been obtained in previous studies involving K6-null mouse models. On the one hand, Wojcik et al. (2000) reported that loss of K6α causes a delay in the epithelialization of partial thickness skin wounds in vivo. In such wounds, keratinocytes originating from the hair follicle outer root sheath participate to epithelialization (Pang et al., 1978). On the other hand, the response of skin tissue to full-thickness injury was not altered in the K6α/K6β-null animals able to survive in the genetic background used (Wojcik et al., 2001). In this instance, the skin response to partial thickness injury was not assessed. Differences in both strain backgrounds and assays likely contribute to the discrepancy between those observations and ours. It could be that the same factors enabling some K6α/K6β double-null mice to survive the life-threatening lesions affecting the oral mucosa (for review see Wojcik et al., 2001) are able to compensate during wound repair. K6hf, which is related to K5, K6α, and K6β, is not a contributing factor given that it is not expressed at the edge of skin wounds (Wojcik et al., 2001; Wang et al., 2003). With these issues notwithstanding, much of the evidence stemming from transgenic mouse models, alongside the evolutionarily conserved regulation of K6, K16, and K17 at the wound edge, argue strongly for an important and specific role provided by these keratins during wound epithelialization.
What is the nature of this role? We propose that the induction of K6, K16, and K17 in keratinocytes located proximal to the wound edge imparts them with mechanical properties that are intermediate between those exhibited by K5/K14-expressing keratinocytes in the basal layer and by K1/K10-expressing cells in the suprabasal layers. We postulate that expression of the K5/K14 pair as primary keratins renders keratinocytes relatively more pliable and, thus, better able to undergo directed cell migration, a notion that is directly supported by our explant culture findings. However, given a lack of K6α/K6β and reduced levels of K16, keratinocytes endowed mainly with K5/K14/K17 (Fig. 1 D) are insufficiently resilient to survive the harshness of the wound site. Rather, they rupture intracellularly in a fashion similar to that seen in keratin-based fragility disorders, as shown by our skin grafting studies. Conversely, abundant expression of the K1/K10 pair in the context of normal epidermal differentiation is expected to promote stronger mechanical resilience at the expense of cellular pliability. In support of this view, differentiating keratinocytes transit more rapidly through the suprabasal compartment of epidermis in K10-null mice (Reichelt and Magin, 2002). The induction of K6α/K6β, K16, and K17 after injury is, thus, likely to reflect a compromise between conflicting needs: retaining enough cellular pliability for migration while acquiring sufficient resilience to survive the wound environment. It is conceivable that a similar role would apply in situations involving constitutive expression of K6, K16, and K17 (e.g., epithelial appendages, palmar/plantar epidermis, and select internal epithelial tissues), although in these instances a specific degree of cellular pliability may be required for purposes other than migration (for review see Swensson et al., 1998; Bernot et al., 2002).
Although the proposed model invokes the major function shared by all types of IF polymers, it does not preclude additional roles for IF proteins during wound repair. For instance, K8/K18, as well as K17, can each promote the survival of epithelial cells under specific circumstances (Caulin et al., 2000; Gilbert et al., 2001; McGowan et al., 2002). Our hypothesis also does not preclude an important role for other keratinocyte constituents during skin wound repair (Martin, 1997). This hypothesis can be generalized to account for the rapid response exhibited by other IF genes after tissue injury, and also may help justify the diversity encountered within the IF protein superfamily. This model makes the key prediction that differential regulation of keratin genes enables epithelial cells to adjust their viscoelastic properties in order to best carry out the demands placed upon them. Whereas there is evidence that type I-type II pairing significantly influences the mechanical properties of keratin filament suspensions (Hofmann and Franke, 1997; Bousquet et al., 2001; Yamada et al., 2002), it is doubtful that such in vitro studies provide a complete account of IF properties in living cells. This said, there is as yet no direct evidence addressing the issue of whether and how IFs influence the cell's viscoelastic properties. Application of particle tracking rheology (Yamada et al., 2000) to genetically modified keratinocytes in culture offers an opportunity to examine this issue. In addition to IF protein composition, key parameters such as filament concentration, organization, posttranslational modifications, and dynamics should all influence how IFs impact on the properties of the cell, viscoelastic and otherwise (Fuchs and Cleveland, 1998; Omary et al., 1998; Coulombe et al., 2000).
Understanding adaptive changes and modifier gene effects in keratinocytes null for K6α/K6β
The increased epithelialization potential exhibited by K6α/K6β-null keratinocytes in the idealized setting of skin explant culture offers an opportunity to identify key effectors of this process. That the keratinocyte cytoskeleton is profoundly affected by the absence of K6 proteins is reflected by alterations in F-actin organization, phosphotyrosine epitopes, and p120ctn. Of relevance to our findings, adenovirus-mediated expression of p120ctn increases the motility of mouse skin keratinocytes in primary culture, and alters F-actin organization (Cozzolino et al., 2003). Changes in F-actin have been observed in fibroblasts carrying null alleles for plectin, an IF-interacting protein, or vimentin, a type III IF protein, correlating with altered migratory properties (Andra et al., 1998; Eckes et al., 2000). Keratin filament and F-actin networks are differentially affected in suprabasal null keratinocytes located at the explant edge and in matrix-attached keratinocytes the leading edge of the outgrowth. This may reflect the occurrence of distinct adaptative responses depending on the immediate surroundings of the cells, or alternatively, the existence of cellular heterogeneity within the epithelial outgrowth. Although the finding of increased stress fibers in leading edge keratinocytes may be perceived to be at odds with an enhanced migratory phenotype, we have no information about the dynamic properties of F-actin in these cells, or whether enhanced outgrowth in null explants is mediated by matrix-attached keratinocytes or by suprabasal keratinocytes.
Participation of IFs to tissue repair: a universal phenomenon with common significance?
Alterations in IF gene expression represent a conserved response after tissue injury. Such changes are manifested as either a switch in the IF genes being expressed or a stimulation of the existing program. Akin to epidermal keratinocytes, neurons respond to injury with the induction of α-internexin concomitant with down-regulation of the type IV neurofilament proteins (Goldstein et al., 1988, Tetzlaff et al., 1988; Muma et al., 1990; McGraw et al., 2002). In addition to its effect on neurons, trauma to the central nervous system (CNS) also triggers a reaction in neuroglia. Surrounding wound-activated astrocytes become mobilized into the site of tissue injury (Galou et al., 1996; Pekny et al., 1999). These cells elevate their expression of type III glial fibrillary acidic protein (GFAP) and reactivate the expression of two other IF proteins, vimentin (type III) and nestin (type VI), which had been expressed as part of their development (Galou et al., 1996; Kaya et al., 1999; Shibuya et al., 2002). In addition, neurofilament-L and -M subunits are transiently expressed in Schwann cells deprived of contacts with injured neurons (Fabrizi et al., 1997). In skeletal muscle, the predominant type III IF protein desmin is quickly down-regulated after injury, whereas vimentin and nestin expression is reactivated (Vaittinen et al., 2001). In simple epithelia such as liver and pancreas, injury results in a rapid and robust stimulation of type I/II IF genes already expressed, K8/K18 (Loranger et al., 1997; Ku et al., 1998; Toivola et al., 2000). And finally, the expression of K6α/K6β, K16, and K17 is either induced or stimulated whenever complex epithelia other than skin, such as the cornea and oral mucosa, are injured (Schermer et al., 1989; Takahashi and Coulombe, 1997).
Inactivation of various types of IF genes in mice compromises the response to injury. Regenerating myelinated neurons from neurofilament-L–null mice exhibit retarded maturation after injury (Zhu et al., 1997). GFAP-null mice show depressed long term potentiation and increased neuronal death subsequent to transient CNS ischemia (Tanaka et al., 2002). Mice lacking both GFAP and vimentin (type III) display defects in glial scar formation after injury to the CNS (Pekny et al., 1999). In primary culture, GFAP/vimentin double-null astrocytes exhibit impaired motility (Lepekhin et al., 2001). Vimentin-null mice, embryonic and adult, are delayed in their ability to heal skin wounds owing to altered mesenchymal contraction and delayed migration of fibroblasts to the wound site, respectively (Eckes et al., 2000). Also, vimentin-null fibroblasts exhibit fragility and lysis when subjected to distending forces. Yet, vimentin-null fibroblasts are competent to migrate in ex vivo culture and exhibit altered actin networks (Eckes et al., 1998). Desmin-null skeletal muscle exhibit defects in stress bearing, proliferation, and myoblast fusion after injury (Li et al., 1997; Smythe et al., 2001). K8-null mice are more likely to die after hepatectomy and other forms of acute challenges to the liver, correlating with their unusual fragility (Loranger et al., 1997; Ku et al., 1998). Although multiple mechanisms could account for these phenotypes, these observations are also consistent with a role of IFs toward promoting an optimal balance between properties of cellular pliability and resilience. Follow-up studies testing this concept will provide insight into the basic mechanisms of tissue repair, and may lead to improved strategies for the therapeutic management of chronic wounds.
Materials and methods
Mouse lines
Mice hemizygous for a null allele at the K6α/K6β (Wong et al., 2000) were backcrossed to wild-type C57Bl/6 (NCI-FCRDC) or 129/SvJ (Jackson Laboratory) mice for at least seven generations. Regardless of strain, mice harboring the K6α/K6β-null mutation appeared normal at birth but died within 10 d without exception. K14-null mice were genotyped as described previously (Lloyd et al., 1995).
Skin explant culture
Ex vivo explant culture of 2–4-d-old mouse skin was performed as described previously (Mazzalupo et al., 2002). Using 4-mm punches (Acuderm, Inc.), circular skin biopsies were obtained and plated with medium in 24-well dishes (Mazzalupo et al., 2002). Explants were cultured for 8 d before fixing (3% PFA for 10 min; 100% methanol for 5 min; room temperature). Immunostaining for K17 (McGowan and Coulombe, 1998b) was used to identify keratinocytes in the cellular outgrowth. Keratinocyte outgrowths were quantitated using two methods yielding similar findings. One consisted of measuring the surface area covered by the K17-positive outgrowth using the MacBas v2.5 software. The other method measured the linear distance extending between the explant biopsy and the distal edge of the cellular outgrowth by randomly applying a pattern of eight radial lines to the K17-stained preparations (Wawersik et al., 2001). Where warranted, stratified statistical analysis was performed as a function of genotype. A subset of explants were treated with mitomycin C (10 μg/ml for 2 h; Sigma-Aldrich) at 48 h after seeding and keratinocyte outgrowth was measured immediately after treatment (day 2) and at days 4, 6, and 8 of culture. Immunofluorescence studies were done on explants cultured on coverslips (Mazzalupo et al., 2002). Primary antibodies used were: rabbit polyclonal antisera directed against K6 or K17 (McGowan and Coulombe, 1998b), K16 (Bernot et al., 2002), and K5 (Covance); and mouse monoclonals directed against K14 (LLOO1; Purkis et al., 1990), BrdU (Sigma-Aldrich), and p120ctn (Transduction Labs). Actin filaments were labeled on PFA-fixed samples using rhodamine-conjugated phalloidin (Sigma-Aldrich). Secondary antibodies used were HRP- (Sigma-Aldrich) and fluorophore-conjugated (Kirkegaard & Perry Laboratories).
Skin grafting studies
Immunocompromised CD-1 nude mice (Charles River) were used as hosts. In brief, hosts were anesthetized with 25 μl/g of body weight of avertin and prepared by scoring the area of backskin where the graft was to be placed. The top layer of the skin was gently removed, leaving the vascular bed intact. 3-d-old pups derived from K6α/K6β hemizygous matings in the C57Bl/6 strain were killed and donor skin was placed on the host vascular bed, sutured (Sofsilk 4–0; United States Surgical), and covered with antibacterial ointment (Alpharma USPD, Inc.). Mice were housed individually thereafter. After normalization, skin grafts (two per genotype) were subjected to full thickness incisional wounding. 3 d later, the wound site and surrounding healthy tissue was biopsied at several locations and processed for morphological analyses. Alternatively, grafted tissue (two per genotype) was subjected to chemical treatment and mechanical trauma as follows. PMA (Sigma-Aldrich) was applied topically using a cotton pad on days 1, 3, and 5, and tissue was harvested on day 7. Mice were injected with 100 μg/g of body weight of BrdU (Sigma-Aldrich) 2 h before killing. Immediately before harvesting tissue for histology, gentle trauma was applied by rubbing with a pencil eraser 10 times across treated and untreated skin graft.
Morphological and protein analyses
For routine histopathology, tissues were fixed in Bouin's, paraffin-embedded, and 5-μm sections cut and stained with hematoxylin and eosin (H&E). Confocal microscopy was done using a dual spinning disk instrument (model Axiovert 200; Carl Zeiss MicroImaging, Inc.) equipped with a 63× oil objective and a digital camera (model Orca–ER; Hamamatsu Co.). Image processing was done via the acquisition software (Ultraview 5.4; Orinda). For plastic embedding, samples were fixed in 2% glutaraldehyde, 1% PFA mixture overnight at 4°C, fixed after with 1% osmium tetroxide, embedded in epoxy resin and semi-thick sections were cut (0.5 μm thick) and stained with toluidine blue. To analyze proteins, cells derived from explants were harvested after 6 d in culture in ice-cold PBS supplemented with protease and phosphatase inhibitors (1 mM EGTA, 20 μM Na3VO4, 10 mM NaF, 1 μg/ml leupeptin, 2 μg/ml antipain, 10 μg/ml aprotinin, 10 μg/ml benzamidine, 1 μg/ml cymostatin, and 1 μg/ml pepstatin-A; Sigma-Aldrich). Skin tissue was removed and outgrown cells were scraped, collected by centrifugation, and lysed in buffer (1% deoxycholate, 1% Triton X-100, 0.1% SDS, 150 mM NaCl, 50 mM Tris, pH 7.5, 0.5 mM EDTA, and 1 mM EGTA) supplemented with inhibitors. Cell scrapings from at least 20 explants from three littermates with the same genotype were pooled. Soluble and insoluble protein fractions were obtained by centrifugation for 10 min at 4°C. Pellets were resuspended in a Tris-buffered 8 M urea solution with inhibitors. 2 μg of protein (Bradford assay) was subjected to SDS-PAGE, transferred onto nitrocellulose in the presence of 1 mM of sodium orthovanadate. Additional antibodies used include mouse monoclonals against K13, K15, and K16 (K8.12; Sigma-Aldrich) or phosphotyrosine (4G10; Upstate Biotechnology). Bound primary antibodies were detected using chemiluminescence (Pierce Chemical Co.).
RT-PCR analyses
Semi-quantitative RT-PCR was performed on total RNA prepared from 6-d-old skin explant cultures using TRIzol (Invitrogen) and reverse transcribed (Advantage RT-for PCR kit; CLONTECH Laboratories, Inc.). K16 (forward, 5′-AACAGCCTAGAAGAGACCAAAGGC-3′; and reverse, 5′-GGTAGGGGAGACAGATGGGGAATGCGC-3′) and β-tubulin (forward, 5′-CAACGTCAAGACGGCCGTGTG-3′; and reverse, 5′-GACAGAGGCAAACTGAGC- ACC-3′) oligonucleotide primers were used. Reactions were sampled after 22, 27, 32, and 37 cycles (94°C for 1 min; 58°C for 1 min; and 68°C for 1 min) to monitor product accumulation.
Acknowledgments
We thank Charles Babinet, Emma Colucci-Guyon, Matthew Wawersik, Angie Lebrun, Brad Harris, Richard Skolasky, Michael Caterina, Kelsie Bernot, and Carole Parent for their assistance.
These studies were supported by grants F32-AR088553 (to P. Wong) and R01-AR42047 (to P.A. Coulombe) from the National Institutes of Health.
Abbreviations used in this paper: CNS, central nervous system; GFAP, glial fibrillary acidic protein; H&E, hematoxylin and eosin; IF, intermediate filament.
References
- Anastasiadis, P.Z., and A.B. Reynolds. 2001. Regulation of Rho GTPases by p120ctn. Curr. Opin. Cell Biol. 13:604–610. [DOI] [PubMed] [Google Scholar]
- Andra, K., B. Nikolic, M. Stocher, D. Drenckhahn, and G. Wiche. 1998. Not just scaffolding: plectin regulates actin dynamics in cultured cells. Genes Dev. 12:3442–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baribault, H., J. Price, K. Miyai, and R.G. Oshima. 1993. Mid-gestational lethality in mice lacking keratin 8. Genes Dev. 7:1191–1202. [DOI] [PubMed] [Google Scholar]
- Baribault, H., J. Penner, R.V. Iozzo, and M. Wilson-Heiner. 1994. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 8:2964–2973. [DOI] [PubMed] [Google Scholar]
- Bernot, K., P.A. Coulombe, and K.M. McGowan. 2002. Keratin 16 expression defines a subset of epithelial cells during skin morphogenesis and the hair cycle. J. Invest. Dermatol. 119:1137–1149. [DOI] [PubMed] [Google Scholar]
- Bousquet, O., L. Ma, S. Yamada, C. Gu, T. Idei, K. Takahashi, D. Wirtz, and P.A. Coulombe. 2001. The nonhelical tail domain of keratin 14 promotes filament bundling and enhances the mechanical properties of keratin intermediate filaments in vitro. J. Cell Biol. 155:747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulin, C., C.F. Ware, T.M. Magin, and R.G. Oshima. 2000. Keratin-dependent, epithelial resistance to tumor necrosis factor- induced apoptosis. J. Cell Biol. 149:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe, P.A., and M.B. Omary. 2002. “Hard” and “soft” principles defining the structure, function and regulation of keratin intermediate filaments. Curr. Opin. Cell Biol. 14:110–122. [DOI] [PubMed] [Google Scholar]
- Coulombe, P.A., O. Bousquet, L. Ma, S. Yamada, and D. Wirtz. 2000. The “ins” and “outs” of intermediate filament organization. Trends Cell Biol. 10:420–428. [DOI] [PubMed] [Google Scholar]
- Cozzolino, M., V. Stagni, L. Spinardi, N. Campioni, C. Fiorentini, E. Salvati, S. Alema, and A.M. Salvatore. 2003. p120 catenin is required for growth factor-dependent cell motility and scattering in epithelial cells. Mol. Biol. Cell. 14:1964–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckes, B., E. Colucci-Guyon, H. Smola, S. Nodder, C. Babinet, T. Krieg, and P. Martin. 2000. Impaired wound healing in embryonic and adult mice lacking vimentin. J. Cell Sci. 113:2455–2462. [DOI] [PubMed] [Google Scholar]
- Eckes, B., D. Dogic, E. Colucci-Guyon, N. Wang, A. Maniotis, D. Ingber, A. Merckling, F. Langa, M. Aumailley, A. Delouvee, et al. 1998. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J. Cell Sci. 111:1897–1907. [DOI] [PubMed] [Google Scholar]
- Estrada, C.M., C.D. Park, M. Castilla, and R.A. Tassava. 1993. Monoclonal antibody WE6 identifies an antigen that is up-regulated in the wound epithelium of newts and frogs. Limb Development and Regeneration. Wiley-Liss Inc., New York. 272–282. [PubMed]
- Fabrizi, C., B.M. Kelly, C.S. Gillespie, W.W. Schlaepfer, S.S. Scherer, and P.J. Brophy. 1997. Transient expression of the neurofilament proteins NF-L and NF-M by Schwann cells is regulated by axonal contact. J. Neurosci. Res. 50:291–299. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., and D.W. Cleveland. 1998. A structural scaffolding of intermediate filaments in health and disease. Science. 279:514–519. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., and K. Weber. 1994. Intermediate filaments: structure, dynamics, function, and disease. Annu. Rev. Biochem. 63:345–382. [DOI] [PubMed] [Google Scholar]
- Galou, M., E. Colucci-Guyon, D. Ensergueix, J.L. Ridet, M. Gimenez y Ribotta, A. Privat, C. Babinet, and P. Dupouey. 1996. Disrupted glial fibrillary acidic protein network in astrocytes from vimentin knockout mice. J. Cell Biol. 133:853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, S., A. Loranger, N. Daigle, and N. Marceau. 2001. Simple epithelium keratins 8 and 18 provide resistance to Fas-mediated apoptosis. The protection occurs through a receptor-targeting modulation. J. Cell Biol. 154:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, M.E., S.R. Weiss, R.A. Lazzarini, P.S. Shneidman, J.F. Lees, and W.W. Schlaepfer. 1988. mRNA levels of all three neurofilament proteins decline following nerve transection. Brain Res. 427:287–291. [DOI] [PubMed] [Google Scholar]
- Hesse, M., T.M. Magin, and K. Weber. 2001. Genes for intermediate filament proteins and the draft sequence of the human genome: novel keratin genes and a surprisingly high number of pseudogenes related to keratin genes 8 and 18. J. Cell Sci. 114:2569–2575. [DOI] [PubMed] [Google Scholar]
- Hofmann, I., and W.W. Franke. 1997. Heterotypic interactions and filament assembly of type I and type II cytokeratins in vitro: viscometry and determinations of relative affinities. Eur. J. Cell Biol. 72:122–132. [PubMed] [Google Scholar]
- Irvine, A.D., and W.H. McLean. 1999. Human keratin diseases: the increasing spectrum of disease and subtlety of the phenotype-genotype correlation. Br. J. Dermatol. 140:815–828. [DOI] [PubMed] [Google Scholar]
- Kaya, S.S., A. Mahmood, Y. Li, E. Yavuz, M. Goksel, and M. Chopp. 1999. Apoptosis and expression of p53 response proteins and cyclin D1 after cortical impact in rat brain. Brain Res. 818:23–33. [DOI] [PubMed] [Google Scholar]
- Ku, N.O., S.A. Michie, R.M. Soetikno, E.Z. Resurreccion, R.L. Broome, and M.B. Omary. 1998. Mutation of a major keratin phosphorylation site predisposes to hepatotoxic injury in transgenic mice. J. Cell Biol. 143:2023–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepekhin, E.A., C. Eliasson, C.H. Berthold, V. Berezin, E. Bock, and M. Pekny. 2001. Intermediate filaments regulate astrocyte motility. J. Neurochem. 79:617–625. [DOI] [PubMed] [Google Scholar]
- Li, Z., M. Mericskay, O. Agbulut, G. Butler-Browne, L. Carlsson, L.E. Thornell, C. Babinet, and D. Paulin. 1997. Desmin is essential for the tensile strength and integrity of myofibrils but not for myogenic commitment, differentiation, and fusion of skeletal muscle. J. Cell Biol. 139:129–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd, C., Q.C. Yu, J. Cheng, K. Turksen, L. Degenstein, E. Hutton, and E. Fuchs. 1995. The basal keratin network of stratified squamous epithelia: defining K15 function in the absence of K14. J. Cell Biol. 129:1329–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loranger, A., S. Duclos, A. Grenier, J. Price, M. Wilson-Heiner, H. Baribault, and N. Marceau. 1997. Simple epithelium keratins are required for maintenance of hepatocyte integrity. Am. J. Pathol. 151:1673–1683. [PMC free article] [PubMed] [Google Scholar]
- Mansbridge, J.N., and A.M. Knapp. 1987. Changes in keratinocyte maturation during wound healing. J. Invest. Dermatol. 89:253–263. [DOI] [PubMed] [Google Scholar]
- Martin, P. 1997. Wound healing - aiming for the perfect skin regeneration. Science. 276:75–81. [DOI] [PubMed] [Google Scholar]
- Mazzalupo, S., M.J. Wawersik, and P.A. Coulombe. 2002. An ex vivo assay to assess the potential of skin keratinocytes for wound epithelialization. J. Invest. Dermatol. 118:866–870. [DOI] [PubMed] [Google Scholar]
- McGraw, T.S., J.P. Mickle, G. Shaw, and W.J. Streit. 2002. Axonally transported peripheral signals regulate alpha-internexin expression in regenerating motoneurons. J. Neurosci. 22:4955–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan, K.M., and P.A. Coulombe. 1998. a. Onset of keratin 17 expression coincides with the definition of major epithelial lineages during skin development. J. Cell Biol. 143:469–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan, K.M., and P.A. Coulombe. 1998b. The wound repair associated keratins 6, 16, and 17: insights into the role of intermediate filaments in specifying cytoarchitecture. Subcellular Biochemistry: Intermediate Filaments. J.R. Harris and H. Herrmann, editors. Plenum Publishing Corp., London. 141–165. [PubMed]
- McGowan, K.M., X. Tong, E. Colucci-Guyon, F. Langa, C. Babinet, and P.A. Coulombe. 2002. Keratin 17 null mice exhibit age- and strain-dependent alopecia. Genes Dev. 16:1412–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll, R., W.W. Franke, D.L. Schiller, B. Geiger, and R. Krepler. 1982. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 31:11–24. [DOI] [PubMed] [Google Scholar]
- Muma, N.A., P.N. Hoffman, H.H. Slunt, M.D. Applegate, I. Lieberburg, and D.L. Price. 1990. Alterations in levels of mRNAs coding for neurofilament protein subunits during regeneration. Exp. Neurol. 107:230–235. [DOI] [PubMed] [Google Scholar]
- Omary, M.B., N.-O. Ku, J. Liao, and D. Price. 1998. Keratin modifications and solubility properties in epithelial cells and in vitro. Subcellular Biochemistry: Intermediate Filaments. Vol. 31. H. Herrman and J.R. Harris, editors. Plenum Press, New York. 105–140. [PubMed]
- Oshima, R.G. 2002. Apoptosis and keratin intermediate filaments. Cell Death Differ. 9:486–492. [DOI] [PubMed] [Google Scholar]
- Paladini, R.D., K. Takahashi, N.S. Bravo, and P.A. Coulombe. 1996. Onset of re-epithelialization after skin injury correlates with a reorganization of keratin filaments in wound edge keratinocytes: defining a potential role for keratin 16. J. Cell Biol. 132:381–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang, S.C., W.H. Daniels, and R.C. Buck. 1978. Epidermal migration during the healing of suction blisters in rat skin: a scanning and transmission electron microscopic study. Am. J. Anat. 153:177–191. [DOI] [PubMed] [Google Scholar]
- Pekny, M., C.B. Johansson, C. Eliasson, J. Stakeberg, A. Wallen, T. Perlmann, U. Lendahl, C. Betsholtz, C.H. Berthold, and J. Frisen. 1999. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J. Cell Biol. 145:503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkis, P.E., J.B. Steel, I.C. Mackenzie, W.B. Nathrath, I.M. Leigh, E.B. Lane. 1990. Antibody markers of basal cells in complex epithelia. J. Cell Sci. 97:39–50. [DOI] [PubMed] [Google Scholar]
- Quinlan, R., C. Hutchinson, and E.B. Lane. 1994. Intermediate filament proteins. Protein Profile. 1:779–911. [PubMed] [Google Scholar]
- Ramirez, A., M. Vidal, A. Bravo, F. Larcher, and J.L. Jorcano. 1995. A 5′-upstream region of a bovine keratin 6 gene confers tissue-specific expression and hyperproliferation-related induction in transgenic mice. Proc. Natl. Acad. Sci. USA. 92:4783–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt, J., and T.M. Magin. 2002. Hyperproliferation, induction of c-Myc and 14-3-3sigma, but no cell fragility in keratin-10-null mice. J. Cell Sci. 115:2639–2650. [DOI] [PubMed] [Google Scholar]
- Schermer, A., J.V. Jester, C. Hardy, D. Milano, and T.T. Sun. 1989. Transient synthesis of K6 and K16 keratins in regenerating rabbit corneal epithelium: keratin markers for an alternative pathway of keratinocyte differentiation. Differentiation. 42:103–110. [DOI] [PubMed] [Google Scholar]
- Shibuya, S., O. Miyamoto, R.N. Auer, T. Itano, S. Mori, and H. Norimatsu. 2002. Embryonic intermediate filament, nestin, expression following traumatic spinal cord injury in adult rats. Neuroscience. 114:905–916. [DOI] [PubMed] [Google Scholar]
- Smythe, G.M., M.J. Davies, D. Paulin, and M.D. Grounds. 2001. Absence of desmin slightly prolongs myoblast proliferation and delays fusion in vivo in regenerating grafts of skeletal muscle. Cell Tissue Res. 304:287–294. [DOI] [PubMed] [Google Scholar]
- Stenn, K.S., and R. Paus. 2001. Controls of hair follicle cycling. Physiol. Rev. 81:449–494. [DOI] [PubMed] [Google Scholar]
- Swensson, O., L. Langbein, J.R. McMillan, H.P. Stevens, I.M. Leigh, W.H. McLean, E.B. Lane, and R.A. Eady. 1998. Specialized keratin expression pattern in human ridged skin as an adaptation to high physical stress. Br. J. Dermatol. 139:767–775. [DOI] [PubMed] [Google Scholar]
- Takahashi, K., and P.A. Coulombe. 1997. Defining a region of the human keratin 6a gene that confers inducible expression in stratified epithelia of transgenic mice. J. Biol. Chem. 272:11979–11985. [DOI] [PubMed] [Google Scholar]
- Takahashi, K., R. Paladini, and P.A. Coulombe. 1995. Cloning and characterization of multiple human genes and cDNAs encoding highly related type II keratin 6 isoforms. J. Biol. Chem. 270:18581–18592. [DOI] [PubMed] [Google Scholar]
- Takahashi, K., B. Yan, K. Yamanishi, S. Imamura, and P.A. Coulombe. 1998. The two functional keratin 6 genes of mouse are differentially regulated and evolved independently from their human orthologs. Genomics. 53:170–183. [DOI] [PubMed] [Google Scholar]
- Tanaka, H., A. Katoh, K. Oguro, K. Shimazaki, H. Gomi, S. Itohara, T. Masuzawa, and N. Kawai. 2002. Disturbance of hippocampal long-term potentiation after transient ischemia in GFAP deficient mice. J. Neurosci. Res. 67:11–20. [DOI] [PubMed] [Google Scholar]
- Tetzlaff, W., M.A. Bisby, and G.W. Kreutzberg. 1988. Changes in cytoskeletal proteins in the rat facial nucleus following axotomy. J. Neurosci. 8:3181–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivola, D.M., H. Baribault, T. Magin, S.A. Michie, and M.B. Omary. 2000. Simple epithelial keratins are dispensable for cytoprotection in two pancreatitis models. Am. J. Physiol. Gastrointest. Liver Physiol. 279:G1343–G1354. [DOI] [PubMed] [Google Scholar]
- Tyner, A.L., and E. Fuchs. 1986. Evidence for posttranscriptional regulation of the keratins expressed during hyperproliferation and malignant transformation in human epidermis. J. Cell Biol. 103:1945–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaittinen, S., R. Lukka, C. Sahlgren, T. Hurme, J. Rantanen, U. Lendahl, J.E. Eriksson, and H. Kalimo. 2001. The expression of intermediate filament protein nestin as related to vimentin and desmin in regenerating skeletal muscle. J. Neuropathol. Exp. Neurol. 60:588–597. [DOI] [PubMed] [Google Scholar]
- Wang, Z., P. Wong, L. Langbein, J. Schweizer, and P.A. Coulombe. 2003. The type II epithelial keratin 6 hf (K6hf) is expressed in the companion layer, matrix, and medulla of anagen-stage hair follicles. J. Invest. Dermatol. In press. [DOI] [PubMed] [Google Scholar]
- Wawersik, M., R.D. Paladini, E. Noensie, and P.A. Coulombe. 1997. A proline residue in the alpha-helical rod domain of type I keratin 16 destabilizes keratin heterotetramers and influences incorporation into filaments. J. Biol. Chem. 272:32557–32565. [DOI] [PubMed] [Google Scholar]
- Wawersik, M.J., S. Mazzalupo, D. Nguyen, and P.A. Coulomb. 2001. Increased levels of keratin 16 alter the epithelialization potential of mouse skin keratinocytes in vivo and ex vivo. Mol. Biol. Cell. 12:3439–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik, S.M., D.S. Bundman, and D.R. Roop. 2000. Delayed wound healing in keratin 6a knockout mice. Mol. Cell. Biol. 20:5248–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik, S.M., M.A. Longley, and D.R. Roop. 2001. Discovery of a novel murine keratin 6 (K6) isoform explains the absence of hair and nail defects in mice deficient for K6a and K6b. J. Cell Biol. 154:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, P., E. Colucci-Guyon, K. Takahashi, C. Gu, C. Babinet, and P.A. Coulombe. 2000. Introducing a null mutation in the mouse K6α and K6β genes reveals their essential structural role in the oral mucosa. J. Cell Biol. 150:921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, S., D. Wirtz, and S.C. Kuo. 2000. Mechanics of living cells measured by laser tracking microrheology. Biophys. J. 78:1736–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, S., D. Wirtz, and P.A. Coulombe. 2002. Pairwise assembly determines the intrinsic potential for self-organization and mechanical properties of keratin filaments. Mol. Biol. Cell. 13:382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Q., S. Couillard-Despres, and J.P. Julien. 1997. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp. Neurol. 148:299–316. [DOI] [PubMed] [Google Scholar]