

Abstract
Signaling from receptor tyrosine kinases (RTKs)* requires the sequential activation of the small GTPases Ras and Rac. Son of sevenless (Sos-1), a bifunctional guanine nucleotide exchange factor (GEF), activates Ras in vivo and displays Rac-GEF activity in vitro, when engaged in a tricomplex with Eps8 and E3b1–Abi-1, a RTK substrate and an adaptor protein, respectively. A mechanistic understanding of how Sos-1 coordinates Ras and Rac activity is, however, still missing. Here, we demonstrate that (a) Sos-1, E3b1, and Eps8 assemble into a tricomplex in vivo under physiological conditions; (b) Grb2 and E3b1 bind through their SH3 domains to the same binding site on Sos-1, thus determining the formation of either a Sos-1–Grb2 (S/G) or a Sos-1–E3b1–Eps8 (S/E/E8) complex, endowed with Ras- and Rac-specific GEF activities, respectively; (c) the Sos-1–Grb2 complex is disrupted upon RTKs activation, whereas the S/E/E8 complex is not; and (d) in keeping with the previous result, the activation of Ras by growth factors is short-lived, whereas the activation of Rac is sustained. Thus, the involvement of Sos-1 at two distinct and differentially regulated steps of the signaling cascade allows for coordinated activation of Ras and Rac and different duration of their signaling within the cell.
Keywords: Sos-1; Eps8; E3b1; Ras; Rac
Introduction
A major mechanism of signal transduction by receptor tyrosine kinases (RTKs)* involves the activation of small GTPases, among which Ras and Rac are pivotal (Scita et al., 2000). There is evidence for hierarchical organization of small GTPases in signal transduction pathways (Van Aelst and D'Souza-Schorey, 1997; Hall, 1998; Scita et al., 2000). In addition, regulators of small GTPase activity, such as the guanine nucleotide exchange factor (GEF) son of sevenless (Sos-1), appear to control different events in the signaling cascade (for review see Bar-Sagi and Hall, 2000; Schlessinger, 2000; Scita et al., 2000). A scenario can thus be envisioned in which coordinated and/or sequential activation of small GTPases is achieved through the action of the same regulator at multiple steps. The molecular mechanisms involved, however, remain yet to be understood. Sos-1 is a well-known Ras-GEF (for review see Bar-Sagi, 1994; Schlessinger, 2000), whose specific catalytic activity is encoded by its Cdc25-like domain (Bar-Sagi, 1994). Elegant biochemical and genetic studies have clarified how the proline-rich COOH-terminal tail of Sos-1 interacts with the SH3-containing adaptor molecule Grb2 (Bar-Sagi, 1994). Grb2 in turn displays a SH2 domain responsible for the recruitment of the Grb2–Sos-1 complex to active, autophosphorylated RTKs (Lowenstein et al., 1992; Ceresa and Pessin, 1998). The relocalization of the complex to the plasma membrane is thought to be sufficient for Sos-1 to catalyze the exchange of guanine nucleotides on Ras, which is also present at the membrane, with ensuing activation of this small GTPase.
Active Ras (Ras-GTP) triggers a number of signaling cascades, among which is the one connecting Ras to Rac, a member of the Rho subfamily of small GTPases. There is evidence that Sos-1 is also active at this step. First, Sos-1 contains a Dbl homology domain in tandem with a Pleckstrin homology domain, a module responsible for GEF activity on Rho GTPases (Cerione and Zheng, 1996; Nimnual et al., 1998). Second, and most importantly, Sos-1 has been shown to form a tricomplex in vivo with two signaling molecules, Eps8 (Fazioli et al., 1993) and E3b1 (also known as Abi-1) (Shi et al., 1995; Biesova et al., 1997). E3b1 contains a SH3 domain and binds Sos-1 (Scita et al., 1999; Fan and Goff, 2000). In addition, E3b1 binds to the SH3 domain of Eps8 (Biesova et al., 1997). Thus E3b1 acts as a scaffold protein, which holds together Sos-1 and Eps8. The tricomplex Sos-1–E3b1–Eps8 (S/E/E8) is endowed with Rac GEF activity in vitro (Scita et al., 1999). Thus, the sum of the above observation raises the possibility that Sos-1 might function at different step in the signaling cascade, acting as a Ras-GEF and a Rac-GEF, respectively. A number of outstanding questions need clarification, however, before such a model could be accepted: does a trimeric S/E/E8 complex exist under physiological conditions? How is the specificity of Sos-1 directed toward Ras or Rac? Does the putative dual function of Sos-1 provide a mechanistic framework for the coordinated activation of Ras and Rac? The present studies were undertaken to elucidate these questions.
Results
The S/E/E8 complex exists under physiological conditions
In previous studies, we showed that Sos-1, E3b1, and Eps8 could form a trimeric complex in vivo upon concomitant overexpression of the three proteins. However, we failed to detect the existence of an endogenous S/E/E8 complex (Scita et al., 1999). We reasoned that this could be due to the low efficiency of the immunoprecipitating antibodies used. We thus sought to exploit the availability of eps8−/− fibroblasts to circumvent this problem. To this end, we performed immunoprecipitation experiments using eps8−/− fibroblasts in which the expression of Eps8 was restored, to physiological levels, with an expression vector encoding a myc epitope-tagged Eps8 (−/− [Eps8myc] cells). We selected transfected clones in which the levels of expression of Eps8myc were very similar to those present in wild-type fibroblasts (Fig. 1 A). Endogenous Sos-1 and E3b1 could be detected in anti-myc immunoprecipitates from lysates of Eps8myc-reconstituted, but not from eps8−/− fibroblasts (Fig. 1 B).
Figure 1.
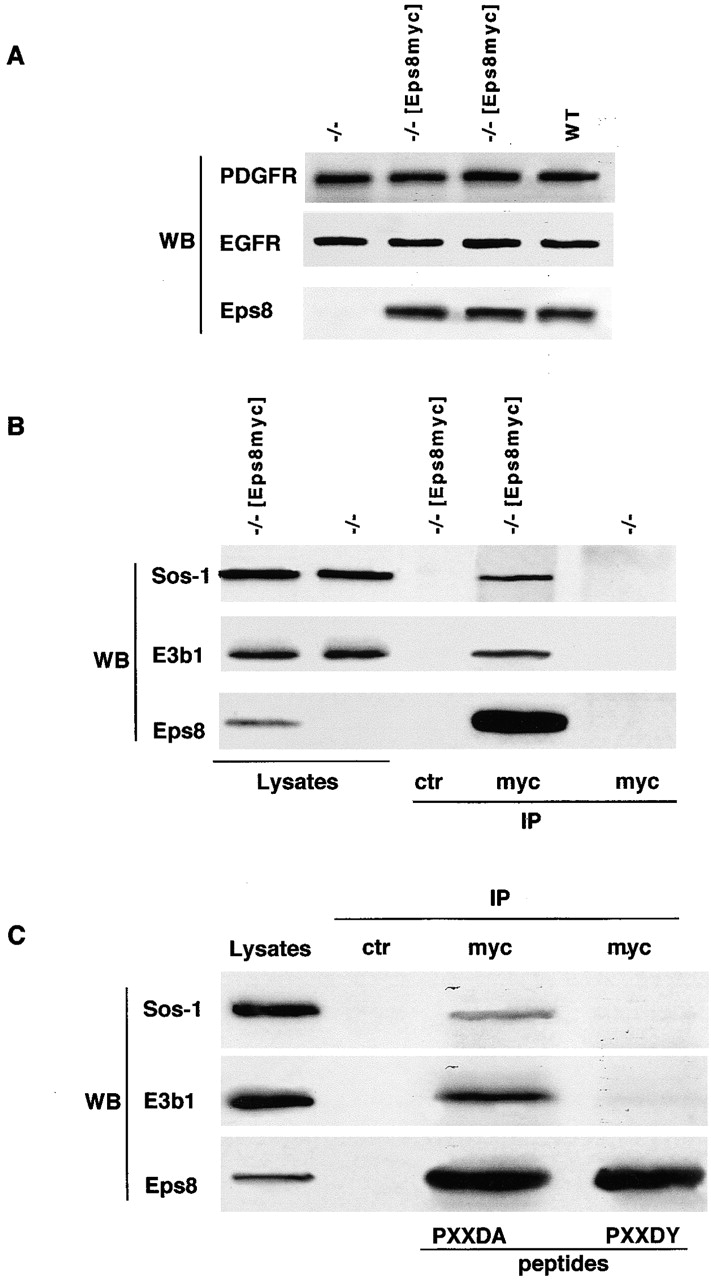
The S/E/E8 complex exists under physiological conditions. (A) Eps8−/− cells were transfected with a control vector (−/− lanes) or a vector coding a myc epitope–tagged Eps8 (−/− [Eps8myc] lanes), both carrying a hygromicin resistance gene. Hygromicin-resistant single-cell clones were established after 10 d of selection with hygromicin (0.2 mg/ml). The levels of Eps8, PDGFR, and EGFR in individual clones of eps8−/−, −/− [Eps8myc], or in wild-type (WT) cells were determined by immunoblotting analysis of equal amounts of total cellular lysates (50 μg) using the indicated antibodies (WB), and clones with levels of expression of Eps8, PDGFR, and EGFR similar to wild-type fibroblasts were used. (B) Total cellular lysates (10 mg), obtained from the transfectants described in A, were immunoprecipitated (IP) with the antibody indicated at the bottom (ctr, irrelevant antibody), followed by immunoblot with indicated antibodies (WB). The indicated lanes (lysates) were loaded with 100 μg of total cellular lysates. (C) Total cellular lysates (10 mg) obtained from −/− [Eps8myc] cells were immunoprecipitated (IP) with the antibody indicated at the top (ctr, irrelevant antibody) in the presence or absence (−) of 40 ng/ml of the indicated peptides (peptides). The peptides used were PPPPPVDYTEDEE (PXXDY) and PPPPPVDATEDEE (PXXDA, used as a control). Immunoblotting was with the indicated antibodies (WB). The indicated lanes (lysates) were loaded with 100 μg of total cellular lysates.
To determine whether E3b1 mediates the interaction between Eps8 and Sos-1, as it would be expected according to the tricomplex model, we performed coimmunoprecipitation experiments under conditions in which the association between Eps8 and E3b1 was disrupted. The binding site of E3b1 to the SH3 domain of Eps8 was previously mapped to the amino acid sequence, PPPPPVDYTEDEE, where the D and the Y residues are critical for efficient binding (Mongiovi et al., 1999). Thus, a peptide encompassing this region should specifically disrupt the Eps8–E3b1 association. Indeed, no E3b1could be recovered in anti-myc immunoprecipitates from lysates of Eps8myc-reconstituted cells, when the immunoprecipitation was performed in the presence of an excess of the competing peptide. The association was, however, preserved when a control peptide, bearing a Y→A substitution and unable to bind to Eps8 (Mongiovi et al., 1999), was used (Fig. 1 C). Similarly, no Sos-1 could be recovered in anti-myc immunoprecipitates in the presence of the competing, but not of the control, peptide (Fig. 1 C). Thus, under physiological conditions, the coimmunoprecipitation of Eps8 and Sos-1 depends on the integrity of the Eps8–E3b1 interaction, pointing to the existence of a physiological S/E/E8 complex. It cannot be formally excluded that Eps8, E3b1, and Sos-1 associate after cell lysis, thus allowing coimmunoprecipitation. However, we have previously demonstrated (Scita et al., 2001) that the three endogenous proteins also colocalize in vivo in dynamic actin structures. Thus, the sum of our results strongly argues in favor of the existence of a physiological S/E/E8 complex.
Similarly, it cannot be formally excluded that other interactors of the SH3 domain of Eps8, besides E3b1, mediate the formation of the endogenous trimeric complex. This is, however, unlikely since no coimmunoprecipitation between Eps8 and Sos-1 could be detected when the two proteins were overexpressed in cell lines with relative low levels of E3b1 (Scita et al., 1999; unpublished data). Moreover, neither RN-tre, nor JIK, two other known interactors of the SH3 domain of Eps8 (Biesova et al., 1997; Mongiovi et al., 1999), were able to bind to Sos-1 (unpublished data).
The adaptor molecules, E3b1 and Grb2, compete for binding to Sos-1 both in vitro and in vivo
Sos-1 has been shown to be part of a signaling complex with Grb2, which mediates the activation of Ras upon RTK stimulation. Our finding that, under physiological conditions, Sos-1 also participates in a complex with Eps8 and E3b1 raises the question of the physical and functional relationships between these two Sos-1–containing complexes. To gain insight into this issue, we initially mapped the region of E3b1 responsible for the interaction with Sos-1 by using a series of deletion mutants of E3b1 fused to glutathione S-transferase (GST). Native Sos-1 could be efficiently recovered onto GST-E3b1 full length (Fig. 2 A, amino acids [aa] 2–480). Further mapping revealed that the SH3 domain of E3b1 (aa 416–480) was necessary and sufficient for binding (Fig. 2 A).
Figure 2.

In vitro competition between E3b1 and Grb2 for binding to Sos-1. (A, Top) schematic of E3b1. The ruler shows amino acid positions. (Bottom) In vitro binding of various fragments of E3b1 (all engineered as GST fusions, amino acid boundaries are indicated on the top) to Sos-1 from lysates of Sos-1–transfected Cos-7 cells. Detection was with anti–Sos-1 antibody. The lane “lysate” was loaded with 50 μg of total cellular lysates. P Rich, proline rich region; SH3, SH3 domain. (B) A His-tagged COOH-terminal fragment (His-Sos, aa 1131–1333) of Sos-1 (10 pmoles) was bound to 60 pmoles of either immobilized GST (GST) or GST-E3b1–331–480 in the presence of increasing concentrations of the NH2-terminal SH3 domain of Grb2 or of Eps8 (SH3s, 0, 10, 100, 1,000 pmoles, as indicated on the top). Detection was with antihistidine antibody. I, input, 10 pmoles of His-Sos-1. (C) His-Sos (10 pmoles) was bound to 60 pmoles of GST fusions encompassing the SH3s of either E3b1 (○ and •) or of Grb2 (□ and ▪). Binding was in the presence of the indicated amounts of the competing peptide NH2-VPVPPPVPPRRR-COOH (derived from the sequence of Sos-1, • and ▪), or of a scrambled control peptide (○ and □). Detection was by immunoblot with anti-His, followed by densitometric scanning. (D) Kinetic analysis and rate constants for the binding of the SH3 domains of Grb2 and E3b1 to the proline-rich COOH-terminal of Sos-1 (aa 1131–1333). Increasing concentrations (10–400 nM) of His-Sos-1 were passed over the immobilized GST fusion proteins (GST-SH3-Grb2 and GST-SH3-E3b1) for 2.5 min at a flow rate of 50 μl/min. The kinetics of binding are shown in the graphs. Coupling of equal amounts of the GST fusion proteins was performed with a goat anti-GST antibody covalently linked to CM5 sensor chip according to the manufacturer's instructions. kon and koff kinetic rates were obtained by simple Langmuir fitting model (biaevaluation software v2.1).
We have previously shown that E3b1 binds to the proline-rich, COOH-terminal tail of Sos-1 (aa 1131–1333) (Scita et al., 1999), which also binds to the SH3 of Grb2 (Li et al., 1993; Cussac et al., 1994). Thus, Grb2 and E3b1 might compete for binding to Sos-1. This hypothesis was validated by a series of experiments performed both in vitro and in vivo. We could demonstrate that (a) the SH3 of Grb2, but not of Eps8, efficiently competed with the SH3 of E3b1 for binding to the proline-rich COOH-terminal tail of Sos-1 in vitro (Fig. 2 B); (b) the Sos-1 peptide VPVPPPVPPRRR, known to constitute a Grb2-SH3 binding site (Li et al., 1993; Cussac et al., 1994), competed equally well the binding of the COOH-terminal tail of Sos-1 to the SH3s of Grb2 or E3b1 (Fig. 2 C); (c) the binding constants of the interaction between the proline rich COOH-terminal tail of Sos-1 and the SH3 domains of Grb2 and E3b1 were very similar (Fig. 2 D); (d) overexpression of Grb2 abolished the ability of GST-E3b1 to bind to native Sos-1 (Fig. 3 A); and (e) overexpression of E3b1 significantly decreased the coimmunoprecipitation between Grb2 and Sos-1 (Fig. 3 B). Thus, Sos-1 binds either E3b1 or Grb2, in a mutually exclusive fashion, suggesting that two distinct pools of Sos-1 exist in the cells.
Figure 3.

In vivo competition between E3b1 and Grb2 for binding to Sos-1. (A, Top) GST-E3b1-331–480, or control GST, was bound to Sos-1 present in Cos-7 cells transfected (Tfx) with either Sos-1 or S/G. Detection was with anti–Sos-1. (Bottom) The same cellular lysates (50 μg) were detected with anti-Grb2. (B) Lysates, from 293T cells transfected (Tfx) with E3b1 or a control vector (ctr), were immunoprecipitated (IP) with anti-Grb2 or an irrelevant antibody (ctr), followed by detection (WB) with anti–Sos-1 (top) or anti-Grb2 (middle). The expression of E3b1 (bottom) was detected with an anti-E3b1 antibody.
To test this possibility in vivo, we set out to perturb the equilibrium between the Sos-1–E3b1 (S/E) and Sos-1–Grb2 (S/G) complexes, by increasing the levels of E3b1 within the cell. If the two complexes are functionally distinct, then the overexpression of E3b1, by competing the S/G interaction, should result in (a) reduced Sos-1 recruitment in vivo to active EGFR, (b) consequent reduction in Ras activation, as measured by reduced Ras-GTP levels, (c) reduced activation of Ras-dependent pathways, as measured by activation of MAPK, and finally (d) reduced proliferative response. All of these predictions were confirmed experimentally (Fig. 4, A–C).
Figure 4.
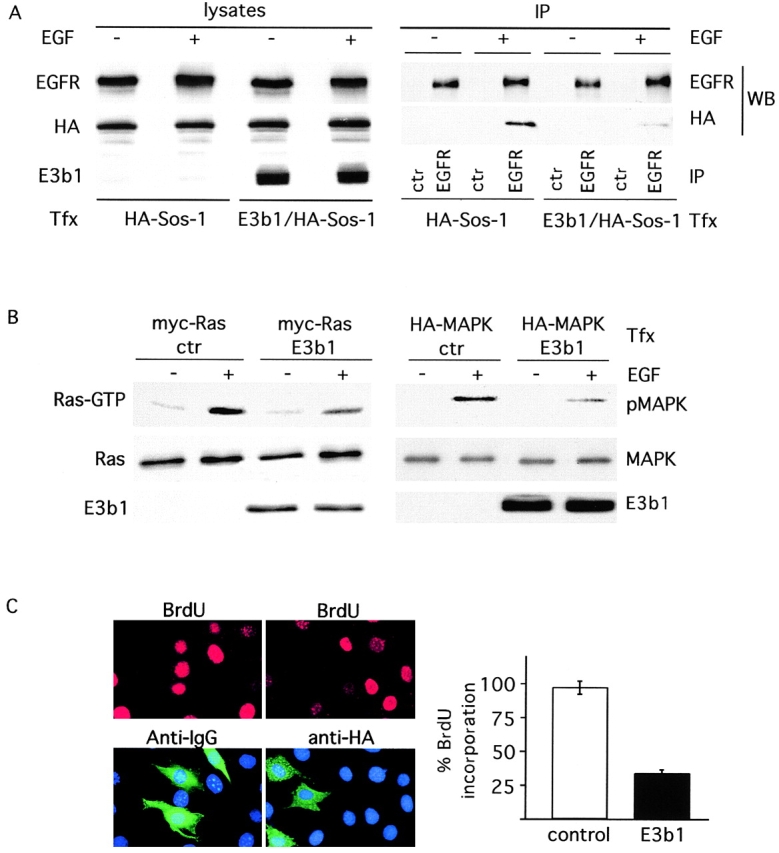
The S/G and the S/E complexes are functionally distinct. (A) Cos-7 cells were transfected with the indicated cDNAs (Tfx), and treated with EGF (3 min, +) or mock treated (−). Immunoprecipitates (IP) with anti-EGFR or preimmune sera (ctr) were analyzed by immunoblotting (WB) with anti–EGFR or HA (for Sos-1 detection). Lysates (50 μg) were analyzed by immunoblotting with anti-EGFR, anti-HA (for Sos-1 detection), or anti-E3b1 antibody. HA-Sos, HA-epitope tagged Sos-1(B) Cos-7 cells were cotransfected (Tfx) with E3b1 (or a control empty vector, ctr) and either a myc-tagged Ras (left) or HA-tagged MAPK (right), followed by treatment with EGF (5 min, +) or mock-treatment (−). (Top, left) Ras-GTP was recovered with the GST-RBD of Raf-1 followed by immunodetection with anti-myc antibodies. (Top, right) Anti-HA immunoprecipitation, followed by immunodetection with an anti–phosphorylated MAPK antibody. The levels of expression of myc-Ras and HA-MAPK (middle) and E3b1 (bottom) are also shown. (C) Quiescent NIH-3T3 fibroblasts were microinjected with an empty vector together with rabbit IgG (left) or with an HA-tagged E3b1 expression vector (right) followed by treatment with 10% serum. After 15 h, BrdU was added and 3 h later the cells were fixed and stained to detect BrdU incorporation (top), E3b1 (anti-HA), or control (anti-IgG). Nuclei were counterstained with Dapi (bottom). A quantitative analysis of BrdU incorporation into control (empty bar) and E3b1 (closed bar) injected cells is shown on the right. The data are the mean ± SEM of three independent experiments. Results are expressed as a percentage of injected cells incorporating BrdU respect to uninjected ones. Serum treatment induces BrdU incorporation in >95% of not injected cells.
We note that our mapping data are at variance with those recently reported by Fan and Goff (2000), who concluded that, in vivo, the interaction between Sos-1 and E3b1–Abi-1 is mediated by the NH2-terminal portion of the latter. Consistently, Fan and Goff (2000) did not observe competition in vivo between Grb2 and E3b1 for binding to Sos-1. Although we cannot exclude that the NH2-terminal region of E3b1–Abi-1 participates in binding, in vivo, we were not able to observe a high-affinity binding surface in that region by in vitro binding experiments (Fig. 2 A; Scita et al., 1999). Conversely, under our conditions of analysis, the efficient competition of the SH3 domains of Grb2 and E3b1 for binding to the same Sos-1 peptide, coupled to the readily detectable biological competition, strongly suggests that the SH3-mediated contact is the major determinant of the interaction between Sos-1 and E3b1.
E3b1 is a limiting factor in Rac activation
The competition between E3b1 and Grb2 for the same binding site on Sos-1 implies that an individual Sos-1 molecule can only be part of one GEF complex at a time. However, different pools of Sos-1, either bound to Grb2 or to E3b1, could exist in vivo. We tried to gain insight into this issue. We showed that BIAcore measurements revealed similar kinetic parameters for the SH3-mediated S/G and S/E interactions (Fig. 2 D). However, when we measured the relative abundance of S/G and S/E complexes within the cell, we found a 10-fold excess of the former (Fig. 5 A), under conditions in which quantitative immunoprecipitation of the adaptor molecules, Grb2 and E3b1, was achieved (Fig. 5 A). Thus, the data indicate that two pools of Sos-1, associated with different adaptors, exist simultaneously in the cell. They further suggest that the pool of E3b1, available for binding to Sos-1, is reduced relative to that of Grb2, and may be rate-limiting. Indeed, overexpression of E3b1 resulted in increased PAK65 activity, an indicator of Rac activation (Manser et al., 1994) (Fig. 5 B), which was abrogated by a dominant negative Rac mutant (Fig. 5 B). On the other hand, overexpression of Sos-1 did not lead to increased PAK65 activity. An additional corollary of this hypothesis is that the overexpression of E3b1, by facilitating the formation of a trimeric complex with Eps8 and Sos-1, should directly lead to activation of Rac. To test this prediction E3b1 or a mutant of E3b1 (E3b1-DY), bearing alanine substitutions of the DY residues critical for the interaction with Eps8 (Mongiovi et al., 1999), were used to infect wild-type and eps8−/− fibroblasts. The levels of GTP-bound Rac were then determined by in vitro binding assays using the immobilized CRIB domain of PAK65 (Manser et al., 1994). E3b1, but not the E3b1 mutant impaired in Eps8 binding, caused a readily detectable increase in Rac-GTP levels in wild type, but not in eps8−/− fibroblasts (Fig. 5 C). Thus, the overexpression of E3b1 is sufficient to activate Rac and this effect is strictly dependent on Eps8.
Figure 5.
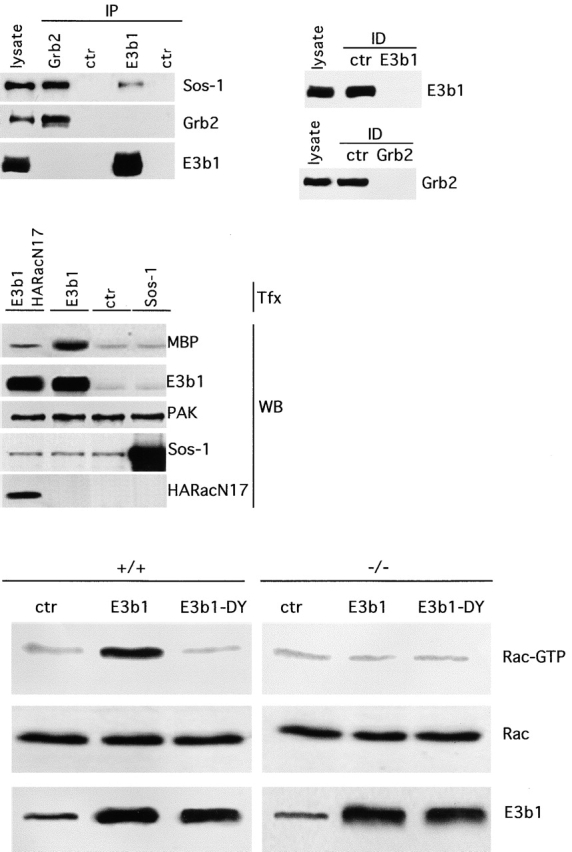
E3b1 is a limiting factor in Rac activation. (Top, left) Lysates of Cos-7 cells were immunoprecipitated (IP) with the antibody indicated on the top and immunoblotted with the antibody indicated on the right. Lysate, 50 μg of total cellular lysate. (Top, right) The efficiency of Grb2 and E3b1 immunoprecipitations was >95%, as determined by immunoblot analysis of the supernatants of the immunoprecipitations performed with either the specific antibody with (Grb2 and E3b1, respectively, as indicated) or with an irrelevant (ctr) antibody. (Middle) Cos-7 cells were transfected (Tfx) with HA-tagged PAK65 (all lanes) together with the plasmids indicated on the top (ctr, empty vector) and serum starved for 24 h. Equal amounts of anti-HA immunoprecipitated PAK65 were subjected to an in vitro kinase assay using myelin basic protein (MBP) as a substrate. The levels of immunoprecipitated PAK65 and the expression of Sos-1, E3b1, and HARacN17 in the various transfectants are also shown. Similar results were obtained using Cos-7 cells. (Bottom) Wild-type (+/+) and eps8−/− (−/−) mouse embryo fibroblasts were infected with retroviral vectors coding for E3b1 (E3b1) or the mutant of E3b1-DY (E3b1-DY), or with an empty vector as a control (ctr). >95% of the cells expressed the infected cDNA, as determined by immunostaining with anti-E3b1 (not shown). Equal amounts of lysates were subjected to in vitro binding assays with the immobilized CRIB domain of PAK65. After washing, bound proteins were resolved by SDS-PAGE and immunoblotted with anti-Rac antibodies (Rac-GTP). The levels of total Rac (Rac) and E3b1 proteins are also shown.
The indispensability of E3b1 in the cascade of events leading to Rac activation and Rac-dependent actin reorganization was further analyzed at the biological level. We have previously shown that interference with E3b1 functions, by microinjection of anti-E3b1 antibodies, inhibited PDGF-induced ruffles (Scita et al. 1999). We also showed that in eps8−/− cells, ruffling induced by PDGF and activated Ras, but not by activated Rac, was inhibited. Ruffling, however, could be restored upon reexpression of Eps8, but not of an Eps8 mutant unable to bind to E3b1 (Scita et al., 1999), suggesting that the Eps8–E3b1-based complex is implicated in the transmission of signal between Ras and Rac. Thus, one might predict that a mutant of E3b1 unable to associate to Eps8 should function as a dominant negative on Ras-induced Rac-dependent ruffling. To test this, we cotransfected the activated versions of either Ras or Rac, together with E3b1 or the E3b1DY mutant. RasV12-induced, but not RacQL-induced, ruffles were efficiently inhibited by the coexpression of E3b1DY, but not of E3b1 wild type (Fig. 6), supporting the contention that the ability of E3b1 to form a complex with Eps8 is required to transmit signals from Ras to Rac. In conclusion, our data show that two pools of Sos-1 exist in the cell, coupled respectively to Grb2 or E3b1. In addition, the availability of E3b1, but not of Sos-1, is indispensable and rate limiting in the pathway leading to Rac activation.
Figure 6.

The E3b1DY mutant inhibits actin remodeling induced by activated Ras, but not by activated Rac. Mouse embryo fibroblasts were transfected, as indicated on the top, with expression vectors for either RasV12 or RacQL, together with ECFP-tagged versions of either E3b1 or the E3b1DY mutant. After serum starvation, cells were fixed and stained with rhodamine-conjugated phalloidine to detect F-actin (top, red). Expression of the transfected proteins was detected (bottom, green) with either anti-Ras or anti-Rac antibodies or by epifluorescence (ECFP). In the cotransfection experiments a molar ratio of 1:3 of the activated GTPases to the ECFP-E3b1 constructs was used, such as that all of the epifluorescent cells expressed also the corresponding GTPase (not shown). The percent of transfected cells (mean ± SEM) undergoing ruffling was as follow: RasV12 = 65 ± 3%; RasV12 + E3b1 WT = 62 ± 4%; RasV12 + E3b1DY = 16 ± 2%; RacQL = 93 ± 3%; RacQL + E3b1 WT = 90 ± 4%; RacQL + E3b1DY = 95 ± 5%. Bar, 10 μm.
The dual GEF activity of Sos-1 depends on its presence in distinct complexes
The existence of Sos-1 in two physically and functionally distinct pools suggests the hypothesis that its presence in a S/G or a S/E/E8 complex could dictate its Ras- or Rac-specific GEF activities, respectively. To explore this possibility, we used an in vitro assay that can score GEF activities in Sos-1–containing immunoprecipitates. Cells were transfected with either Sos-1 alone (SosTfx) or a combination of S/E/E8 (Triple Tfx) (Fig. 7 A). When Sos-1 was immunoprecipitated from SosTfx, it displayed Ras-GEF, but not Rac-GEF, activity (Fig. 7 B). Of note, little E3b1 and no Eps8 could be detected in Sos-1 immunoprecipitates (Fig. 7 A). Conversely, Grb2 was readily recovered from the same immunoprecipitates (unpublished data). We then used the Triple Tfx and coimmunoprecipitated Sos-1 with anti-Eps8 antibodies (Fig. 7, A and B). Under these conditions, all of the coimmunoprecipitated Sos-1 is present in the S/E/E8 tricomplex (Scita et al., 1999). Despite lower levels of Sos-1 in the coimmunoprecipitate (with respect to a Sos-1 immunoprecipitate from SosTfx), Rac-GEF activity was now present (Fig. 7 B). Conversely, no Ras-GEF activity could be detected (Fig. 7 B). The lack of Ras-GEF activity was not due to the low levels of Sos-1. To prove this point, we immunoprecipitated from SosTfx an amount of Sos-1 comparable to that present in anti-Eps8 immunoprecipitates from Triple Tfx (Fig. 7, A and B, lanes SosTfx1/10). Under these conditions, Ras-GEF activity was readily detectable (Fig. 7 B). The detection of Rac-specific GEF activity in anti-Eps8 immunoprecipitates strictly required the presence of Sos-1, since no GEF activity was detected in Eps8 immunoprecipitates, in the absence of coexpressed Sos-1 (Scita et al., 1999; unpublished data). Furthermore, no Rac-GEF activity could be detected in Eps8 immunoprecipitates from lysates of cells transfected with Sos-1, E3b1, and an Eps8 mutant that is impaired in its ability to bind E3b1 and thereby cannot form a trimeric complex (Scita et al., 1999).
Figure 7.
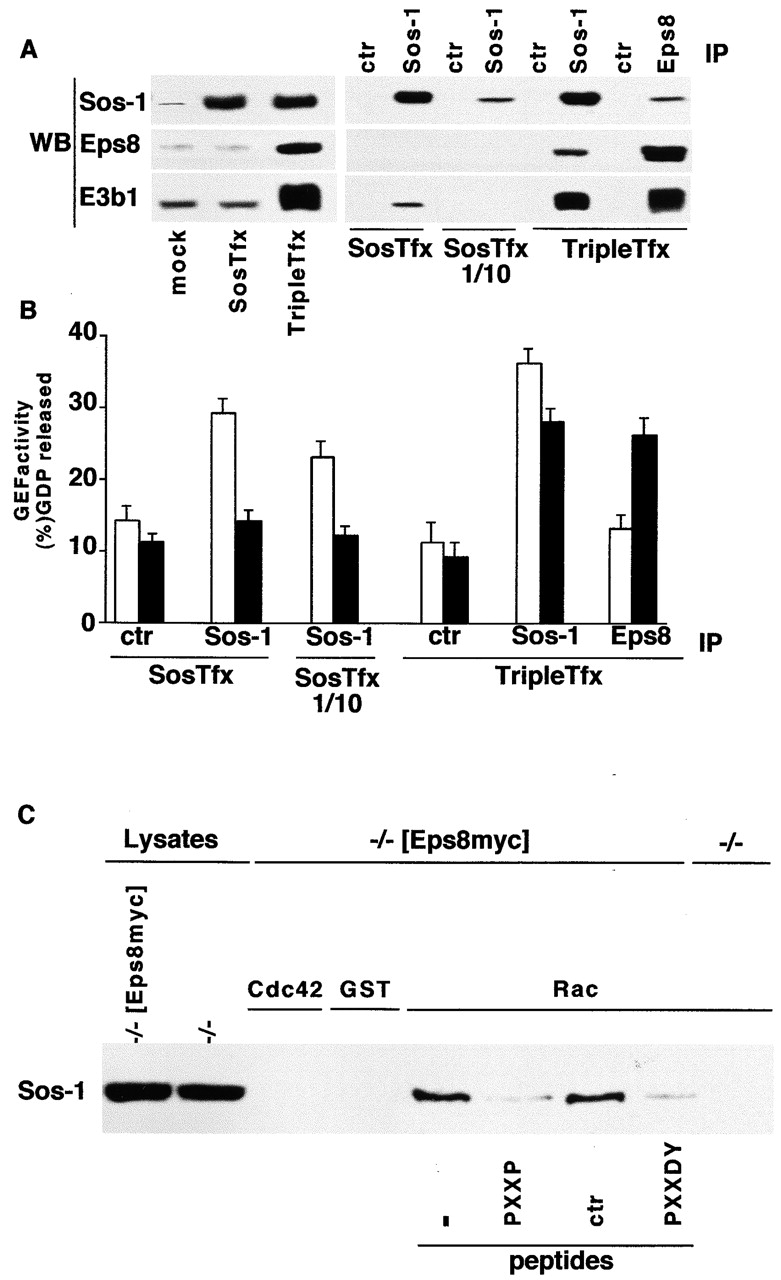
The bifunctional guanine nucleotide exchange activity of Sos-1 is regulated by its assembly into distinct molecular complexes. (A) Cos-7 cells were transfected with expression vectors for Sos-1 (SosTfx) or for Sos-1, E3b1, and Eps8 (TripleTfx) or with the corresponding empty vectors (mock). Total cellular lysates (100 μg, left), or immunoprecipitates (IP, right) with the antibody indicated on the top (ctr, irrelevant antibody), were immunoblotted (WB) with the indicated antibody. 5 mg of total cellular lysates were used for the immunoprecipitations, except in the SosTfx 1/10 lanes, in which 0.5 mg were used. (B) Ras-GEF (empty bars) and Rac-GEF (closed bars) activities in aliquots of the same immunoprecipitates (IP) shown in B. Data are expressed as described in Material and methods. (C) Immobilized, purified GST-Rac (Rac) and GST-Cdc42 (Cdc42) were depleted of guanine nucleotide by extensive washing in 10 mM EDTA as described in Material and methods. Equal amounts of the nucleotide-free GST-GTPases or GST alone (GST) were incubated with total cellular lysates from eps8−/− fibroblasts (−/−) or eps8−/− stably expressing Eps8myc (−/−[Eps8myc]) in the presence or absence (−) of the 40 ng/ml of the indicated peptides (peptides). The peptide used were PPPPPPVDATEDEE (PXXDA), used as control (see text and Fig. 1); PPPPPVDYTEDEE (PXXDY), which corresponds to the E3b1 binding site for the SH3 domain of Eps8 and prevents Eps8-E3b1 association (refer to Fig. 1 B); VPVPPPVPPRRR (PXXP), which corresponds to the Sos-1 binding site for the SH3 domain of E3b1 and readily competes the interaction between Sos-1 and E3b1 (refer to Fig. 2 C). Bound proteins were resolved by SDS-PAGE. Detection was with anti–Sos-1 antibody (Sos-1). The indicted lanes (lysate) were loaded with 50 μg of total cellular lysates.
At this stage, we cannot formally exclude that an unknown GEF, coprecipitating with Sos-1, is responsible for the observed activity. However, such a hypothetical protein should not only be associated with Sos-1, but also be active only in the presence of Eps8 and E3b1, which are both required for the formation of the trimeric complex endowed with Rac-specific GEF activity (Scita et al., 1999), making this possibility unlikely. It is reasonable, therefore, to propose that Sos-1 alone (or complexed with proteins, such as Grb2) is endowed with Ras-GEF activity. However, upon interaction with Eps8 and E3b1, its specificity is switched toward Rac.
A common feature of proteins endowed with GEF catalytic activity is their ability to bind to their specific nucleotide-depleted GTPase with relative high affinities. Thus, one might postulate that Sos-1 associates with the nucleotide-free form of Rac exclusively when engaged in the S/E/E8 complex. Purified and nucleotide depleted, GST-Rac and GST-Cdc42 proteins were therefore used to test their ability to interact with Sos-1. Native Sos-1, present in lysates of −/− [Eps8myc] fibroblasts, could be specifically recovered with nucleotide-depleted immobilized GST-Rac, but not with GST-Cdc42 or GST alone (Fig. 7 C). Conversely, no Sos-1 could be recovered with GST-Rac from lysates of eps8−/− cells. Thus, Eps8 is required for the association between Sos-1 and nucleotide-free Rac. Furthermore, under conditions in which the Eps8–E3b1 or the E3b1–Sos-1 interactions were disrupted by specifically competing peptides, a reduction of >80% in the amount of Sos-1 bound to nucleotide-depleted Rac was observed (Fig. 7 C). This latter result strongly suggests that an intact S/E/E8 complex is required for binding to Rac, providing a molecular basis for the catalytic specificity of the complex.
RTK activation differentially regulates the S/G and S/E/E8 complexes
Since S/G and S/E complexes are present in the cells simultaneously, then both Sos-1-dependent Ras- and Rac-GEF activities are present at the same time. The question remains as to how these two complexes, and the ensuing Ras and Rac activities, are coordinated to achieve propagation of signals. We therefore looked for evidence of differential regulation of the S/G and S/E complex upon RTK stimulation. Upon PDGF stimulation, we observed decreased coimmunoprecipitation between Grb2 and Sos-1 and consequently between PDGFR and Sos-1 (Fig. 8, A and B), which correlated with the appearance of a mobility-retarded form of Sos-1. This likely represents a hyperphosphorylated form of Sos-1, as previously demonstrated (Baltensperger et al., 1993; Cherniack et al., 1994; Pronk et al., 1994). Conversely, under identical conditions, the coimmunoprecipitation between E3b1 and Sos-1 was not affected (Fig. 8 A). Similarly, the stability of the endogenous trimeric complex, Sos-1/Eps8/E3b1 was not affected by treatment of cells with growth factors (Fig. 8 C).
Figure 8.
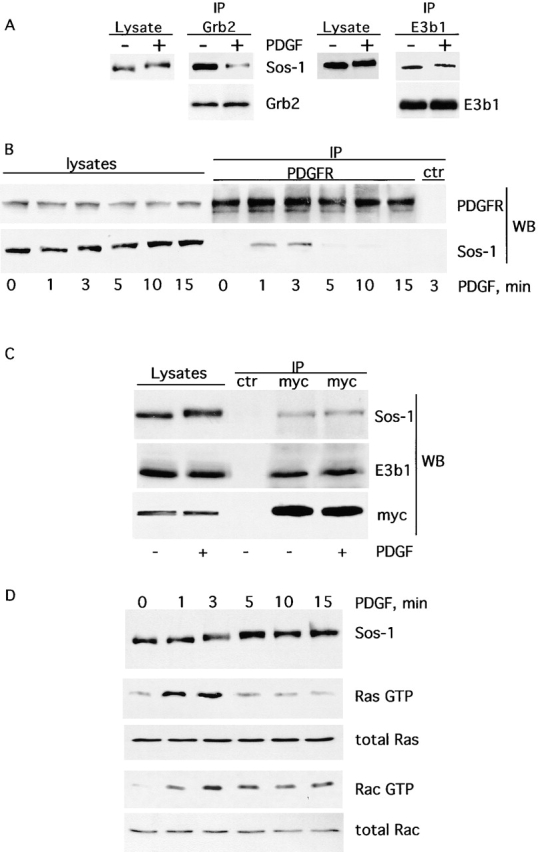
The differential regulation of the S/G and S/E/E8 complexes by RTKs correlates with the kinetics of activation of Ras and Rac. (A) Mouse embryo fibroblasts cells were treated with PDGF for 15 min (+) or mock treated (−). Lysates were immunoprecipitated (IP) with anti-Grb2 (left) or anti-E3b1 (right) antibody and detected with anti–Sos-1 antibody (top) or anti-Grb2 (bottom, left), or anti-E3b1 (bottom, right). The indicated lanes (lysate) (50 μg) were detected with anti–Sos-1 antibody to show the mobility shift after PDGF stimulation. Similar results were obtained upon EGF stimulation in Cos-7 cells (not shown). (B) Fibroblasts were treated with PDGF for the indicated lengths of time. Lysates were immunoprecipitated (IP) with anti-PDGFR (PDGFR) or an irrelevant antibody as a control (ctr) and detected with the indicated antibodies (WB). (C) −/− [Eps8myc] cells were treated with 20 ng/ml of PDGF (+) or mock treated (−), and total cellular lysates were prepared. Immmunoprecipitations (IP) were performed with anti-myc antibody or with an irrelevant (ctr) antibody, followed by immunoblot with the antibody indicated on the right (WB). The indicated lanes (lysates) were loaded with 100 μg of total cellular lysates. (D) Fibroblasts were treated with PDGF for the indicated lengths of time. The levels of Ras-GTP and Rac-GTP in total cellular lysates were determined by affinity precipitation using GST-RBD and GST-CRIB, as described in Materials and methods. Elevated levels of Rac-GTP could be observed ≤30 min of RTK stimulation (not shown). The expression levels of Ras (total Ras) and Rac (total Rac), in the same lysates, were determined by immunoblotting with anti-Ras and anti-Rac antibodies, respectively. The kinetic of the mobility shift of Sos-1, after PDGF stimulation, was determined by immunoblotting with anti–Sos-1 antibodies.
The above data indicate that, as a consequence of activation of RTKs, the S/G complex is disrupted, whereas the S/E/E8 complex persists in the cell. This might lead to a transient peak in Ras activity vis a vis a more prolonged activation of Rac. Therefore, we measured the kinetic of RTK-induced activation of Ras and Rac. As shown in Fig. 8 D, activation of Ras was rapid and short lived, whereas activation of Rac was sustained over a longer period of time, compatible with the differential regulation of the corresponding activating complexes, S/G and S/E/E8.
Discussion
Previously, we have demonstrated that the genetically engineered removal of Eps8 from mouse fibroblasts led to the abrogation of the activation of the small GTPase Rac by RTKs and/or activated Ras (Scita et al., 1999). Biochemically, this effect was mirrored by the absence of Rac-specific GEF activity in extracts of Eps8-null cells (Scita et al., 1999). Sos-1 might be the GEF involved, as witnessed by the observations that it forms a tricomplex with Eps8 and the scaffolding protein E3b1 in vivo and that this tricomplex is endowed with Rac-GEF activity in vitro (Scita et al., 1999). A model was therefore proposed in which Sos-1 acted as a dual GEF, involved in both Ras and Rac activation. In this study, after showing that a S/E/E8 complex can exist under physiological conditions, we investigated the molecular mechanisms through which Sos-1 coordinates the activation of these two small GTPases after RTK stimulation.
These results show that when Sos-1 is engaged in a multimolecular complex containing at least Eps8, E3b1, and Sos-1 itself (the contribution of other proteins cannot be excluded), it displays exclusive Rac specificity. This is mirrored biochemically by the finding that Sos-1 binds to nucleotide-depleted Rac only when its association with Eps8 and E3b1 is preserved. On the contrary, Sos-1 acts as a Ras-specific GEF when alone or in association with Grb2 (Fig. 7; Buday and Downward, 1993b). Thus, in light of these results and previous finding, we conclude that a S/G complex directs Ras activation through binding to RTKs, whereas a S/E complex directs Rac activation by entering into a tricomplex with Eps8 (Fig. 9).
Figure 9.
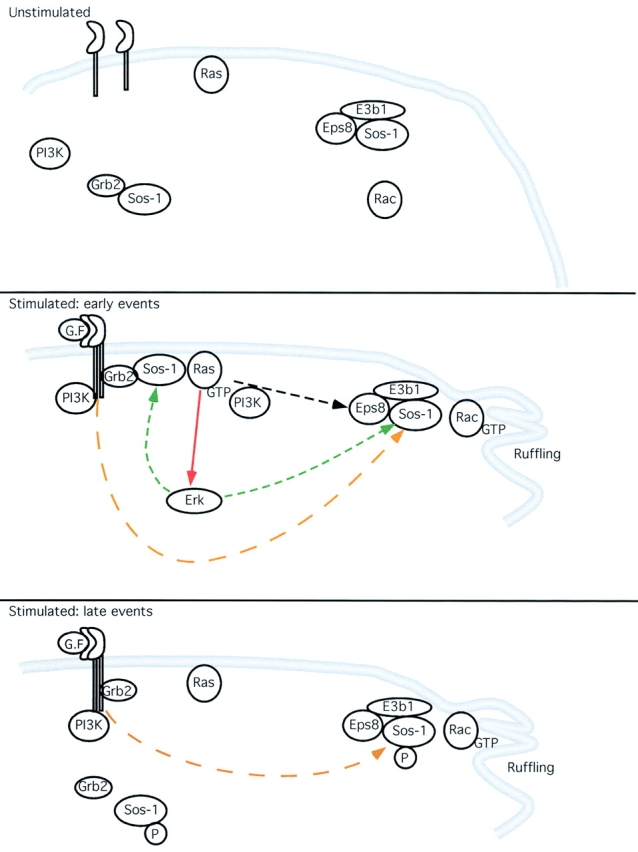
Proposed model for the roles of Sos-1 in the signaling pathway leading to activation of Rac. Under unstimulated conditions (unstimulated), Sos-1-based complexes, PI3K, and the small GTPases, Ras and Rac, are present in the cell in an inactive state. Immediately after stimulation (Stimulated: early events) by growth factor (G.F.), the S/G complex is recruited to the activated RTK at the plasma membrane, leading to Ras activation. GTP-loaded Ras, in turn, mediates signal propagation with the ensuing activation of Rac, through the trimeric complex S/E/E8 (dashed black arrow). In this pathway, PI3K, which binds to GTP-Ras, plays a key role, although it cannot be formally excluded that other Ras effectors may participate in the activation of Rac. Activation of Rac can also be achieved through Ras-independent pathways, among which the one involving the direct recruitment of PI3K to activated RTK (dashed orange arrow). All these pathways converge on the S/E/E8 Rac GEF complex. Activated Rac finally mediates actin remodelling. Concomitantly, activated Ras leads to stimulation of the ERK kinase via the RAF-MEK-ERK cascade (solid red arrow). ERK can phosphorylate Sos-1 (dashed green arrow) with ensuing reduced affinity for Grb2, but not for E3b1. This results (Stimulated: late events) in the disassembly of the RTK-Grb2-Sos-1 complex contributing to the termination of Ras activation. Conversely, the trimeric complex S/E/E8 is stable, thus allowing sustained Rac activation through Ras-independent pathways (dashed orange arrow), which are most likely PI3K dependent, but could also be PI3K dependent.
The finding that Sos-1 participates in both Ras and Rac activation does not immediately explain the temporal sequence by which the activation of Ras precedes that of Rac. This latter concept, however, is largely based on the fact that ectopic expression of a constitutively active Ras mutant is sufficient to cause membrane ruffles, indicative of Rac activation (Bar-Sagi and Feramisco, 1986; Rodriguez-Viciana et al., 1997; Scita et al., 1999). Current thinking holds therefore that active Ras, through binding and activation of phosphatidylinositol 3-kinase, leads to the formation of phosphatidylinositol 3, 4, 5 triphosphate, which in turn regulates Rac-specific GEFs, like Vav-1, and possibly Sos-1 (Kotani et al., 1994; Rodriguez-Viciana et al., 1994, 1996, 1997; Han et al., 1998; Nimnual et al., 1998; Das et al., 2000) (Fig. 9).
Our data and published literature highlight a much more complex scenario. First, it is difficult to conceive how Rac activation could be exclusively Ras dependent. In some cell lines, such as Swiss 3T3, RTK-induced Rac activation is apparently Ras independent (Ridley et al., 1992). In addition, even in those cell lines in which a dominant negative Ras mutant can suppress ruffle formation, the inhibition is never quantitative (a maximum of 65 ± 5% SEM inhibition of PDGF-induced ruffling was observed in mouse embryo fibroblasts microinjected with a Ras dominant negative mutant) (Scita, G., personal communication). Second, our kinetic data revealed that Rac activation persists long after the return to basal levels of Ras-GTP, a finding not immediately reconcilable with a simple and unique Ras→Rac pathway. It seems therefore that multiple mechanisms could lead to Rac activation from stimulated RTK (Fig. 9).
Nevertheless, signals appear to converge on a S/E/E8 complex, as suggested by the fact that genetic and biochemical interference with this complex leads to abrogation of Rac activation induced by either active RTKs or dominant active Ras and phosphatidylinositol 3-kinase mutants (Scita et al., 1999). Our demonstration of differential regulation of the stability of the S/G and S/E complexes sheds new light on the molecular understanding of this circuitry. A dynamic interaction between Sos-1 and Grb2 after RTK stimulation has been previously reported (Buday and Downward, 1993a; Egan et al., 1993; Rozakis-Adcock et al., 1993; Cherniack et al., 1994; Waters et al., 1995; Hu and Bowtell, 1996). Moreover, it is well established that upon EGF treatment, Sos-1 is rapidly phosphorylated, mainly as a consequence of MAPK activation (Cherniack et al., 1994; Rozakis-Adcock et al., 1995; Corbalan-Garcia et al., 1996; Porfiri and McCormick, 1996), leading to a decrease in its affinity for Grb2 (Corbalan-Garcia et al., 1996). Thus, the interaction between Sos-1 and Grb2 is dynamically regulated by RTK and might contribute to the rapid, but transient, activation of Ras after RTK stimulation (Fig. 8, A, B, and D). Conversely, the S/E/E8 complex is stable under the same conditions, and may account, at least in part, for the prolonged activation of Rac signaling (Fig. 8, A–D) and ruffling activity (Ridley et al., 1992; unpublished data) (Fig. 9 shows a proposed mode of action of Sos-1)
The shift in catalytic specificity of Sos-1, upon interaction with Eps8 and E3b1 does not exclude the possibility that additional mechanisms might participate in its action on Rac in vivo. This is supported by the observation that the Rac-specific GEF activity, detected in total cellular lysates and in the immunoprecipitated trimeric S/E/E8 complex, is not apparently affected by RTK activation (unpublished data). GTP-bound active Rac accumulates on the plasma membrane at sites where ruffling takes place (Kraynov et al., 2000). Similarly, upon growth factor stimulation, Eps8, E3b1, and Sos-1 are localized at the plasma membrane and colocalize with F-actin within membrane ruffles (Provenzano et al., 1998; Scita et al., 1999, 2001), whereas Grb2 cannot be detected in these structures (Scita et al. 2001). This provides a potential docking system for the Rac-based signaling machinery and argues that differential localization of adaptor molecules may further participate in the regulation of Sos-1 biological activities. Although more work will be needed to define the exact interplay of all components of the circuitry, our findings of differential regulation of the S/G and S/E complexes suggest an efficient mechanism for a wave-like propagation of signals in which upstream signaling is rapidly switched off after stimulation, whereas downstream signaling is still permitted (Fig. 9).
Materials and methods
Expression vectors and antibodies
CMV-based eukaryotic expression vectors and GST fusion or His-tagged bacterial expression vectors were generated by recombinant PCR. Where indicated, epitope tagging (either myc or HA) was also obtained by recombinant PCR. The mutant of E3b1 (E3b1-DY), in which the D and Y residues were replaced by A, was generated by PCR-based site directed mutagenesis and cloned in the retroviral pBabe vector. All constructs were sequence verified. Details are available upon request. HA–Sos-1 was from Dr. D. Bar-Sagi (Yang et al., 1995). Antibodies were anti-Eps8 (Fazioli et al., 1993), anti-E3b1 (Biesova et al., 1997), and anti-EGFR sera (Di Fiore et al., 1990); rabbit polyclonals anti–Sos-1, anti-Grb2, and anti-ERK-1; anti-PAK65, anti-PDGFR (Santa Cruz Biotechnology); monoclonals anti-phosphoMAPK (New England BioLabs), anti–v-H-Ras (Oncogene Science), anti–Histidine (Sigma Aldrich), anti-BrdU (Becton and Dickinson), anti-HA11, and anti-myc 9E10 (Babco); and anti-Rac (Transduction Laboratories).
Biochemical and functional assays
Protein analysis.
Immunoprecipitation and coimmunoprecipitation were performed as described (Fazioli et al., 1993). Briefly, cells were lysed on ice with buffer containing 1% Triton X-100, 50 mM Hepes (pH 7.5), 150 mM NaCl, 5 mM EGTA, 1.5 mM MgCl2, 10% glycerol, 0.1 mM sodium orthovanadate, 2 mM phenylmethylphosphate fluoride, 2 mM sodium pyrophosphate 10 μg/ml aprotinin, and 10 μg/ml leupeptin. Immunoprecipitations were performed for 2 h at 4°C, and immunocomplexes were recovered by adsorption to protein A–Sepharose. After several washes with buffer containing 0.1% Triton X-100, 20 mM Hepes (pH 7.6), 10% glycerol, and 150 mM NaCl (washing buffer), immunoprecipiates were resolved be SDS-PAGE and analyzed by immunoblotting.
In vitro binding assays were performed as described (Fazioli et al., 1993). GST fusion proteins were purified by affinity chromatography on agarose-bound glutathione. His-tagged COOH-terminal tail of Sos-1 was purified by affinity chromatography on agarose-bound Ni. For the in vitro binding experiments, 5–60 pmol of purified immobilized GST fusion proteins, or wild-type GST, were incubated with total cellular proteins (1–2 mg), prepared as described above. After several washing with washing buffer, proteins were resolved by SDS-PAGE and analyzed by immunoblotting.
The in vitro binding to recombinant purified nucleotide-free GST-Cdc42 and GST-Rac was performed as described (Fan et al., 1998). Briefly, Rac1 and Cdc42, prepared as GST fusion proteins (20 μg) and immobilized on glutathione–agarose beads (∼1 mg protein/ml resin), were rendered free of nucleotide by incubation for 20 min at 23°C in nucleotide-depleting (20 mM Tris-HCl, pH 7.5, 20 mM NaCl, 5% glycerol, 1 mM dithiothreitol, 0.1% Triton X-100) supplemented with 10 mM EDTA. Clarified lysates were prepared from eps8−/− or −/− [Eps8myc] cells in 20 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1.0 mM PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin. After incubation with total cellular lysates for 3 h at 4°C, beads were washed extensively with nucleotide-depleting buffer. Bound Sos-1 was determined by immunoblotting with anti–Sos-1 antibody.
The levels of Ras-GTP and Rac GTP were measured by affinity precipitation using GST-RBD (Ras-binding domain) of Raf (Taylor and Shalloway, 1996) or GST-CRIB (Cdc42 and Rac Interactive Region) of PAK65 (Manser et al., 1994), respectively, as previously described (Scita et al., 1999). The activity of PAK65 was also measured as described (Manser et al., 1995). Whenever indicated, treatment was performed with EGF (100 ng/ml) or PDGF (20 ng/ml), after serum starvation, for the indicated lengths of time.
In vitro GEF activity toward Ras or Rac was performed as described (Scita et al., 1999). Data are the mean ± SEM of at least three independent experiments performed in triplicate. Results are expressed as the [3H]GDP released after 20 min relative to time 0, after subtracting the background counts released in control reactions (obtained in the presence of a mock immunoprecipitate).
Fibroblasts, seeded on gelatine, were microinjected with 100 ng/ml of an empty vector together with rabbit IgG or of an HA-tagged E3b1 expression vector followed by treatment with 10% serum. After 15 h, BrdU was added, three hours later the cells were fixed in 4% paraformaldheyde for 10 min, permeabilized in 0.1% Triton X-100 and 2% BSA for 10 min, blocked with 2% BSA for 30 min and stained to detect BrdU incorporation with anti-BrdU antibody, E3b1, or control microinjected cells with anti-E3b1 and anti-rabbit IgG, respectively.
Acknowledgments
We thank Dr. D. Bar-Sagi for the gift of HA-Sos-1; Giuseppina Giardina for technical assistance; Enrica Migliaccio, Pascale Romano, Veronica Raker, and members of the Di Fiore laboratory for helpful discussion. The microinjector Axiovert 100 (ZEISS) was donated by the Lattanzio family.
This work was supported by a grant from Associazione Italiana Ricerca sul Cancro.
Footnotes
Abbreviations used in this paper: aa, amino acids; GEF, guanine nucleotide exchange factor; GST, glutathione S-transferase; RTK, receptor tyrosine kinase; S/E/E8; Sos-1–E3b1–Eps8; S/E, Sos-1–E3b1; S/G, Sos-1–Grb2; Sos-1, son of sevenless.
References
- Baltensperger, K., L.M. Kozma, A.D. Cherniack, J.K. Klarlund, A. Chawla, U. Banerjee, and M.P. Czech. 1993. Binding of the Ras activator son of sevenless to insulin receptor substrate-1 signaling complexes. Science. 260:1950–1952. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi, D. 1994. The Sos (Son of Sevenless) protein. Trends Endocrin. Metab. 5:164–169. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi, D., and J.R. Feramisco. 1986. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 233:1061–1068. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi, D., and A. Hall. 2000. Ras and Rho GTPases: a family reunion. Cell. 103:227–238. [DOI] [PubMed] [Google Scholar]
- Biesova, Z., C. Piccoli, and W.T. Wong. 1997. Isolation and characterization of e3B1, an eps8 binding protein that regulates cell growth. Oncogene. 14:233–241. [DOI] [PubMed] [Google Scholar]
- Buday, L., and J. Downward. 1993. a. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell. 73:611–620. [DOI] [PubMed] [Google Scholar]
- Buday, L., and J. Downward. 1993. b. Epidermal growth factor regulates the exchange rate of guanine nucleotides on p21ras in fibroblasts. Mol. Cell. Biol. 13:1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceresa, B.P., and J.E. Pessin. 1998. Insulin regulation of the Ras activation/inactivation cycle. Mol. Cell. Biochem. 182:23–29. [PubMed] [Google Scholar]
- Cerione, R.A., and Y. Zheng. 1996. The Dbl family of oncogenes. Curr. Opin. Cell Biol. 8:216–222. [DOI] [PubMed] [Google Scholar]
- Cherniack, A.D., J.K. Klarlund, and M.P. Czech. 1994. Phosphorylation of the Ras nucleotide exchange factor son of sevenless by mitogen-activated protein kinase. J. Biol. Chem. 269:4717–4720. [PubMed] [Google Scholar]
- Corbalan-Garcia, S., S.S. Yang, K.R. Degenhardt, and D. Bar-Sagi. 1996. Identification of the mitogen-activated protein kinase phosphorylation sites on human Sos1 that regulate interaction with Grb2. Mol. Cell. Biol. 16:5674–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cussac, D., M. Frech, and P. Chardin. 1994. Binding of the Grb2 SH2 domain to phosphotyrosine motifs does not change the affinity of its SH3 domains for Sos proline-rich motifs. EMBO J. 13:4011–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, B., X. Shu, G.J. Day, J. Han, U.M. Krishna, J.R. Falck, and D. Broek. 2000. Control of intramolecular interactions between the pleckstrin homology and Dbl homology domains of Vav and Sos1 regulates Rac binding. J. Biol. Chem. 275:15074–15081. [DOI] [PubMed] [Google Scholar]
- Di Fiore, P.P., O. Segatto, F. Lonardo, F. Fazioli, J.H. Pierce, and S.A. Aaronson. 1990. The carboxy-terminal domains of erbB-2 and epidermal growth factor receptor exert different regulatory effects on intrinsic receptor tyrosine kinase function and transforming activity. Mol. Cell. Biol. 10:2749–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan, S.E., B.W. Giddings, M.W. Brooks, L. Buday, A.M. Sizeland, and R.A. Weinberg. 1993. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature. 363:45–51. [DOI] [PubMed] [Google Scholar]
- Fan, P.D., and S.P. Goff. 2000. Abl interactor 1 binds to sos and inhibits epidermal growth factor- and v-abl-induced activation of extracellular signal-regulated kinases. Mol. Cell. Biol. 20:7591–7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, W.T., C.A. Koch, C.L. de Hoog, N.P. Fam, and M.F. Moran. 1998. The exchange factor Ras-GRF2 activates Ras-dependent and Rac-dependent mitogen-activated protein kinase pathways. Curr. Biol. 8:935–938. [DOI] [PubMed] [Google Scholar]
- Fazioli, F., L. Minichiello, V. Matoska, P. Castagnino, T. Miki, W.T. Wong, and P.P. Di Fiore. 1993. Eps8, a substrate for the epidermal growth factor receptor kinase, enhances EGF-dependent mitogenic signals. EMBO J. 12:3799–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, A. 1998. Rho GTPases and the actin cytoskeleton. Science. 279:509–514. [DOI] [PubMed] [Google Scholar]
- Han, J., K. Luby-Phelps, B. Das, X. Shu, Y. Xia, R.D. Mosteller, U.M. Krishna, J.R. Falck, M.A. White, and D. Broek. 1998. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 279:558–560. [DOI] [PubMed] [Google Scholar]
- Hu, Y., and D.D. Bowtell. 1996. Sos1 rapidly associates with Grb2 and is hypophosphorylated when complexed with the EGF receptor after EGF stimulation. Oncogene. 12:1865–1872. [PubMed] [Google Scholar]
- Kotani, K., K. Yonezawa, K. Hara, H. Ueda, Y. Kitamura, H. Sakaue, A. Ando, A. Chavanieu, B. Calas, F. Grigorescu, et al. 1994. Involvement of phosphoinositide 3-kinase in insulin- or IGF-1-induced membrane ruffling. EMBO J. 13:2313–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraynov, V.S., C. Chamberlain, G.M. Bokoch, M.A. Schwartz, S. Slabaugh, and K.M. Hahn. 2000. Localized Rac activation dynamics visualized in living cells. Science. 290:333–337. [DOI] [PubMed] [Google Scholar]
- Li, N., A. Batzer, R. Daly, V. Yajnik, E. Skolnik, P. Chardin, D. Bar-Sagi, B. Margolis, and J. Schlessinger. 1993. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature. 363:85–88. [DOI] [PubMed] [Google Scholar]
- Lowenstein, E.J., R.J. Daly, A.G. Batzer, W. Li, B. Margolis, R. Lammers, A. Ullrich, E.Y. Skolnik, D. Bar-Sagi, and J. Schlessinger. 1992. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 70:431–442. [DOI] [PubMed] [Google Scholar]
- Manser, E., C. Chong, Z.S. Zhao, T. Leung, G. Michael, C. Hall, and L. Lim. 1995. Molecular cloning of a new member of the p21-Cdc42/Rac-activated kinase (PAK) family. J. Biol. Chem. 270:25070–25078. [DOI] [PubMed] [Google Scholar]
- Manser, E., T. Leung, H. Salihuddin, Z.S. Zhao, and L. Lim. 1994. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 367:40–46. [DOI] [PubMed] [Google Scholar]
- Mongiovi, A.M., P.R. Romano, S. Panni, M. Mendoza, W.T. Wong, A. Musacchio, G. Cesareni, and P.P. Di Fiore. 1999. A novel peptide-SH3 interaction. EMBO J. 18:5300–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimnual, A.S., B.A. Yatsula, and D. Bar-Sagi. 1998. Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science. 279:560–563. [DOI] [PubMed] [Google Scholar]
- Porfiri, E., and F. McCormick. 1996. Regulation of epidermal growth factor receptor signaling by phosphorylation of the ras exchange factor hSOS1. J. Biol. Chem. 271:5871–5877. [DOI] [PubMed] [Google Scholar]
- Pronk, G.J., A.M. de Vries-Smits, L. Buday, J. Downward, J.A. Maassen, R.H. Medema, and J.L. Bos. 1994. Involvement of Shc in insulin- and epidermal growth factor-induced activation of p21ras. Mol. Cell. Biol. 14:1575–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano, C., R. Gallo, R. Carbone, P.P. Di Fiore, G. Falcone, L. Castellani, and S. Alema. 1998. Eps8, a tyrosine kinase substrate, is recruited to the cell cortex and dynamic F-actin upon cytoskeleton remodeling. Exp. Cell Res. 242:186–200. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J., H.F. Paterson, C.L. Johnston, D. Diekmann, and A. Hall. 1992. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 70:401–410. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana, P., P.H. Warne, R. Dhand, B. Vanhaesebroeck, I. Gout, M.J. Fry, M.D. Waterfield, and J. Downward. 1994. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 370:527–532. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana, P., P.H. Warne, A. Khwaja, B.M. Marte, D. Pappin, P. Das, M.D. Waterfield, A. Ridley, and J. Downward. 1997. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 89:457–467. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana, P., P.H. Warne, B. Vanhaesebroeck, M.D. Waterfield, and J. Downward. 1996. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- Rozakis-Adcock, M., R. Fernley, J. Wade, T. Pawson, and D. Bowtell. 1993. The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature. 363:83–85. [DOI] [PubMed] [Google Scholar]
- Rozakis-Adcock, M., P. van der Geer, G. Mbamalu, and T. Pawson. 1995. MAP kinase phosphorylation of mSos1 promotes dissociation of mSos1-Shc and mSos1-EGF receptor complexes. Oncogene. 11:1417–1426. [PubMed] [Google Scholar]
- Schlessinger, J. 2000. Cell signaling by receptor tyrosine kinases. Cell. 103:211–225. [DOI] [PubMed] [Google Scholar]
- Scita, G., J. Nordstrom, R. Carbone, P. Tenca, G. Giardina, S. Gutkind, M. Bjarnegard, C. Betsholtz, and P.P. Di Fiore. 1999. EPS8 and E3B1 transduce signals from Ras to Rac. Nature. 401:290–293. [DOI] [PubMed] [Google Scholar]
- Scita, G., P. Tenca, E. Frittoli, A. Tocchetti, M. Innocenti, G. Giardina, and P.P. Di Fiore. 2000. Signaling from Ras to Rac and beyond: not just a matter of GEFs. EMBO J. 19:2393–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scita, G., P. Tenca, L.B. Areces, A. Tocchetti, E. Frittoli, G. Giardina, I. Ponzanelli, P. Sini, M. Innocenti, and P.P. Di Fiore. 2001. An effector region in Eps8 is responsible for the activation of the Rac-specific GEF activity of Sos-1 and for the proper localization of the Rac-based actin-polymerizing machine. J. Cell Biol. 154:1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Y., K. Alin, and S.P. Goff. 1995. Abl-interactor-1, a novel SH3 protein binding to the carboxy-terminal portion of the Abl protein, suppresses v-abl transforming activity. Genes Dev. 9:2583–2597. [DOI] [PubMed] [Google Scholar]
- Taylor, S.J., and D. Shalloway. 1996. Cell cycle-dependent activation of Ras. Curr. Biol. 6:1621–1627. [DOI] [PubMed] [Google Scholar]
- Van Aelst, L., and C. D'Souza-Schorey. 1997. Rho GTPases and signaling networks. Genes Dev. 11:2295–2322. [DOI] [PubMed] [Google Scholar]
- Waters, S.B., K. Yamauchi, and J.E. Pessin. 1995. Insulin-stimulated disassociation of the SOS-Grb2 complex. Mol. Cell. Biol. 15:2791–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S.S., L. Van Aelst, and D. Bar-Sagi. 1995. Differential interactions of human Sos1 and Sos2 with Grb2. J. Biol. Chem. 270:18212–18215. [DOI] [PubMed] [Google Scholar]