

Abstract
Disruption of the BPAG1 (bullous pemphigoid antigen 1) gene results in progressive deterioration in motor function and devastating sensory neurodegeneration in the null mice. We have previously demonstrated that BPAG1n1 and BPAG1n3 play important roles in organizing cytoskeletal networks in vivo. Here, we characterize functions of a novel BPAG1 neuronal isoform, BPAG1n4. Results obtained from yeast two-hybrid screening, blot overlay binding assays, and coimmunoprecipitations demonstrate that BPAG1n4 interacts directly with dynactin p150Glued through its unique ezrin/radixin/moesin domain. Studies using double immunofluorescent microscopy and ultrastructural analysis reveal physiological colocalization of BPAG1n4 with dynactin/dynein. Disruption of the interaction between BPAG1n4 and dynactin results in severe defects in retrograde axonal transport. We conclude that BPAG1n4 plays an essential role in retrograde axonal transport in sensory neurons. These findings might advance our understanding of pathogenesis of axonal degeneration and neuronal death.
Keywords: cytoskeleton; BPAG1n4–dynactin interaction; axonal transport; neurodegeneration; ERM domain
Introduction
The BPAG1 (bullous pemphigoid antigen 1) null mouse has a fascinating and complex phenotype, displaying sensory ataxia and devastating sensory neurodegeneration (Guo et al., 1995). This degeneration is characterized by neurofilament disorganization, microtubule network abnormality, and intermittent accumulation of membranous organelles along sensory axons. The BPAG1 gene encodes multiple isoforms with BPAG1e expressed in the epidermis and the others (BPAG1n1, BPAG1n2, and BPAG1n3) expressed in neurons. Throughout normal development, mRNAs of BPAG1 neuronal isoforms are present in a variety of neurons. Postnatally, this expression is absent or reduced in motor neurons and restricted to sensory neurons (Brown et al., 1995; Dowling et al., 1997). It has been demonstrated that BPAG1n1 links intermediate filaments to actin microfilaments and that BPAG1n3 can stabilize microtubules (Yang et al., 1996, 1999). However, the mechanisms underlying the devastating sensory neuron degeneration in BPAG1 null mice await further characterization.
Impaired axonal transport in neurons has long been implicated as a mechanism underlying axonal degeneration and neuronal death. In neurons, motor protein of kinesin superfamily drives anterograde transport, whereas cytoplasmic dynactin/dynein powers retrograde transport. The mutations discovered in critical components of transport pathways provide evidence for the notion that axonal transport is essential for neuronal survival. Transgenic mice carrying mutant superoxide dismutase-1, mouse models of amyotrophic lateral sclerosis, show deficits in slow axonal transport early in the disease course (Williamson and Cleveland, 1999). Additionally, in some superoxide dismutase-1 mutant lines, an early up-regulation of the kinesin superfamily motor protein KIF1A was detected in spinal motor neurons (Dupuis et al., 2000). Mutations in the gene encoding KIF1Bβ cause an axonal form of a hereditary neuropathy (Zhao et al., 2001). A recent work has linked mutations in cytoplasmic dynein heavy chain with defects in retrograde transport that lead to motor neuron degeneration (Hafezparast et al., 2003). Furthermore, a mutation in the p150Glued subunit of dynactin has been identified in a family with a slowly progressive, autosomal-dominant form of lower motor neuron disease in which sensory symptoms are absent (Puls et al., 2003).
In this work, we analyze the functions of BPAG1n4, the fourth neuronal isoform of the BPAG1 gene family. BPAG1n4, referred to as BPAG1a by Leung et al. (2001), harbors a structurally and functionally unique ezrin/radixin/moesin (ERM) domain (Burridge and Mangeat, 1984; Anderson and Marchesi, 1985; Lankes and Furthmayr, 1991). We provide biochemical evidence that the ERM domain of BPAG1n4 directly interacts with dynactin, and show that this isoform physiologically colocalizes with the dynactin–dynein complex in vivo in sensory axons. Finally, we demonstrate that disruption of the interaction between p150Glued dynactin and BPAG1n4 leads to a failure of retrograde axonal transport in sensory neurons.
Results and discussion
BPAG1n4 localizes to vesicle-like structures
Although BPAG1n4 and BPAG1n1 share a conserved NH2-terminal actin-binding domain, BPAG1n4's COOH terminus harbors a distinctive functional microtubule-binding domain (Leung et al., 1999; unpublished data). This domain features two EF-hand Ca2+-binding motifs (Ikura, 1996) immediately upstream of a short stretch of GAS2 motif (EF-hand/GAS2; Schneider et al., 1988). Furthermore, a central ERM domain (Burridge and Mangeat, 1984; Anderson and Marchesi, 1985; Lankes and Furthmayr, 1991) is flanked by two functional cytoskeleton-binding domains (Yang et al., 1996; Leung et al., 1999; Fig. 1 A). Human BPAG1n4 shares ∼82% sequence identity with a recently published mouse form, BPAG1a (Leung et al., 2001). The expression of BPAG1n4 was detected at both the mRNA and protein levels in wild type (WT), but not in null tissues (Fig. 1, B and C). The specificity of anti-BPiso4 was confirmed in cells transiently expressing a FLAG epitope-tagged BPAG1n4 NH2-terminal segment (Fig. 1 C, lanes 3 and 4) that was also recognized by anti-FLAG (not depicted). Immunohistochemical studies using anti-BPiso4 antibody recapitulated our previously published staining pattern characteristic of sensory neurons using anti-BPAG1n antibody, which recognizes all isoforms (Yang et al., 1996; Dowling et al., 1997). No significant staining was found in motor neurons of postnatal animals (Yang et al., 1996).
Figure 1.
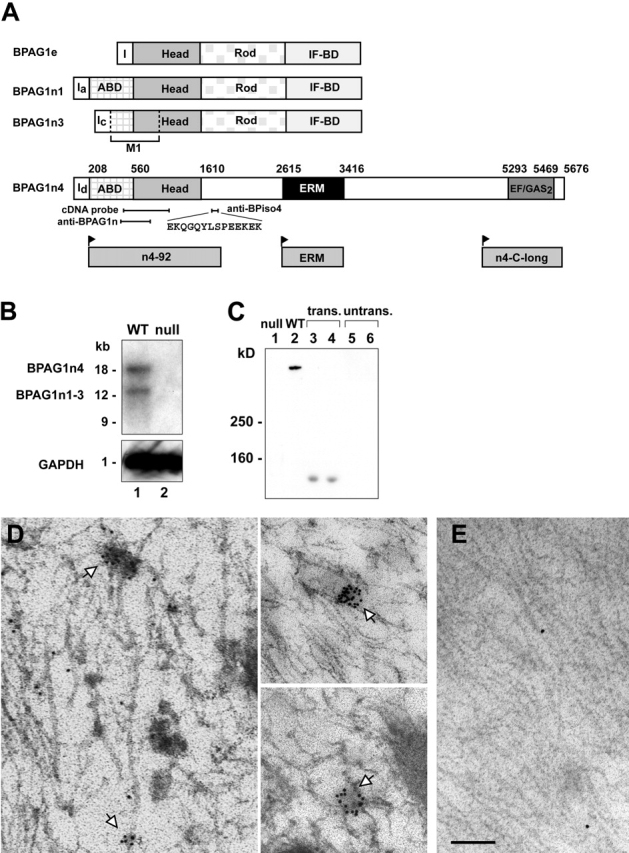
Characterization of BPAG1n4. (A) Schematic of the domain structures of BPAG1 isoforms. I, first exon of each isoform; BPAG1e, the epithelial isoform; ABD, actin-binding domain; IF-BD, intermediate filament-binding domain; M1, microtubule-binding domain. Amino acid residue numbers denote functional domain boundaries in BPAG1n4. Individual domains and regions used in this work are illustrated below. Anti-BPAG1n recognizes all BPAG1 isoforms. (B) Northern blot analysis of mouse dorsal root ganglion (DRG) RNA. (probes) A 1.6-kb cDNA head domain fragment (top; 4-d exposure); GAPDH as internal control (bottom; 1-h exposure). (C) Protein expression of BPAG1n4 in mouse brain. Blot was probed with anti-BPiso4. COS-7 (lane 3) and NIH 3T3 cells (lane 4) were transfected with an expression construct for n4-92 (A, bottom) encompassing the epitope for anti-BPiso4. (untrans.) Untransfected COS-7 (lane 5) and NIH cells (lane 6). (D–E) ImmunoEM reveals subcellular localization of BPAG1n4 in dorsal roots. (D) BPAG1n4 labeling with anti-BPiso4 was visualized with 12-nm gold particles conjugated with secondary antibody. White arrows indicate BPAG1n4 on vesicle-like structures in association with microtubules. (E) Negative control, secondary antibodies alone. Bar: (D) 300 nm; (E) 400 nm.
As a first step toward understanding the function of this new isoform, anti-BPiso4 antibody was used to determine the subcellular localization of BPAG1n4 in the spinal cord dorsal column by immunoelectron microscopy (immunoEM). Intriguingly, ∼72% of BPAG1n4-associated particles localized to vesicle-like structures associated with microtubules (Fig. 1 D, gold particles). No significant labeling was detected in negative controls using secondary antibodies only (Fig. 1 E).
BPAG1n4 interacts directly with dynactin p150Glued
ERM domain containing proteins are known to modify the interaction of cytoskeletal and integral membrane proteins (Burridge and Mangeat, 1984; Anderson and Marchesi, 1985; Lankes and Furthmayr, 1991). We used the ERM domain of BPAG1n4 as bait in a yeast two-hybrid screen (Ding et al., 2002) of a human brain cDNA library. 28 putative positive clones exclusively identified the COOH-terminal region of dynactin p150Glued (Gill et al., 1991) as the binding partner for BPAG1n4. Consistent with our two-hybrid results, HA-tagged ERM could be coimmunoprecipitated by FLAG-tagged p150Glued when coexpressed in cells (Fig. 2 C, lane 4). Additional assays using tissue extracts corroborated the interaction of BPAG1n4 and p150Glued, including in vitro blot overlay (Fig. 2, A and B) and coimmunoprecipitations (co-IPs; Fig. 2, D–F). In brief, by in vitro blot overlay binding assay, p150Glued bound only to the immobilized HA-tagged ERM domain (Fig. 2 A, lane 4) or to BPAG1n4 (Fig. 2 B, lane 3), but not to control proteins (Fig. 2 A, lanes 5 and 6; and Fig. 2 B, lanes 4, 6, and 7). In co-IP experiments from tissues, BPAG1n4 was specifically detected in the complex coimmunoprecipitated by anti-p150Glued (Fig. 2 D, lane 3). Conversely, p150Glued and p50, another dynactin subunit, as well as DIC but not a control protein, were specifically coimmunoprecipitated by anti-BPiso4 (Fig. 2 E, lane 2 and 5, respectively). Neither the secondary alone nor a sham antibody pulled down BPAG1n4 (Fig. 2 D, lanes 5 and 6) or dynactin (Fig. 2 E, lane 4 and 6). Neuronal isoforms BPAG1n1–3 were detected in total lysates (Fig. 2 F, lane 1) using anti-BProd antibody, which recognizes the domain common to these isoforms, but not in complexes precipitated by either anti-p150Glued or anti-BPiso4 (Fig. 2 D, lanes 2 and 3). Together, our results provide strong evidence that BPAG1n4 associates with dynactin p150Glued via its ERM domain.
Figure 2.
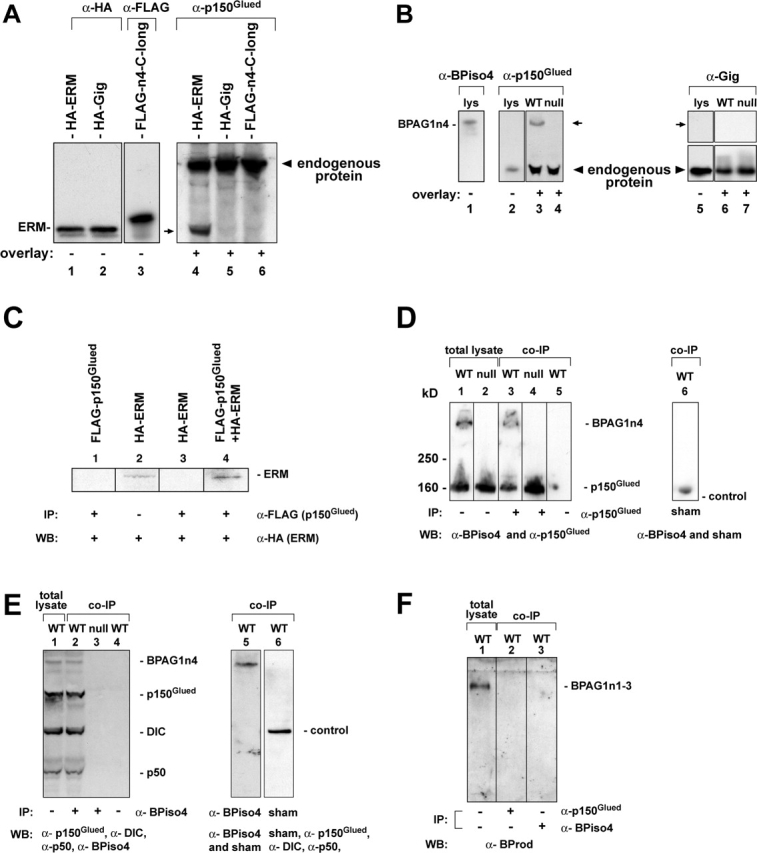
Blot overlay and co-IP assays show BPAG1n4–dynactin interaction. (A) Blot overlay assays with ERM domain. 85-kD epitope-tagged ERM (HA-ERM), 78-kD gigaxonin (HA-Gig), or 90-kD BPAG1n4 COOH terminus (FLAG-n4-C-long) from transfected CHO cells were immobilized on membranes. Membranes were incubated with (+) or without (−) total lysates of mouse spinal cord and sciatic nerve and probed with anti-HA (HA.11; CRP, Inc.), anti-FLAG (Sigma-Aldrich), and anti-p150Glued, respectively. In addition to its endogenous band (arrowhead), p150Glued was also detected at the position of HA-ERM (lane 4, arrow) as a result of its binding to the ERM domain of BPAG1n4. (B) Blot overlay assays with BPAG1n4. BPAG1n4 was immobilized on membrane and overlaid with (+) or without (−) total lysates of spinal cord and sciatic nerve. Blots were probed with anti-p150Glued and antigigaxonin (Gig, control). (lys) total brain lysates. p150Glued (lane 3), but not gigaxonin (lane 6), binds to full-length BPAG1n4 (arrows). (C) Co-IP of ERM domain and p150Glued in COS-7 cells. (WB) Western blotting; (IP) immunoprecipitation. Cell lysates of COS-7 transfected with pHA-ERM (lanes 2 and 3), or pFLAG-p150Glued (lane 1), or both (lane 4) were incubated with anti-FLAG M2 affinity gel. Protein samples were analyzed by immunoblotting with anti-HA antibody. (D–E) co-IP of BPAG1n4 and the dynactin–dynein complex from mouse nerve tissue extracts. A rabbit anti–p150Glued (Santa Cruz Biotechnology, Inc.) antibody, but not the sham antibody, was able to coimmunoprecipitate BPAG1n4 (D, lane 3). Conversely, anti-BPiso4 but not the sham antibody was able to coimmunoprecipitate the dynactin complex (E, lane 2). Antibodies against BPAG1n4, p150Glued, p50, DIC, and control protein (sham) were used for immunoblotting. (F) Neither anti-p150Glued nor anti-BPiso4 could coimmunoprecipitate other BPAG1 neuronal isoforms (BPAG1n1–3). Immunoblot was probed with rabbit antibody against the common rod domain shared by BPAG1n1–3 (anti-BProd).
The dynactin complex is required for functional cytoplasmic dynein (Waterman-Storer et al., 1997; Holleran et al., 1998; King and Schroer, 2000; Deacon et al., 2003), a motor protein that powers retrograde transport. co-IP results suggested that, through interaction with p150Glued, BPAG1n4 might indirectly associate with dynein in vivo. We performed double immunofluorescence staining on cultured dorsal root ganglion (DRG) neurons and observed striking colocalizations of BPAG1n4 with p150Glued (Fig. 3, A–C) and with DIC (Fig. 3 D) in neurites. Double immunoEM labeling on mouse sciatic nerve provided additional in vivo evidence for colocalization of BPAG1n4 and dynein. Approximately 71% of the BPAG1n4-associated gold particles colocalized with 44% of the dynein-associated particles (Fig. 3 E).
Figure 3.
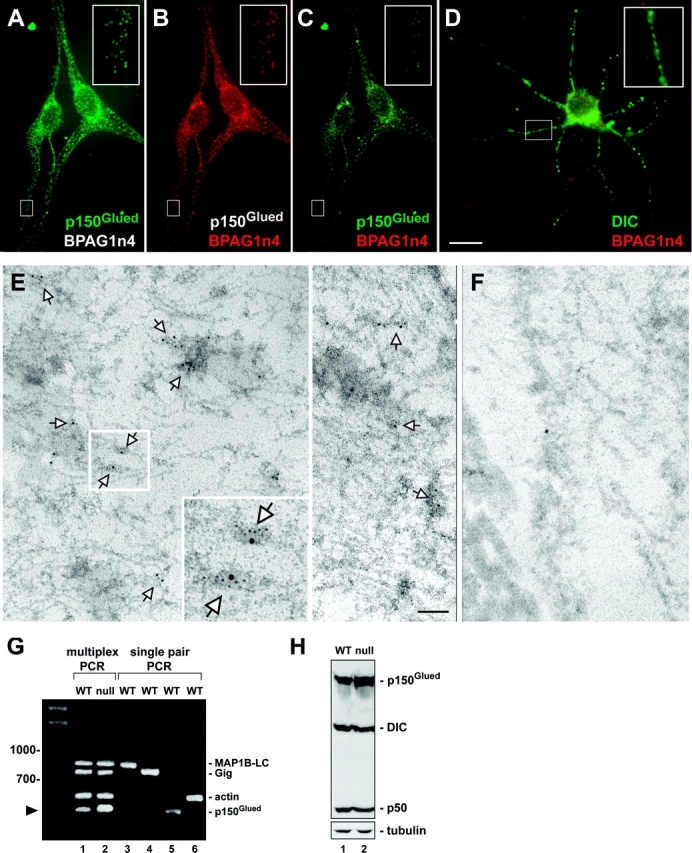
Colocalization of BPAG1n4 with dynactin/dynein and up-regulation of p150 Glued in BPAG1 null mouse. (A–D) Double immunofluorescence staining of cultured DRG neurons shows colocalization of BPAG1n4 with dynactin/dynein. Cells were stained with anti-BPiso4/anti-p150Glued (BD Biosciences) in A–C and anti-BPiso4 /anti-DIC (Chemicon) in D. AlexaFluor-conjugated secondary antibodies (Molecular Probes) were used for detection. Insets show colocalizations at higher magnification. (E) Double ImmunoEM labeling of mouse sciatic nerve sections with anti-BPiso4 (18 nm) and anti-DIC (6 nm). Arrows indicate colocalization of BPAG1n4 and dynein. (F) Negative control with secondary antibodies only. (G) RT-PCR analysis of DRG mRNAs. Multiplex gene-specific primers (lanes 1 and 2) were used on cDNAs from WT and null DRG. Lanes 3–6 indicate each individually amplified primer pair product from WT: p150Glued (410 bp), microtubule associated protein 1B light chain (MAP1B-LC; 850 bp), gigaxonin (Gig, 780 bp), and actin (560 bp). Arrowhead indicates up-regulation of p150Glued in BPAG1 null (lane 2) versus WT (lane 1). (H) Protein analysis on mouse spinal cord and sciatic nerve lysates using antibodies to p150Glued, DIC, and p50 (BD Biosciences). Antitubulin served as loading control. Bars: (A–D) 10 μm; (E and F) 400 nm.
Intriguingly, quantitative multiplex RT-PCR analysis on DRG extracts from WT and BPAG1 null mice revealed a specific up-regulation of p150Glued mRNA expression in null mice (Fig. 3 G, lane 2). Actin, gigaxonin (Bomont et al., 2000; Ding et al., 2002), and MAP1B-LC were used as controls and showed no corresponding up-regulation (Fig. 3 G, lane 2). Similarly, analysis of protein extracts from spinal cord and sciatic nerves revealed a substantial increase of p150Glued expression (but not of p50 or DIC) in BPAG1 null mice (Fig. 3 H). Specific up-regulation of p150Glued expression in the null mice might represent a compensatory response for loss of BPAG1n4 as an interaction partner.
BPAG1n4 is required for retrograde axonal transport in sensory neurons
At the ultrastructural level, vesicles, multivesicular bodies, mitochondria, and other membranous organelles were found to accumulate in BPAG1 null sensory axons, a phenotype characteristic of impaired axonal transport (Fig. 4 A). In contrast, WT axons displayed no such accumulations (Fig. 4 B). We analyzed axonal transport by double-ligation of sciatic nerves (Fig. 4 C), monitoring a sensory neuron protein, p75NTR (Raivich et al., 1991), for any subsequent accumulation. Immunofluorescence staining revealed that in BPAG1 null mice p75NTR failed to accumulate at distal sites 6 h after ligation (Fig. 4 D, bottom, distal), but exhibited proximal accumulations (Fig. 4 D, bottom, proximal) surprisingly similar to those observed in WT (Fig. 4 D, top). Quantitative biochemical assays were conducted on ∼3-mm segments proximal and distal to the double ligatures at 3- and 6-h time points. In WT mice, a substantial accumulation of p75NTR was observed bidirectionally at both time points (Fig. 4 E, lanes 1 and 5, and lanes 3 and 7, respectively). In contrast, in the null mice, the accumulation of p75NTR was barely detectable at both directions 3 h after ligation (Fig. 4 E, lanes 2 and 4). Interestingly, at the 6-h time point the retrograde accumulation remained undetectable in the null mice (Fig. 4 E, lane 8), but at anterograde direction the difference appears less marked (Fig. 4 E, lane 6). These results suggest that, whereas the axonal transport in BPAG1 null mice is bidirectionally impaired, the retrograde direction is apparently more severely affected.
Figure 4.

Retrograde axonal transport is disrupted in BPAG1 null mice. (A and B) EM of longitudinal sections of spinal cord dorsal column reveals accumulation of vesicles and other organelles in BPAG1 null (A), but not in WT (B). (C–E) Proteins failed to accumulate on the distal side of a double ligature (schematic shown in C) of BPAG1 null sciatic nerves. Shown are confocal images of nerve sections stained with anti-p75NTR (D), and immunoblots of p75NTR from ∼3-mm sections proximal or distal to double ligatures (E). Anti–α-tubulin served as loading control. Bar: (A and B) 500 nm; (D) 140 μm.
The gene targeting strategy in BPAG1 null mice ablated all isoforms (Guo et al., 1995), all of which could collectively play important roles bidirectionally in axonal transport. However, the specific interaction of BPAG1n4 and dynactin/dynein in vivo pointed to a potential role for the BPAG1n4 isoform in retrograde axonal transport. To test this hypothesis, we overexpressed an ERM-GFP fusion protein in cultured mouse DRG neurons. ERM-GFP and GFP proteins were efficiently transported to the neurite tips in transfected neurons (Fig. 5 A–B). To examine retrograde transport, the neurons were incubated for 2 h with media containing the tracer transferrin conjugated with Texas red (Tf-TR). The media were replaced with tracer-free media, and retrograde transport of Tf-TR was subsequently monitored at 2-h time points. In the first 2 h after removing Tf-TR, no significant differences were observed in the cultured neurons (Fig. 5, E–H). However, after 4–6 h, the WT and GFP-expressing neurons had begun to accumulate Tf-TR in their cell bodies (Fig. 5, K and J, respectively). After 12–14 h, ∼94% of these neurons had completed retrograde transport of the Tf-TR pulse (Fig. 5, N and O). In contrast, in three independent experiments, 82% of the ERM-GFP–overexpressing neurons showed severe defects identical to those seen in 91% of BPAG1 null neurons, namely, a nearly complete failure to transport Tf-TR to the cell bodies (PFig. 5, I and M, and L and P, respectively). Together, these results demonstrate that the isolated ERM is sufficient to disrupt BPAG1n4's function in sensory neurons in a dominant-negative fashion, leading to a failure of retrograde axonal transport that could account for the impaired retrograde transport found in the BPAG1 null mouse. We conclude that BPAG1n4 is essential for retrograde axonal transport in sensory neurons. Because the postnatal expression of BPAG1 neuronal isoforms is restricted to sensory neurons (Yang et al., 1996), it is likely that a related molecule expressed in motor neurons, such as ACF7 (Byers et al., 1995; Karakesisoglou et al., 2000), may play a corresponding role in axonal transport in those cells. Alternatively, motor neurons may rely on different mechanisms for such a process.
Figure 5.

Disruption of retrograde transport is BPAG1n4 isoform specific. Mouse DRG neurons were transfected with pEGFP-ERM-N1 or pEGFP-N1. Retrograde transport of transferrin conjugated with Texas red (Tf-TR) was examined as described in Materials and methods. Cells expressing ERM-GFP (A) or GFP (B) were first incubated with Tf-TR for 2 h. After removal of the Tf-TR, the cells were observed and pictured every 2 h. Shown are the cells after 2 h (E–H), 6 h (I–L), and 12 h (M–P). Untransfected DRG neurons of WT (C, G, K, and O) and BPAG1 null mice (D, H, L, and P) served as controls. The ERM-GFP–expressing neurons mimicked BPAG1 null neurons in their failure to transport Tf-TR retrogradely into the cell bodies. Bar, 23 μm.
Defects in axonal transport have been implicated in many human neurodegenerative disorders (Williamson and Cleveland, 1999; Hafezparast et al., 2003; Puls et al., 2003). The extraordinary length of axons and unique features of neurons may require a more complex system to meet the unusual transport challenges that are far beyond those of nonneuronal cells. Our findings pertaining to BPAG1n4 may contribute to a better understanding of relations between neurodegeneration and axonal transport. The next challenge will be to understand how the interactions of BPAG1n4 with dynactin/dynein and cytoskeletons facilitate retrograde axonal transport in sensory neurons.
Materials and methods
Molecular cloning of BPAG1n4
Complementary DNAs of BPAG1n4 were amplified by rapid amplification of cDNA ends–PCR in human brain Marathon-Ready cDNA (CLONTECH Laboratories, Inc.). Sequencing information from three overlapping clones of long-range PCR products (A, 8.1 kb; B, 7.2 kb; and C, 7.8 kb) confirmed that we obtained the full-length cDNA sequence of BPAG1n4.
Blot overlay binding assays and co-IPs
Nerve tissues (spinal cord and sciatic nerves) were homogenized in PBS with protease inhibitors (Roche), and then briefly centrifuged at 2,300 g for 30 s to remove tissue debris and any particulate materials. The supernatants were divided into two portions, one for further centrifugation at 109,000 g to collect pellets, and two for directly serving as total lysate for overlay and immunoprecipitations. Pellets were resolved on 5% SDS-PAGE and transferred on polyvinylidene fluoride membranes to immobilize the full-length BPAG1n4. The blots were incubated with the total lysates overnight, followed by immunoblot analyses using the indicated antibodies. For co-IP, the rabbit anti-dynactin (H-300; Santa Cruz Biotechnology, Inc.) was used to coimmunoprecipitate BPAG1n4, and anti-BPiso4 was used to coimmunoprecipitate p150Glued. The lysates were incubated with different antibodies and protein A–Sepharose 4B beads (Zymed Laboratories) at 4°C overnight. Beads were washed a few times with PBS. Bound proteins were eluted with SDS sample buffer. Protein samples were resolved through 4–15% gradient SDS-PAGE (Bio-Rad Laboratories) and analyzed by immunoblotting.
ImmunoEM
WT animals were killed by intravenous perfusion with 2% PFA and 0.05% glutaraldehyde. The dissected samples of dorsal roots and sciatic nerves were postembedded as described previously (Yang et al., 1999). The antibody incorporations on ultrathin sections were visualized with 12 nm anti–rabbit for single labeling or 18 nm anti–rabbit and 6 nm anti–mouse gold-conjugated particles (Jackson ImmunoResearch Laboratories). After staining with uranyl acetate, followed by lead citrate, the sections were analyzed under an electron microscope (model CM10; Philips).
Double ligation and immunostaining of sciatic nerves
WT control and BPAG1 null mice (13–17 postnatal days) were anesthetized with a mixture of xylamine/ketamine. On the right sciatic nerve of each mouse, two ligatures 5 mm apart were placed at mid-thigh. For immunostaining, 6 h after ligature, mice were perfused with 10 ml of 0.1 M phosphate buffer, pH 7.4, and 40 ml of fixation solution (4% PFA in PB). Sciatic nerves were postfixed for 2 h and placed overnight in a cryoprotective solution (PB with 15% sucrose). After cryoprotection, sciatic nerves were embedded in OTC and frozen at −80°C. 8–10-μm sections were cut in a cryostat at −20°C, mounted on glass slides, and stained with anti-p75NTR (Promega), followed by fluorescent dye-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). Samples were analyzed and images were captured using a confocal microscope (model Radiance200; Bio-Rad Laboratories).
Primary sensory neuron transfection and transferrin transport
DRG neurons from newborn WT mice were transfected with pEGFP-ERM-N1 or the control construct pEGFP-N1. Transfection was performed using the Mouse Neuron Nucleofector™ kit and the Nucleofector™ device (Amaxa). Transfection rates were ∼20%. Cells were cultured in Neurobasal™ complete medium (Invitrogen) on collagen-coated glass coverslips. For the transferrin transport assay, cells were incubated with medium containing 50 μg/ml of human transferrin conjugated with Texas red (Tf-TR; Molecular Probes) for 2 h at 37°C to allow for uptake. After removal of the transferrin-containing medium, coverslips were rinsed with PBS and incubated with Tf-TR–free medium at 37°C. Confocal fluorescent images were taken every 2 h with a confocal laser scanning microscope (model LSM 510; Carl Zeiss MicroImaging, Inc.) to monitor the transport of Tf-TR.
Acknowledgments
J.-J. Liu is supported by Dean's, McCormick, and Berry fellowships from Stanford Medical School. This work was supported by Basil O'Connor Starter Scholar Research Award (March of Dimes) and National Institutes of Health grants NS42791 and NS43281 to Y. Yang; AR27883 to E.V. Fuchs; and NS24054, NS38869, and AG16999 to W. Mobley. E.V. Fuchs is an investigator of Howard Hughes Medical Institute.
Abbreviations used in this paper: BPAG1, bullous pemphigoid antigen 1; co-IP, coimmunoprecipitation; DRG, dorsal root ganglion; ERM, ezrin/radixin/moesin; immunoEM, immunoelectron microscopy; Tf-TR, transferrin conjugated with Texas red; WT, wild type.
References
- Anderson, R.A., and V.T. Marchesi. 1985. Regulation of the association of membrane skeletal protein 4.1 with glycophorin by a polyphosphoinositide. Nature. 318:295–298. [DOI] [PubMed] [Google Scholar]
- Bomont, P., L. Cavalier, F. Blondeau, C. Ben Hamida, S. Belal, M. Tazir, E. Demir, H. Topaloglu, R. Korinthenberg, B. Tuysuz, et al. 2000. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat. Genet. 26:370–374. [DOI] [PubMed] [Google Scholar]
- Brown, A., G. Dalpe, M. Mathieu, and R. Kothary. 1995. Cloning and characterization of the neural isoforms of human dystonin. Genomics. 29:777–780. [DOI] [PubMed] [Google Scholar]
- Burridge, K., and P. Mangeat. 1984. An interaction between vinculin and talin. Nature. 308:744–746. [DOI] [PubMed] [Google Scholar]
- Byers, T.J., A.H. Beggs, E.M. McNally, and L.M. Kunkel. 1995. Novel actin crosslinker superfamily member identified by a two step degenerate PCR procedure. FEBS Lett. 368:500–504. [DOI] [PubMed] [Google Scholar]
- Deacon, S.W., A.S. Serpinskaya, P.S. Vaughan, M. Lopez Fanarraga, I. Vernos, K.T. Vaughan, and V.I. Gelfand. 2003. Dynactin is required for bidirectional organelle transport. J. Cell Biol. 160:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, J., J.J. Liu, A.S. Kowal, T. Nardine, P. Bhattacharya, A. Lee, and Y. Yang. 2002. Microtubule-associated protein 1B: a neuronal binding partner for gigaxonin. J. Cell Biol. 158:427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling, J., Y. Yang, R. Wollmann, L.F. Reichardt, and E. Fuchs. 1997. Developmental expression of BPAG1-n: insights into the spastic ataxia and gross neurologic degeneration in dystonia musculorum mice. Dev. Biol. 187:131–142. [DOI] [PubMed] [Google Scholar]
- Dupuis, L., M. de Tapia, F. Rene, B. Lutz-Bucher, J.W. Gordon, L. Mercken, L. Pradier, and J.P. Loeffler. 2000. Differential screening of mutated SOD1 transgenic mice reveals early up-regulation of a fast axonal transport component in spinal cord motor neurons. Neurobiol. Dis. 7:274–285. [DOI] [PubMed] [Google Scholar]
- Gill, S.R., T.A. Schroer, I. Szilak, E.R. Steuer, M.P. Sheetz, and D.W. Cleveland. 1991. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J. Cell Biol. 115:1639–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, L., L. Degenstein, J. Dowling, Q.C. Yu, R. Wollmann, B. Perman, and E. Fuchs. 1995. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell. 81:233–243. [DOI] [PubMed] [Google Scholar]
- Hafezparast, M., R. Klocke, C. Ruhrberg, A. Marquardt, A. Ahmad-Annuar, S. Bowen, G. Lalli, A.S. Witherden, H. Hummerich, S. Nicholson, et al. 2003. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 300:808–812. [DOI] [PubMed] [Google Scholar]
- Holleran, E.A., S. Karki, and E.L. Holzbaur. 1998. The role of the dynactin complex in intracellular motility. Int. Rev. Cytol. 182:69–109. [DOI] [PubMed] [Google Scholar]
- Ikura, M. 1996. Calcium binding and conformational response in EF-hand proteins. Trends Biochem. Sci. 21:14–17. [PubMed] [Google Scholar]
- Karakesisoglou, I., Y. Yang, and E. Fuchs. 2000. An epidermal plakin that integrates actin and microtubule networks at cellular junctions. J. Cell Biol. 149:195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, S.J., and T.A. Schroer. 2000. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat. Cell Biol. 2:20–24. [DOI] [PubMed] [Google Scholar]
- Lankes, W.T., and H. Furthmayr. 1991. Moesin: a member of the protein 4.1-talin-ezrin family of proteins. Proc. Natl. Acad. Sci. USA. 88:8297–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, C.L., D. Sun, M. Zheng, D.R. Knowles, and R.K. Liem. 1999. Microtubule actin cross-linking factor (MACF): a hybrid of dystonin and dystrophin that can interact with the actin and microtubule cytoskeletons. J. Cell Biol. 147:1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung, C.L., M. Zheng, S.M. Prater, and R.K. Liem. 2001. The BPAG1 locus: alternative splicing produces multiple isoforms with distinct cytoskeletal linker domains, including predominant isoforms in neurons and muscles. J. Cell Biol. 154:691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls, I., C. Jonnakuty, B.H. LaMonte, E.L. Holzbaur, M. Tokito, E. Mann, M.K. Floeter, K. Bidus, D. Drayna, S.J. Oh, et al. 2003. Mutant dynactin in motor neuron disease. Nat. Genet. 33:455–456. [DOI] [PubMed] [Google Scholar]
- Raivich, G., R. Hellweg, and G.W. Kreutzberg. 1991. NGF receptor-mediated reduction in axonal NGF uptake and retrograde transport following sciatic nerve injury and during regeneration. Neuron. 7:151–164. [DOI] [PubMed] [Google Scholar]
- Schneider, C., R.M. King, and L. Philipson. 1988. Genes specifically expressed at growth arrest of mammalian cells. Cell. 54:787–793. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer, C.M., S.B. Karki, S.A. Kuznetsov, J.S. Tabb, D.G. Weiss, G.M. Langford, and E.L. Holzbaur. 1997. The interaction between cytoplasmic dynein and dynactin is required for fast axonal transport. Proc. Natl. Acad. Sci. USA. 94:12180–12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, T.L., and D.W. Cleveland. 1999. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 2:50–56. [DOI] [PubMed] [Google Scholar]
- Yang, Y., J. Dowling, Q.C. Yu, P. Kouklis, D.W. Cleveland, and E. Fuchs. 1996. An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell. 86:655–665. [DOI] [PubMed] [Google Scholar]
- Yang, Y., C. Bauer, G. Strasser, R. Wollman, J.P. Julien, and E. Fuchs. 1999. Integrators of the cytoskeleton that stabilize microtubules. Cell. 98:229–238. [DOI] [PubMed] [Google Scholar]
- Zhao, C., J. Takita, Y. Tanaka, M. Setou, T. Nakagawa, S. Takeda, H.W. Yang, S. Terada, T. Nakata, Y. Takei, et al. 2001. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 105:587–597. [DOI] [PubMed] [Google Scholar]