

Abstract
A thiol-reactive membrane-associated protein (TRAP) binds covalently to the cytoplasmic domain of the human insulin receptor (IR) β-subunit when cells are treated with the homobifunctional cross-linker reagent 1,6-bismaleimidohexane. Here, TRAP was found to be phospholipase C γ1 (PLCγ1) by mass spectrometry analysis. PLCγ1 associated with the IR both in cultured cell lines and in a primary culture of rat hepatocytes. Insulin increased PLCγ1 tyrosine phosphorylation at Tyr-783 and its colocalization with the IR in punctated structures enriched in cortical actin at the dorsal plasma membrane. This association was found to be independent of PLCγ1 Src homology 2 domains, and instead required the pleckstrin homology (PH)–EF-hand domain. Expression of the PH–EF construct blocked endogenous PLCγ1 binding to the IR and inhibited insulin-dependent phosphorylation of mitogen-activated protein kinase (MAPK), but not AKT. Silencing PLCγ1 expression using small interfering RNA markedly reduced insulin-dependent MAPK regulation in HepG2 cells. Conversely, reconstitution of PLCγ1 in PLCγ1 −/− fibroblasts improved MAPK activation by insulin. Our results show that PLCγ1 is a thiol-reactive protein whose association with the IR could contribute to the activation of MAPK signaling by insulin.
Keywords: receptors; PLC; signal transduction; cultured cells; mass spectrometry
Introduction
The pleiotropic actions of insulin are initiated by binding of the hormone to the extracellular domain of the insulin receptor (IR) and activation of its intrinsic tyrosine kinase activity. Insulin signal transduction requires IR autophosphorylation and phosphorylation of a number of intracellular molecules, including insulin receptor substrate 1 (IRS-1) and Shc proteins (Saltiel and Pessin, 2002). Many of these molecules contain modular domains (e.g., Src homology 2 [SH2] domain and/or phosphotyrosine-binding domains) that allow interaction with the tyrosine-phosphorylated IR. The phosphotyrosine-binding domain of IRS-1 has been shown to bind to the NPXpY motif of the IR after insulin stimulation, which leads to the recruitment of various cytosolic signaling intermediates to the cell surface (Virkamaki et al., 1999). The SH2-containing protein tyrosine phosphatase (PTP) 2 binds to the COOH-terminal phosphotyrosines of the activated IR (Rocchi et al., 1996), whereas Grb10 isoforms play a negative role in insulin signaling by binding with the tyrosine kinase loop of the activated IR via the BPS (between the pleckstrin homology [PH] domain and the SH2 domain) region (He et al., 1998; Kasus-Jacobi et al., 2000). Thus, the level of IR autophosphorylation may serve a crucial function in controlling both the phosphorylation of endogenous substrates and the interaction between the IR β-subunit and a number of proteins that regulate receptor-based signals.
The cytoplasmic domain of the IR β-subunit contains reactive cysteine thiol(s) that can modulate the receptor catalytic activity (Li et al., 1991; Bernier et al., 1995; Schmid et al., 1998). The importance of the IR cytoplasmic cysteines for the association between this receptor and intracellular effectors has been investigated in intact cells using 1,6-bismaleimidohexane (BMH), an irreversible thiol-specific homobifunctional cross-linking reagent (Garant et al., 2000). This approach has led to the identification of a complex between the human IR and a thiol-reactive membrane-associated protein (TRAP). The IR–TRAP complex migrates as an ∼250 kD protein on SDS-PAGE under reducing conditions and does not contain the receptor α-subunit as assessed by immunoblot analysis. In the same report, point-mutation analyses have shown that cysteine 981 of the cytoplasmic domain of the human IR β-subunit is the nucleophilic thiol responsible for the covalent binding to TRAP after BMH-induced cross-linking (Garant et al., 2000).
To further our understanding of the biological importance of TRAP in insulin signaling, we purified the IR–TRAP complex and identified TRAP as PLCγ1 using matrix-assisted laser desorption/ionization (MALDI) analysis. Here, our coimmunoprecipitation assays demonstrated constitutive and insulin-inducible association of PLCγ1 with the IR in a number of cultured cell lines and a primary culture of rat hepatocytes, which reflects the potential for physiological significance. Structurally, the catalytic region of PLCγ1 contains an insert with two SH2 domains and an SH3 domain. It has been proposed that the two SH2 domains are essential for association of PLCγ1 with activated growth factor receptor tyrosine kinases (Middlemas et al., 1994, Ji et al., 1999), whereas the SH3 domain directs PLCγ1 to bind to the cytoskeleton (Park et al., 1999). Whether these and other motifs play an important function in the recruitment of PLCγ1 to the IR remains unknown.
The dynamic association between PLCγ1 and the IR must depend on specific domains within both proteins. In an attempt to identify some of these motifs, we have expressed mutant forms of PLCγ1 and analyzed the pattern of IR–PLCγ1 association in intact cells. Now, we report on the identification of a domain of PLCγ1 containing the PH and EF-hand (PH–EF) that is required for interaction with the IR. Overexpression of the PH–EF fragment or reduction of PLCγ1 expression using small interfering RNA (siRNA) abrogates MAPK regulation by insulin, strengthening the notion that PLCγ1 plays an important role in insulin signaling (Kayali et al., 1998; Eichhorn et al., 2002).
Results
Insulin promotes formation of the IR–TRAP complex
CHO cells expressing the human IR were incubated with insulin and then subjected to a cross-linking reaction with BMH before cell lysis and Western blotting with an antibody against the IR β-subunit (Fig. 1 A, top). The IR–TRAP complex was detected in lysates from unstimulated cells upon BMH addition. Insulin increased the recruitment of TRAP to the IR with a concomitant reduction in the free IR β-subunit (Fig. 1 A, lane 4 vs. lane 3), but not in the IR α-subunit (Fig. 1 A, bottom). A second protein band was detected just below the IR–TRAP complex (Fig. 1 A, lane 4); however, it contained a much smaller amount of the conjugated IR β-subunit. Thus, TRAP can interact with the IR both in a constitutive and insulin-inducible manner. Of significance, association between the activated IR and TRAP was also observed in NIH3T3-IR cells and the human HepG2 cell line after incubation with BMH (unpublished data). The formation of the IR–TRAP complex was then assessed in anti-IR immunoprecipitates. Metabolically labeled CHO-IR cells were left untreated or incubated with insulin to induce IR autophosphorylation, followed by cross-linking reaction with BMH. Analysis of IR immunoprecipitates from BMH-treated cells demonstrated insulin's ability to increase the extent of IR–TRAP covalent association with concomitant decrease in the amount of free IR β-subunit (Fig. 1 B).
Figure. 1.
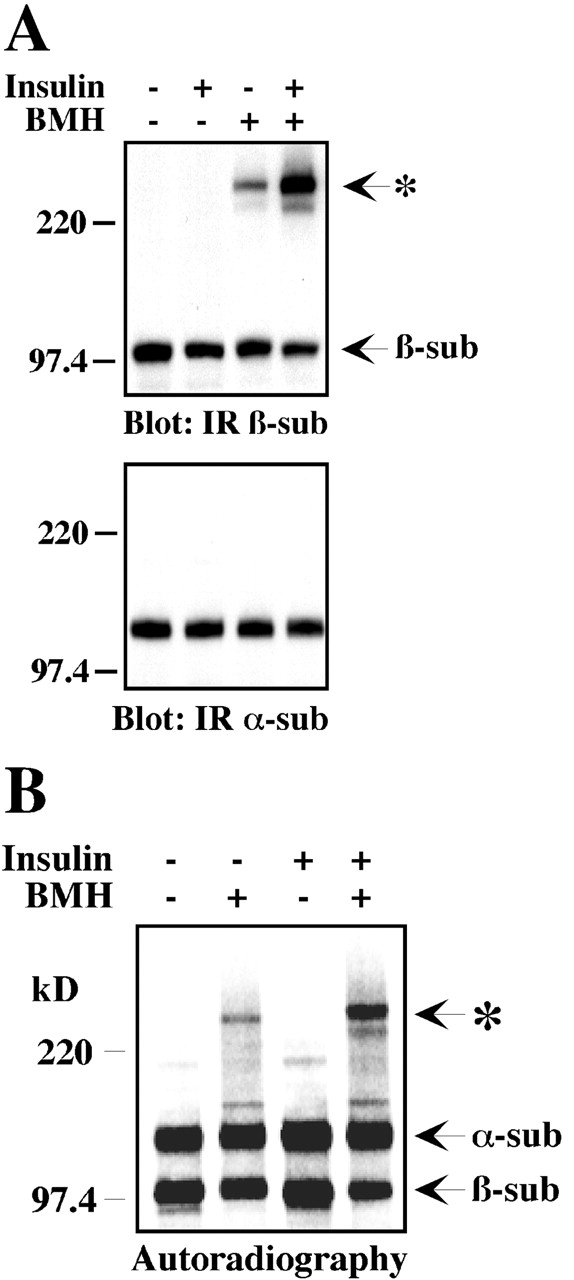
TRAP recruitment to the human IR in intact cells. (A) CHO-IR cells were serum starved before stimulation with 100 nM insulin for 5 min at 37°C. After a cross-linking reaction with 100 μM BMH, cell lysates were prepared and then immunoblotted with anti-IR β-subunit or anti-IR α-subunit antibodies. (B) CHO-IR cells were labeled with [35S]Met/Cys for 16 h before stimulation with 100 nM insulin for 5 min at 37°C. After a cross-linking reaction, endogenous IR was immunoprecipitated with anti-IR antibodies, resolved by SDS-PAGE, and detected by autoradiography. Right margin, α- and β-subunits of the IR; left margin, Mr × 10−3; asterisk, TRAP/IR β-subunit complex.
To ascertain whether the length of the cross-linker spacer arm dictates the extent of IR–TRAP covalent association, insulin-stimulated CHO-IR cells were incubated either with bismaleimidoethane (BMOE), bismaleimidobutane, or BMH, which are three related thiol-specific homobifunctional cross-linking reagents whose maleimido groups are separated with flexible spacer arms of 8.0, 10.9, and 16.1 Å, respectively. Insulin promoted IR–TRAP complex formation irrespective of the cross-linker used (unpublished data), indicating that the nucleophilic thiols (on TRAP and the IR β-subunit) may be separated by at least 8 Å.
Characterization of TRAP
The silver-stained gel of the anti-IR immunoprecipitates resolved four major bands that corresponded to the TRAP/β-subunit complex, IR proreceptor (αβ-dimer), and α- and β-subunit, respectively, with apparent molecular masses ranging between ∼100 kD (β-subunit) and ∼275 kD (TRAP/β-subunit) (Fig. 2). The IR β-subunit and TRAP/β-subunit protein bands were subjected to in-gel digestion with trypsin, followed by peptide mass fingerprinting and MALDI analysis of the eluted peptides to provide tentative identification of each protein species. 17 and 15 peptide masses covering 17 and 9% of the IR β-subunit, respectively, were found in both protein bands (estimated z value of 2.16 and 2.38, respectively), whereas 12 peptide masses within the TRAP/β-subunit band matched the 155-kD PLCγ1 (estimated z value of 2.39), corresponding to 10% of the molecule. These peptides covered various regions of PLCγ1. Analysis of recombinant GST-tagged PLCγ1 SH2/SH3 domain fusion protein by MALDI returned 18 peptide masses (estimated z value of 1.82), many of which were strong matches with those found in the TRAP/β-subunit protein band. Subsequent immunoblot analyses revealed the presence of PLCγ1 in the IR–TRAP complex (see below).
Figure 2.

Purification and characterization of TRAP. Endogenous IR was immunoprecipitated from insulin-stimulated CHO-IR cells that were treated with BMH, and the immunoprecipitates were size fractionated before SDS-PAGE and gel staining (left). A duplicate sample was blotted with anti-IR β-subunit antibody (right). M, size markers.
The cross-linking of PLCγ1 with the IR upon cell treatment with BMH indicates that both proteins must contain reactive cysteines. Therefore, the ability of PLCγ1 to react with maleimidobutyrylbiocytin was investigated in HEK293 cells transfected with vector alone or HA-tagged PLCγ1. In this thiol-specific biotinylation assay (Bernier et al., 1995), recombinant as well as endogenous PLCγ1 were readily modified (unpublished data), supporting the notion that PLCγ1 contains reactive thiol group(s).
Immunodetection of the PLCγ1–IR complex
The association of PLCγ1 with the IR was evaluated in CHO-IR cells that were left untreated or exposed to a saturating concentration of insulin (100 nM) for 3–30 min. Immunoblotting the anti-IR immunoprecipitates with anti-PLCγ1 antibody showed a time-dependent increase in PLCγ1 association with the IR in response to insulin that persisted throughout the 30 min of the experiment (Fig. 3 A). The interaction is stimulated by insulin in a dose-dependent manner, with detectable levels at 5 nM insulin (Fig. 3 B). Similar results were obtained by probing anti-PLCγ1 immunoprecipitates with anti-IR antibody (unpublished data). When immunoprecipitation was performed with a control IgG, no cosedimentation of the IR with PLCγ1 was detectable (unpublished data).
Figure 3.

Insulin elicits recruitment of PLCγ1 to the IR. (A) Serum-starved CHO-IR cells were left untreated or stimulated with 100 nM insulin for 3, 10, or 30 min. (B) CHO-IR cells were incubated with the indicated concentrations of insulin for 15 min. Anti-IR immunoprecipitates were blotted using anti-PLCγ1PH and anti-IR α-subunit antibodies. (C) CHO-IR cells were pretreated for 30 min with 200 μM orthovanadate before stimulation with the indicated concentrations of insulin for 15 min. Lysates were blotted with antibodies against PLCγ1 phosphorylated at Tyr-783 (pPLCγ1) or PLCγ1PH. (D) Vanadate-pretreated CHO-IR cells were stimulated with 100 nM insulin for 15 min. Anti-PLCγ1 immunoprecipitates were subsequently probed with anti-IR β-subunit, pPLCγ1, or PLCγ1PH antibodies. Preincubation of anti-pPLCγ1 antibody with antigenic peptide abolishes pPLCγ1 signals (not depicted). Results shown are representative of several independent experiments.
PLCγ1 is a member of the phosphoinositide (PI)-specific PLC family whose phosphorylation by many activated nonreceptor and receptor tyrosine kinases results in its subsequent activation (Rhee 2001). To test the predictions that PLCγ1 tyrosine phosphorylation could occur with insulin, CHO-IR cells were left untreated or treated with insulin for 15 min both in the absence or presence of orthovanadate, a PTP inhibitor. The extent of PLCγ1 phosphorylation at Tyr-783 was then determined in total cell lysates by Western blot analysis using a commercially available phosphospecific antibody. In the absence of vanadate, the levels of tyrosine-phosphorylated PLCγ1 were barely detectable under basal conditions and after insulin stimulation (unpublished data). However, PLCγ1 tyrosine phosphorylation was increased in a dose-dependent manner after the addition of insulin to vanadate-pretreated CHO-IR cells (Fig. 3 C), peaking within 10–30 min (unpublished data). Thus, insulin was found to induce tyrosine phosphorylation of PLCγ1, and this effect was clearly sensitive to PTP inhibition.
To further evaluate the role of insulin in mediating tyrosine phosphorylation and association of PLCγ1 with the activated IR, anti-PLCγ1 immunoprecipitates from vanadate-treated CHO-IR cells were probed with anti-IR. Insulin stimulation led to a significant increase (6.4 ± 1.6-fold; n = 6) in IR cosedimentation with PLCγ1 and in phosphoPLCγ1 levels (Fig. 3 D).
Physiological significance of the IR–PLCγ1 association
Next, we investigated the role of insulin in the recruitment of PLCγ1 to the endogenous IR in insulin-responsive HepG2 cells. These cells were pretreated with orthovanadate and then left untreated or exposed to 100 nM insulin for 15 min. Fig. 4 A shows the results of a typical experiment analyzing PLCγ1 immunoprecipitates that were blotted with the IR β-subunit. In agreement with our previous results with CHO-IR cells from this report, a constitutive and insulin-inducible cosedimentation of the IR with PLCγ1 was observed, suggesting that insulin could promote the recruitment of PLCγ1 to the IR in a number of cell types. Higher tyrosine phosphorylation of PLCγ1 was also noted in response to insulin when PLCγ1 was immunoprecipitated and then visualized with either anti-phosphoPLCγ1 (pTyr-783) or phosphotyrosine (clone RC20) antibody (Fig. 4 A). Next, we determined that endogenous PLCγ1 interacted with the IR in primary culture of rat hepatocytes (Fig. 4 B). These results strongly support a physiological role for the PLCγ1 association to the IR in insulin signaling.
Figure 4.
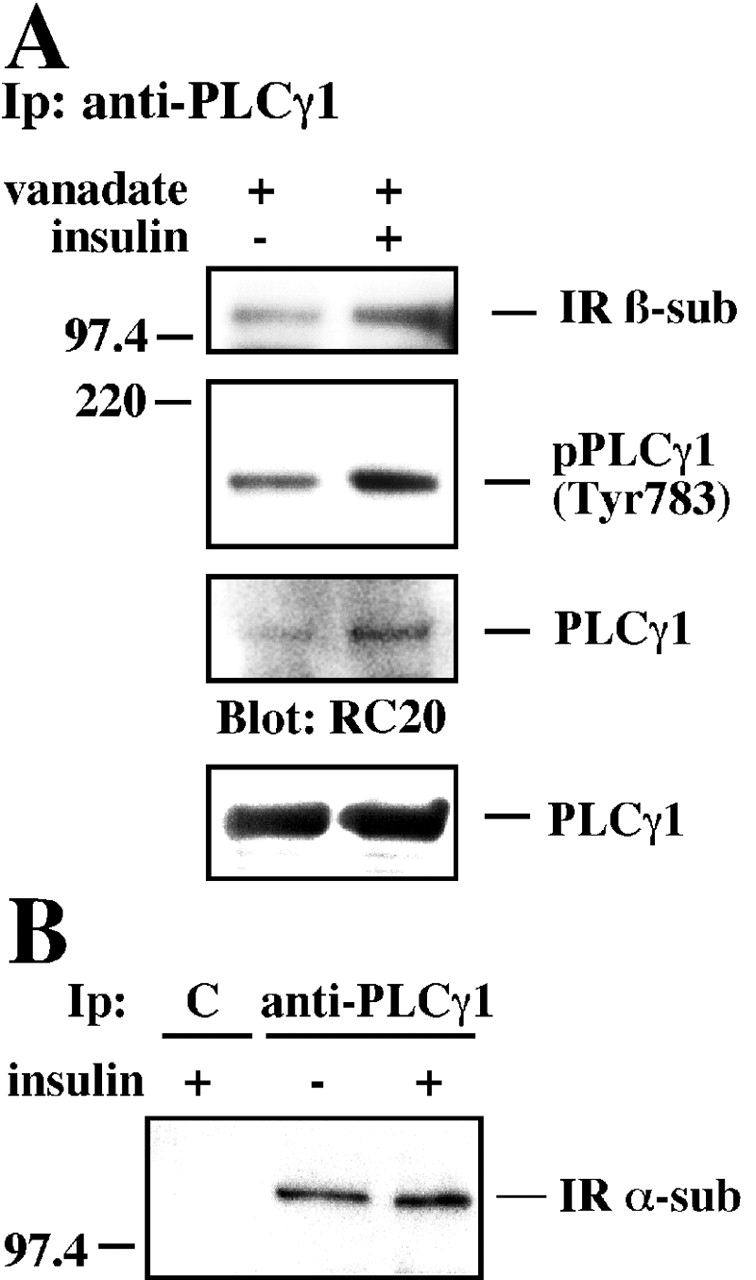
Insulin induces tyrosine phosphorylation and association of PLCγ1 with the IR in HepG2 cells and rat hepatocytes. (A) Serum-starved HepG2 cells were pretreated with 200 μM orthovanadate for 30 min before stimulation with 100 nM insulin for 15 min. Anti-PLCγ1PH immunoprecipitates were blotted with the indicated antibodies. Equal loading was confirmed by reprobing the membranes with anti-PLCγ1PH. Results shown are representative of three independent observations. (B) A primary culture of rat hepatocytes was treated (or not treated) with 100 nM insulin for 10 min. Lysates were incubated with anti-PLCγ1 antibody or a control mAb (C), and the immunoprecipitates were blotted with anti-IR α-subunit antibody.
PLCγ1 colocalizes with the IR at the plasma membrane
Immunofluorescence microscopy was used to test whether the subcellular localization of the IR, PLCγ1, and tyrosine-phosphorylated PLCγ1 is affected after stimulation of CHO-IR cells with insulin. The IR was primarily found at the plasma membrane when cells were left untreated or incubated with insulin for 10 min (Fig. 5, left panels). The distribution of PLCγ1 throughout the cytosolic space was not affected by the addition of insulin (Fig. 5, right panels). In contrast, a strong tyrosine-phosphorylated PLCγ1 signal was found at the plasma membrane of insulin-stimulated cells (Fig. 5, middle panels). Confocal sectioning showed that the ventral side of the cells (point of attachment to the substratum) was largely devoid of IR and PLCγ1 (unpublished data), whereas the apical side was decorated with both the IR and tyrosine-phosphorylated PLCγ1 in the form of small clusters surrounding the cell membrane that are likely to be derived from the cortical cytoskeleton (Fig. 5, bottom panels).
Figure 5.
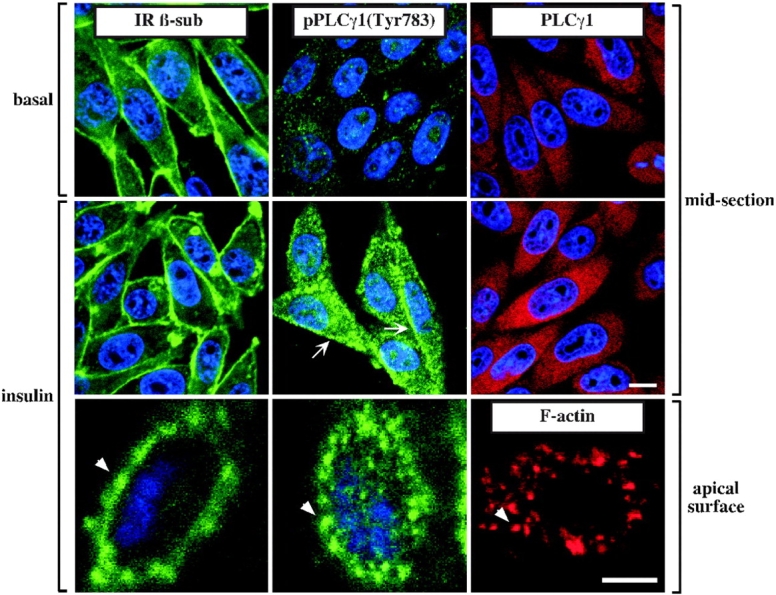
Cellular localization of IR and tyrosine-phosphorylated PLCγ1. CHO-IR cells were left untreated or were stimulated with 100 nM insulin for 10 min before fixation and permeabilization. Cells were stained with antibodies against IR β-subunit (left panels), pPLCγ1 (middle panels), and total PLCγ1 (top two right panels). Bound primary antibodies were detected with Alexa® 488–conjugated (green) or Alexa® 568–conjugated (red) secondary antibody, and DNA was stained blue by TO-PRO®-3. In some instances, cells were stained only for F-actin using Alexa® 568–conjugated phalloidin (red). Confocal sectioning in mid area (bar, 10 μm) and apical surface (bar, 5 μm) of representative cells is shown. Similar results were obtained in at least three independent experiments. Arrows indicate localization of tyrosine-phosphorylated PLCγ1 to the plasma membrane. Arrowheads indicate punctate signal coalescence at the plasma membrane.
Role of PI 3-kinase in mediating PLCγ1 recruitment to the IR
Recently, it has been shown that the generation of PI 3,4,5-trisphosphate by PI 3-kinase may serve to target PLCγ1 to the plasma membrane via its PH domain (Falasca et al., 1998). Therefore, we sought to examine the potential role of the PI 3-kinase pathway in the modulation of PLCγ1 binding to the IR. To address this issue, CHO-IR cells were pretreated with wortmannin, a pharmacological inhibitor of PI 3-kinase, followed by the addition of insulin. Blocking insulin-dependent phosphorylation of AKT on Ser 473 with wortmannin failed to inhibit PLCγ1 association with the IR (Fig. 6 A). Moreover, anti-phosphoPLCγ1 (pTyr-783) immunoprecipitates did not display a reduction in BMH-induced IR–PLCγ1 cross-linking after pretreatment of cells with wortmannin (Fig. 6 B), suggesting that PLCγ1 recruitment to the ligand-activated IR is independent of the PI 3-kinase pathway.
Figure 6.
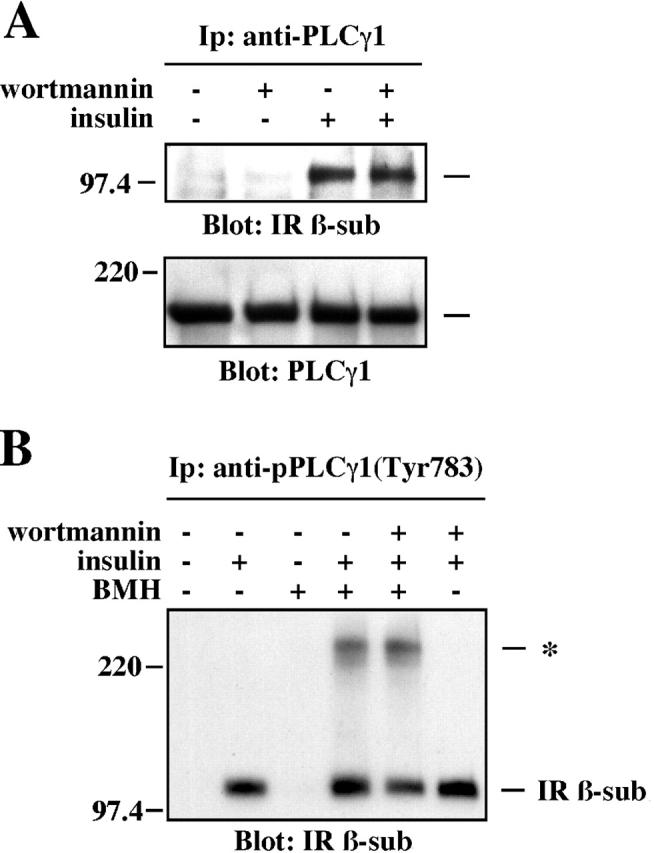
The effect of PI 3-kinase inhibition on insulin-dependent recruitment of PLCγ1 to the IR. (A) Serum-starved CHO-IR cells were pretreated with 200 μM orthovanadate for 20 min before addition of either vehicle (DMSO) or 100 nM wortmannin for 30 min and a 10-min stimulation with 100 nM insulin. Anti-PLCγ1 immunoprecipitates were blotted using anti-IR β-subunit and anti-PLCγ1PH antibodies. (B) Cells were treated as in A before cross-linking reaction with BMH. Anti-pPLCγ1 immunoprecipitates were probed for the IR β-subunit. Asterisk, IR β-subunit/PLCγ1 complex. Results shown are representative of at least two separate experiments.
SH2 domain–independent association of PLCγ1 with the IR
In addition to its catalytic subdomains, PLCγ1 has a region that contains two adjacent SH2 domains and an SH3 domain (Rhee, 2001). It has been proposed that the two SH2 domains are prerequisite for the association of PLCγ1 with activated receptors for PDGF and EGF. The R586K and R694K mutations within the rat PLCγ1 SH2 domains (N−C−) block the ability of PLCγ1 to associate with activated PDGF receptors and to become tyrosine phosphorylated (Ji et al., 1999). To test the importance of SH2 domains in mediating PLCγ1 association with the IR, HEK293 cells were transiently cotransfected with the IR or EGF receptor along with either the HA-tagged PLCγ1 wild-type or N−C− double SH2 domain mutant. After stimulation with insulin or EGF, total cell lysates were prepared and analyzed by immunoblotting. Both the wild-type and mutant PLCγ1 proteins were expressed at comparable levels (Fig. 7 A, middle panels). As anticipated, the mutant PLCγ1 protein was not tyrosine phosphorylated upon the addition of insulin or EGF, despite marked autophosphorylation of these receptors (Fig. 7 A). However, the expressed N−C− PLCγ1 mutant was coprecipitated with the IR, but not with the liganded EGF receptor (Fig. 7 B).
Figure 7.
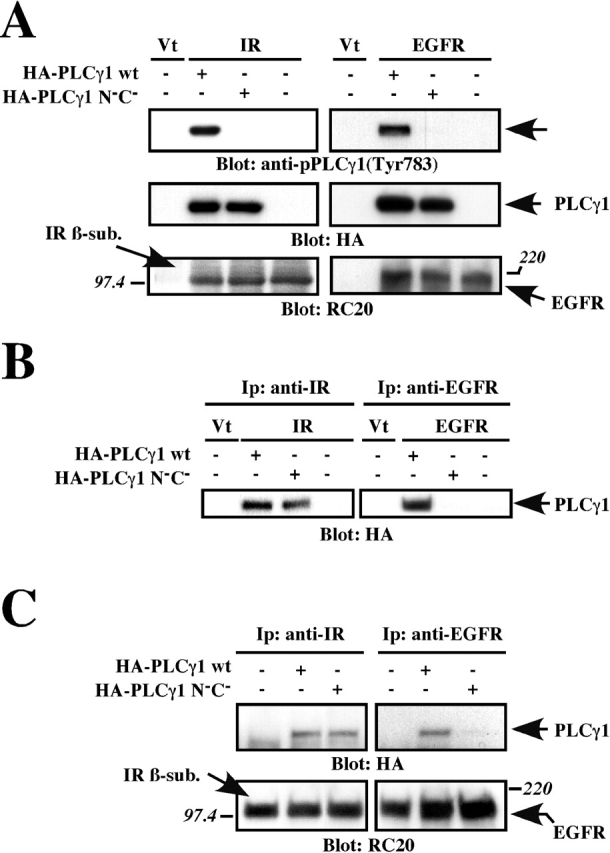
SH2 domain–independent association of PLCγ1 with the IR. (A and B) HA-tagged wild-type (wt) or N−C− mutant PLCγ1 was coexpressed with either the IR or EGF receptor in HEK293 cells. Cells were serum starved before stimulation with 100 nM insulin (left panels) or 20 nM EGF (right panels). Lysates were blotted with the indicated antibodies (A). Exogenous IR or EGF receptors were immunoprecipitated with specific antibodies and blotted with anti-HA mAb (B). Vt, empty vector. (C) CHO cells stably expressing human IR and EGF receptors were transfected with vector control and either wild-type or N−C− mutant PLCγ1. Lysates from insulin- or EGF-treated cells were immunoprecipitated as illustrated in B, and blotted with anti-HA and RC20 antibodies. Shown are blots of a representative experiment that was repeated three times with identical results.
To further test the selectivity of PLCγ1 interaction with these receptors, we transfected CHO cells stably expressing both the IR and EGF receptors (CHO-EI) with wild-type PLCγ1 or the N−C− mutant. Stimulation of CHO-EI cells in response to insulin or EGF resulted in the cosedimentation of wild-type PLCγ1 with activated IR or EGF receptors (Fig. 7 C). In contrast, the N−C− PLCγ1 mutant was recruited to the liganded IR, but not to EGF receptors (Fig. 7 C). Together, these data show the SH2 domain–independent association of PLCγ1 with the IR.
A number of IR-interacting proteins, including Gab-1 and IRS, contain a PH domain that allows their membrane association. To assess the importance of this domain in the recruitment of PLCγ1 to the IR in intact cells, various experiments were performed using HA-tagged PH–EF domain (aa 1–301) of rat PLCγ1. This construct was readily detected as a 40-kD protein upon transient transfection in HEK-293 cells and upon immunoprecipitation using anti-HA or an antibody against the PLCγ1 PH domain (Fig. 8 A). Expression of the HA-tagged PH–EF construct led to a 60 ± 11% decrease (P < 0.01; n = 4) in the ability of insulin to stimulate recruitment of cellular PLCγ1 to the activated IR (Fig. 8 B, top left). To determine if a PLCγ1 mutant lacking the PH–EF motif could also interfere with this interaction, an NH2-terminal truncation of 301 amino acids was performed to generate the ΔPH–EF PLCγ1 mutant. HEK293 cells expressing HA-tagged ΔPH–EF displayed no reduction in the binding of endogenous PLCγ1 with the IR (Fig. 8 B, top right), but markedly abrogated the PLCγ1–EGF receptor interaction (Fig. 8 C, middle right). Importantly, expression of the PH–EF construct did not block PLCγ1 association with the activated EGF receptor in HEK293 cells (Fig. 8 C, middle left) or HepG2 cells (unpublished data). Ligand-mediated phosphorylation of the EGF receptors was normal in all conditions tested (Fig. 8 C, top panels). These results are consistent with the PH–EF domain being required for PLCγ1 interaction with the IR.
Figure 8.
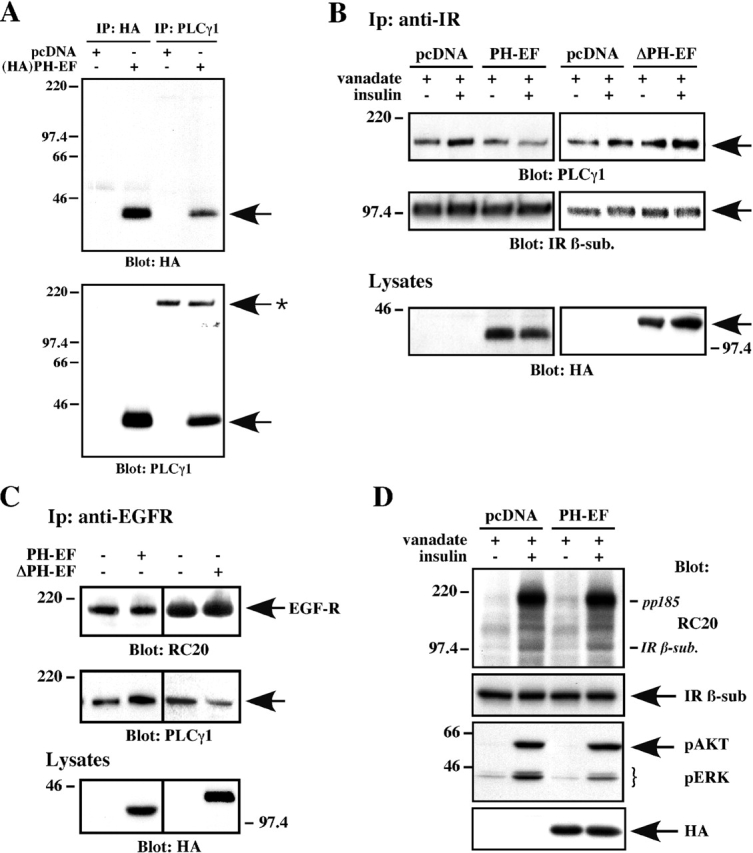
Ectopic expression of the PH–EF domain alters PLCγ1 interaction with the IR. (A) Empty vector (pcDNA) or HA-tagged PH–EF domain of PLCγ1 was expressed in HEK293 cells for 48 h. Lysates were immunoprecipitated with either anti-HA or anti-PLCγ1PH antibody and blotted as indicated. Asterisk, endogenous PLCγ1. (B) HEK293 cells transfected with either control pcDNA vector, PH–EF, or ΔPH–EF PLCγ1 mutant were treated with 200 μM orthovanadate before stimulation with 100 nM insulin for 10 min. Cosedimentation of endogenous PLCγ1 in anti-IR immunoprecipitates was detected with anti-PLCγ1PH antibody, and the membrane was then reprobed with anti-IR β subunit antibody. (C) HEK293 cells transfected with either pcDNA, PH–EF, or ΔPH–EF PLCγ1 mutant were serum starved and then stimulated with 20 nM EGF for 10 min. Endogenous EGF receptors were immunoprecipitated and then blotted with anti-RC20 and PLCγ1PH antibodies. An aliquot of total cell lysates was probed with anti-HA antibody to confirm expression of each construct (B and C). (D) Serum-starved HEK293 cells transfected with either control pcDNA vector or PH–EF were treated with 200 μM orthovanadate before stimulation with 100 nM insulin for 10 min. Cell lysates were blotted with the indicated antibodies. Shown are representative experiments that were repeated at least three times.
Overexpression of PH–EF had no effect on the stimulation of IR and IRS tyrosine phosphorylation in response to insulin (Fig. 8 D, top), and it did not inhibit insulin stimulation of AKT phosphorylation. However, the levels of p42/44 MAPK (ERK) phosphorylation elicited by insulin were reduced by ectopic expression of the PH–EF construct. (Fig. 8 D, third panel).
To further test the requirement of PLCγ1 for insulin signaling, we used PLCγ1 −/− fibroblasts reconstituted with the IR alone or together with wild-type PLCγ1. After serum withdrawal, cells were stimulated in the absence or presence of insulin, then in the phosphorylation of endogenous ERK, and AKT phosphorylation was measured in total cell lysates using phosphospecific antibodies. Insulin-stimulated ERK phosphorylation was activated to a greater extent in cells reconstituted with wild-type PLCγ1, whereas there was only an ∼20% increase in AKT phosphorylation levels by insulin (Fig. 9 A). Lastly, the role of PLCγ1 in insulin action was determined using siRNA methodology. HepG2 cells transfected with a control siRNA duplex had no reduction in PLCγ1 expression (Fig. 9 B, second panel). However, with a PLCγ1-specific siRNA duplex targeting to the 2979–2999 region of the human PLCγ1 mRNA-coding sequence, the expression of PLCγ1 was dropped to ∼30% of the levels of siRNA controls. Exposure of these cells to insulin activated the phosphorylation of IRSs and AKT to levels equivalent to those in insulin-stimulated cells transfected with control siRNA (Fig. 9 B). More significantly, incubation with PLCγ1 siRNA attenuated ERK phosphorylation elicited by insulin (Fig. 9 B, fifth panel). These results demonstrate the efficiency of the siRNA template and indicate the pathway of insulin signaling that PLCγ1 may relate to.
Figure 9.
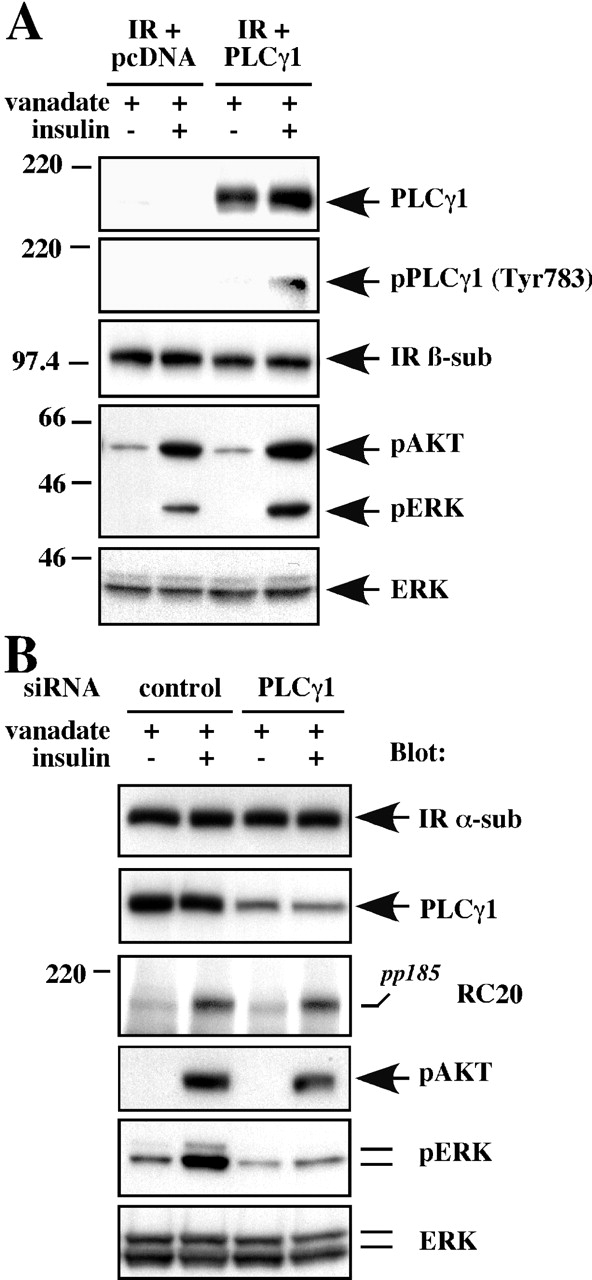
PLCγ1 is required for ERK activation by insulin. (A) Empty vector (pcDNA) or HA-tagged PLCγ1 was coexpressed with the IR in PLCγ1 −/− mouse embryonic fibroblasts for 24 h. (B) HepG2 cells were transfected with either control or PLCγ1 siRNA duplex for 48 h. In both cases, serum-starved cells were preincubated with 200 μM vanadate for 30 min before stimulation with 100 nM insulin for 10 min. Cell lysates were blotted with the indicated antibodies. Similar results were obtained in three independent experiments.
Discussion
We have identified and characterized a signaling complex between the IR and PLCγ1 in a number of cultured cell lines and in a primary culture of rat hepatocytes. The results originate from our initial efforts aimed at identifying a thiol-reactive protein that covalently associates with the IR upon cell treatment with the cross-linking agent BMH. The IR-associated protein was found to be PLCγ1 by mass spectrometry analysis, and was independently confirmed by reciprocal immunoprecipitation experiments. Insulin increases the binding of PLCγ1 to the activated IR in an SH2 domain–independent manner. Using various PLCγ1 constructs, we found that the NH2-terminal region of PLCγ1 encompassing the PH and EF-hand domain is necessary for binding the IR. Additional experiments demonstrated that PLCγ1 and its interaction with the IR play an important role in ERK activation in response to insulin.
Increase in PLCγ1-mediated PI(4,5)-bisphosphate hydrolysis has been reported in anti-IR immunoprecipitates from insulin-stimulated 3T3-L1 adipocytes (Eichhorn et al., 2001). However, whether the binding of PLCγ1 to the IR was direct or through an accessory protein remains unclear. It should be noted that c-Cbl tyrosine phosphorylation by insulin requires the adaptor protein APS, which coordinates interaction between c-Cbl and the activated IR (Liu et al., 2002). Our data show the direct interaction between PLCγ1 and the IR using cross-linking methodology in intact cells. A significant conformational change of the cytoplasmic region of the receptor β-subunit occurs as the result of IR autophosphorylation (Baron et al., 1992; Lee et al., 1997). Hence, the mechanism by which PLCγ1 is recruited to the IR in response to insulin may involve change in conformational flexibility at the interface between the two proteins, which brings the pair of reactive thiols (Cys 981 of the IR [Garant et al., 2000] and that of PLCγ1) in close proximity. The inter-thiol distance could be separated by as much as 8 Å, as the BMH analogue (BMOE) was efficient at promoting the formation of a covalent IR–PLCγ1 complex.
Our results show insulin-stimulated phosphorylation of a positive regulatory residue (Tyr-783) on PLCγ1 both in CHO-IR and HepG2 cells, as well as in HEK293 cells and PLCγ1 −/− fibroblasts transiently expressing wild-type PLCγ1. A commercially available phosphoPLCγ1 antibody (pTyr-783) was used, and the results were confirmed with anti-phosphotyrosine. By contrast, no PLCγ1 tyrosine phosphorylation was detected upon addition of insulin in 3T3-L1 adipocytes (Eichhorn et al., 2001). It has been suggested that kinases of the Src family have the ability to phosphorylate and activate PLCγ1 (Nakanishi et al., 1993). Src-related kinases are abundant in caveolin-rich raft preparations of adipocytes (Mastick and Saltiel, 1997; Müller et al., 2001) and CHO-IR cells (unpublished data), and are believed to play a role during insulin signaling (Sun et al., 1996). Because the IR appears to be incapable of directly phosphorylating PLCγ1 (Nishibe et al., 1990), it is possible that upon insulin stimulation, PLCγ1 is repositioned for phosphorylation by raft-associated Src-family kinases. PLCγ1 contains several tyrosine residues that are targets of receptor and nonreceptor tyrosine kinases and whose phosphorylation may contribute to positive or negative regulation of PLCγ1 (Kim et al., 1991; Plattner et al., 2003). However, a subset of these phosphotyrosine moieties may function as a docking site for SH2 domain–containing proteins during signal transduction (Pei et al., 1997) rather than participating directly in the regulation of PLCγ1.
PLCγ1 accumulates preferentially to cortical actin structures in EGF-stimulated A431 cells (Diakonova et al., 1995), where it binds to actin-binding proteins via its SH3 domain (Park et al., 1999). Furthermore, interaction between the COOH-terminal SH2 domain of PLCγ1 and the actin cytoskeleton has been demonstrated in an in vitro binding assay (Pei et al., 1996). Our data show that upon insulin stimulation, the IR and tyrosine-phosphorylated PLCγ1 colocalize with the actin clusters that ringed the plasma membrane. These results are consistent with the important role played by PLCγ1 in cytoskeletal reorganization and membrane ruffling after cell activation (Yu et al., 1998). Similarly, PI 3-kinase is linked to cytoskeletal reorganization (Vanhaesebroeck et al., 2001) and for full activation of PLCγ1 in some models (Rhee, 2001). Inhibition of PI 3-kinase activity by wortmannin has provided an opportunity to assess the mechanism of PLCγ1 binding to membrane-associated IR in response to insulin. We found that the insulin-stimulated formation of PI (3,4,5)-trisphosphate does not act as a targeting signal for PLCγ1 interaction with the IR.
A principal conclusion of this report is that SH2 domains have little role, if any, in promoting PLCγ1 recruitment to the IR. In contrast, disabling both SH2 domains was found to prevent the N−C− PLCγ1 mutant to associate with ligand-activated receptors for PDGF (Ji et al., 1999) and EGF (this paper). In this regard, Grb14 has been proposed to interact with the IR in an SH2-independent manner, with the BPS domain being the main interacting region (Kasus-Jacobi et al., 2000). It is noteworthy that the binding of the N−C− PLCγ1 mutant to the IR occurs even though the mutant is not phosphorylated at Tyr-783 in response to insulin, indicating that efficient PLCγ1 association with the IR may not require this phosphorylation event. We established that the NH2-terminal region of PLCγ1 encompassing the PH–EF domain is able to bind to the IR, as is the full-length protein, thereby selectively blocking recruitment of endogenous PLCγ1 to the activated IR, but not EGF receptors. Importantly, our data show that a truncated PLCγ1 mutant lacking the PH–EF region fails to bind to the IR, which is consistent with the notion that the PH–EF-hand domain is necessary for PLCγ1 association with the IR. Mutations in the PH domain of PLCγ1 did not affect recruitment of PLCγ1 to the EGF receptor (Matsuda et al., 2001). It is now believed that PH domains can interact specifically with a subset of signaling molecules rather than exerting promiscuous effects. For example, the IRS-1 PH domain has recently been shown to bind to a protein ligand referred to as PHIP (Farhang-Fallah et al., 2000), and interaction of F-actin with proteins that contain PH domains directs them to sites of cytoskeletal rearrangement at the plasma membrane (Yao et al., 1999). On the other hand, the β-adrenergic receptor kinase PH domain must bind to heterotrimeric G-protein βγ subunits and with PI (4,5)-bisphosphate to promote effective membrane targeting (Pitcher et al., 1995). The importance that EF-hand alone has in modulating IR–PLCγ1 association will be the subject of future investigations.
Our findings suggest that PH–EF overexpression may exert selective effects in insulin action through alteration in PLCγ1 signaling. Expression of PH–EF has been found to inhibit endogenous PLCγ1 association with the IR with concomitant reduction in ERK (but not AKT) phosphorylation in response to insulin. Similarly, increase in ERK phosphorylation by insulin was markedly reduced after blocking PLCγ1 expression in HepG2 cells using siRNA methodology. Additionally, reconstitution of PLCγ1 in PLCγ1 −/− fibroblasts significantly elevates the ability of insulin to promote ERK activation. PLCγ1 has been implicated in the regulation of MAPK activation in some systems (Zhang et al., 2000; Jacob et al., 2002). Together, our results support the hypothesis that PLCγ1 association with the IR is necessary for ERK regulation in response to insulin. This may be of physiological significance, as the unique structure of PLCγ1 with its PH, SH2, and SH3 domains may allow scaffolding of effector proteins harboring phosphotyrosine residues or proline-rich domains near the activated IR. The SH3 domain of PLCγ1 has been shown to be involved in SOS-mediated Ras activation (Kim et al., 2000) and to interact with c-Cbl (Tvorogov and Carpenter, 2002). The finding that the activated hybrid receptor encompassing the tyrosine kinase domain of the IR requires PLCγ1 for efficient calcium mobilization is potentially important (Telting et al., 1999). On the other hand, a PLCγ1 mutant lacking the lipase activity can induce DNA synthesis (Smith et al., 1994), indicating that the products of PLCγ1 activation and its associated mobilization of intracellular calcium may not be required for all aspects of PLCγ1 signaling. In view of the fact that PLCγ1 can fulfill functions that are not necessarily dependent on its enzymatic activity, this raises the possibility of a unique activation mechanism whereby PLCγ1 acts as an adaptor protein. To what extent the findings reported here relate to the role of PLCγ1 in insulin action remains to be elucidated.
Materials and methods
Materials
The anti–human IR mAbs for immunoprecipitation (clones 29B4 and CII 25.3) were purchased from Calbiochem. The anti-IR β-subunit antibody as well as HRP-linked phosphotyrosine (clone RC20) antibody for Western blot were purchased from Transduction Laboratories. The phospho-p42/44 MAPK and phospho-AKT antibodies were purchased from Cell Signaling Technology. The anti-phosphoPLCγ1(Tyr-783) antibody for immunoprecipitation and immunofluorescence experiments (sc-12943R), PLCγ1 SH2/SH3 domain fusion protein (residues 530–850), and anti-IR α-subunit antibody were purchased from Santa Cruz Biotechnology, Inc. The anti-phosphoPLCγ1(Tyr-783) antibody for Western blot was purchased from Biosource International. The anti-PLCγ1PH mAb (generated against a 19-aa sequence within the PH domain) was purchased from CHEMICON International, and a mixture of anti-PLCγ1 mAbs (05–163) was obtained from Upstate Biotechnology. The HA epitope antibodies were purchased from Covance. Alexa Fluor® secondary antibodies, Alexa Fluor® 568–conjugated phalloidin, and TO-PRO®-3 were purchased from Molecular Probes, Inc. FuGENE™ 6 and LipofectAMINE™ 2000 were purchased from Roche and Invitrogen, respectively. Recombinant human insulin and EGF were purchased from Calbiochem and Upstate Biotechnology, respectively. BMH, BMOE, and bismaleimidobutane were purchased from Pierce Chemical Co. Wortmannin, sodium orthovanadate, and DMSO were purchased from Sigma-Aldrich. The commercial sources for electrophoresis reagents, culture media, sera, films, HRP-linked secondary antibodies, and the ECL detection system for immunoblot detection have been described previously (Garant et al., 2000).
Plasmids and mutagenesis
The pRK5 vector containing cDNA for the HA-tagged rat PLCγ1 wild-type and the PLCγ1 SH2 domain double mutant (N−C−) were obtained from Graham Carpenter (Vanderbilt University, Nashville, TN). The plasmid encoding the human EGF receptor (pXER) was provided by Alexander Sorkin (University Colorado Health Science Center, Denver, CO). The HA-tagged PH–EF domain of rat PLCγ1 (1–301) was amplified from the pRK5/HA-PLCγ1 plasmid using PCR-based site-directed mutagenesis with primers to introduce a HindIII site between EF-hand and catalytic domain “X” of PLCγ1. A 2,961-bp HindIII–HindIII fragment was excised, and the linearized pRK5/HA-tagged PH–EF plasmid was then self-ligated. An HA-tagged truncated PLCγ1 mutant lacking the PH–EF domain (ΔPH–EF) was created using PCR-based site-directed mutagenesis with primers to introduce EcoRI sites both at the junction between HA epitope and PH domain and between EF-hand and catalytic domain “X”. A 903-bp EcoRI–EcoRI fragment was excised and the linearized pRK5/HA-tagged ΔPH–EF plasmid was then self-ligated. The constructs were verified by DNA sequence analysis.
Cell culture and metabolic labeling
CHO cells stably expressing wild-type human IR or both the IR and EGF receptors (CHO-EI cells) have been described previously (Kole et al., 1996). HEK293 and liver-derived HepG2 cells were purchased from American Type Culture Collection (Manassas, VA), and PLCγ1 −/− mouse embryonic fibroblasts were gifts from Dr. G. Carpenter (Ji et al., 1998). All CHO cell lines were expanded and maintained in Ham's F12 supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, whereas HepG2 and HEK293 cells were maintained in DME and McCoy's 5A medium containing 10% FBS and antibiotics. Cells were incubated in a humidified atmosphere of 5% CO2 at 37°C.
For metabolic labeling experiments, confluent monolayers of CHO-IR were incubated for 16 h with 60 μCi/ml Trans 35S-label (ICN Biomedicals) in methionine- and cysteine-free RPMI 1640 medium containing 3% FCS. After a series of PBS washes, cells were serum starved for 3 h and were then subjected to treatments as described below.
Isolation and culture of rat hepatocytes
Hepatocytes were isolated from 5-mo-old male Fischer 344 rats by the collagenase perfusion method of Seglen (Ikeyama et al., 2002). The isolated cells were seeded onto Biocoat Collagen I cellware (BD Discovery Labware) in William's E medium supplemented with 5% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin for 2 h in 5% CO2 at 37°C to allow attachment to the dishes. The medium was then replaced with serum-free William's E medium plus the above supplements, and cells were cultured for an additional 16 h before treatment. This procedure results in <5% contamination with nonhepatocyte cells.
Transient transfection assays
HEK293 cells were cultured for 24 h until 60–80% confluence was reached. Transient transfection was performed according to the manufacturer's protocol for the use of FuGENE™ 6. In brief, empty expression vector (pcDNA3.1) and expression plasmids encoding HA-tagged PLCγ1 [wild type or N−C−] together with recombinant human IR or human EGF receptor were mixed with the transfection reagent and directly added into the culture plates at a ratio of 1.5 μg of each plasmid per 60-mm dish. Both CHO-EI cells and PLCγ1 −/− mouse embryonic fibroblasts were transfected using LipofectAMINE™ 2000 according to the manufacturer's protocol. 24 h after transfection, cells were serum starved for 18 h and then subjected to a 30-min treatment with 200 μM orthovanadate followed by stimulation with 100 nM insulin or 20 nM EGF for 5–10 min at 37°C. Transfection efficiency was monitored using a plasmid DNA encoding eGFP.
siRNA preparation and cell transfection
The siRNA sequence targeting human PLCγ1 (GenBank/EMBL/DDBJ accession no. NM_002660) was from position 2979–2999 relative to the start codon. This PLCγ1 sequence was reversed and used as unspecific siRNA control. 21-nt RNAs were purchased from Dharmacon in deprotected and desalted form, and the formation of siRNA duplex (annealing) was performed according to the manufacturer (Dharmacon). Subconfluent HepG2 cells were transiently transfected with siRNAs using Oligofectamine™ according to the manufacturer's protocol (Life Technologies). In brief, 100 μl Opti-MEM® I medium and 10 μl Oligofectamine™ per 60-mm dish were preincubated for 5 min at RT. During the time for this incubation, 100 μl Opti-MEM® I medium was mixed with 20 μl of 20 μM siRNA. The two mixtures were combined and incubated for 20 min at RT for complex formation. The entire mixture was then added to the cells in one dish resulting in a final concentration of 100 nM for the siRNAs. Cells were usually assayed 48–72 h after transfection. Specific silencing was confirmed by at least three independent experiments.
Immunofluorescence and confocal microscopy
Cells grown on coverslips were fixed in fresh 4% PFA in PBS for 10 min and permeabilized in 0.1% Triton X-100 in PBS for 10 min at RT. The cells were incubated with blocking buffer (8% BSA in PBS) for 20 min at RT, washed in PBS supplemented with 0.5% BSA and 0.05% Tween 20, and incubated with anti-IR β-subunit (1:100; 06–492, Upstate Biotechnology), phosphoPLCγ1(1:50; Tyr-783), or PLCγ1 (1:200; Upstate Biotechnology) antibody for 16 h at 4°C. After washing, cells were stained with Alexa Fluor® secondary antibody (1:1,000). For immunolocalization of F-actin, fixed cells were incubated with Alexa Fluor® 568–conjugated phalloidin. Nuclear counterstaining was performed by incubating coverslips with TO-PRO®-3 in PBS for 5 min before mounting slides with Vectashield® (Vector Laboratories). Images were acquired using an inverted confocal microscope (LSM-410; Carl Zeiss MicroImaging, Inc.) with a 63× oil-immersed objective, and processed using the MetaMorph® software (Universal Imaging Corp.). No fluorescent staining was observed when the primary antibody was omitted.
IR–TRAP cross-linking in intact cells
Serum-starved cells were washed twice in PBS, and were then incubated in Krebs Ringer phosphate buffer for 5 min at 37°C. 100 nM insulin was added for 5 min and cells were then transferred to thermoregulated aluminum cooling plates set at 6°C. The cross-linking reaction was initiated by the addition of 100 μM BMH or vehicle (DMSO) and quenched 10 min later with 4 mM l-cysteine. In some instances, cross-linking was performed in the presence of 100 μM BMOE or BMB. For wortmannin treatment, 100 nM wortmannin was added to the cells 30 min before insulin stimulation.
Immunoprecipitation and immunoblotting
Cells were lysed in immune precipitation buffer (20 mM Tris-HCl, pH 7.5, 137 mM NaCl, 1 mM orthovanadate, 100 mM NaF, 0.1% SDS, 0.5% deoxycholate, 1% Triton X-100, 0.02% sodium azide, 0.25 mM Pefabloc-SC [Boehringer], 1 mM benzamidine, 8 μg/ml aprotinin, and 2 μg/ml leupeptin) for 20 min on ice, and then centrifuged at 10,000 g for 20 min at 4°C to sediment insoluble materials. The clarified lysates were incubated with the indicated antibodies for 16 h at 4°C with rocking. Then, protein A/G-agarose (Oncogene Research Products) beads were added and the incubation was continued at 4°C for 2 h. The beads were pelleted by centrifugation and washed twice in the same buffer and twice in 50 mM Hepes, pH 7.4, and 0.1% Triton X-100 before solubilization in Laemmli sample buffer supplemented with 5% 2-mercaptoethanol. In some experiments, cells were lysed directly in Laemmli sample buffer containing 5% 2-mercaptoethanol and 1 mM orthovanadate. After heating at 70°C for 10 min, proteins were separated by SDS-PAGE and were electrotransferred onto polyvinylidene difluoride membranes. Detection of individual proteins was performed by immunoblotting with specific primary antibodies and visualized by ECL. Signals were quantitated by densitometry coupled with the ImageQuant software (Molecular Dynamics). Where indicated, membranes from 35S-labeling experiments were dried and autoradiography was performed.
Purification of the IR–TRAP complex
10 × 150-mm dishes of CHO-IR cells were incubated with 100 nM insulin for 5 min and were then subjected to cross-linking reaction with BMH as shown above. After immunoprecipitation of the cell lysates with anti-IR antibodies prebound to protein G-agarose, the immune pellets were washed extensively and then incubated with 1 ml 1.5× Laemmli sample buffer without 2-mercaptoethanol for 60 min at RT. The eluted proteins were then concentrated down to 50 μl using an Ultrafree® centrifugal filter (molecular weight cut-off of 100 kD, Millipore). The concentrated material was incubated with 2-mercaptoethanol (7.5% final concentration) for 10 min at 70°C, and was then resolved by SDS-PAGE.
TRAP identification by MALDI mass spectrometry
Colloidal blue–stained bands were cut out of the gels for in-gel digestion as follows. The gel pieces were equilibrated for 20 min in 200 μl 25 mM ammonium bicarbonate, 50% acetonitrile. The supernatant was decanted and the same procedure was repeated until full decoloration of the gel. The gel pieces were dried, rehydrated for digestion with 5 μg/ml porcine trypsin (Roche) in 25 mM ammonium bicarbonate, and incubated at 37°C overnight. The reaction was stopped by adding 1 vol of 50% acetonitrile, 0.5% trifluoroacetic acid. The peptides were extracted from the gel matrix by sonication for 0.5–1 h. Peptide mass fingerprinting was performed using a mass spectrometer (Voyager-DE STR; PerkinElmer) operating in delayed reflector mode at an accelerating voltage of 20 kV. The peptide samples were cocrystallized with matrix on a gold-coated sample plate using 1 μl matrix (α-cyano-4-hydroxy-transcinnamic acid) and 1 μl sample. After internal calibration with protein standards (renin, angiotensin, and adrenocorticotropic hormone), the monoisotope peptide masses were assigned and then used in database searches with ProFound (http://prowl.rockefeller.edu/profound_bin/webProFound.exe). Cysteines were modified by acrylamide, and methionine was considered to be oxidized. One missed cleavage was allowed.
Acknowledgments
We thank Madgalena Juhaszova for her valuable assistance with confocal experiments and Gertrude Kokkonen for the preparation of rat hepatocytes. We would like to thank Graham Carpenter and Alexander Sorkin for providing us with reagents. We gratefully acknowledge the contribution of Dr. Yu Sam Kim from Proteome Tech Inc. (Seoul, Korea) for the MALDI-TOF analyses.
Abbreviations used in this paper; BMH, 1,6-bismaleimidohexane; BMOE, bismaleimidoethane; IR, insulin receptor; IRS-1, insulin receptor substrate 1; MALDI, matrix-assisted laser desorption/ionization; PH, pleckstrin homology; PI, phosphoinositide; PTP, protein tyrosine phosphatase; SH2, Src homology 2; siRNA, small interfering RNA; TRAP, thiol-reactive membrane-associated protein.
References
- Baron, V., P. Kaliman, N. Gautier, and E. van Obberghen. 1992. The insulin receptor activation process involves localized conformational changes. J. Biol. Chem. 267:23290–23294. [PubMed] [Google Scholar]
- Bernier, M., O. Nadiv, and H.K. Kole. 1995. Thiol-specific biotinylation of the insulin receptor in permeabilized cells enhances receptor function. Biochemistry. 34:8357–8364. [DOI] [PubMed] [Google Scholar]
- Diakonova, M., B. Payrastre, A.G. van Velzen, W.J. Hage, P.M. van Bergen en Henegouwen, J. Boonstra, F.F. Cremers, and B.M. Humbel. 1995. Epidermal growth factor induces rapid and transient association of phospholipase C-γ 1 with EGF-receptor and filamentous actin at membrane ruffles of A431 cells. J. Cell Sci. 108:2499–2509. [DOI] [PubMed] [Google Scholar]
- Eichhorn, J., A.G. Kayali, D.A. Austin, and N.J.G. Webster. 2001. Insulin activates phospholipase C-γ 1 via a PI-3 kinase dependent mechanism in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 282:615–620. [DOI] [PubMed] [Google Scholar]
- Eichhorn, J., A.G. Kayali, L. Resor, D.A. Austin, D.W. Rose, and N.J.G. Webster. 2002. PLC-γ 1 enzyme activity is required for insulin-induced DNA synthesis. Endocrinology. 143:655–664. [DOI] [PubMed] [Google Scholar]
- Falasca, M., S.K. Logan, V.P. Lehto, G. Baccante, M.A. Lemmon, and J. Schlessinger. 1998. Activation of phospholipase C γ by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 17:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhang-Fallah, J., X. Yin, G. Trentin, A.M. Cheng, and M. Rozakis-Adcock. 2000. Cloning and characterization of PHIP, a novel insulin receptor substrate-1 pleckstrin homology domain interacting protein. J. Biol. Chem. 275:40492–40497. [DOI] [PubMed] [Google Scholar]
- Garant, M.J., E. Maksimova, C. Montrose-Rafizadeh, W. Lee-Kwon, S. Kole, and M. Bernier. 2000. Cysteine 981 of the human insulin receptor is required for covalent cross-linking between beta-subunit and a thiol-reactive membrane-associated protein. Biochemistry. 39:7178–7187. [DOI] [PubMed] [Google Scholar]
- He, W., D.W. Rose, J.M. Olefsky, and T.A. Gustafson. 1998. Grb10 interacts differentially with the insulin receptor, insulin-like growth factor I receptor, and epidermal growth factor receptor via the Grb10 Src homology 2 (SH2) domain and a second novel domain located between the pleckstrin homology and SH2 domains. J. Biol. Chem. 273:6860–6867. [DOI] [PubMed] [Google Scholar]
- Ikeyama, S., G. Kokkonen, S. Shack, X.T. Wang, and N.J. Holbrook. 2002. Loss in oxidative stress tolerance with aging linked to reduced extracellular signal-regulated kinase and Akt kinase activities. FASEB J. 16:114–116. [DOI] [PubMed] [Google Scholar]
- Jacob, A., D. Cooney, M. Pradhan, and K.M. Coggeshall. 2002. Convergence of signaling pathways on the activation of ERK. J. Biol. Chem. 277:23420–23426. [DOI] [PubMed] [Google Scholar]
- Ji, Q.S., S. Ermini, J. Baulida, F.L. Sun, and G. Carpenter. 1998. Epidermal growth factor signaling and mitogenesis in Plcγ1 null mouse embryonic fibroblasts. Mol. Biol. Cell. 9:749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, Q.S., A. Chattopadhyay, M. Vecchi, and G. Carpenter. 1999. Physiological requirement for both SH2 domains for phospholipase C-γ 1 function and interaction with platelet-derived growth factor receptors. Mol. Cell. Biol. 19:4961–4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasus-Jacobi, A., V. Bereziat, D. Perdereau, J. Girard, and A.F. Burnol. 2000. Evidence for an interaction between the insulin receptor and Grb7. A role for two of its binding domains, PIR and SH2. Oncogene. 19:2052–2059. [DOI] [PubMed] [Google Scholar]
- Kayali, A.G., J. Eichhorn, T. Haruta, A.J. Morris, J.G. Nelson, P. Vollenweider, J.M. Olefsky, and N.J. Webster. 1998. Association of the insulin receptor with phospholipase C-γ (PLCgamma) in 3T3-L1 adipocytes suggests a role for PLC γ in metabolic signaling by insulin. J. Biol. Chem. 273:13808–13818. [DOI] [PubMed] [Google Scholar]
- Kim, H.K., J.W. Kim, A. Zilberstein, B. Margolis, J.G. Kim, J. Schlessinger, and S.G. Rhee. 1991. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-γ 1 phosphorylation on tyrosine residues 783 and 1254. Cell. 65:435-441. [DOI] [PubMed] [Google Scholar]
- Kim, M.J., J.S. Chang, S.K. Park, J.I. Hwang, S.H. Ryu, and P.G. Suh. 2000. Direct interaction of SOS1 Ras exchange protein with the SH3 domain of phospholipase C-γ 1. Biochemistry. 39:8674–8682. [DOI] [PubMed] [Google Scholar]
- Kole, H.K., A.S. Liotta, S. Kole, J. Roth, C. Montrose-Rafizadeh, and M. Bernier. 1996. A synthetic peptide derived from a COOH-terminal domain of the insulin receptor specifically enhances insulin receptor signaling. J. Biol. Chem. 271:31619–31626. [DOI] [PubMed] [Google Scholar]
- Lee, J., P.F. Pilch, S.E. Shoelson, and S.F. Scarlata. 1997. Conformational changes of the insulin receptor upon insulin binding and activation as monitored by fluorescence spectroscopy. Biochemistry. 36:2701–2708. [DOI] [PubMed] [Google Scholar]
- Li, C.H., M.L. Moule, and C.C. Yip. 1991. Insulin receptors prepared with iodoacetamide show enhanced autophosphorylation and receptor kinase activity. J. Biol. Chem. 266:7051–7057. [PubMed] [Google Scholar]
- Liu, J., A. Kimura, C.A. Baumann, and A.R. Saltiel. 2002. APS facilitates c-Cbl tyrosine phosphorylation and GLUT4 translocation in response to insulin in 3T3-L1 adipocytes. Mol. Cell. Biol. 22:3599–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastick, C.C., and A.R. Saltiel. 1997. Insulin-stimulated tyrosine phosphorylation of caveolin is specific for the differentiated adipocyte phenotype in 3T3-L1 cells. J. Biol. Chem. 272:20706–20714. [DOI] [PubMed] [Google Scholar]
- Matsuda, M., H.F. Paterson, R. Rodriguez, A.C. Fensome, M.V. Ellis, K. Swann, and M. Katan. 2001. Real time fluorescence imaging of PLC γ translocation and its interaction with the epidermal growth factor receptor. J. Cell Biol. 153:599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middlemas, D.S., J. Meisenhelder, and T. Hunter. 1994. Identification of TrkB autophosphorylation sites and evidence that phospholipase C-γ 1 is a substrate of the TrkB receptor. J. Biol. Chem. 269:5458–5466. [PubMed] [Google Scholar]
- Müller, G., C. Jung, S. Wied, S. Welte, H. Jordan, and W. Frick. 2001. Redistribution of glycolipid raft domain components induces insulin-mimetic signaling in rat adipocytes. Mol. Cell. Biol. 21:4553–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi, O., F. Shibasaki, M. Hidaka, Y. Homma, and T. Takenawa. 1993. Phospholipase C-γ 1 associates with viral and cellular src kinases. J. Biol. Chem. 268:10754–10759. [PubMed] [Google Scholar]
- Nishibe, S., M.I. Wahl, P.B. Wedegaertner, J.W. Kim, S.G. Rhee, G. Carpenter, and J.W. Kim. 1990. Selectivity of phospholipase C phosphorylation by the epidermal growth factor receptor, the insulin receptor, and their cytoplasmic domains. Proc. Natl. Acad. Sci. USA. 87:424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S.Y., E. Barron, P.G. Suh, S.H. Ryu, and E.P. Kay. 1999. FGF-2 facilitates binding of SH3 domain of PLC-γ 1 to vinculin and SH2 domains to FGF receptor in corneal endothelial cells. Mol. Vis. 5:18. [PubMed] [Google Scholar]
- Pei, Z., L. Yang, and J.R. Williamson. 1996. Phospholipase C-γ 1 binds to actin-cytoskeleton via its C-terminal SH2 domain in vitro. Biochem. Biophys. Res. Commun. 228:802–806. [DOI] [PubMed] [Google Scholar]
- Pei, Z., J.A. Maloney, L. Yang, and J.R. Williamson. 1997. A new function for phospholipase C-γ1: coupling to the adaptor protein GRB2. Arch. Biochem. Biophys. 345:103–110. [DOI] [PubMed] [Google Scholar]
- Pitcher, J.A., K. Touhara, E.S. Payne, and R.J. Lefkowitz. 1995. Pleckstrin homology domain-mediated membrane association and activation of the beta-adrenergic receptor kinase requires coordinate interaction with Gβγ subunits and lipid. J. Biol. Chem. 270:11707–11710. [DOI] [PubMed] [Google Scholar]
- Plattner, R., B.J. Irvin, S. Guo, K. Blackburn, A. Kazlauskas, R.T. Abraham, J.D. York, and A.M. Pendergast. 2003. A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-γ1. Nat. Cell Biol. 5:309–319. [DOI] [PubMed] [Google Scholar]
- Rhee, S.G. 2001. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 70:281–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocchi, S., S. Tartare-Deckert, D. Sawka-Verhelle, A. Gamha, and E. van Obberghen. 1996. Interaction of SH2-containing protein tyrosine phosphatase 2 with the insulin receptor and the insulin-like growth factor-I receptor: studies of the domains involved using the yeast two-hybrid system. Endocrinology. 137:4944–4952. [DOI] [PubMed] [Google Scholar]
- Saltiel, A.R., and J.E. Pessin. 2002. Insulin signaling pathways in time and space. Trends Cell Biol. 12:65–71. [DOI] [PubMed] [Google Scholar]
- Schmid, E., J. El Benna, D. Galter, G. Klein, and W. Droge. 1998. Redox priming of the insulin receptor β-chain associated with altered tyrosine kinase activity and insulin responsiveness in the absence of tyrosine autophosphorylation. FASEB J. 12:863–870. [DOI] [PubMed] [Google Scholar]
- Smith, M.R., Y.L. Liu, N.T. Matthews, S.G. Rhee, W.K. Sung, and H.F. Kung. 1994. Phospholipase C-γ 1 can induce DNA synthesis by a mechanism independent of its lipase activity. Proc. Natl. Acad. Sci. USA. 91:6554–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, X.J., S. Pons, T. Asano, M.G. Myers, Jr., E. Glasheen, and M.F. White. 1996. The Fyn tyrosine kinase binds Irs-1 and forms a distinct signaling complex during insulin stimulation. J. Biol. Chem. 271:10583–10587. [DOI] [PubMed] [Google Scholar]
- Telting, D., R.L. Smeets, P.H. Willems, G.C. van der Zon, W.S. Frankhuizen, and J.A. Maassen. 1999. The insulin receptor tyrosine kinase domain in a chimaeric epidermal growth factor-insulin receptor generates Ca2+ signals through the PLC-γ1 pathway. Biochim. Biophys. Acta. 1431:421–432. [DOI] [PubMed] [Google Scholar]
- Tvorogov, D., and G. Carpenter. 2002. EGF-dependent association of phospholipase C-γ 1 with c-Cbl. Exp. Cell Res. 277:86–94. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck, B., S.J. Leevers, K. Ahmadi, J. Timms, R. Katso, P.C. Driscoll, R. Woscholski, P.J. Parker, and M.D. Waterfield. 2001. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 70:535–602. [DOI] [PubMed] [Google Scholar]
- Virkamaki, A., K. Ueki, and C.R. Kahn. 1999. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J. Clin. Invest. 103:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, L., P. Janmey, L.G. Frigeri, W. Han, J. Fujita, Y. Kawakami, J.R. Apgar, and T. Kawakami. 1999. Pleckstrin homology domains interact with filamentous actin. J. Biol. Chem. 274:19752–19761. [DOI] [PubMed] [Google Scholar]
- Yu, H., K. Fukami, T. Itoh, and T. Takenawa. 1998. Phosphorylation of phospholipase C γ1 on tyrosine residue 783 by platelet-derived growth factor regulates reorganization of the cytoskeleton. Exp. Cell Res. 243:113–122. [DOI] [PubMed] [Google Scholar]
- Zhang, W., R.P. Trible, M. Zhu, S.K. Liu, C.J. McGlade, and L.E. Samelson. 2000. Association of Grb2, Gads, and phospholipase C-γ 1 with phosphorylated LAT tyrosine residues. Effect of LAT tyrosine mutations on T cell antigen receptor-mediated signaling. J. Biol. Chem. 275:23355–23361. [DOI] [PubMed] [Google Scholar]