

Abstract
The rat homologue of a mitochondrial ATP-dependent protease Lon was cloned from cultured astrocytes exposed to hypoxia. Expression of Lon was enhanced in vitro by hypoxia or ER stress, and in vivo by brain ischemia. These observations suggested that changes in nuclear gene expression (Lon) triggered by ER stress had the potential to impact important mitochondrial processes such as assembly and/or degradation of cytochrome c oxidase (COX). In fact, steady-state levels of nuclear-encoded COX IV and V were reduced, and mitochondrial-encoded subunit II was rapidly degraded under ER stress. Treatment of cells with cycloheximide caused a similar imbalance in the accumulation of COX subunits, and enhanced mRNA for Lon and Yme1, the latter another mitochondrial ATP-dependent protease. Furthermore, induction of Lon or GRP75/mtHSP70 by ER stress was inhibited in PERK (−/−) cells. Transfection studies revealed that overexpression of wild-type or proteolytically inactive Lon promoted assembly of COX II into a COX I–containing complex, and partially prevented mitochondrial dysfunction caused by brefeldin A or hypoxia. These observations demonstrated that suppression of protein synthesis due to ER stress has a complex effect on the synthesis of mitochondrial-associated proteins, both COX subunits and ATP-dependent proteases and/or chaperones contributing to assembly of the COX complex.
Keywords: hypoxia; ER stress; protein synthesis; ATP-dependent protease; molecular chaperone
Introduction
Hypoxia/ischemia imposes severe metabolic stress on intracellular organelles (Paschen, 2000). Previous studies from our and other laboratories have demonstrated that several ER-associated proteins or ER stress-related proteins such as GRP78, GRP94, ORP150, RA410, Sec61 complex, and SERP1/Ramp4 were induced under such conditions (Lowenstein et al., 1994; Kuwabara et al., 1996; Matsuo et al., 1997; Yamaguchi et al., 1999). Suppression of GRP78 or ORP150 transcripts increased vulnerability to hypoxic/ischemic stress, whereas overexpression of these genes resulted in cytoprotection (Liu et al., 1997; Ozawa et al., 1999; Tamatani et al., 2001). The crucial roles of ER-resident proteins under stress were also reported in a range of pathological conditions such as diabetes, cancer, and neurodegenerative diseases (Katayama et al., 1999; Harding et al., 2001; Ozawa et al., 2000). These findings strongly support the hypothesis that events in the ER might have an important impact on cellular adaptation to environmental challenges.
The ER is the organelle in which membrane and secretory proteins achieve correct folding and oligomerization. Once cells are exposed to stresses such as glucose starvation, inhibition of protein glycosylation, disturbance of Ca2+ homeostasis (ER stress), or oxygen deprivation, unfolded proteins accumulate in the ER and eukaryotic cells respond by several mechanisms: (a) transcriptional induction; (b) translational attenuation; and (c) degradation (Mori, 2000). Attenuation of protein synthesis in response to ER stress occurs to lessen the load of protein entering the ER, and this pathway requires an activation of the ER-resident protein kinase, PERK (Harding et al., 1999). Although general suppression of protein synthesis improved cell viability under ER stress (Harding et al., 2000), the detailed mechanisms involved, especially at the level of translational products destined for particular organelles, have not been elucidated.
Here we describe a novel signaling pathway from the ER to mitochondria through suppression of protein synthesis. Under ER stress, expression and assembly of cytochrome c oxidase (COX)*are disturbed, whereas mitochondrial ATP-dependent proteases/chaperones are induced, at least in part, to improve assembly of these complexes, and, potentially, to sustain mitochondrial function.
Results
Identification and molecular cloning of a rat mitochondrial ATP-dependent protease Lon
Cultured rat astrocytes were exposed to hypoxia, and seven differentially expressed genes were identified using the equalization of cDNAs, subtractive hybridization, and differential display (ESD) (Suzuki et al., 1996). One of these genes, GT1, showed 85% homology with human mitochondrial Lon protease (Wang et al., 1993; Amerik et al., 1994). To obtain a full-length cDNA for rat Lon, a rat brain cDNA library was screened using GT1 as a probe. A cDNA (2.97 kb) comprising the entire ORF was cloned. Rat Lon is a polypeptide of 950 amino acids (aa) with 95 and 86% homology to the mouse and human counterparts, respectively, at the protein level. The key structural features of Lon protease were all conserved in the rat homologue: the NH2-terminal sequence met criteria for a mitochondrial targeting signal (von Heijne et al., 1989); a Walker-type ATP binding domain (aa 513–520) was observed; and a serine residue was found at the active site of the putative protease domain (aa 845). The aa sequences of rat, mouse, and human Lon are available from GenBank/EMBL/DDBJ and SWISS-PROT under the accession numbers AB064323, AK004820, and P36776, respectively.
Expression of Lon in response to cell stress
Northern blot analysis using total RNA (10 μg) from cultured astrocytes subjected to oxygen deprivation showed an increase in the level of Lon transcript after 16–22 h (4.5-fold increase at the latter time point), and a subsequent decrease in Lon mRNA within 4 h of reoxygenation (Fig. 1 A). The temporal pattern of Lon mRNA expression in hypoxia paralleled that of GRP78 mRNA, a molecular chaperone in the ER, although GRP78 was induced to a greater extent than Lon (Fig. 1 A). Prompted by the results of these studies, the expression of Lon under conditions of hypoxia or other stimuli was investigated in general cell lines. When HeLa cells were exposed to hypoxia, Lon mRNA level was increased by 3.0–3.5-fold (unpublished data). Treatment of HeLa cells with tunicamycin (Tm), brefeldin A (BFA) or thapsigargin (Tg), all of which cause accumulation of unfolded polypeptides in the ER (ER stress), resulted in 3.8–4.3-fold increases in Lon transcript levels (Fig. 1 B, lanes 1–4). In contrast, heat shock (HS) or sodium arsenite (AS) had no effect (Fig. 1 B, lanes 5–7). Modulation of the expression of Lon mRNA in response to ER stress was compared with that of other molecular chaperones associated with the ER or mitochondria (GRP78, GRP75/mtHSP70, and HSP60), and with another mitochondrial ATP-dependent protease, Yme1. Transcripts of GRP75/mtHSP75, but not HSP60, were enhanced moderately (twofold increase) in response to ER stress, consistent with a previous report (Mizzen et al., 1989). In addition, Yme1 mRNA was also enhanced by ER stress (3.6–4.5-fold increase; Fig. 1 B).
Figure 1.
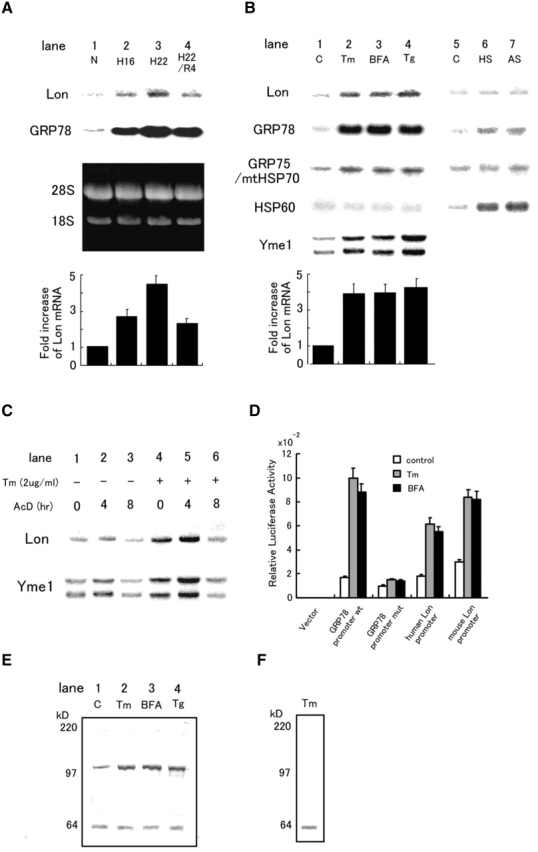
Expression of Lon in response to cell stress. (A) Expression of Lon and GRP78 mRNA in cultured astrocytes subjected to various stresses for the indicated periods: N, normoxia for 22 h; H16 and H22, hypoxia for 16 and 22 h, respectively; R, hypoxia for 22 h followed by reoxygenation for 4 h. The third (dark) panel shows the ethidium bromide stained gel. Quantification of relative intensities for Lon mRNA was accomplished by laser densitometry, and is shown as fold increase (normoxic controls were arbitrarily assigned a value of 1). The values shown are means ± SD of three experiments. (B) Expression of Lon, GRP78, GRP75/mtHSP70, HSP60, and Yme1 mRNA in HeLa cells subjected to other forms of stress. Tm (2 μg/ml for 16 h); BFA (5 μg/ml for 16 h); Tg (no treatment). (C) Effects of actinomycin D on the expression of Lon and Yme1. HeLa cells were exposed to actinomycin D (AcD; 5 μg/ml) for the indicated times in the presence/absence of tunicamycin (cells were pretreated with the latter for 16 h) and Northern blotting was performed. (D) Relative luciferase activity of Lon promoter–luciferase constructs. Human or mouse Lon promoter, wild-type or mutant GRP78 promoter in the pGL3 basic vector, or pGL3basic vector alone was transfected with pRL-SV40 into HeLa cells. Relative luciferase activity was measured as described in the text. (E) Expression of Lon protein in response to ER stress. HeLa cells were treated with Tm, BFA, or Tg as described above, and Western blotting was performed with anti-Lon antibody 665. Migration of simultaneously run molecular weight standards is indicated on the far left in kD. (F) Competition of Lon antigen with a 100-fold excess of the synthetic Lon peptide was examined using cell extracts from HeLa cells treated with Tm as described above.
To determine whether the enhanced levels of Lon mRNA were due to an increase in the rate of transcription or a change in mRNA stability, HeLa cells exposed to tunicamycin or medium alone were treated with actinomycin D (Fig. 1 C). Stability of Lon or Yme1 transcripts was not changed in the presence or absence of tunicamycin; in each case, ∼50% of the original mRNA level was present after 8 h of treatment with actinomycin D. When actinomycin D was added to cultures prior to tunicamycin treatment, there was no enhancement of Lon or Yme1 transcript level by Tm (unpublished data). To directly assess activation of transcription at the Lon promoter, promoter–reporter (luciferase) constructs were transiently transfected into HeLa cells (Fig. 1 D). Transient transfection of such constructs bearing promoters for either murine Lon (−2559/+24) or human Lon (−584/+24) demonstrated increased luciferase activity in each case with either tunicamycin or BFA as the agent for induction of ER stress (Fig. 1 D). Similar results were obtained with a GRP78 (−304/+7) promoter–luciferase construct under the same conditions (Fig. 1 D). Mutational inactivation of the ER stress response element (ERSE) in the GRP78 promoter blocked its induction in response to ER stress, as reported previously (Yoshida et al., 1998). Induction of transcription from the human Lon, murine Lon or GRP78 promoters (wild-type) in the presence of tunicamycin corresponded to 3.2-, 2.9-, and 5.4-fold, respectively. Consistent with these results, nuclear runoff analysis displayed an increase in the rate of Lon transcription in the presence of ER stress-inducing agents (unpublished data).
To analyze Lon expression at the protein level, Western blotting was performed using anti-Lon antibody (#665) generated against an 18-aa Lon-derived polypeptide, which recognizes both human and rat Lon antigens. Immunoblotting of cultured cell extracts with this antibody displayed an immunoreactive band with Mr. of ∼100 kD as well as a band of ∼64 kD (Fig. 1 E). The appearance of the 100- kD band, but not the 64-kD band, was blocked by an excess amount of Lon peptide immunogen (Fig. 1 F), suggesting that only the 100-kD band represented Lon antigen. ER stress-induced elevation of Lon mRNA was accompanied by a 3.1–3.5-fold increase in the level of Lon antigen in HeLa cells (Fig. 1 E) and in astrocytes (unpublished data).
The results of the above in vitro studies were extended to the in vivo setting using the rat middle cerebral artery (MCA) occlusion model. 8 h after MCA occlusion, Northern blotting of total RNA extracted from the ischemic brain showed increased Lon transcript level, compared with the RNA from the brain after a sham procedure (Fig. 2 A). In situ hybridization indicated increased Lon mRNA level in the ipsilateral cerebral cortex in response to ischemia, especially in neurons (Fig. 2 B). Western blotting with anti-Lon antibody 665 also confirmed the enhancement of Lon antigen in the ischemic rat brain (Fig. 2 C).
Figure 2.

Expression of Lon in the ischemic rat brain. (A) Total RNA (10 μg) from rat brains after MCA occlusion (8 h; I, ischemia) or sham operation (8 h; C, control) was subjected to Northern blotting (top). The bottom panel shows the ethidium bromide stained gel. (B) Brain slices after MCA occlusion (8 h) were studied by in situ hybridization with riboprobes derived from the Lon cDNA. Bar, 200 μm. (C) Expression of Lon antigen after MCA occlusion. Brains homogenates (50 μg protein) after MCA operation (8 h, I) or sham operation (8 h, C) were subjected to Western blotting with anti-Lon antibody 665. Migration of simultaneously run molecular weight standards is indicated on the far left in kD.
ER stress and expression of COX subunits
To assess the contribution of Lon to mitochondrial adaptation to ER stress, COX was employed as a model system. HeLa cells were treated with Tm, BFA, or Tg. Steady-state levels of nuclear-encoded COX IV and COX V were reduced to 25–35% of those observed in untreated controls, whereas levels of mitochondrial-encoded COX I and COX II were not affected (Fig. 3, A and B). These findings concerning COX IV and V were clearly evident when cultures underwent a 16-h period ER stress (Fig. 3 C). After 24 h, level of COX II, but not of COX I, was also reduced to 76% (Fig. 3 C). Further evaluation of the degradation of COX subunits under ER stress was performed with pulse-chase analysis applied to cell lysis followed by immunoprecipitation with antibodies against each subunit and autoradiography (Fig. 3 D). As predicted by the above results, translation of COX IV protein was reduced in response to ER stress (∼10% of control; compare with Fig, 3 D, lane 1 and lane 5, bottom row), whereas stability was unchanged (Fig. 3 D, lanes 5–8, bottom column). In contrast, COX II, but not COX I, was rapidly degraded in response to ER stress (the half-life of COX II antigen was 8 and 3 h, under control and stress-induced conditions, respectively; Fig. 3 D, top and middle rows). To address the assembly state of COX II, solubilized mitochondria from HeLa cells treated with or without Tm were incubated with different concentrations of trypsin for 30 min on ice, followed by Western blotting (Rep et al., 1996). COX II protein was more sensitive to degradation by trypsin in tunicamycin-treated cells; 45% of COX II antigen harvested from control HeLa cultures was degraded by trypsin (250 μg/ml), whereas 78% of COX II antigen from Tm-treated cells was degraded (Fig. 3 E). In contrast, COX I and COX IV proteins displayed similar levels of resistance to trypsin whether Tm was present or not (∼60 and 80% of antigens were resistant to 250 μg/ml of trypsin under both conditions, respectively) (Fig. 3 E). Similar results were obtained when HeLa cells were exposed to BFA or Tg (unpublished data). Taken together, these observations indicated that ER stress results in the presence of a pool of COX II displaying increased sensitivity to trypsin degradation, potentially due to accumulation of free COX II subunits. The latter might occur because of the inability of COX II produced under conditions of ER stress to assemble effectively into the multisubunit structure of COX.
Figure 3.

Effects of ER stress on the expression of COX subunits. (A) HeLa cells were treated with Tm, BFA, Tg, or cultured in medium alone (C) for 16 h as described in the legend for Fig. 1. Western blotting was then performed with antibodies against COX I, II, IV, V, or KDEL. Sites of primary antibody binding were identified using alkaline phosphatase-conjugated secondary antibodies. (B) Quantification of band intensities in A was accomplished by laser densitometry. Percent band intensities under ER stress (A, lanes 2–4) compared with control (A, lane1) are shown. The values shown are means ± SD of three experiments. (C) Time course of changes in COX subunit expression in response to Tm treatment. Western blotting was performed as described in A. (D) Turnover of COX subunits under ER stress. Pulse-chase analysis was performed using HeLa cells in the presence of Tm (16 h pretreatment with Tm) or in medium alone, as described in the text, and immunoprecipitation was performed with the indicated antibodies followed by autoradiography. (E) Effects of Tm treatment (16 h) on the trypsin sensitivity of COX I and II. Mitochondria were isolated as described in the text and protein extracts were incubated with the indicated concentration of trypsin for 30 min on ice. Western blotting was then performed and sites of primary antibody binding were determined by the ECL method.
Suppression of protein synthesis and expression of COX subunits and Lon. To analyze whether the effects of ER stress on COX and Lon were mediated by suppression of protein synthesis (which also accompanies hypoxia), cycloheximide, an inhibitor of protein synthesis in the cytosol, was used. In our system, treatment of cultures cycloheximide, either 0.2 or 2 μg/ml, decreased protein synthesis to 52 and 5% of control levels, respectively, based on the analysis of TCA precipitable radioactivity (Hori et al., 1994). Steady-state levels of COX IV antigen were reduced in a dose-dependent manner when HeLa cells were treated with cycloheximide for 4 h (Fig. 4 A, lanes 1–3). After 8 h of cycloheximide treatment, levels of COX II also decreased slightly (Fig. 4 A, lane 6). By pulse-chase analysis, it was evident that COX II was rapidly degraded in cells exposed to cycloheximide (Fig. 4 B). When cells were treated with cycloheximide (Fig. 4 C) or another protein synthesis inhibitor, anisomycin (unpublished data), Lon and Yme1 transcript levels were elevated by ∼4.5-fold. In contrast, GRP78 transcript levels, which are directly regulated by the unfolded protein response (UPR) (Yoshida et al., 1998), increased by only ∼1.4-fold after 16 h of treatment with cycloheximide (Fig. 4 C). Further studies with mouse embryonic fibroblasts revealed that PERK, a protein kinase in the ER (Harding et al., 1999), is required for the induction of Lon and GRP75/mtHSP70 under ER stress (Fig. 4 D).
Figure 4.

The effects of suppression of protein synthesis on expression of COX subunits and Lon. (A) Western blotting was performed as described in Fig. 3 A after exposure of HeLa cells to cycloheximide for 4 h (lanes 1–3) or 8 h (lanes 4–6). (B) Turnover of COX subunits in the presence of cycloheximide. Pulse-chase analysis was performed with HeLa cells exposed to cycloheximide (4 h pretreatment with cycloheximide) or in medium alone. Immunoprecipitation was then performed as described in the legend to Fig. 3 D. (C) Expression of Lon, Yme1, and GRP78 in the presence of cycloheximide. Northern blotting was performed using total RNA (10 μg) from HeLa cells exposed to cycloheximide (2 μg/ml) for the indicated times. (D) Requirement of PERK for the expression of Lon and GRP75/mtHSP75. Northern blotting was performed with total RNA from wild-type or PERK(−/−) mouse embryonic fibroblasts after treating cells with Tm or Tg for the indicated times. The bottom panel shows the amounts of β-actin transcript as a control.
The effects of Lon overexpression on the assembly and degradation of COX II. To study the effects of enhanced expression of Lon on the stability and degradation of COX subunits, wild-type Lon and two mutants were generated by site-directed mutagenesis of serine at the protease active site (S845N) and lysine at the ATP binding site (K519N) (Rep et al., 1996). The latter two mutants have been characterized previously; mutation of Lon at the ATP binding site inhibits both the ATPase and protease activity, whereas mutation of the protease active site maintains ATPase activity (van Dijl et al., 1998). For our experiments, all constructs were tagged with FLAG so that expression could be monitored with both anti-Lon and anti-FLAG antibodies. Lon was expressed along with the hygromycin resistance gene under control of the SRα promoter in the plasmid pME18Sf(+)/hygro.
When wild-type or mutant forms (S845N and K519N) of Lon were transiently transfected into 293T cells, wild-type and S845N Lon antigens were detected as a single band in the mitochondrial matrix fraction (Fig. 5 A). however, K519N Lon antigen was detected in both mitochondrial matrix and cytosol fractions, and there was a poorly defined smear of FLAG immunoreactivity in the upper portion of the membrane, suggesting that mitochondrial import and oligomerization of K519N Lon may be impaired (Fig. 5 A). Therefore, wild-type and S845N Lon were used for further studies. Transfection efficiency into 293T cells was ∼70%, and the level of Lon antigen was 4-5-fold higher in transfectants compared with mock-transfected and nontransfected controls (Fig. 5 B). Resistance of COX I and II antigens, isolated from mitochondria of 293T cells, to degradation by trypsin was employed to monitor their assembly into COX. After transfection, cells were treated with Tm and protease degradation were performed. Trypsin resistance of COX II was enhanced in 293T cells transfected with the plasmid encoding wild-type or S845N compared to mock-transfected controls (Fig. 5 C). Quantative analysis of data from three experiments with Tm-treated cultures demonstrated that the percentages of COX II antigen resistant to tyrpsin (250 μg/ml) were 52, 76, and 19% in wild-type, S845N, and mock-transfected cells, respectively (Fig. 5 D).
Figure 5.
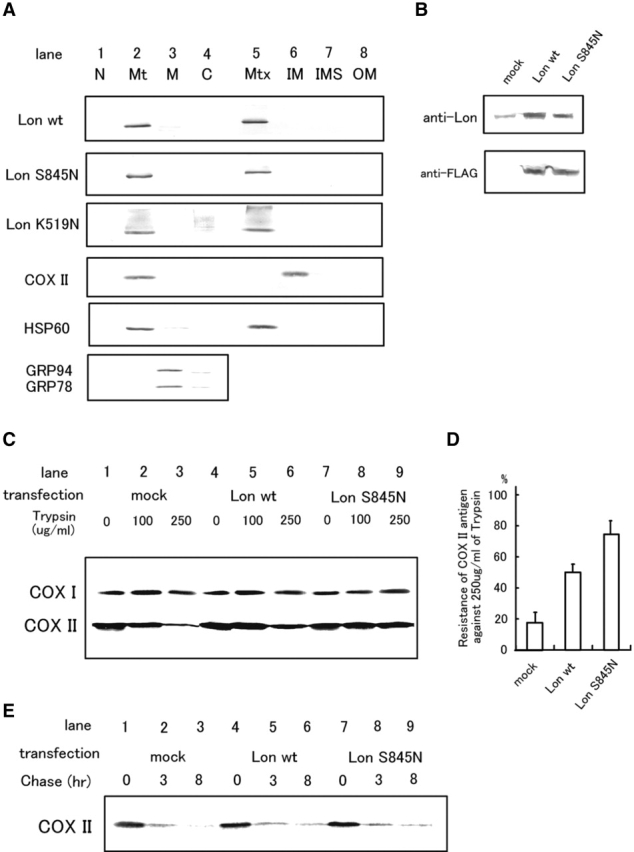
The effects of transient overexpression of Lon on the assembly and degradation of COX II. (A) Expression and localization of ectopically expressed Lon antigens. Each construct (wild-type, S845N, or K519N Lon) was inserted in pME18Sf+/hygro and transfected into 293T cells. Cultures were harvested after 48 h, and crude and submitochondrial fractions were obtained as described in the text. Western blotting was then performed with anti-FLAG (for ectopically expressed Lon), anti-COX II, anti-HSP60, or anti-KDEL antibodies. C, cytosol; IM, inner membrane; IMS, inter-membrane space; M, microsome; Mt, mitochondria; Mtx, matrix; N, nucleus; and OM, outer membrane. (B) Detection of endogenous and ectopically expressed Lon antigens. Western blotting using whole-cell lysates from mock, wild-type, or S845N- transfected 293T cells was performed with anti-FLAG antibody. (C) Trypsin sensitivity of COX I and II in the presence of tunicamycin was assessed as described in Fig. 3 E. (D) Quantification of band intensities in (B) was accomplished by laser densitometry. Levels/intensities of COX II antigen after digestion with trypsin (250 μg/ml) are shown as percentage intensity versus COX II antigen in the absence of trypsin. The values shown are means ± SD of three experiments. (E) Turnover of COX II in the presence of tunicamycin. Pulse-chase analysis was performed with 293T cells overexpressing Lon in the presence of tunicamycin (pretreatment with Tm for 16 h) followed by immunoprecipitation with anti-COX II antibody.
To analyze the degradation of COX II, pulse-chase analysis, followed by immunoprecipitation with antibody to COX II, was performed using 293T cells transfected to overexpress wild-type Lon in the presence of tunicamycin. Under these conditions, ∼95% of COX II antigen was degraded after an 8-h chase period in mock-transfected cells, and 85% of COX II antigen was degraded in both wild-type and S845N Lon-transfected cells (Fig. 5 E). These data suggest that overexpression of Lon did not promote degradation of COX II.
Based on these observations, we assessed the trypsin resistance of COX II in wild-type and S845N Lon-transfected cells using stably transfected HeLa cells. The level of Lon antigen was 3.5–4.5 times higher in stable transfectants expressing each of the Lon constructs as compared with mock-transfected controls (Fig. 6 A). Trypsin digestion assays, performed after each clone was subjected to Tm treatment, revealed that resistance of COX II to trypsin proteolysis was enhanced in transfectants overexpressing wild-type and S845N Lon (Fig. 6 B). The percentages of COX II antigen resistant to trypsin (250μg/ml) were 41, 56, 48, and 17% in wild-type, S845N-1, S548N-2 (the latter represent two different clones expressing the S845N variant), and mock-transfected clones, respectively (Fig. 6 C). To exclude the possibility that enhanced trypsin resistance of COX II was due to the aggregation of unassembled (free) COX II, the binding of COX II to COX I, which occurs at an early stage in COX synthesis (Nijtmans et al., 1998), was assessed by immunoprecipitation followed by Western blotting (Fig. 6 D). The percentage of COX II bound to COX I (lanes 2, 4, 6, 8) compared with total COX II (lanes 3, 5, 7, and 9) was increased in clones overexpressing Lon (32, 40, 30, and 16% in wild-type, S845N-1, S845N-2, and mock-transfected clones, respectively; Fig. 6 E). Immunoprecipitation of mitochondria with nonimmune IgG did not show COX II antigen (Fig. 6 D, lane 1).
Figure 6.

The effects of stable overexpression of Lon on the assembly of COX II. (A) Expression of wild-type or S845N Lon by stably transfected HeLa cells. Western blotting was performed with anti-Lon 665 or anti-FLAG antibody. (B) Trypsin sensitivity of COX II in the presence of Tm was assessed in stably transfected HeLa cells as described in Fig. 3 E. (C) Quantification of band intensities in (B) was accomplished by laser densitometry. Levels/intensities of COX II antigen after digestion with trypsin (250 μg/ml) are shown as the percentage of intensity versus COX II antigen in the absence of trypsin. The values shown are means ± SD of three experiments. (D) Detection of COX II bound to COX I and total COX II antigen was performed by immunoprecipitation (IP) with the indicated antibodies followed by Western blotting (Blot) with anti-COX II antibody. (E) Percentage of COX II bound to COX I. The values shown are means ± SD of three experiments.
The effects of Lon overexpression on mitochondrial dysfunction caused by BFA treatment or hypoxia. To analyze whether enhanced trypsin resistance of COX II was associated with promotion of mitochondrial function in the context of stress response, mitochonrial membrane potential was measured. These studies were performed with the Lon S845N-1 stably transfected HeLa cells, as these cells showed the highest level of COX assembly. Lon S845N-1 HeLa cells or mock-transfected HeLa cells were exposed to BFA for 16 h or to hypoxia for 48 h. In the mock-transfected HeLa clone, the number of cells that showed signals derived from mitochondrial membrane potential was reduced to 10–14% under stress condition, whereas this figure was 34–39% in Lon S845N-1cells (Fig. 7), suggesting that mitochondrial dysfunction caused by stress was partially prevented in the latter transfectants. A protective effect was also observed in other HeLa clones overexpressing Lon, although to a lesser extent.
Figure 7.

Effects of Lon overexpression on mitochondrial dysfunction caused by BFA treatment or hypoxia. (A) Lon S845–1 (IV, V, and IV) or mock-transfected cells (I, II, and III) were treated with BFA for 16 h (II and V), exposed to hypoxia for 48 h (III and VI), or cultured under normoxia (I and III). Mitochondrial membrane potential was studied microscopically after treating cells with MitosensorTM for 15 min. Bar, 15 μm. (B) The numbers of cells that showed signals derived from mitochondrial membrane potential were counted and indicated as the percentage of the total cell population. The values shown are means ± SD of three experiments. *P < 0.01.
Discussion
Lon, a protein highly conserved from bacteria to mammals, is located in mitochondrial matrix of yeast and higher species. There are three salient structural features of Lon: a mitochondrial matrix targeting sequence at the NH2 terminus (in yeast and higher species), a protease domain, and a Walker-type ATP binding motif (ATPase domain), the latter raising the possibility that Lon may function as a protease and as a molecular chaperone. In fact, yeast Lon, also referred to as Pim1, was shown to be required for intramitochondrial proteolysis (Suzuki et al., 1994; van Dyck et al., 1994), and the proteolytically inactive variant of Lon was shown to facilitate mitochondrial membrane complex assembly (Rep et al., 1996).
In the present study, we first confirmed the increased expression of Lon in cells subjected to hypoxia or ER stress using several approaches: (a) increased Lon promoter activation, based on enhanced luciferase activity following transient transfection with human or murine Lon promoter–reporter constructs; (b) increased mRNA level with no change in its stability; and (c) increased Lon antigen level. Another mitochondrial ATP-dependent protease, Yme1, and the molecular chaperone GRP75/mitochondrial HSP70, also displayed increased transcript levels in response to ER stress (Fig. 1 B). Taken together, these observations suggested the existence of a connection, direct and/or indirect, between ER stress and mitochondrial properties.
COX provided a useful model system in which to study the effects of ER stress on mitochondrial proteins because of its multisubunit structure, comprised of both mitochondrial- (I, II, and III) and nuclear-derived (IV, Va, Vb, VIa, VIb, VIc, VIIa, VIIb, VIIc, and VIII in human) components, and as expression/assembly of COX complex has been studied extensively (Nijtmans et al., 1998). Although the enzymatic core of COX consists of subunits I and II (mitochondrial-derived), COX IV (nuclear-derived), is essential for assembly of the enzyme (Dowhan et al., 1985) and COX II and III, but not COX I, were rapidly degraded in COX IV-deleted yeast (Nakai et al., 1994). In this setting, Yme1, also referred to Osd1, was required for the degradation of COX II (Nakai et al., 1995). Although yeast Lon did not affect the degradation of COX subunits, as a molecular chaperone, it promoted the assembly of COX complex (Rep et al., 1996).
In the current study, we demonstrated that ER stress reduced steady-state levels of nuclear-derived COX IV and V subunits, and caused rapid degradation of COX II, the latter of which was likely due to accumulation of free COX II subunits (Fig. 3). In contrast, the apparent stability of COX I under the same conditions may reflect its interaction with heme A (Wielburski and Nelson, 1984). Treatment of cells with cycloheximide mimicked the altered expression of COX subunits with ER stress, and enhanced expressions of Lon and Yme1 at the mRNA level (Fig. 4, A–C). Induction of Lon or GRP75/mtHSP70 was not observed in cells without PERK, a protein kinase that medicates suppression of protein synthesis in response to ER stress (Harding et al., 1999; Fig. 4 D). These observations suggested that suppression of cytosolic protein synthesis under ER stress is the common denominator linking these events.
Overexpression of wild-type Lon or a proteolytically inactive Lon mutant facilitates assembly of COX II into COX I-containing complexes under ER stress in experimental systems with transiently or stably transfected cells (Figs. 5 and 6). From the vantage point of maintenance of mitochondrial function, stably transfected HeLa cells overexpressing the proteolytically inactive Lon mutant were relatively resistant to changes in mitochondrial membrane potential induced by brefeldin A or hypoxia (Fig. 7).
Under apoptotic conditions, there have been several reports concerning mechanisms of communication between the ER and mitochondria via Ca2+ (Häcki et al., 2000; Nakamura et al., 2000). However, the results of our present study provided insight into a pathway involving ER stress in which suppression of cytosolic protein synthesis has a central role. Several recent findings have emphasized the potentially important role of mitochondrial ATP-dependent proteases or molecular chaperones in pathological-stressful conditions. Expression and activity of Lon were increased in rats bearing Zajdela hepatoma or in T3-treated hypothyroid animals (Luciakova et al., 1999). Mutations in Paraplegin, another mitochondrial ATP-dependent protease, cause hereditary spastic paraplegia (Casari et al., 1998). Preservation of the mitochondrial electron transport chain, by ischemic preconditioning or overexpression of HSP60 and HSP10, facilitated restoration of energy stores and cellular functions after reperfusion (Kobara et al., 1996; Lin et al., 2001).
Although the detailed mechanism underlying the cytoprotective effect(s) of Lon and the relevance of its protease activity under ER stress remain to be elucidated, our results emphasized the importance of the chaperone activity of this molecule for ensuring assembly of protein complexes and optimal functioning of mitochondria during the cellular response to environmental challenges.
Materials and methods
Cell culture and conditions for hypoxia/reoxygenation and other stresses
Astrocytes were isolated from the cerebral cortex of E18 rat embryos and cultured in MEM (10% FCS), as described (Yamaguchi et al., 1999). When cells reached confluence, cultures were subjected to hypoxia (H) for the indicated time (up to 22 h) in an incubator equipped with an hypoxia chamber (Coy Laboratory Products). In some experiments, cells were returned to the ambient atmosphere after H and incubated for 4 h (reoxygenation; R). In other experiments, cells were maintained in normoxia and treated with Tm (2 μg/ml; Sigma-Aldrich), Tg (300 nM; Sigma-Aldrich), BFA (2.5 μg/ml; Sigma-Aldrich), sodium arsenite (100 μM; Sigma), cycloheximide (0.2 or 2 μg/ml; Sigma-Aldrich), anisomycin (0.2 or 2 μg/ml; Sigma-Aldrich), actinomycin D (5 μg/ml; Sigma), doxycycline (5, 10, or 15 μg/ml) or exposed to heat shock (43°C) for the indicated times. HeLa cells and 293T cells were cultured in DME (10% FCS). Wild-type and PERK(−/−) embryonic fibroblasts were cultured in DME (10% FCS) as described previously (Harding et al., 2000).
ESD screening method for differentially expressed genes
The ESD screening for isolating genes upregulated in rat astrocytes exposed to hypoxia was performed as described (Suzuki et al., 1996). In brief, cDNA fragments derived from cultured astrocytes exposed to hypoxia for 22 h (H′-tracer) were equalized and subtracted with samples from normoxia (N-driver) or hypoxia (H-driver) up to three times (H′-N and H′-H, respectively). PCR was then carried out using [α32P] dCTP (>3,000 Ci/mlM) and products were separated on a sequencing gel followed by autoradiography. Candidate DNA bands were cut out of the gel and cloned into pGEM-T vector (Promega). DNA sequencing was performed using a 377 DNA sequencer (Applied Biosystems) and differential expression of candidate genes in hypoxia versus normoxia was confirmed by Northern blotting using [32P]-radiolabeled cDNAs as probes. Sequence searches and comparisons were carried out using several databases, including the National Center for Biotechnology Information, FASTA, and BLAST molecular analysis systems, and the DNA Data Bank of Japan.
Cloning of rat Lon cDNA
One of the cDNA fragments obtained by ESD was 346 bp in length and 85% identical to human Lon. This fragment was used to screen a rat cDNA library (lambda ZapII cDNA library; Stratagene) and a cDNA of 2.97 kb was obtained spanning the entire open reading frame. Both strands were sequenced using a 377 DNA sequencer.
Northern blot analysis
Total RNA (10 μg), isolated from cultured astrocytes, HeLa cells, 293T cells or the ischemic rat brain was separated on agarose/formaldehyde (1%) gels and transferred onto Immobilon N membranes (Millipore). cDNA fragments for probes were generated as follows: rat Lon cDNA was obtained as described above; human GRP78, human GRP75/mitochondrial HSP70, human HSP60, and human Yme1 cDNAs were produced by PCR with specific primers. Each fragment was labeled with 32P by the random hexamer procedure (specific activity; 0.5–3 × 109 cpm/μg DNA) and was used to probe membranes with immobilized RNA. Primers used for PCR were: ATG GTA TTC TCC GAG TGA CA and TTG GCT TTA AAG TCT TCA AT for GRP78; GGA TGG CTG GAA TGG CCT TAG and CCA ACA AGT CGC TCC CCA TCT for GRP75/mtHSP70; ATG CTG TGG CCG TTA CAA TG and CTC CTG ATT TCC ACT GGA TT for HSP60; and AGA TAC AGT TCC TGA GCA TGA and GCT AGA GGA ACG TAA TGT ACT for Yme1. After washing in 2 × SSC and 0.5 × SDS for 1 h, membranes were subjected to autoradiography.
Construction of Lon promoter–luciferase reporters and measurement of relative luciferase activity
A 608-bp fragment of the human Lon promoter (−584 to +24) was amplified by PCR using primers CAA CAT CAG CAT CGA CTT GG and ATA CTG GCG GCT CAC ACA ACT and cloned into Sac I-Hind III sites of pGL3 basic vector (Promega) after adding enzyme sites at both ends. Both strands were sequenced using a 377 DNA sequencer. Mouse Lon genomic DNA was cloned by screening a mouse BAC library using PCR primers AGC ACA GGC TAC GTG CGG CTC T and CTC GGA GGT CTC GTC GCT GCC A (Incyte Genomics). A 2.58-kb fragment (−2559 to +24; the numbers are tentative because the transcriptional start site was yet to be defined) was subcloned into Kpn I-Sma I sites of pGL3 basic vector after adding enzyme sites at both ends. Wild-type GRP78 promoter and a promoter mutant defective in the ERSE of GRP78 (−304 to +7) was inserted in pGL3 basic vector, provided by Dr. Mori (Kyoto University, Kyoto, Japan) (Yoshida et al., 1998). A reference plasmid (pRL-SV40) was purchased from Promega. Transfection was carried out by lipofection and, after 24 h, cells were exposed to Tm (2 μg/ml) or BFA (2.5 μg/ml) for 16 h to induce ER stress. Firefly and Renilla luciferase activities were measured using the Ascent system (Labsystems) and relative luciferase activity was obtained by dividing the former values by the latter. Fold induction was defined as the ratio of induced-to-basal levels of relative luciferase activity.
Generation of antibodies and immunoblotting
To obtain antibodies reactive with Lon, a peptide with the sequence CEKDDKDAIEEKFRERLKE was synthesized (an extra C was introduced at its NH2 terminus) and conjugated to keyhole limpet hemocyanin. Rabbits were immunized with this peptide by conventional methods. Once high titer antibodies were obtained, the IgG fraction was purified by chromatography on protein G columns (GIBCO BRL). For detection of Lon and other proteins, cultured astrocytes, HeLa cells, or 293T cells were lysed in buffer containing 10 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 1% Deoxycholate, 1 mM PMSF, 1 μg/ml Aprotinin, 1 μg/ml leupeptin, and 1 μg/ml Pepstatin. Immunoblotting was performed using antibodies to Lon, FLAG (Sigma-Aldrich), KDEL (StressGen; Biotechnologies Corp.), and COX subunits I, II, IV, or Vb (Molecular Probes). Sites of primary antibody binding were determined by alkaline phosphatase-conjugated secondary antibodies or the ECL method.
Expression of Lon in the ischemic brain
Unilateral permanent MCA occlusion was performed in male Sprague-Dawley rats (250 g) as described (Yamaguchi et al., 1999). After 8 h of ischemia, rats were sacrificed and brains were frozen at –80°C. Northern blotting was performed to detect Lon mRNA as described above. Serial coronal sections were also cut and the distribution of Lon mRNA was examined by in situ hybridization using digoxigenin-labeled cRNA probes as described (Hori et al., 1995). In brief, sense and antisense riboprobes for Lon were in vitro transcribed from the rat Lon cDNA inserted into the pGEM T vector. After linearizing the vector with NcoI (for the sense probe) or NdeI (for the antisense probe), reaction mixtures were incubated with digoxigenin-UTP and SP6 or T7 RNA polymerase (Roche Diagnostics Corporation). Brain sections were then hybridized with either the sense or antisense probe. For detection of hybridized cRNA probes, alkaline phosphatase–conjugated antibody to digoxigenin was used and the color was developed with NBT and X-phosphate solution. Lon antigen was also detected by Western blotting with antibody to Lon using brain homogenates after MCA occlusion (8 h) or sham operation (8 h) as described above.
Pulse-chase analysis of COX subunits. HeLa cells (5 × 106cells /condition) treated with Tm for 16 h, or maintained in medium alone (as control) were labeled with [35S]-methionine (200 μCi/ml; Amersham Pharmacia Biotech) for 30 min in methionine-free medium and chased for the indicated times (up to 8 h) in regular medium. Cell extracts in lysis buffer (see above) were immunoprecipitated with anti-COX subunit I, II, or IV antibodies, and subjected to SDS-PAGE followed by autoradiography.
Isolation of mitochondria and assembly of COX II
HeLa cells (107 cells) exposed to Tm or incubated in medium alone were homogenized by nitrogen bomb cavitation, and mitochondria were isolated by sequential centrifugation (Evans, 1992). After solubilizing mitochondria in 4% Na-cholate, 50 mM NaHPO4, 0.9% NaCl, and 1 mM EDTA, assembly of COX was assessed by monitoring trypsin resistance of COX I and II antigens as described (Rep et al., 1996). Binding of COX I and COX II subunits was monitored by immunoprecipitation of solubilized mitochondria (in 3% Na-cholate, 50 mM NaHPO4, 0.9% NaCl, 1 mM EDTA, 1 mM PMSF, 1 μg/ml Aprotinin, 1 μg/ml Leupeptin, and 1 μg/ml Pepstatin) with anti-COX I antibody followed by Western blotting with anti-COX II antibody.
Submitochondrial fractionation
Crude fractionation of 293T cells (107cells) transfected with Lon cDNA was performed by sequential centrifugation as described above, and submitochondrial fractions were prepared as described previously (Schnaitman and Greenawalt, 1968). In brief, isolated mitochondria were dissolved in isolation buffer containing 2 mM Hepes, 220 mM D-mannitol, 70 mM sucrose, 0.5 mg/ml BSA, and 0.5% digitonin, and centrifuged at 8,000 g for 10 min. Both the supernatant and pellet were treated with Lubrol or 1% digitonin for 15 min on ice and centrifuged at 144,000 g for 1 h.
Plasmid construction and overexpression of Lon
Rat Lon cDNA encoding the complete open reading frame was tagged with FLAG epitope at the COOH terminus and cloned into pcDNA3.1(+) (Invitrogen) or pME18Sf+, a gift from Dr. Maruyama (Tokyo Medical and Dental University, Tokyo, Japan) containing the hygromycin resistance gene, provided from Dr. Ohishi (Osaka University, Osaka, Japan). Lon mutants were constructed using LA PCR in vitro mutagenesis primer set (Takara Bichemicals). Primers used for creating Lon K519N and S845N were AAT GCT GGT GTT GCC GCC CAC ACC TGG T and ATG GCC CTA ATG CAG GTT GCA CCA TT, respectively. All constructs were sequenced prior to transfection studies. Wild-type or mutant Lon was transiently transfected into 293T cells (4 μg of DNA/107cells) and stably transfected into HeLa cells (25 μg of DNA/107cells) by lipofection (Lipofectamine; GIBCO BRL) or electroporation (GenePulser II; Bio-Rad Laboratories), respectively. HeLa cells overexpressing Lon were selected with hygromycin (300 μg/ml; Sigma-Aldrich) for 2 wk.
Measurement of mitochondrial membrane potential. Mitochondrial membrane potential was assessed by microscopically (excitation wavelength, 510–560 nm) after treating cells with MitosensorTM (CLONTECH Laboratories, Inc.). The number of cells showing positive signals in a mitochondria-derived dot-like pattern was counted and indicated as the percentage of total cells (100 cells). Statistical analysis was performed using Student's t test.
Laser densitometric analysis
Laser densitometric analysis was performed to standardize the results of Western and Northern blotting using Quality One software (pdi) as previously (Yamaguchi et al, 1999).
Acknowledgments
We are grateful to Dr. Yutaka Suzuki (TANABE SEIYAKU Co., Ltd.) for advice regarding the ESD method. We thank Dr. Kazutoshi Mori (Kyoto University, Kyoto, Japan) for providing GRP78 promoters, Dr. Kazuo Maruyama (Tokyo Medical and Dental University, Tokyo, Japan) for the pMESf(+) vector, and Dr Kazuhito Ohishi (Osaka University, Osaka, Japan) for hygromycin resistance gene.
Experiments in the Ron lab were supported by the National Institutes of Health (ES08681 and DK47119). D. Ron is an Ellison Medical Foundation Senior Scholar on Aging.
Footnotes
Abbreviations used in this paper: aa, amino acid(s); AS, sodium arsenite; BFA, brefeldin A; COX, cytochrome c oxidase; ERSE, ER stress response element; ESD, equalization of cDNAs, subtractive hybridization, and differential display; GRP, glucose-regulated protein; HSP, heat shock protein; MCA, middle cerebral artery; ORP, oxygen-regulated protein; Tg, thapsigargin; Tm, tunicamycin; UPR, unfolded protein response.
References
- Amerik, A.Y., G.V. Petukhova, V.G. Grigorenko, I.P. Lykov, S.V. Yarovoi, V.M. Lipkin, and A.E. Gorbalenya. 1994. Cloning and sequence analysis of cDNA for a human homologue of eubacterial ATP-dependent Lon proteases. FEBS Lett. 340:25–28. [DOI] [PubMed] [Google Scholar]
- Casari, G., M. De Fusco, S. Ciarmatori, M. Zeviani, M. Mora, P. Fernandez, G. De Michele, A. Filla, S. Cocozza, R. Marconi, A. Dürr, B. Fontaine, and A. Ballabio. 1998. Spastic paraplegia and OXPHOS impairment caused by mutation in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 93:973–983. [DOI] [PubMed] [Google Scholar]
- Dowhan, W., C.R. Bibus, and G. Schatz. 1985. The cytoplasmically-made subunit IV is necessary for assembly of cytochrome c oxidase in yeast. EMBO J. 4:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, W.H., 1992. Preparative Centrifugation. A Practical Approach. Rickwood, D., editor. Oxford University Press, Oxford, UK. 399 pp.
- Kobara, M., T. Tatsumi, S. Matoba, Y. Yamahara, C. Nakagawa, B. Ohta, T. Matumoto, D. Inoue, J. Asayama, and M. Nakagawa. 1996. Effect of ischemic preconditioning on mitochondrial oxidative phosphorylation and high energy phosphates in rat hearts. J. Mol. Cell. Cardiol. 28:417–428. [DOI] [PubMed] [Google Scholar]
- Häcki, J., L. Egger, L. Monney, S. Conus, T. Rossé, I. Fellay, and C. Borner. 2000. Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene. 19:2286–2295. [DOI] [PubMed] [Google Scholar]
- Harding, H.P, Y. Zhang, and D. Ron. 1999. Translation and protein folding are coupled by an endoplasmic reticulum resident kinase. Nature. 397:271–274. [DOI] [PubMed] [Google Scholar]
- Harding, H.P, I. Novoa, Y. Zhang, H. Zeng, R. Wek, M. Schapira, and D. Ron. 2000. Regulated translation inhibition controls stress-induced gene expression in mammalian cells. Mol. Cell. 6:1099–1108. [DOI] [PubMed] [Google Scholar]
- Harding H.P., H. Zeng, Y. Zhang, R. Jungries, P. Chung, H. Plesken, D.D. Sabatini, and D. Ron. 2001. Diabetes mellitus and exocrine pancreatic dysfunction in PERK−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell. 7:1153–1163. [DOI] [PubMed] [Google Scholar]
- Hori, O., M. Matsumoto, Y. Maeda, H. Ueda, T. Kinoshita, D. Stern, S. Ogawa, and T. Kamada. 1994. Metabolic and biosynthetic alteration in cultured astrocytes exposed to hypoxia/reoxygenation. J. Neurochem. 62:1489–1495. [DOI] [PubMed] [Google Scholar]
- Hori, O., J. Bred, T. Slattery, R. Cao, J. Zhang, J.X. Chen, M. Nagashima, E.R. Lundh, S. Vijay, D. Nitechi, J. Morser, D. Stern, and A.M. Schmidt. 1995. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. J. Biol. Chem. 270:25752–25761. [DOI] [PubMed] [Google Scholar]
- Katayama, T., K. Imaizumi, N. Sato, K. Miyoshi, T. Kudo, J. Hitomi, T. Morihara, T. Yoneda, F. Gomi, Y. Mori, Y. Nakano, J. Takeda, T. Tsuda, Y. Itoyama, O. Murayama, A. Takashima, P. St George-Hyslop, M. Takeda, and M. Tohyama. 1999. Presenilin-1 mutations downregulate the signaling pathway of the unfolded-protein response. Nat. Cell. Biol. 1:479–485. [DOI] [PubMed] [Google Scholar]
- Kuwabara, K., M. Matsumoto, J. Ikeda, O. Hori, S. Ogawa, Y. Maeda, K. Kitagawa, N. Imuta, T. Kinoshita, D. Stern, H. Yanagi, and T. Kamada. 1996. Purification and characterization of a novel stress protein, the 150-kD oxygen-regulated protein (ORP150), from cultured rat astrocytes and its expression in ischemic mouse brain. J. Biol. Chem. 27:5025–5032. [DOI] [PubMed] [Google Scholar]
- Lin, K.M., B. Lin, I.Y. Lian, R. Mestril, I.E., Scheffler, W.H. Dillmann. 2001. Combined and individual mitochondrial HSP60 and HSP10 expression in cardiac myocytes protects mitochondrial function and prevents apoptotic cell deaths induced by simulated ischemia-reoxygenation. Circulation. 103:1787–1792. [DOI] [PubMed] [Google Scholar]
- Liu H., R.C. Bowes III, B. van de Water, C. Sillence, J.F. Nagelkerke, and J.L. Stevens. 1997. Endoplasmic reticulum chaperons (GRP78 and calreticulin) prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J. Biol. Chem. 272:21751–21759. [DOI] [PubMed] [Google Scholar]
- Lowenstein, D.H., R.P. Gwinn, and M.S. Seren. 1994. Increased expression of mRNA encoding calbindin-D28K, the glucose-regulated proteins, or 72 kD heat shock protein in three models of acute CNS injury. Mol. Brain Res. 22:299–308. [DOI] [PubMed] [Google Scholar]
- Luciakova, K., B. Sokolikova, M. Chloupkova, and B.D. Nelson. 1999. Enhanced mitochondrial biogenesis is associated with increased expression of the mitochondrial ATP-dependent Lon protease. FEBS Lett. 444:186–188. [DOI] [PubMed] [Google Scholar]
- Matsuo N., S. Ogawa, T. Takagi, D. Stern, A. Wanaka, and M. Tohyama. 1997. Cloning of a putative vesicle transport-related protein, RA410, from cultured rat astrocytes and its expression in ischemic rat brain. J. Biol. Chem. 272:16438–16444. [DOI] [PubMed] [Google Scholar]
- Mizzen, L.A., C. Chang, J.I. Garrels, and W.J. Welch. 1989. Identification, characterization, and purification of two mammalian stress proteins present in mitochondria, grp75, a member of the hsp 70 family and hsp58, a homologue of the bacterial groEL protein. J. Biol. Chem. 264:20664–20675. [PubMed] [Google Scholar]
- Mori, K. 2000. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 101:451–454. [DOI] [PubMed] [Google Scholar]
- Nakai, T., Y. Mera, T. Yasuhara, and A. Ohashi. 1994. Divalent metal ion-dependent mitochondrial degradation of unassembled subunit 2 and 3 of cytochrome c oxidase. J. Biochem. 116:756–758. [DOI] [PubMed] [Google Scholar]
- Nakai, T., T. Yasuhara, Y. Fujiki, and A. Ohashi. 1995. Multiple genes, including a member of the AAA family, are essential for degradation of unassembled subunit 2 of cytochrome c oxidase in yeast mitochondria. Mol. Cell. Biol. 15:4441–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, K., E. Bossy-Wetzel, K. Burns, M.P. Fadel, M. Lozyk, I.S. Goping, M. Opas, C. Bleackley, D.R. Green, and M. Michalak. 2000. Changes in endoplasmic reticulum luminal environment after cell sensitivity to apoptosis. J. Cell Biol. 150:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijtmans, L.G.J., J. Taanman, A.O. Muijsers, D. Speijer, and Van Den Bogert. 1998. Assembly of cytochrome-c oxidase in cultured human cells. Eur. J. Biochem. 254:389–394. [DOI] [PubMed] [Google Scholar]
- Ozawa, K., K. Kuwabara, M. Tamatani, K. Takatsuji, Y. Tsukamoto, S. Kaneda, H. Yanagi, D. Stern, Y. Eguchi, Y. Tsujimoto, S. Ogawa, and M. Tohyama. 1999. 150 kD Oxygen-regulated protein (ORP150) suppressed hypoxia-induced apoptotic cell death. J. Biol. Chem. 274:6397–6404. [DOI] [PubMed] [Google Scholar]
- Ozawa, K., Y. Tsukamoto, O. Hori, Y. Kitao, H. Yanagi, D. Stern, and S. Ogawa. 2000. Regulation of tumor angiogenesis by oxygen-regulated protein 150, an inducible endoplasmic reticulum chaperone. Cancer Res. 61:4206–4213. [PubMed] [Google Scholar]
- Paschen, W. 2000. Role of calcium in neuronal cell injury: which subcellular compartment is involved? Brain Res. Bul. 53:409–413. [DOI] [PubMed] [Google Scholar]
- Rep, M., J.M. van Dijl, K. Suda, G. Scahtz, L.A. Grivell, and C.K. Suzuki. 1996. Promotion of mitochondrial membrane complex assembly by a proteolytically inactive yeast Lon. Science. 274:103–106. [DOI] [PubMed] [Google Scholar]
- Schnaitman, C., and J.W. Greenawalt. 1968. Enzymatic properties of the inner and outer membranes of rat liver mitochondria. J. Cell Biol. 38:158–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, C.K., K. Suda, N. Wang, and G. Schatz. 1994. Requirement of the yeast gene LON in intramitochondrial proteolysis and maintenance of respiration. Science. 264:273–276 [DOI] [PubMed] [Google Scholar]
- Suzuki, Y., N. Sato, M. Tohyama, A. Wanaka, and T. Takagi. 1996. Efficient isolation of differentially expressed genes by means of a newly established method, “ESD.” Nucleic Acids Res. 24:797–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamatani, M., T. Matsuyama, A. Yamaguchi, N. Mitsuda, Y. Tsukamoto, M. Taniguchi, Y.H. Che, K. Ozawa, O. Hori, H. Nishimura, A. Yamashita, M. Okaba, H. Yanagi, D. Stern, S. Ogawa, and M. Tohyama. 2001. ORP150 protects against hypoxia/ischemia-induced neuronal death. Nat. Med. 7:317–323. [DOI] [PubMed] [Google Scholar]
- van Dijl, J.M., E. Kutejova, K. Suda, D. Perecko, G. Schatz, and C.K. Suzuki. 1998. The ATPase and protease domains of yeast mitochondrial Lon: roles in proteolysis and respiration-dependent growth. Proc. Natl. Acad. Sci. USA. 95:10584–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyck, L., D.A. Pearce, and F. Sherman. 1994. PIM1 encodes a mitochondrial ATP-dependent protease that is required for mitochondrial function in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 269:238–242. [PubMed] [Google Scholar]
- von Heijne, G., J. Steppuhn, and R.G. Herrmann. 1989. Domain structure of mitochondrial and chloroplast targeting peptides. Eur. J. Biochem. 180:535–545. [DOI] [PubMed] [Google Scholar]
- Wang, N., S. Gottesman, M.C. Willingham, and M.M. Gottesman. 1993. A human mitochondrial ATP-dependent protease that is highly homologous to bacterial Lon protease. Proc. Natl. Acad. Sci. USA. 90:11247–11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielburski, A., and D. Nelson. 1984. Heme a induces assembly of rat liver cytochrome c oxidase subunits I-III in isolated mitochondria. FEBS Lett. 177:291–294. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, A., O. Hori, D. Stern, E. Hartmann, S. Ogawa, and M. Tohyama. 1999. Stress-associated endoplasmic reticulum protein 1 (SERP)/ribosome-associated membrane protein 4 (RAMP4) stabilizes membrane proteins during stress and facilitates subsequent glycosylation. J. Cell Biol. 147:1195–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, H., K. Haze, H. Yanagi, T. Yura, and K. Mori. 1998. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins J Biol. Chem. 273:33741–33749. [DOI] [PubMed] [Google Scholar]