

Abstract
Ezrin, radixin, and moesin (ERM) regulate cortical morphogenesis and cell adhesion by connecting membrane adhesion receptors to the actin-based cytoskeleton. We have studied the interaction of moesin and ezrin with the vascular cell adhesion molecule (VCAM)-1 during leukocyte adhesion and transendothelial migration (TEM). VCAM-1 interacted directly with moesin and ezrin in vitro, and all of these molecules colocalized at the apical surface of endothelium. Dynamic assessment of this interaction in living cells showed that both VCAM-1 and moesin were involved in lymphoblast adhesion and spreading on the endothelium, whereas only moesin participated in TEM, following the same distribution pattern as ICAM-1. During leukocyte adhesion in static or under flow conditions, VCAM-1, ICAM-1, and activated moesin and ezrin clustered in an endothelial actin-rich docking structure that anchored and partially embraced the leukocyte containing other cytoskeletal components such as α-actinin, vinculin, and VASP. Phosphoinositides and the Rho/p160 ROCK pathway, which participate in the activation of ERM proteins, were involved in the generation and maintenance of the anchoring structure. These results provide the first characterization of an endothelial docking structure that plays a key role in the firm adhesion of leukocytes to the endothelium during inflammation.
Keywords: ERM; VCAM-1; ICAM-1; leukocyte adhesion and transendothelial migration; docking structure
Introduction
Leukocyte extravasation across the endothelial barrier is a fundamental requirement in a wide variety of physiological and pathological scenarios, including immunity and inflammation. This phenomenon is an active, multistep process that requires drastic morphological changes involving cytoskeletal-directed clustering of adhesion receptors in both leukocytes and endothelial cells (Butcher, 1991). Among adhesion receptors, the integrins α4β1 (VLA-4) and αLβ2 (LFA-1) play a major role in the tight adhesion of leukocytes to endothelium (González-Amaro and Sánchez-Madrid, 1999). Their main ligands on endothelium are vascular cell adhesion molecule (VCAM)*-1 and intercellular adhesion molecule (ICAM)-1, respectively (Marlin and Springer, 1987; Elices et al., 1990). ICAM-1 but not VCAM-1 is basally expressed in resting cells, and both molecules are induced upon activation by proinflammatory cytokines such as IL-1 and TNF-α (Carlos and Harlan, 1994). Although it has been described that both VLA-4/VCAM-1 and LFA-1/ICAM-1 interactions mediate the firm adhesion of leukocytes, only the latter molecular pair seems to be required for lymphocyte diapedesis (Oppenheimer-Marks et al., 1991).
The interaction of adhesion molecules with cytoskeletal components is of critical importance for cell–cell and cell–substratum adhesion as well as for receptor internalization. The cortical cytoskeleton regulates the membrane localization of several adhesion receptors, such as ICAMs, CD43, and CD44, through one or more members of the ezrin, radixin, and moesin (ERM) family of proteins (Serrador et al., 1997; Heiska et al., 1998; Yonemura et al., 1998). These molecules function as membrane-actin cytoskeleton linkers regulating cortical morphogenesis and cell adhesion. Accordingly, they play a key role in the formation of protrusive plasma membrane structures such as filopodia, microspikes, or microvilli (Mangeat et al., 1999; Yonemura and Tsukita, 1999). Structurally, they are closely related to each other and seem to be functionally redundant, as suggested by the apparently normal phenotype of moesin knockout mice (Doi et al., 1999). Their NH2-terminal domains interact with integral membrane proteins, whereas their COOH-terminal domains bind F-actin (Turunen et al., 1994). These functions are conformationally regulated by reversible changes from inactive to functionally active forms. Binding of phosphatidylinositol 4,5-biphosphate (PI[4,5]P2) and phosphorylation of a specific COOH-terminal threonine residue unmask the F-actin and membrane binding sites and stabilize the active conformation (Nakamura et al., 1999; Barret et al., 2000). The Rho/p160 ROCK signaling pathway and the phosphatidylinositol turnover are the major regulatory mechanisms for ERM activation, inducing their phosphorylation and translocation into apical membrane/actin protrusions (Hirao et al., 1996; Matsui et al., 1998; Shaw et al., 1998). On the other hand, activated ERM proteins can sequestrate Rho-GDI to permit Rho activation, providing a positive feedback pathway (Takahashi et al., 1997).
These proteins have been studied in a variety of cellular types, but thus far their functions have not been addressed in endothelial cells. Herein, we describe the direct interaction of VCAM-1 with moesin and ezrin, the two members of the ERM family most highly expressed in endothelium (Menager et al., 1999). We also describe the dynamic distribution of VCAM-1, ICAM-1, and moesin during leukocyte adhesion and lymphoblast transendothelial migration (TEM), using live time-lapse fluorescence confocal microscopy. Our data indicate that an endothelial docking structure is formed during leukocyte-endothelium interaction in static and under flow conditions. VCAM-1, ICAM-1, and activated ERM proteins participate in this novel anchoring structure together with structural and regulatory molecules typical of nascent phagosomes.
Results
VCAM-1 interacts with moesin and ezrin at the apical surface of activated endothelial cells
The subcellular distribution of VCAM-1, ezrin, and moesin, was analyzed by confocal microscopy in TNF-α–activated human umbilical vein endothelial cells (HUVEC). VCAM-1 colocalized with ezrin (Fig. 1, a–c) and moesin (Fig. 1, e–g) in the microspikes and microvilli that protrude from the apical surface of these cells. Colocalization analysis confirmed these observations (Fig. 1, d and h). By contrast, VE-cadherin did not colocalize with ERM proteins (Fig. 1, i–l).
Figure 1.
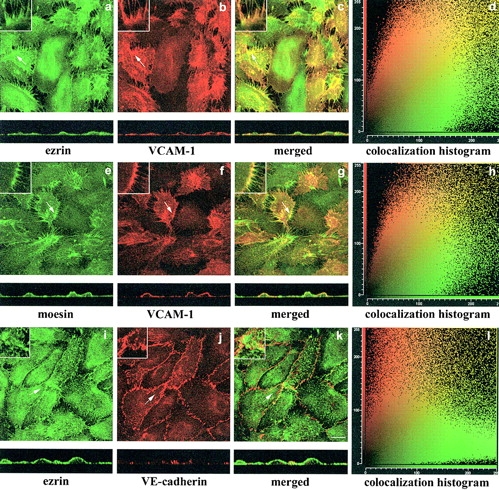
VCAM-1 colocalizes with moesin and ezrin at the apical surface of activated HUVEC. Confluent HUVEC were activated with 20 ng/ml TNF-α for 20 h. Thereafter, cells were fixed, permeabilized, and stained with the anti-ezrin pAb 90/3 (a and i, green), anti-moesin pAb 95/2 (e, green), anti–VCAM-1 mAb P8B1 (b and f, red), or anti–VE-cadherin mAb Tea1/31 (j, red). Merged images are shown in c, g, and k, where colocalizations are observed (yellow). Images represent confocal laser scanning micrographs showing horizontal projections or the corresponding orthogonal section of the same field. Insets correspond to the amplified image of the zones pointed to by arrows. Colocalization histograms of green and red signals corresponding to these images are shown on the right (d, h, and l). The corresponding colocalization percentages are 65.7% (d), 67.6% (h), and 13.3% (l). Bar, 20 μm.
To assess whether this codistribution in activated HUVEC is correlated with the formation of complexes between VCAM-1 molecules and ERM proteins, immunoprecipitation assays were performed. As shown in Fig. 2 A, moesin and ezrin coimmunoprecipitated with VCAM-1. To determine whether these interactions are direct, binding assays were performed using a glutathione-S-transferase (GST) fusion protein containing the cytoplasmic tail of VCAM-1 (GST–VC). The fusion protein containing the cytoplasmic tail of ICAM-3, GST–IC3, which has been demonstrated to bind ERM, as well as the partially truncated form GST-Y9, which shows a considerable reduced binding to moesin and ezrin (Serrador et al., 2002), were used as positive and negative controls, respectively. 35S-Met–labeled NH2-terminal domain of moesin, N-moesin, and the NH2-terminal domain of ezrin, N-ezrin, were added to Sepharose beads coupled to the GST fusion proteins. Strong binding of VCAM-1 to ezrin and moesin was observed, which was much higher in comparison to ICAM-3 (Fig. 2 B). Altogether, these data demonstrate that VCAM-1 can directly associate with ezrin and moesin in vitro and presumably also at the apical membrane sites of endothelial cells.
Figure 2.
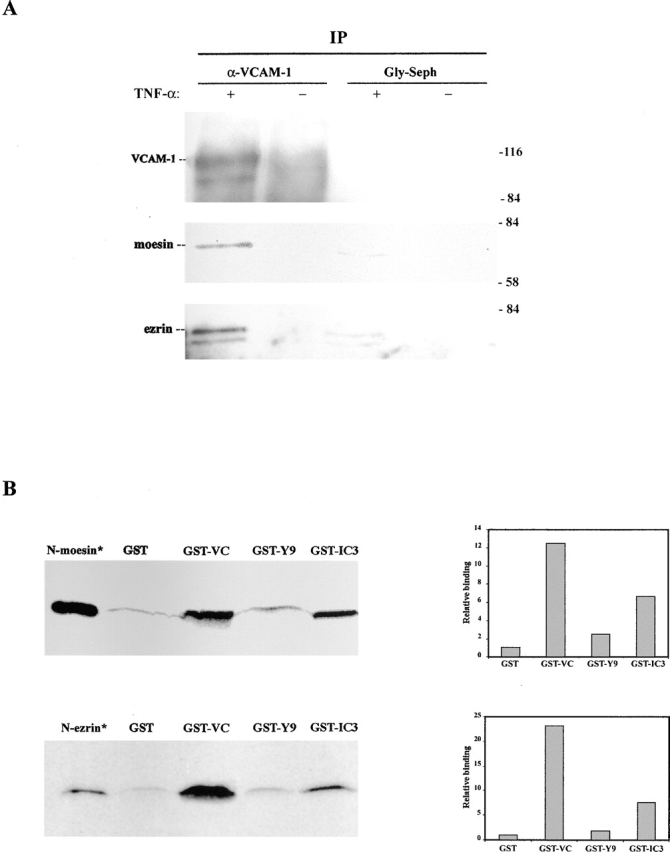
Association of VCAM-1 with moesin and ezrin. (A) Cytokine-activated HUVEC were lysed and immunoprecipitated with the anti–VCAM-1 mAb 4B9 or Gly-Sepharose. Immunoprecipitates were then resolved on a 10% SDS-PAGE, and sequentially immunoblotted with the anti-VCAM-1 mAb 4B9, the anti-moesin pAb 95/2, and the anti-ezrin pAb 90/3. Molecular weights (kD) are indicated on the right side. (B) GST or the GST fusion proteins GST–VC, GST–IC3 and GST–Y9 were bound to glutathione-Sepharose beads and incubated with 35S-Met-N-moesin or 35S-Met-N-ezrin (top and bottom, respectively). After incubation, beads were boiled in sample buffer and eluted proteins were analyzed by 10% SDS-PAGE, autoradiography, and fluorography. Lanes with isotope-labeled N-moesin and N-ezrin (*) indicate the molecular mass of these truncated proteins. Densitometric diagrams normalized to the loading controls of GST proteins are shown on the right.
Differential contribution of VCAM-1, ICAM-1, and ERM proteins to the extravasation process
The functional role of the VCAM-1/ERM association in the endothelial cell–lymphocyte interaction was analyzed and compared with another adhesion receptor that also interacts with ERM proteins, namely ICAM-1 (Heiska et al., 1998). For this purpose, we used T lymphoblasts, that express high levels of VLA-4 and LFA-1, ligands of VCAM-1 and ICAM-1, respectively (Fig. 3 A). Both integrins were active since mAb against them blocked T lymphoblast TEM (Fig. 3 B). These cells were allowed to adhere and migrate across an activated HUVEC monolayer and confocal microscopic analysis of endogenous endothelial VCAM-1 and ICAM-1 distribution was performed. When lymphoblasts were spread on the apical surface of endothelium, VCAM-1 clustered around these cells (Fig. 3 C, a). However, such VCAM-1 clusters were neither observed during the passage of lymphoblasts across the endothelium nor after transmigration (Fig. 3 C, b). The orthogonal section showed two lymphoblasts migrating across adjacent endothelial cells, where clustered VCAM-1 molecules colocalized with endothelial ezrin only at the apical surface of the lymphoblast-endothelial cell contact area (Fig. 3 C, c). On the other hand, ICAM-1 was clustered around lymphoblasts during all the lymphoblast adhesion and transmigration processes (Fig. 3 C, d and e), colocalizing with endothelial ezrin (Fig. 3 C, f).
Figure 3.

Distribution of endogenous VCAM-1, ICAM-1, and ezrin during lymphoblast TEM. (A) Expression of α4-integrin (thick line), and αL-integrin (thin line) on T lymphoblasts as determined by flow cytometry analysis. P3×63 (dotted line) was used as negative control. (B) Transendothelial migration assay of T lymphoblasts pretreated with the blocking anti-α4 mAb HP2/1, the blocking anti-αL mAb TS1/11, the mixture of anti-α4 plus αL mAb, or the anti–ICAM-3 mAb TP1/24 as negative control. Values correspond to the arithmetic mean ± SD of a representative experiment run by duplicate out of three independent ones. Statistically significant values, as defined by unpaired Student's t test, are indicated with * (P < 0.05) or ** (P < 0.015) compared with no mAb treatment. (C) T lymphoblasts were allowed to transmigrate across an activated HUVEC monolayer, and then cells were fixed, permeabilized, and stained with the anti–VCAM-1 mAb P8B1 (a–c, red), the anti-ICAM-1 mAb Hu5/3 (d–f, red), or the anti-ezrin pAb 90/3 (c and f, green). Representative confocal horizontal images showing an apical section of endothelium with adhered lymphoblasts on top (a and d) and a basal section with transmigrated lymphoblasts beneath the endothelium (b and e) are presented. DIC images are shown overlaid with VCAM-1 (a and b) or ICAM-1 staining (d and e). Arrows point to VCAM-1 or ICAM-1 clusters at the contact area. Representative orthogonal sections corresponding to the white line in panel a or panel d are shown in panels c and f, respectively. Green signal corresponds to ezrin staining both in lymphoblasts and endothelium. Arrowheads point to the sites of VCAM-1/ezrin or ICAM-1/ezrin clustering at the apical surface of endothelial cells. Bars: (a and b) 20 μm; (d and e) 8 μm.
To dynamically assess the changes in distribution of VCAM-1, ICAM-1, and ERM proteins, HUVEC transiently transfected with VCAM-1–, ICAM-1–, or moesin–green fluorescent protein (GFP) were separately monitored by live time-lapse confocal microscopy after addition of lymphoblasts. As observed previously for endogenous molecules, VCAM-1– and moesin–GFP fusion proteins redistributed to sites of contact during lymphoblast initial adhesion and spreading. However, only moesin was concentrated in the transmigration cleft and apparently participated in subsequent interactions, when lymphoblasts migrated beneath the activated endothelium (Fig. 4 A). Interestingly, moesin–GFP exhibited a dynamic behavior similar to ICAM-1–GFP (Fig. 4 B), presumably because this adhesion molecule binds to ERM proteins to actively participate in TEM. Digital movies showing more clearly the differential dynamic distribution of these molecules are included as additional material.
Figure 4.
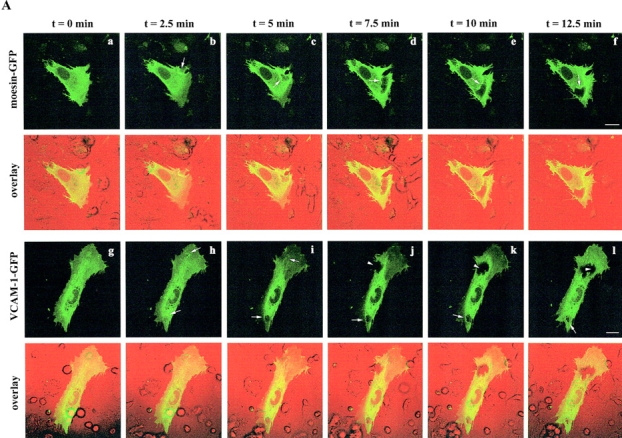
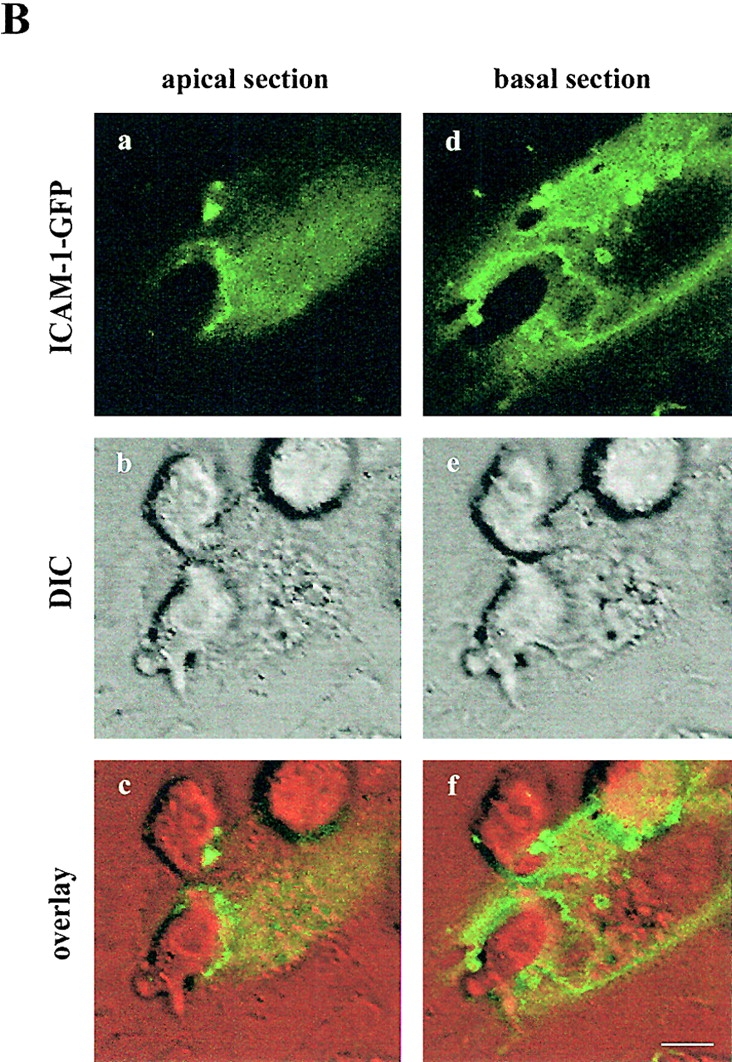
Dynamic changes in the localization of VCAM-1, ICAM-1, and moesin during lymphoblast TEM. (A) Lymphoblasts were allowed to transmigrate across activated HUVEC transfected with moesin- or VCAM-1–GFP. Video sequences tracking the spatial and temporal distribution of moesin (a–f) and VCAM-1 (g–l) were obtained using live time-lapse fluorescence confocal microscopy. Each image represents a projection of several representative horizontal sections of a confocal image stack depicted from the video sequence at the specified times. DIC and fluorescence images are merged and presented at the lower side of each panel. Arrows point to the GFP proteins clustering during the lymphoblast–endothelium interaction. Arrowheads indicate the absence of VCAM-1–GFP from the contact area between the transfected endothelial cell and a migrated lymphoblast placed beneath the endothelial monolayer. Corresponding digital video sequences are available at http://www.jcb.org/cgi/content/full/jcb.200112126/DC1. Bars, 20 μm. (B) Lymphoblast transmigration across activated HUVEC transfected with ICAM-1–GFP was analyzed by live time-lapse fluorescence confocal microscopy. Two representative horizontal sections from the apical and the basal side of the endothelial cell belonging to the same confocal stack depicted from the video sequence are presented. ICAM-1–GFP signal is shown in panels a and d. DIC images and the overlaid images are presented in panels b and e, and c and f, respectively. The corresponding video sequence is available at http://www.jcb.org/cgi/content/full/jcb.200112126/DC1. Bar, 5 μm.
Redistribution of VCAM-1, ICAM-1, and ERM proteins at a docking structure during endothelium–leukocyte interaction
To focus our study on the role of ERM interaction with endothelial adhesion receptors during the initial adhesion of leukocytes to the endothelium, to which VCAM-1 involvement was mainly restricted, we used K562 cells stably transfected with α4 integrin (4M7 cells) (Muñoz et al., 1996). These cells expressed high levels of VLA-4, but negligible amounts of LFA-1 (Fig. 5 A). In addition, their adhesion to activated endothelium was dependent on VLA-4, as it was inhibited by the blocking anti-α4 HP2/1 mAb, and induced by the activating anti-β1 TS2/16 mAb (Fig. 5 B). 4M7 cells adhered to activated endothelium mainly via VLA-4, but these cells were unable to progress to TEM (unpublished data). When the distribution of endogenous endothelial VCAM-1 was examined during 4M7 cell adhesion, we found that it was strongly concentrated around attached leukocytes (Fig. 5 C, c). Endogenous ezrin colocalized with VCAM-1 at the endothelial–leukocyte contact area (Fig. 5 C, d), and antibodies to ezrin also stained the leukocyte membrane (Fig. 5 C, b). Furthermore, a three-dimensional reconstruction of the site of adhesion showed that VCAM-1 and ezrin were contained in a unique docking structure that was raised above the level of the endothelial cell surface and that surrounded the adherent leukocyte in a cup-like fashion. Both proteins were preferentially concentrated in microspikes of this structure that presumably served to anchor the attached leukocyte (Fig. 5 C, e–g). The redistribution of VCAM-1 and moesin to the leukocyte–endothelium contact area was tracked separately by live time-lapse fluorescence confocal microscopy during the interaction of 4M7 cells with VCAM-1– or moesin–GFP transfected HUVEC. The dynamic studies demonstrated that VCAM-1/VLA-4 engagement was associated with the progressive concentration of VCAM-1 and moesin at the specialized anchoring structure formed between the two interacting cells, which was progressively strengthened and sustained with time (Fig. 5 D).
Figure 5.
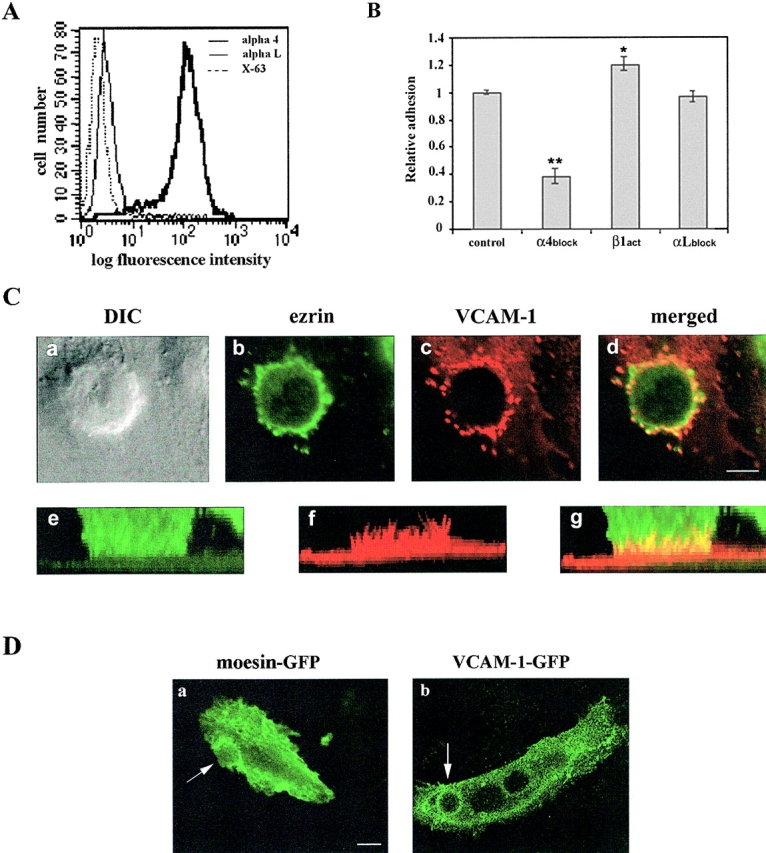
Localization of VCAM-1 and ezrin at the contact area of activated HUVEC with leukocytes. (A) Expression of α4-integrin (thick line), and αL-integrin (thin line) on 4M7 cells as determined by flow cytometry analysis. P3×63 (dotted line) was used as negative control. (B) Adhesion to activated HUVEC of 4M7 cells pretreated with the blocking anti-α4 mAb HP2/1, the activating anti-b1 mAb TS2/16, or the blocking anti-αL mAb TS1/11. Values correspond to the arithmetic mean ± SD of a representative experiment run by triplicate out of three independent ones. Statistically significant values, as defined by unpaired Student's t test, are indicated with * (P < 0.005) or ** (P < 0.0001), compared with no mAb treatment. (C) 4M7 cells interacting with activated endothelial cells were fixed, permeabilized, and stained with the anti-ezrin pAb 90/3 (green) and the anti–VCAM-1 mAb P8B1 (red). Representative horizontal sections of confocal laser scanning images (b and c) are merged in d. The corresponding DIC image is shown in a. The corresponding three-dimensional reconstruction is presented in e–g. Bar, 5 μm. (D) 4M7 cells were allowed to adhere to activated HUVEC transfected with moesin- or VCAM-1–GFP. GFP staining was monitored using live time-lapse fluorescence confocal microscopy. Horizontal sections showing the staining of moesin- (a) or VCAM-1–GFP (b) after 60 min of leukocyte–endothelium interaction. Arrows point to the GFP proteins clustered in the anchoring structure. Bar, 20 μm.
To ascertain the physiological relevance of this docking structure, we analyzed the redistribution of VCAM-1, ICAM-1, and ezrin to the contact area of migrating peripheral blood lymphocytes (PBLs) allowed to adhere under fluid shear conditions, using a physiological wall shear stress (1.8 dyn/cm2) for perfusion periods from 30 s to 10 min. We found these endothelial molecules colocalizing in clusters at the docking structures formed around spread PBLs (Fig. 6 A), at early time points during the arrest of lymphocytes. This structure was also observed during the interaction of activated HUVEC with T lymphoblasts in static conditions (Fig. 6 B; unpublished data). Similarities in three-dimensional VCAM-1 distribution around 4M7 cells, T lymphoblasts or peripheral blood lymphocytes supported the generality of the docking structure (Fig. 6 B).
Figure 6.

Formation of the endothelial docking structure for adhered lymphocytes under flow. (A) Transendothelial migration assay of peripheral blood lymphocytes under fluid shear conditions. After 10 min of perfusion, cells were fixed and stained for ICAM-1 (b and d, green), VCAM-1 (c, d, g, and h, red), and ezrin (f and h, green). The corresponding DIC images are shown in panels a and e. Merged images are shown in panels d and h. Bar, 3.5 μm. (B) Three-dimensional reconstruction of VCAM-1 staining during 4M7 cell (a), T lymphoblast (b), or PBL under flow (c) adhesion.
Relocation of cytoskeletal components to the endothelial docking structure
The cytoskeletal components involved in the generation of this endothelial structure were analyzed. Samples of 4M7 cells adhered to activated HUVEC and stained for VCAM-1 and F-actin revealed a considerable enrichment of endothelial actin within the docking structure (Fig. 7 A, a–b). Interestingly, this apical actin scaffold appeared not to be connected to basal stress fibers (Fig. 7 A, c–d). On the contrary, tubulin was not present with F-actin at the anchoring structure (Fig. 7 A, e–h). Vinculin and α-actinin–GFP also redistributed to this structure with a punctuate pattern (Fig. 7 B, c and d, and C, a–c). Likewise, VASP-GFP colocalized with VCAM-1 (Fig. 7 C, f–g), whereas talin and paxillin-GFP were only found colocalizing with VCAM-1 at some adhesion structures (Fig. 7 B, a and b, and C, d and e). These data indicate that the endothelial docking structure was supported by the actin cytoskeleton, actin bundling proteins such as α-actinin, actin-nucleating proteins such as VASP, and focal adhesion proteins such as vinculin, talin or paxillin. The formation of this structure was associated with a remarkable change in distribution of several of these proteins, in particular vinculin, talin, and paxillin, from their normal subcellular localization at focal adhesions at the basal surface to the docking structure at the apical surface.
Figure 7.
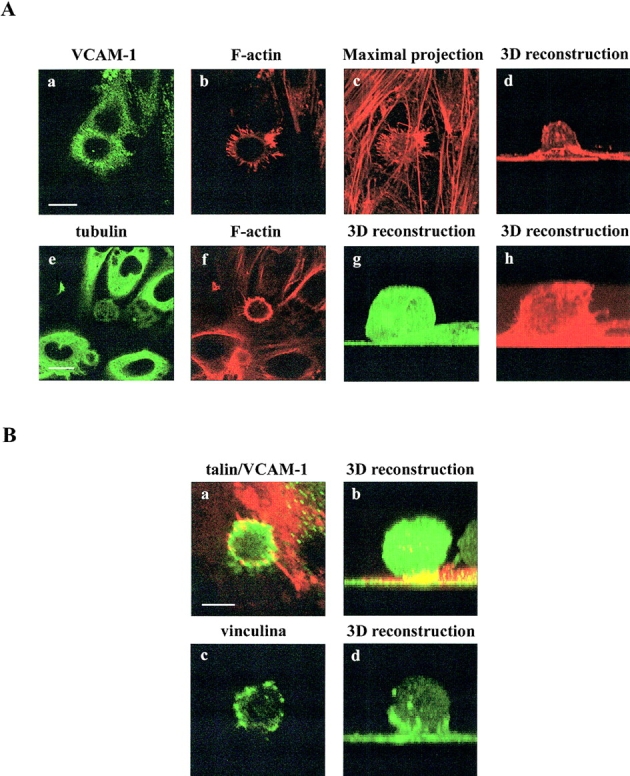
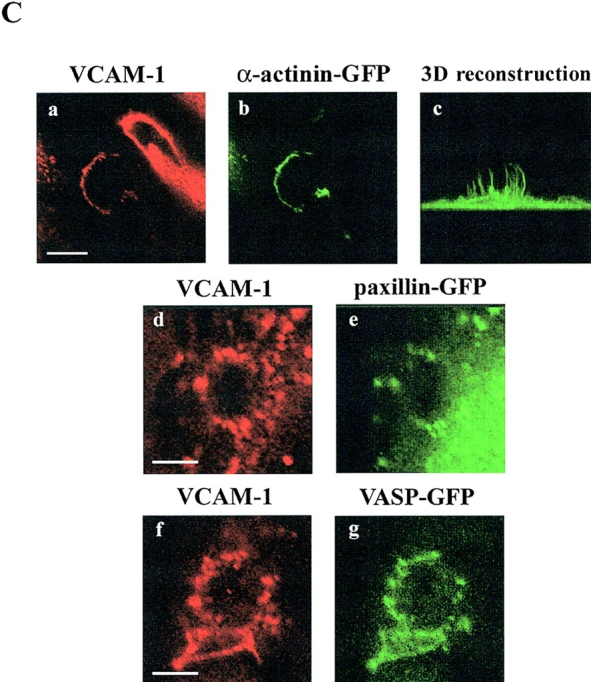
Characterization of the endothelial docking structure formed during leukocyte adhesion. (A) 4M7 cells were allowed to adhere to activated HUVEC cells, then fixed, permeabilized and stained for VCAM-1 (a, green), F-actin (b–d, f, and h, red), and tubulin (e and g, green). Representative horizontal sections of confocal image-stacks are presented in a and b and e and f. The panel c shows the projection of all the horizontal sections corresponding to the image presented in a and b. Three-dimensional reconstructions of F-actin (d and h) and tubulin (g) stainings are also shown. Bars, 20 μm. (B) 4M7 cells adhered to activated HUVEC were fixed, permeabilized, and stained for talin (a and b, green), VCAM-1 (a and b, red), or vinculin (c and d, green). Representative horizontal sections of confocal images are presented in a and c. Three-dimensional reconstructions are shown in b and d. Bar, 5 μm. (C) 4M7 cells were allowed to adhere to activated HUVEC transfected with α-actinin, paxillin-, and VASP-GFP. Thereafter, cells were fixed and stained with the anti-VCAM-1 mAb P8B1 (a, d, and f, red). Green signal corresponds to GFP fusion proteins (b, c, e, and g). Representative horizontal sections of confocal image stacks are presented in all panels except for c, which shows the three-dimensional reconstruction of α-actinin–GFP signal. Bars, 5 μm.
The docking structure is regulated by PI(4,5)P2 and Rho/p160 ROCK
To assess whether the clustering of ERM proteins at the endothelial-leukocyte contact area is associated with an activated state of these proteins, immunofluorescence studies using the 297S mAb, which recognizes a COOH-terminal threonine phosphorylated in ERM proteins (Matsui et al., 1998), were conducted in the 4M7 cell adhesion model. A high concentration of phosphorylated ERM proteins was evident at the endothelial anchoring structure generated after VCAM-1/VLA-4 interaction (Fig. 8 A). We have also studied two important regulators of ERM activation, namely phosphoinositides and components of the Rho/p160 ROCK pathway. The subcellular localization of different phosphoinositides was determined by using as probes the PH domain of PLCδ, which binds PI(4,5)P2, and that of GRP1, which binds PI(3,4,5)P3 and PI(3,4)P2, fused to GFP (Gray et al., 1999; Várnai et al., 1999). Upon endothelial cell transfection, both probes colocalized with VCAM-1 at the endothelial docking structure (Fig. 8 B, a and b; d and e). PLCδ-PH–GFP showed a higher concentration at microspike tips (Fig. 8 B, c), whereas GRP1-PH–GFP appeared to be more diffusely distributed and localized throughout the entire structure (Fig. 8 B, f).
Figure 8.
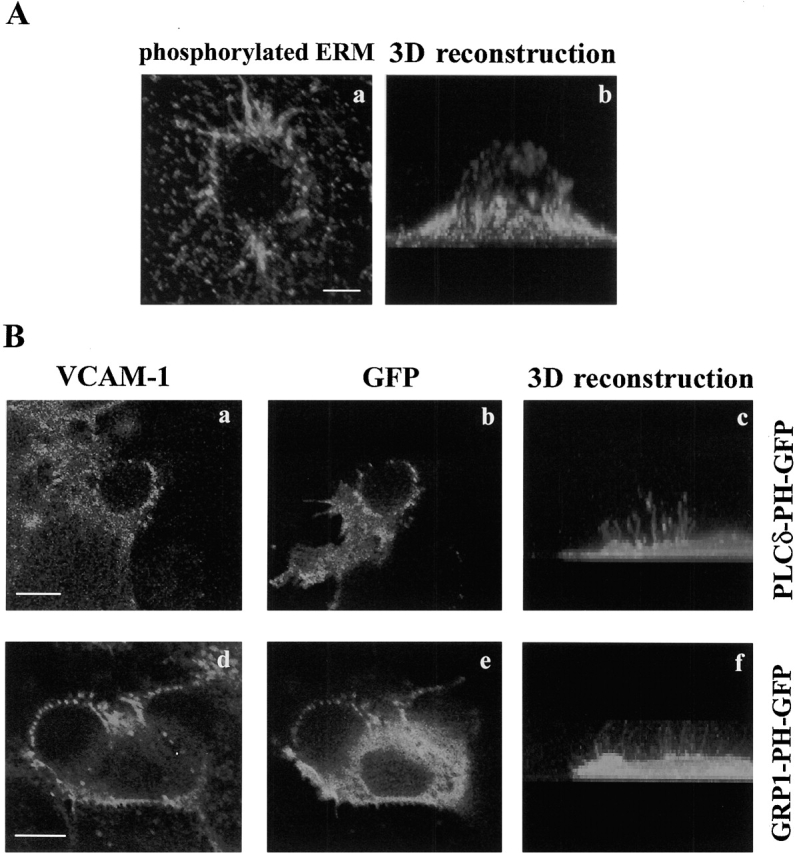
Localization of phosphorylated ERM proteins and phosphoinositides at the anchoring structure. (A) 4M7 cells adhered to activated HUVEC were fixed, permeabilized and stained with the mAb 297S. Representative horizontal section of a confocal micrograph (a), and a three-dimensional reconstruction of a series of horizontal sections (b) are shown. Bar, 5 μm. (B) HUVEC cells were transfected with PLCs-PH- and GRP1-PH-GFP and then, 4M7 cells were allowed to adhere. Thereafter, cells were fixed, permeabilized and stained with the anti-VCAM-1 mAb P8B1 (a and d). Green signal corresponds to GFP fusion proteins (b and e). Panels c and f show three-dimensional reconstructions of horizontal sections corresponding to the green signal. Bars, 5 μm.
Next, blocking studies with chemical inhibitors were performed before and after HUVEC-4M7 cell adhesion. The p160 ROCK inhibitor Y-27632 strongly inhibited the generation and maintenance of the anchoring structure (Fig. 9 A). On the other hand, the PI3K inhibitor Ly 294002 only had a minor effect on the generation of this structure, but moderately inhibited its maintenance (Fig. 9 A). The classical, PKCs inhibitor Gö6976 did not exert a significant inhibitory effect neither on the generation nor in the maintenance of the anchoring structure (Fig. 9 A). Dynamic studies using VCAM-1–GFP transfected HUVEC showed that the p160 ROCK inhibitor Y-27632 acted by destroying the docking structure formed around 4M7 cells, as observed in Fig. 9 B. Finally, the effect of this inhibitor was assayed on endothelium during peripheral blood lymphocyte adhesion and transmigration under flow conditions. We found a diminished lymphocyte rolling and adhesion after Y-27632 treatment with regard to control conditions. Furthermore, some cells that were initially adhered, began to roll and finally detached from the monolayer, indicating an abnormal adhesion process. Accordingly, all these events led to a significant inhibition of transmigration. The effect of the inhibitor Y-27632 was analyzed at different time periods (3, 6, 8, and 10 min of perfusion), being persistent at any time tested, but only the last time is shown, as representative (Fig. 9 C). Furthermore, immunofluorescence analysis revealed that most of adhered lymphocytes were not tightly anchored by an endothelial docking structure (unpublished data). In conclusion, these findings suggest that both, PI(4,5)P2 and the Rho/p160 ROCK pathway, are important for the generation as well as for the maintenance of the endothelial docking structure.
Figure 9.
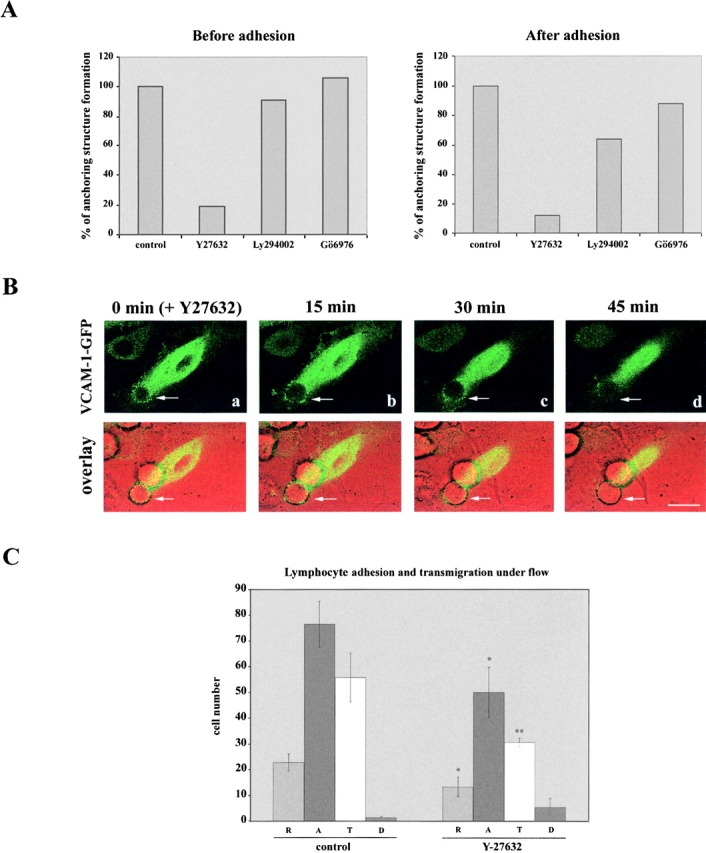
Rho/p160 ROCK pathway regulates the generation and maintenance of the VCAM-1–mediated docking structure. (A) Activated HUVEC were pretreated with Y27632 (30 μM), Ly294002 (20 μM), or Gö6976 (1 μM) for 20 min before or 30 min after the addition of 4M7 cells. Total adhesion time was in both cases 60 min. Quantification of leukocyte adhesion and endothelial docking structure formation was carried out by staining with the mAb anti–VCAM-1 P8B1 and counting 300 adhered cells of each treatment. A representative experiment out of four independent ones is presented. (B) Kinetics of the endothelial anchoring structure dissolution after the addition of the p160 ROCK inhibitor Y-27632. The inhibitor Y-27632 (30 μM) was added after the formation of the docking structure in a 4M7 adhesion assay performed as above. Representative horizontal sections captured every 15 min are shown in a–d. DIC and fluorescence images are merged and presented in the lower side of each panel. Arrows indicate the clustering of GFP proteins. Bar, 10 μm. (C) Effect of Y-27632 on peripheral blood lymphocyte adhesion and TEM under flow conditions. Activated endothelium was pretreated or not with Y-27632 (30 μM) for 30 min. Thereafter, PBLs were allowed to adhere and transmigrate under flow conditions for 10 min. Quantification of rolling (R), adhesion (A), transmigration (T), and detachment (D) events during the last minute of perfusion was carried out. Values correspond to the arithmetic mean ± SD of four different fields belonging to a representative experiment. Statistically significant values, as defined by unpaired Student's t test, are indicated with *(P < 0.01) or **(P < 0.002) compared with no inhibitory treatment.
Discussion
VCAM-1 is one of the major endothelial receptors that mediates leukocyte adhesion to the vascular endothelium (Carlos and Harlan, 1994). Recent data obtained with neonatally deficient VCAM-1 mice have strongly suggested that VCAM-1 plays an important role for lymphocyte homing and for T cell-dependent humoral immune responses (Koni et al., 2001; Leuker et al., 2001). In addition, VCAM-1 may play an important role in the pathogenesis of diseases such as atherosclerosis (Cybulsky et al., 2001), rheumatoid arthritis (Carter and Wicks, 2001), and multiple sclerosis (Alon, 2001). Thus, the elucidation of VCAM-1 function in leukocyte adhesion and transmigration is crucial, as this molecule could constitute a molecular target for therapeutic intervention.
The cytoskeletal components involved in the redistribution of VCAM-1 at the leukocyte-endothelial cell contact area have not been studied previously. Among potential candidates, ERM proteins seemed likely to mediate this process as these molecules play an important role in the remodelling of the plasma membrane, as reported for ICAMs (Helander et al., 1996; Serrador et al., 1997; Heiska et al., 1998). We found that endogenous VCAM-1 colocalizes and is physically associated with moesin and ezrin in microspikes and microvilli of the apical surface of cytokine-activated endothelial cells. Furthermore, the cytoplasmic tail of VCAM-1 and the active NH2-terminal domain of moesin or ezrin are capable to directly bind in vitro. These data strongly suggest that ERM proteins are directly involved in the redistribution of VCAM-1. However, it cannot be ruled out completely that adaptor proteins such as EBP50 or E3KARP (Bretscher et al., 2000), also play a role. It has been reported that ERM proteins bind to a positively charged amino acid cluster in the juxta-membrane cytoplasmic domain of CD44, CD43, and ICAM-2 (Yonemura et al., 1998). In addition, we have found recently that a novel serine-rich motif within the cytoplasmic tail of ICAM-3 is critical for its interaction with ERM proteins (Serrador et al., 2002). The amino acid sequence comparison of VCAM-1 and ICAM-3 cytoplasmic tails suggests that VCAM-1 contains a similar serine-rich motif, likely accounting for ERM association.
To study the VCAM-1/ERM interaction during the extravasation of lymphoblasts, we have made extensive use of a live cell system in combination with time-lapse fluorescence microscopy and GFP fusion proteins. This afforded us with information on dynamic relationships of the two molecules during early adhesion and later stages of TEM. Although both VCAM-1 and moesin clustered around spreading lymphoblasts on the apical endothelial surface, only moesin remained at lymphoblast-endothelial contacts during the passage of lymphoblasts across the endothelium and their subsequent migration beneath the endothelial monolayer. However, it has been reported that VCAM-1 actively participates in T lymphoblast transmigration across high endothelial venules and monocyte extravasation (Meerschaert and Furie, 1995; Faveeuw et al., 2000). These discrepancies point to a differential role of VCAM-1 that might be both leukocyte and endothelial cell-type specific. Our data on moesin dynamics clearly indicate that another receptor linked to moesin could drive this migration. In this regard, we have found that the dynamic behavior of moesin is similar to that of ICAM-1 during lymphoblast transmigration, a finding that is in accordance with previous reports describing the distribution of ICAM-1 on the lumenal and basal surfaces of the endothelium and its involvement in TEM (Oppenheimer-Marks et al., 1991; Randolph and Furie, 1996). The differential dynamic behavior of VCAM-1 and ICAM-1 during lymphoblast adhesion and transmigration on the one hand, and of moesin on the other, could suggest a mechanism by which ERM proteins are able to regulate the distribution of both molecules independently.
As VCAM-1/ERM interaction was mostly restricted to leukocyte tight adhesion and spreading, we took advantage of a cellular model based mainly on VLA-4/VCAM-1. In this model, leukocytes are restricted to a sustained tight adhesion and are unable to progress to TEM, allowing a detailed study of the endothelial VCAM-1/ERM interaction. In this cell model, VCAM-1 and both, moesin and ezrin, clustered around adherent leukocytes, participating in the formation of an actin-rich docking structure that was attached to and partially engulfed the leukocyte. Similar docking structures were formed around spreading lymphoblasts in static conditions or PBLs adhered to endothelium under flow, in which VCAM-1 and ICAM-1 were concentrated together with ERM proteins. The fact that both adhesion receptors actively participate in such endothelial structure would reinforce the concept of cross-talk between their ligands VLA-4 and LFA-1 during lymphocyte adhesion (Chan et al., 2000; Rose et al., 2001). Under these physiological conditions, docking structures rapidly vanished as lymphoblasts or lymphocytes began to migrate through the endothelial monolayer.
Our findings highlight the remarkable active role played by the endothelium during leukocyte adhesion. Thus, the initial VCAM-1 and ICAM-1 engagement trigger their clustering and the subsequent activation and clustering of endothelial moesin and ezrin at the site of cell–cell contact. In turn, phosphorylated active ERM proteins would participate in concert with α-actinin, an actin-bundling protein, in the rearrangement of the actin cytoskeleton to create the docking structure. Several focal adhesion proteins such as vinculin, talin, and paxillin seem to be involved as well. Interestingly, the endothelial docking structure is reminiscent of nascent receptor-mediated phagosomes, in that the subcellular distribution of all these structural proteins is similar in both structures (Allen and Aderem, 1996). In this regard, it has been proposed that focal adhesions and complement receptor-mediated phagosomes could share some conserved mechanisms requiring the same molecules (May and Machesky, 2001). A similar argument could be made to functionally link the former structures and the novel leukocyte–endothelium docking structure described here. On the other hand, VASP is concentrated in the docking structure. This protein is present in actin-based protrusions, where it cooperates with WASp-Arp2/3 complex, as occurs in phagosomes (Castellano et al., 2001). Its presence at the docking structure could indicate the existence of actin polymerization supporting this endothelial structure, and suggests common mechanisms for de novo actin assembly in all these structures. Interestingly, ERM proteins have been also implicated in the de novo actin assembly on mature Fc receptor-mediated phagosomes (Defacque et al., 2000).
As ERM activation occurred during the formation of the anchoring structure, we studied the regulatory mechanisms involved. These proteins are regulated by the interplay of PI(4,5)P2 metabolism and components of the Rho GTPase signaling pathway, thereby linking events occurring at the plasma membrane with cytoskeletal remodelling (Sechi and Wehland, 2000). Interestingly, these regulatory molecules are also important in phagocytosis (Botelho et al., 2000; Chimini and Chavrier, 2000). We found that PI(3,4)P2, PI(3,4,5)P3, and more abundantly PI(4,5)P2, colocalized with VCAM-1 and ERM proteins in the endothelial docking structure. Notably, PI(4,5)P2 was preferentially concentrated at the microspike tips, whereas the other phosphoinositides exhibited a more diffuse pattern. These observations point to a prominent role of PI(4,5)P2 in regulating molecular events during endothelial docking of leukocytes. One such event could be the activation of moesin and ezrin through its binding to their NH2-terminal domain. PI(4,5)P2 production could be mediated by Rho, as PI4P5K is a down-stream RhoGTPase effector. On the other hand, the presence of PI(3,4)P2 and PI(3,4,5)P3 could be related to PI3K activity. Inhibitors of PI3K only mildly affected the generation and maintenance of the docking structure, a finding that is in agreement with its role in phagocytosis, namely to mediate phagosome closure (Cox et al., 1999). In our experimental model, the endothelial anchoring structure does not progress to engulf the leukocyte; therefore, it is less PI3K dependent. In contrast, the p160 ROCK inhibitor Y27632 inhibited the formation of the anchoring structure and induced its dissolution as well. Abnormal formation of the endothelial docking structure was also observed under flow conditions after the treatment with Y-27632, which rendered an inhibitory effect in lymphocyte adhesion and transmigration. In addition, the phenomenon of rolling was also decreased. This finding is in agreement with previous reports describing the existence of an E-selectin/actin cytoskeleton adhesion complex induced by leukocyte adhesion (Yoshida et al., 1996; Lorenzon et al., 1998), which it is likely to be also affected by the inhibition of the Rho/p160 ROCK pathway. Our results concur with the previously described regulation of the VCAM-1, ICAM-1, and E-selectin clustering by the GTPase Rho during monocyte adhesion (Wojciak-Stothard et al., 1999). Furthermore, the key role of the Rho/p160 ROCK signaling pathway in the regulation of the adhesion receptor/ERM/actin cytoskeleton interaction and remodelling, which results in the formation of this protrusive structure, is further strengthened by the implication of this pathway in the regulation of complement receptor–mediated phagosomes (Caron and Hall, 1998).
In conclusion, our results provide novel insights into the links between the actin cytoskeleton and adhesion receptors involved in leukocyte adhesion and TEM during inflammation. Further analysis will be focused on molecules involved in the regulation of the endothelial docking structure disruption to allow diapedesis, and the signaling pathways that interconnect both processes.
Materials and methods
Cells and cell cultures
HUVEC were obtained and cultured as previously described (Yáñez-Mó et al., 1998). Cells were used up to the third passage in all assays. To activate HUVEC, TNF-α (20 ng/m; R&D Systems) was added to the culture media 20 h before the assays were performed. Human PBLs and T lymphoblasts were obtained and cultured as described elsewhere (Serrador et al., 1997). K562 cells stably transfected with the α4 integrin chain gene (4M7 cells) were grown in RPMI 1640 medium (GIBCO BRL) supplemented with 10% FCS, 50 IU/ml penicillin, 50 μg/ml streptomycin, and 1 mg/ml of G418 (Calbiochem).
Antibodies and reagents
The TEA1/31 anti-VE cadherin and TS2/16 anti-β1 integrin (Yáñez-Mó et al., 1998), the HP2/1 anti-α4 integrin, TS1/11 anti-αL integrin, and TP1/24 anti-ICAM-3 (Serrador et al., 1997) mAb have been described elsewhere. The 4B9 and P8B1 (anti–VCAM-1), Hu5/3 (anti–ICAM-1), and 297S (antiphosphorylated forms of ERM proteins) mAb were provided by Dr. R.R. Lobb (Biogen Inc.), Dr. E.A. Wayner (Fred Hutchinson Cancer Research Center, Seattle, WA), Dr. F.W. Luscinskas (Brigham and Women's Hospital and Harvard Medical School, Boston, MA), and Dr. S. Tsukita (Faculty of Medicine, Kyoto University, Japan), respectively. The moesin-specific polyclonal antiserum 95/2 and the ezrin-polyclonal antiserum 90/3 have been previously described (Serrador et al., 2002). The anti-tubulin and -vinculin mAb were purchased from Sigma-Aldrich. The anti-talin polyclonal antibody (pAb) was a gift of Dr. K. Burridge (University of North Carolina, Chapel Hill, NC). The monoclonal IgG1,κ from the P3×63 myeloma cell line was used as negative control. Recombinant human fibronectin was purchased from Sigma-Aldrich. The p160 ROCK inhibitor Y-27632, the PI3K inhibitor Ly 294002, and the classical PKCs inhibitor Gö6976 were purchased from Calbiochem.
Recombinant DNA constructs and cell transfections
The fusion protein GST–VC, containing the cytoplasmic tail of VCAM-1, was obtained by PCR amplification using as template the human VCAM-1 cDNA, and [TATGGATCCAGAAAAGCCAACATGAAG] and [TGGAATTCATAGATGGGCATTTC] as 5′ and 3′ primers, respectively. The PCR product was cloned as a BamHI/EcoRI fragment into pGEX-4T (Pharmacia LKB Biotechnology). The cytoplasmic tail of ICAM-3 fused to GST (GST-IC3), and a truncated form of it (GST-Y9, Y490stop) have been described elsewhere (Serrador et al., 2002). Expression of GST fusion proteins in BL21 bacteria and purification were performed following the manufacturer's instructions.
VCAM-1–GFP and ICAM-1–GFP were obtained using the corresponding human cDNAs as templates to amplify by PCR the complete encoding region of these molecules without the stop codon. A Xho I site was added to the 5′ end and a Xma I site at the 3′ end of VCAM-1 cDNA. Likewise, HindIII and BamHI sites were added to the 5′ and 3′ ends of ICAM-1 cDNA, respectively. The PCR products were then cloned into pEGFP-N1 (CLONTECH Laboratories, Inc.) resulting in an in-frame fusion of enhanced GFP to the COOH terminus of VCAM-1 and ICAM-1. The VCAM-1- and ICAM-1–GFP proteins behaved similarly to the corresponding endogenous proteins in terms of their binding to ezrin and moesin. The generation of the GFP fusion construct containing the GFP cDNA inserted at the COOH-terminal end of the rat full-length moesin (moesin-GFP, residues 1–577) has been previously described (Amieva et al., 1999). α-actinin–GFP and paxillin-GFP were gifts of Dr. A.F. Horwitz (University of Virginia, Charlottesville, VA). VASP-GFP, PLCδ-PH-GFP, and GRP1-PH-GFP, were provided by Dr. J.V. Small (Institute of Molecular Biology, Salzburg, Austria), Dr. T. Balla (National Institutes of Health, Bethesda, MA), and Dr. A. Gray (University of Dundee, Dundee, U.K.), respectively.
Transiently transfected HUVEC were generated by electroporation at 200 V and 975 μF using a Gene Pulser (Bio-Rad Laboratories) and adding 20 μg of each DNA construct. These cells were used 24–48 h after transfection.
Flow cytometry analysis, immunofluorescence, and confocal microscopy
For flow cytometry analysis and immunofluorescence experiments, cells were treated as previously described (Yáñez-Mó et al., 1998). Rhodamine Red-X-Affinipure goat anti–mouse IgG (H+L), Alexa Fluor 488 rabbit anti–mouse or goat anti–rabbit IgG (H + L) conjugate highly crossadsorbed, Alexa Fluor 488 streptavidin, and Phalloidin Alexa Fluor 568 were used as fluorescent reagents (Molecular Probes). Staining with the 297S mAb was performed as previously described (Hayashi et al., 1999). Series of optical sections were obtained with a Leica TCS-SP confocal laser scanning unit equipped with Ar and He/Ne laser beams and attached to a Leica DMIRBE inverted epifluorescence microscope (Leica Microsystems), using a 63× oil immersion objective. Colocalization histograms were obtained using the Leica Confocal Software.
Immunoprecipitation, Western blot, in vitro translation, and protein binding assays
Lysates from activated HUVEC, immunoprecipitation, and Western blot were performed as described (Serrador et al., 2002). The pCR3 plasmids carrying the inserts of untagged moesin and ezrin NH2-terminal regions (amino acid residues 1–310) were transcribed, translated, and isotope labeled in vitro using a TNT-coupled rabbit reticulocyte lysate system (Promega). Then, binding assays using these isotope-labeled recombinant proteins and the GST fusion proteins (GST–VC, GST–IC3, GST–Y9, and GST alone) were performed as previously described (Serrador et al., 2002).
Adhesion and transendothelial migration assays
For cellular adhesion assays, HUVEC were grown to confluence in 96-microwell plates (Costar) and activated with TNF-α for 20 h. K562 cells were labeled with 1 μM of BCECF-AM for 15 min at 37°C, preincubated with different purified mAb, and allowed to adhere to activated HUVEC for 15 min at 37°C as previously described (Yáñez-Mó et al., 1998). Fluorescence intensity was measured in a microplate reader (Biotek FL500). T lymphoblast migration through a confluent monolayer of activated HUVEC was assayed in 3-μm pore Transwell cell culture chambers (Costar). HUVEC were seeded and grown to confluence on these Transwell inserts precoated with 1% gelatin and activated with TNF-α for 20 h. Cultured lymphoblasts (2 × 105 in 100 μl of complete 199 medium/well) were incubated with 10 μg/ml of different purified mAbs for 20 min at 4°C, and then added to the upper chambers. In the lower well, 600 μl of complete 199 medium were poured. Cells were incubated for 90 min at 37°C, and migrated cells were recovered from the lower chamber. The relative number of migrated cells was estimated by flow cytometry.
Time-lapse fluorescence confocal microscopy
HUVEC transfected with different GFP constructs were grown to confluence on glass-bottom dishes (WillCo Wells) precoated with Fn (20 μg/ml). Then, cells were activated with TNF-α for 20 h, and placed on the microscope stage. 4M7 cells or T lymphoblasts resuspended in 500 μl of complete 199 medium were added. Plates were maintained at 37°C in a 5% CO2 atmosphere using an incubation system (La-con GBr Pe-con GmbH). Confocal series of fluorescence and differential interference contrast (DIC) images, distanced 0.4 μm in the z axis, were simultaneously obtained at 30-s or 1-min intervals, with a 63× oil immersion objective. Images were processed and assembled into movies using the Leica Confocal Software.
Parallel plate flow chamber analysis of endothelial-PMN interactions
The parallel plate flow chamber used for leukocyte adhesion and transmigration under defined laminar flow has been described in detail (Luscinskas et al., 1994). PBLs (106/ml) were drawn across activated confluent monolayers at an estimated wall shear stress of 1.8 dynes/cm2 for perfusion times from 30 s to 10 min. Lymphocyte rolling on the endothelium were easily visualized as they travelled more slowly than free-flowing cells. Lymphocytes were considered to be adherent after 20 s of stable contact with the monolayer. Transmigrated lymphocytes were determined as being beneath the endothelial monolayer. Lymphocytes were considered to be detached when they returned to free-flowing after having been completely arrested on endothelium. The number of rolling, adhered, transmigrated, and detached cells was quantified by direct visualization of 4 different fields (40× phase-contrast objective) at each time point of every independent experiment. Coverslips were fixed immediately in PFA 4% at room temperature for 10 min, washed with HBSS, and stained for VCAM-1, ICAM-1, or ezrin.
Online supplemental material
All videos are available at http://www.jcb.org/cgi/content/full/jcb.200112126/DC1. Video 1 (corresponding to Fig. 4 A, a–f) shows a lymphoblast that contacts with a moesin–GFP-transfected endothelial cell, and immediately transmigrates and moves beneath the endothelium. Moesin is clustered around the lymphoblast along the whole process. Video 2 (corresponding to Fig. 4 A, g–l), shows a lymphoblast (upper site) that adheres to, spreads on, and moves toward a lateral junction of the VCAM-1–GFP-transfected endothelial cell. It then transmigrates and moves beneath the endothelium, whereas another lymphoblast (lower site) remains spread on the endothelial cell. VCAM-1 is clustered around lymphoblasts adhered to the apical surface of endothelium (arrows), but it is not concentrated around migrating lymphocytes beneath the endothelium (arrowheads). Video 3 (corresponding to Fig. 4 B), shows a lymphoblast that adheres to, spreads on, and moves toward a lateral junction of the ICAM-1–GFP-transfected endothelial cell (black arrows). It then transmigrates and moves beneath the endothelium (white arrows). ICAM-1 is clustered around the lymphoblast along the whole process. In all videos, the apical endothelial surface is at a plane remote from the observer.
Supplemental Material
Acknowledgments
We thank Drs. R. González-Amaro, D. Sancho, and P. Roda-Navarro for critical reading of the manuscript.
This work was supported by grants SAF99-0034-C01 and FEDER 2FD97-068-C02-02 from the Ministerio de Educación, and grant QLRT-1999-01036 from the European Community to F. Sánchez-Madrid, and a fellowship from the Ministerio de Educación to O. Barreiro.
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: ERM, ezrin, radixin, and moesin; DIC, differential interference contrast; GFP, green fluorescent protein; GST, glutathione-S-transferase; HUVEC, human umbilical vein endothelial cells; ICAM, intercellular adhesion molecule; pAb, polyclonal antibody; PBL, peripheral blood lymphocyte; PI(4,5)P2, phosphatidylinositol 4,5-biphosphate; TEM, transendothelial migration; VCAM, vascular cell adhesion molecule.
References
- Allen, L.-A.H., and A. Aderem. 1996. Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor-mediated phagocytosis in macrophages. J. Exp. Med. 184:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon, R. 2001. Encephalitogenic lymphoblast recruitment to resting CNS microvasculature: a natural immunosurveillance mechanism? J. Clin. Invest. 108:517–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieva, M.R., P. Litman, L. Huang, E. Ichimaru, and H. Furthmayr. 1999. Disruption of dynamic cell surface architecture of NIH3T3 fibroblasts by the N-terminal domains of moesin and ezrin: in vivo imaging with GFP fusion proteins. J. Cell Sci. 112:111–125. [DOI] [PubMed] [Google Scholar]
- Barret, C., C. Roy, P. Montcourrier, P. Mangeat, and V. Niggli. 2000. Mutagenesis of the phosphatidilinositol 4,5-biphosphate (PIP[2]) binding site in the NH(2)-terminal domain of ezrin correlates with its altered cellular distribution. J. Cell Biol. 151:1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho, R.J., M. Teruel, R. Dierckman, R. Anderson, A. Wells, J.D. York, T. Meyer, and S. Grinstein. 2000. Localized biphasic changes in phosphatidylinositol-4,5-biphosphate at sites of phagocytosis. J. Cell Biol. 151:1353–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher, A., D. Chambers, R. Nguyen, and D. Reczek. 2000. ERM-Merlin and EBP50 protein families in plasma membrane organization and function. Annu. Rev. Cell Dev. Biol. 16:113–143. [DOI] [PubMed] [Google Scholar]
- Butcher, E.C. 1991. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 67:1033–1036. [DOI] [PubMed] [Google Scholar]
- Carlos, T.M., and J.M. Harlan. 1994. Leukocyte-endothelial adhesion molecules. Blood. 84:2068–2101. [PubMed] [Google Scholar]
- Caron, E., and A. Hall. 1998. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 282:1717–1721. [DOI] [PubMed] [Google Scholar]
- Carter, R.A., and I.P. Wicks. 2001. Vascular cell adhesion molecule 1 (CD 106). A multifaceted regulator of joint inflammation. Arthritis Rheum. 44:985–994. [DOI] [PubMed] [Google Scholar]
- Castellano, F., C. Le Clainche, D. Patin, M.-F. Carlier, and P. Chavrier. 2001. A WASp-VASP complex regulates actin polymerization at the plasma membrane. EMBO J. 20:5603–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, J.R., S.J. Hyduk, and M.I. Cybulsky. 2000. α4β1 integrin/VCAM-1 interaction activates αLβ2 integrin-mediated adhesion to ICAM-1 in human T cells. J. Immunol. 164:746–753. [DOI] [PubMed] [Google Scholar]
- Cox, D., C.C. Tseng, G. Bjekic, and S. Greenberg. 1999. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J. Biol. Chem. 274:1240–1247. [DOI] [PubMed] [Google Scholar]
- Cybulsky, M.I., K. Iiyama, H. Li, S. Zhu, M. Chen, M. Iiyama, V. Davis, J.C. Gutierrez-Ramos, P.W. Connelly, and D.S. Milstone. 2001. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Invest. 107:1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimini, G., and P. Chavrier. 2000. Function of Rho family proteins in actin dynamics during phagocytosis and engulfment. Nat. Cell Biol. 2:E191–E196. [DOI] [PubMed] [Google Scholar]
- Defacque, H., M. Egeberg, A. Habermann, M. Diakonova, C. Roy, P. Mangeat, W. Voelter, G. Marriot, J. Pfannstiel, H. Faulstich, and G. Griffiths. 2000. Involvement of ezrin/moesin in de novo actin assembly on phagosomal membranes. EMBO J. 19:199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi, Y., M. Itoh, S. Yonemura, S. Ishihara, H. Takano, T. Noda, and S. Tsukita. 1999. Normal development of mice and unimpaired cell adhesion/cell motility/actin-based cytoskeleton without compensatory up-regulation of ezrin or radixin in moesin gene knockout. J. Biol. Chem. 274:2315–2321. [DOI] [PubMed] [Google Scholar]
- Elices, M.J., L. Osborn, Y. Takada, C. Crouse, S. Luhowskyj, M.E. Hemler, and R.R. Lobb. 1990. VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell. 60:577–584. [DOI] [PubMed] [Google Scholar]
- Faveeuw, C., M.E. Di Mauro, A.A. Price, and A. Ager. 2000. Roles of alpha(4) integrins/VCAM-1 and LFA-1/ICAM-1 in the binding and transendothelial migration of T lymphocytes and T lymphoblasts across high endothelial venules. Int. Immunol. 12:241–251. [DOI] [PubMed] [Google Scholar]
- González-Amaro, R., and F. Sánchez-Madrid. 1999. Cell adhesion molecules: selectins and integrins. Crit. Rev. Immunol. 19:389–429. [PubMed] [Google Scholar]
- Gray, A., J. van der Kaay, and C.P. Downes. 1999. The pleckstrin homology domains of protein kinase B and GRP1 (general receptor for phosphoinositides-1) are sensitive and selective probes for the cellular detection of phosphatidylinositol 3,4-bisphosphate and/or phosphatidylinositol 3,4,5-triphosphate in vivo. Biochem. J. 344:929–936. [PMC free article] [PubMed] [Google Scholar]
- Hayashi, K., S. Yonemura, T. Matsui, and S. Tsukita. 1999. Immunofluorescence detection of ezrin/radixin/moesin (ERM) proteins with their carboxil-terminal threonine phosphorylated in cultured cells and tissues. J. Cell Sci. 112:1149–1158. [DOI] [PubMed] [Google Scholar]
- Heiska, L., K. Alfthan, M. Gronholm, P. Vilja, A. Vaheri, and O. Carpen. 1998. Association of ezrin with intercellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2). Regulation by phosphatidylinositol 4, 5-bisphosphate. J. Biol. Chem. 273:21893–21900. [DOI] [PubMed] [Google Scholar]
- Helander, T.S., O. Carpen, O. Turunen, P.E. Kovanen, A. Vaheri, and T. Timonen. 1996. ICAM-2 redistributed by ezrin as a target for killer cells. Nature. 382:265–268. [DOI] [PubMed] [Google Scholar]
- Hirao, M., N. Sato, T. Kondo, S. Yonemura, M. Monden, T. Sasaki, Y. Takai, and S. Tsukita. 1996. Regulation mechanism of ERM (ezrin/radixin/moesin) protein/plasma membrane association: possible involvement of phosphatidylinositol turnover and Rho-dependent signaling pathway. J. Cell Biol. 135:37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koni, P.A., S.K. Joshi, U.A. Temann, D. Olson, L. Burkly, and R.A. Flavell. 2001. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J. Exp. Med. 193:741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuker, C.E., M. Labow, W. Müller, and N. Wagner. 2001. Neonatally induced inactivation of the vascular cell adhesion molecule 1 gene impairs B cell localization and T cell-dependent humoral immune response. J. Exp. Med. 193:755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzon, P., E. Vecile, E. Nardon, E. Ferrero, J.M. Harlan, F. Tedesco, and A. Dobrina. 1998. Endothelial cell E- and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors. J. Cell Biol. 142:1381–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscinskas, F.W., G.S. Kansas, H. Ding, P. Pizcueta, B.E. Schleiffenbaum, T.F. Tedder, and M.A. Gimbrone Jr. 1994. Monocyte rolling, arrest and spreading on IL-4 activated vascular endothelium under flow is mediated via sequential action of L-selectin, beta-1-integrins, and beta-2-integrins. J Cell Biol. 125:1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat, P., C. Roy, and M. Martin. 1999. ERM proteins in cell adhesion and membrane dynamics. Trends Cell Biol. 9:187–192. [DOI] [PubMed] [Google Scholar]
- Marlin, S.D., and T.A. Springer. 1987. Purified inter-cellular adhesion molecule (ICAM-1) is a ligand for lymphocyte-associated antigen 1 (LFA1). Cell. 51:813–819. [DOI] [PubMed] [Google Scholar]
- Matsui, T., M. Maeda, Y. Doi, S. Yonemura, M. Amano, K. Kaibuchi, and S. Tsukita. 1998. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J. Cell Biol. 140:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May, R.C., and L.M. Machesky. 2001. Phagocytosis and the actin cytoskeleton. J. Cell Sci. 114:1061–1077. [DOI] [PubMed] [Google Scholar]
- Meerschaert, J., and M.B. Furie. 1995. The adhesion molecules used by monocytes for migration across endothelium include CD11a/CD18, CD11b/CD18, and VLA-4 on monocytes and ICAM-1, VCAM-1, and other ligands on endothelium. J. Immunol. 154:4099–4112. [PubMed] [Google Scholar]
- Menager, C., J. Vassy, C. Doliger, Y. Legrand, and A. Karniguian. 1999. Subcellular localization of RhoA and ezrin at membrane ruffles of human endothelial cells: differential role of collagen and fibronectin. Exp. Cell Res. 249:221–230. [DOI] [PubMed] [Google Scholar]
- Muñoz, M., J. Serrador, F. Sanchez-Madrid, and J. Teixido. 1996. A region of the integrin VLA alpha 4 subunit involved in homotypic cell aggregation and in fibronectin but not vascular cell adhesion molecule-1 binding. J. Biol. Chem. 271:2696–2702. [DOI] [PubMed] [Google Scholar]
- Nakamura, F., L. Huang, K. Pestonjamasp, E.J. Luna, and H. Furthmayr. 1999. Regulation of F-actin binding to platelet moesin in vitro by both phosphorylation of threonine 558 and polyphosphatidylinositides. Mol. Biol. Cell. 10:2669–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer-Marks, N., L.S. Davis, D.T. Bogue, J. Ramberg, and P.E. Lipsky. 1991. Differential utilization of ICAM-1 and VCAM-1 during the adhesion and transendothelial migration of human T lymphocytes. J. Immunol. 147:2913–2921. [PubMed] [Google Scholar]
- Randolph, G.J., and M.B. Furie. 1996. Mononuclear phagocytes egress from an in vitro model of the vascular wall by migrating across endothelium in the basal to apical direction: role of intercellular adhesion molecule 1 and the CD11/CD18 integrins. J. Exp. Med. 183:451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, D.M., V. Grabovsky, R. Alon, and M.H. Ginsberg. 2001. The affinity of integrin α4β1 governs lymphocyte migration. J. Immunol. 167:2824–2830. [DOI] [PubMed] [Google Scholar]
- Sechi, A.S., and J. Wehland. 2000. The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P2 influences cytoskeletal protein activity at the plasma membrane. J. Cell Sci. 113:3685–3695. [DOI] [PubMed] [Google Scholar]
- Serrador, J.M., J.L. Alonso-Lebrero, M.A. del Pozo, H. Furthmayr, R. Schwartz-Albiez, J. Calvo, F. Lozano, and F. Sánchez-Madrid. 1997. Moesin interacts with the cytoplasmic region of intercellular adhesion molecule-3 and is redistributed to the uropod of T lymphocytes during cell polarization. J. Cell Biol. 138:1409–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrador, J.M., M. Vicente-Manzanares, J. Calvo, O. Barreiro, M.C. Montoya, R. Schwartz-Albiez, H. Furthmayr, F. Lozano, and F. Sanchez-Madrid. 2002. A novel serine-rich motif in the intercellular adhesion molecule-3 is critical for its ERM-directed subcellular targeting. J. Biol Chem. 277:10400–10409. [DOI] [PubMed] [Google Scholar]
- Shaw, R.J., M. Henry, F. Solomon, and T. Jacks. 1998. RhoA-dependent phosphorylation and relocalization of ERM proteins into apical membrane/actin protrusions in fibroblasts. Mol. Biol. Cell. 9:403–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, K., T. Sasaki, A. Mammoto, K. Takaishi, T. Kameyama, S. Tsukita, and Y. Takai. 1997. Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J. Biol. Chem. 272:23371–23375. [DOI] [PubMed] [Google Scholar]
- Turunen, O., T. Wahlstrom, and A. Vaheri. 1994. Ezrin has a COOH-terminal actin-binding site that is conserved in the ezrin protein family. J. Cell Biol. 126:1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Várnai, P., K.I. Rother, and T. Balla. 1999. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton's tyrosine kinase pleckstrin homology domain visualized in single living cells. J. Biol. Chem. 274:10983–10989. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard, B., L. Williams, and A.J. Ridley. 1999. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J. Cell Biol. 145:1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez-Mó, M., A. Alfranca, C. Cabañas, M. Marazuela, R. Tejedor, M.A. Ursa, L.K. Ashman, M.O. de Landázuri, and F. Sánchez-Madrid. 1998. Regulation of endothelial cell motility by complexes of tetraspan molecules CD81/TAPA-1 and CD151/PETA-3 with alpha3beta1 integrin localized at endothelial lateral junctions. J. Cell Biol. 141:791–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura, S., M. Hirao, Y. Doi, N. Takahashi, T. Kondo, and S. Tsukita. 1998. Ezrin/radixin/moesin (ERM) proteins bind to a positively charged amino acid cluster in the juxta-membrane cytoplasmic domain of CD44, CD43, and ICAM-2. J. Cell Biol. 140:885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura, S., and S. Tsukita. 1999. Direct involvement of ezrin/radixin/moesin (ERM)-binding membrane proteins in the organization of microvilli in collaboration with activated ERM proteins. J. Cell Biol. 145:1497–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, M., W.F. Westlin, N. Wang, D.E. Ingber, A. Rosenzweig, N. Resnick, and M.A.J. Gimbrone. 1996. Leukocyte adhesion to vascular endothelium induces E-selectin linkage to the actin cytoskeleton. J. Cell Biol. 133:445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.