

Abstract
We here describe the structural requirements for Golgi localization and a sequential, localization-dependent activation process of protein kinase C (PKC)μ involving auto- and transphosphorylation. The structural basis for Golgi compartment localization was analyzed by confocal microscopy of HeLa cells expressing various PKCμ–green fluorescent protein fusion proteins costained with the Golgi compartment–specific markers p24 and p230. Deletions of either the NH2-terminal hydrophobic or the cysteine region, but not of the pleckstrin homology or the acidic domain, of PKCμ completely abrogated Golgi localization of PKCμ. As an NH2-terminal PKCμ fragment was colocalized with p24, this region of PKCμ is essential and sufficient to mediate association with Golgi membranes. Fluorescence recovery after photobleaching studies confirmed the constitutive, rapid recruitment of cytosolic PKCμ to, and stable association with, the Golgi compartment independent of activation loop phosphorylation. Kinase activity is not required for Golgi complex targeting, as evident from microscopical and cell fractionation studies with kinase-dead PKCμ found to be exclusively located at intracellular membranes. We propose a sequential activation process of PKCμ, in which Golgi compartment recruitment precedes and is essential for activation loop phoshorylation (serines 738/742) by a transacting kinase, followed by auto- and transphosphorylation of NH2-terminal serine(s) in the regulatory domain. PKCμ activation loop phosphorylation is indispensable for substrate phosphorylation and thus PKCμ function at the Golgi compartment.
Keywords: PKCμ; Golgi localization; activation; phosphorylation; green fluorescent protein
Introduction
The PKCs comprise a family of intracellular serine/threonine kinases which are expressed in a cell type–specific pattern. PKCs have been shown to be involved in signal transduction of a wide range of biological responses including changes in cell morphology, proliferation, and differentiation (Toker, 1998; Black, 2000). Typically, PKCs are lipid-activated kinases that can be distinguished by different lipid-dependent activation modes.
Two novel lipid-activated kinases, sharing significant homology to PKCs as well as to calmodulin-dependent kinases were identified in man and mouse and named PKCμ (Johannes et al., 1994) and PKD (Valverde et al., 1994), respectively. PKC homologies reside particularly in the NH2-terminal cysteine-rich zinc finger region, comprising the structural basis for lipid-mediated activation and the COOH-terminal kinase domain which exerts even closer homologies to the calmodulin kinases. However, PKCμ/PKD differ from the three major groups of PKC isozymes by the presence of a pleckstrin homology (PH)* domain within the regulatory region (Gibson et al., 1994), an acidic domain (Gschwendt et al., 1997), and an NH2-terminal hydrophobic region. A PKC-typical pseudosubstrate site could not be identified. More recent work reported on novel PKCμ/PKD-related isotypes termed PKCν (Hayashi et al., 1999) and PKD2 (Sturany et al., 2001), together defining a novel PKC-like kinase family.
PKCμ is ubiquitously expressed and apparently involved in diverse cellular functions, probably in a cell type–specific manner. For example, PKCμ shows particularly high expression in thymus and hematopoietic cells, suggesting a potential role in immune functions (Rennecke et al., 1996; Matthews et al., 2000b). In accordance with these studies is the finding that PKCμ is recruited together with the tyrosine kinase Syk and phospholipase Cγ to the B cell receptor complex upon B cell receptor stimulation and negatively regulates PLCγ activity (Sidorenko et al., 1996). Our previous studies further suggested a function in antiapoptotic signaling (Johannes et al., 1998). Probably the most intriguing finding is the Golgi compartment localization of PKCμ and involvement in constitutive transport processes in epithelial cells (Prestle et al., 1996). Indeed, very recent data point to a fundamental importance of PKCμ in G protein–mediated regulation of Golgi organization (Jamora et al., 1999) and initiation of vesicular transport processes at the TGN (Liljedahl et al., 2001).
In accordance with cell type–specific functions, PKCμ/PKD location and activation appears to differ in different cell types and may involve different upstream regulators, including conventional PKCs (Zugaza et al., 1996; Matthews et al., 2000b). For example, PKD activation by exogenous stimuli is mediated via a PKC-dependent pathway in murine mast cells and B cells (Matthews et al., 2000b). Localization studies in the lymphocytic cell line A20 indicated a reversible, antigen receptor–triggered membrane translocation independent of the PKD PH domain (Matthews et al., 2000a).
We have performed the present study to analyze in detail structural requirements for constitutive PKCμ localization at the Golgi compartment using the epithelial-derived HeLa cell line. We show that the NH2-terminal domain is essential for localization of PKCμ at the Golgi compartment and that intrinsic kinase activity is not necessary for this localization. Golgi complex attachment of PKCμ is required for phosphorylation of activation loop serines 738/742 and subsequent NH2-terminal phosphorylation at different serines. Overexpression of PKCμ–green fluorescent protein (GFP) mutants comprised of the Golgi localization domains only or of a kinase-dead variant, both acting as dominant negative inhibitors of endogenous PKCμ function, severely affected PKCμ localization, showing in addition to Golgi localization a localization in/at vesicle-like structures.
Results
Characterization of PKCμ–GFP expression constructs
To analyze cellular localization of PKCμ in living cells, a set of plasmids was constructed expressing PKCμ mutants as COOH-terminal GFP fusion proteins. The mutants used in this study are schematically shown in Fig. 1 (for details see Materials and methods). Mutants were transiently expressed in HEK293 cells to check expression by Western blot analyses and activity pattern by in vitro autophosphorylation of PKCμ immunoprecipitates (Fig. 2). GFP-tagged wild-type PKCμ and the kinase-dead PKCμK612W-GFP mutant migrated with the expected relative molecular weight of ∼140 kD (Fig. 2, top), showing either basal autophosphorylation (wild-type) or a complete lack of kinase activity (PKCμK612W-GFP, Fig. 2, bottom) as shown previously for wild-type PKCμ and the K612W kinase-dead mutant (Johannes et al., 1998). Several deletion mutants lacking either the PH domain (PKCμΔPH-GFP), the cysteine finger region (PKCμΔCI-GFP, PKCμΔCII-GFP, PKCμΔCRD-GFP), or the acidic region (PKCμΔAD-GFP) were expressed and analyzed in the same way by immunoprecipitation and autophosphorylation (Fig. 2). Interestingly, deletion of the acidic domain, which was predicted to be involved in regulation of PKCμ kinase activity (Gschwendt et al., 1997), resulted in enhancement of constitutive kinase activity, which was similar to that shown for the PKCμΔPH-GFP mutant (Fig. 2, bottom). Deletion of each of the cysteine fingers (Cys1: H147-C196, Cys2: H271-C320) or both did not affect PKCμ kinase activity (Fig. 2, bottom). NH2-terminal deletion mutants lacking either the first 78 amino acids, representing the hydrophobic region (PKCμΔ1–78-GFP), or the 340 NH2-terminal amino acids (PKCμΔ1–340-GFP), largely covering the zinc-finger region, were constructed and analyzed for protein expression as well as for kinase activity. PKCμΔ1–78-GFP displayed a weak reduction in autophosphorylation efficiency, whereas enzyme activity of PKCμΔ1–340-GFP was comparable to wild-type kinase activity (Fig. 2, bottom). As a control for localization and phosphorylation studies, the PH domain and the NH2-terminal domain (amino acids 1–325) were each separately expressed as a GFP fusion protein. As expected, these fragments, which lack the kinase domain, showed no autophosphorylation (Fig. 2, right lane, top and bottom; see Fig. 6 A).
Figure 1.
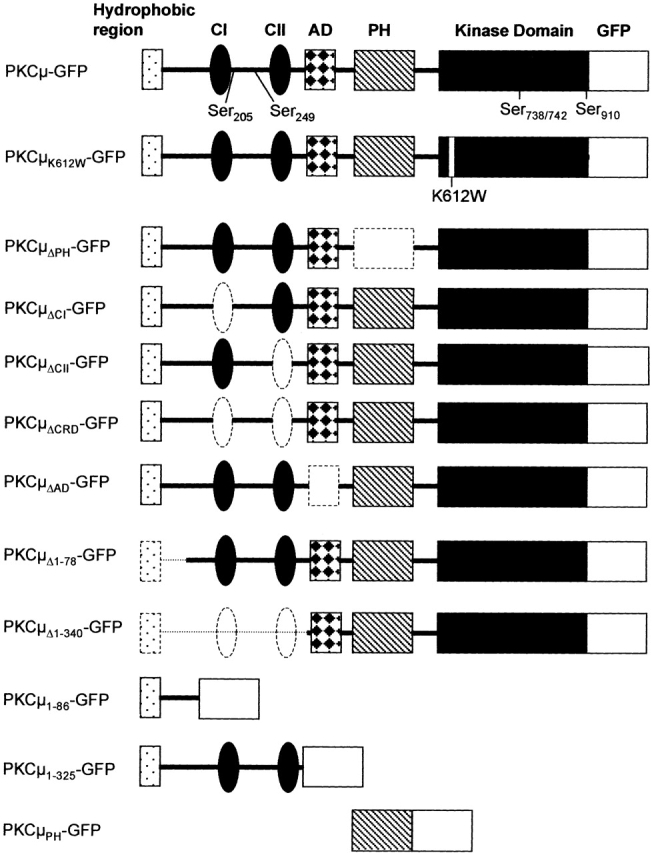
Schematic view of the PKCμ-GFP mutants used in this study. The hydrophobic region (amino acids M1–D86) and the cysteine-rich region (CI and CII; amino acids H147–C196 and amino acids H271–C320) are located in the NH2-terminal domain of PKCμ. The PH domain (V409–T552) is located between CII and the COOH-terminal kinase domain. PKCμΔPH-GFP lacks the PH domain, whereas PKCμΔCI-GFP and PKCμΔCII-GFP lack the first or the second cysteine-rich region. In PKCμΔCRD-GFP both cysteine rich regions were deleted. PKCμΔ1–78-GFP lacks the hydrophobic region (M1-R78), whereas PKCμ1–86-GFP contains only the hydrophobic regions of wild-type PKCμ. PKCμ1–325-GFP consists of 325 NH2-terminal amino acids. PKCμPH contains the PH domain (V409–T552). The acidic domain includes amino acids E336–D391. All mutants used in this study were expressed as COOH-terminal GFP fusion proteins as schematically indicated. Deleted domains are indicated by dashed lines. Phosphorylatable serine residues are indicated in wild-type PKCμ-GFP. AD, acidic domain; CRD, cysteine-rich domain; WT, wild-type. K612W indicates a point mutation in the ATP-binding site.
Figure 2.

Expression and in vitro phosphorylation of PKCμ-GFP fusion proteins. HEK293 cells were transfected with the indicated constructs. 40 h after transfection cells were lysed and PKCμ-GFP was immunoprecipitated using an anti-GFP antibody and subjected to Western blotting (top) and in vitro autophosphorylation (bottom). Shown are autoradiographs after overnight exposition.
Figure 6.
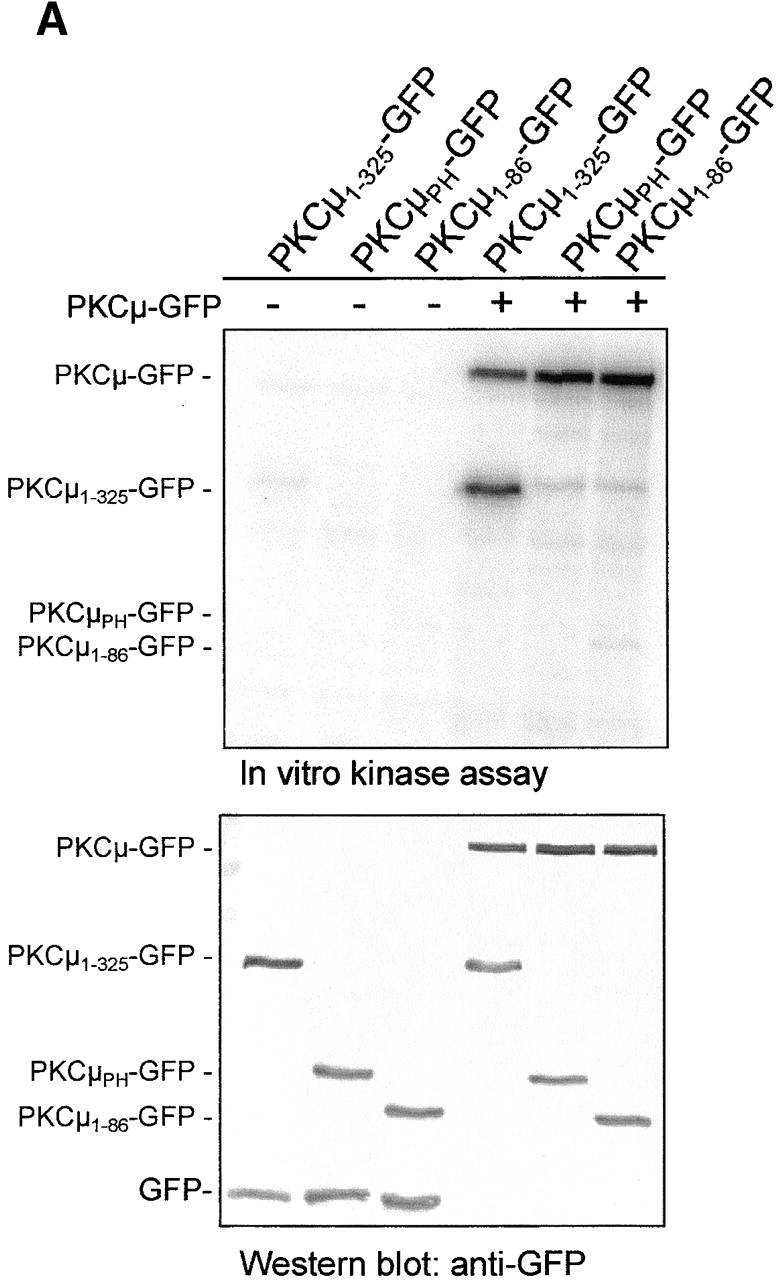

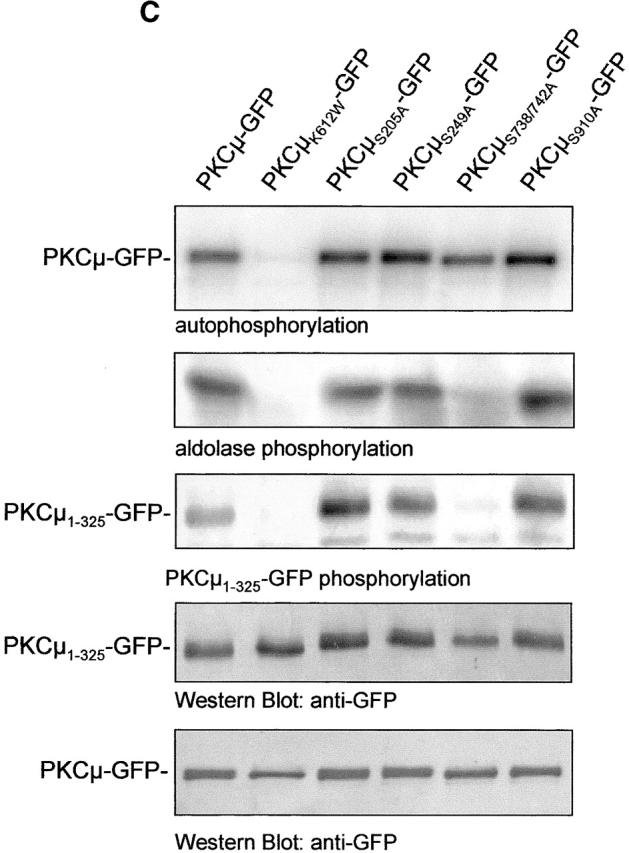
In vitro transphosphorylation of NH2-terminal PKCμ domains. (A) Shown are in vitro kinase assays of GFP immunoprecipitates (top) after expression of the indicated PKCμ domains in HEK293 cells together with PKCμ-GFP or with vector controls. Expression of proteins was verified by Western blot analysis (bottom). (B) The NH2-terminal PKCμ domain is phosphorylated in intact cells. PKCμ1–325 was coexpressed with the indicated PKCμ-GFP mutants. Total cell lysates were analyzed for PKCμ1–325 expression using a mouse antiserum directed against the NH2-terminal domain (top) or a GFP antibody detecting PKCμ-GFP expression levels (bottom). (C) Ser738/742 activation loop phosphorylation is essential for NH2-terminal transphosphorylation. Wild-type PKCμ and the indicated mutants were expressed as GFP fusion proteins, immunoprecipitated, and subjected to in vitro kinase assays measuring either auto-, aldolase, or phosphorylation of PKCμ1–325-GFP (top). Expression of the PKCμ-GFP mutants was measured by Western blot analysis using an anti-GFP antibody (bottom).
Identification of the binding domain mediating Golgi membrane association of PKCμ
To specify determinants and functional activities relevant for subcellular localization, wild-type PKCμ-GFP and kinase-dead PKCμK612W-GFP were expressed in HeLa cells and analyzed by confocal microscopy. For PKCμ-GFP a diffuse signal was revealed throughout the cell, but a clear enrichment in perinuclear structures was noted (Fig. 3 A, top rows). Staining of endogenous PKCμ in HEK293 cells with PKCμ-specific antibodies verified that the PKCμ-GFP fusion protein does not differ in localization from endogenous PKCμ (Fig. 3 D). These data are in accordance with previous studies (Prestle et al., 1996; Liljedahl et al., 2001). As a Golgi compartment–specific marker served p24, a vesicle and Golgi compartment–associated protein (Gommel et al., 1999). P24 appeared perinuclear and in vesicular structures throughout the cell (Fig. 3, middle). Partial colocalization of PKCμ-GFP with p24-positive compartments was verified by overlay (Fig. 3 A, right). Further, costaining with antibodies specific for p230 (Kjer-Nielsen et al., 1999) and GM130 (unpublished data), independent markers of the trans- and cis Golgi network, respectively, verified Golgi compartment association of PKCμ (Fig. 3 A). Interestingly, kinase-dead PKCμK612W-GFP, although still partially colocalized with the Golgi markers at a perinuclear region, was found in structures dispersed throughout the cell with appearance of long tubuli and large vesicular structures (Fig. 3 A, see enlargements). Of note, these PKCμ-positive structures did no longer costain with any of the three applied Golgi markers (Fig. 3 A, right).
Figure 3.
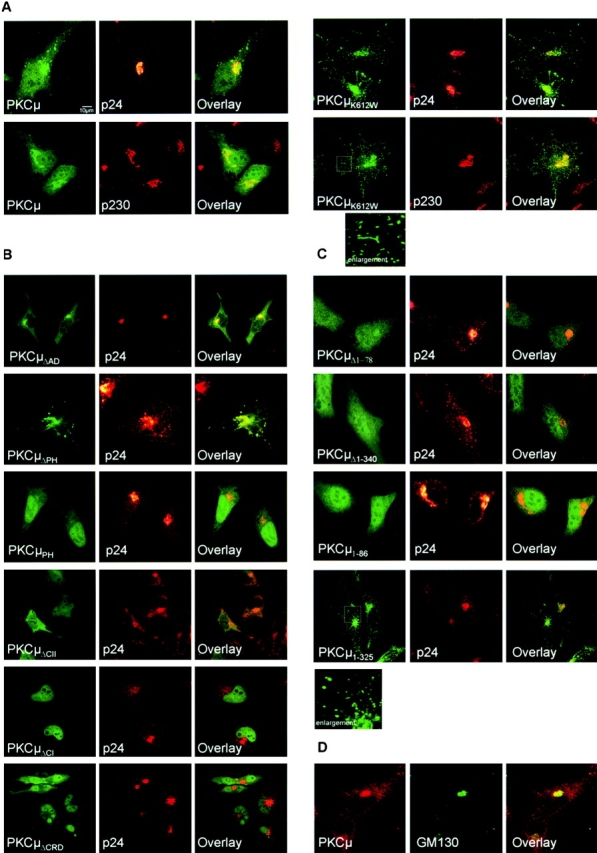
Subcellular localization of PKCμ-GFP mutants. The indicated PKCμ-GFP mutants were transiently expressed in HeLa cells and analyzed by confocal laser scanning microscopy. 40 h after transfection cells were fixed and stained for p24 or p230 with an anti-p24 rabbit antiserum or an anti-p230 monoclonal antibody followed by an incubation with Alexa 546–labeled anti–rabbit or anti–mouse antibodies. Intact cell morphology was controlled by transmission light microscopy. PKCμ-GFP (green) and p24/p230 (red) stains were combined (right). The overlay is indicated by the yellow color. (A) Localization of wild-type PKCμ and a kinase-dead K612W mutant. (B) Localization of deletion mutants and selective domains. (C) Localization of NH2-terminal PKCμ deletion mutants and the respective NH2-terminal domains. Enlargement of the indicated section is shown. (D) Localization of endogenous PKCμ in HEK293 cells. Cells were stained with a PKCμ-specific antibody and with anti-GM130 as a Golgi compartment–specific marker.
Overexpression of a PKCμ mutant lacking kinase activity was shown recently to disrupt normal Golgi morphology, pointing to an essential role of kinase activity in maintaining Golgi structure (Liljedahl et al., 2001). Further corroborating this finding, we can show that not only expression of a kinase-dead PKCμK612W-GFP, but also of the Golgi binding NH2-terminal fragment PKCμ1–325-GFP provoked changes in normal PKCμ localization, with signs of tubulation and/or large vesicle formation of PKCμ-positive structures (Fig. 3, A and B), suggesting a dominant negative action of both constructs on endogenous wild-type PKCμ. Using PKCμK612W-GFP, we analyzed whether the observed morphological changes and segregation of PKCμ from p24-positive structures might also be associated with a relocation of PKCμ to different membrane compartments or other intracellular structures. Costaining of PKCμK612W-GFP with various vesicular markers was analyzed. To this end, we could not detect any colocalization with EEA-1, Rab5, and Rab8 as endosomal markers. Furthermore, no colocalization with TGN38, BIP, Caveolin-1, Clathrin, or Lamp1 was detectable (unpublished data).
Although PH domains are frequently responsible for membrane association (Falasca et al., 1998), the deletion mutant of PKCμ showed no apparent differences in intracellular localization from the wild-type, and Golgi structure appeared normal (Fig. 3 B). Moreover, analysis of the isolated PH domain expressed as a GFP fusion protein (PKCμPH-GFP) revealed complete segregation from p24 staining and cytosolic/nuclear location (Fig. 3 B). Contrary to the expectations, these data show that the PH domain is apparently not required for PKCμ association with the Golgi compartment. Likewise, a deletion of the acidic domain of PKCμ (PKCμΔAD-GFP) displayed enhanced basal kinase activity (Fig. 2) and did not interfere with Golgi compartment localization of PKCμ (Fig. 3 B). Together, these data suggest that the PH and the acidic domain play a role in negative regulation of kinase activity rather than in localization.
The deletion of NH2-terminal regions affected Golgi compartment localization of PKCμ. As shown in Fig. 3 C, expression of PKCμ-GFP mutants lacking either 78 (PKCμΔ1–78-GFP) or 340 (PKCμΔ1–340-GFP) NH2-terminal amino acids led to a complete cytosolic distribution of PKCμ. No colocalization with p24-staining structures was detectable (Fig. 3 C). Deletion of the complete kinase domain did not affect Golgi compartment localization (unpublished data). An NH2-terminal PKCμ fragment (PKCμ1–86-GFP) was found to be located completely in the cytosol, whereas the entire NH2-terminal region covering both cysteine fingers (PKCμ1–325-GFP) showed partial colocalization with p24 staining structures (Fig. 3 C). These data already suggest that the NH2-terminal hydrophobic region itself is not sufficient, but might be required in concert with the cysteine-rich domains to mediate Golgi complex association of PKCμ. The supposed important role of the cysteine-rich region was verified by expressing the respective deletion mutants. Deletion of either the second cysteine finger (PKCμΔCII-GFP) or the complete cysteine rich region (PKCμΔCRD-GFP); each resulted in cytosolic and nuclear distribution. In the case of PKCμΔCI-GFP, an exclusive nuclear localization was detected (Fig. 3 C). These data identify the NH2-terminal hydrophobic domain and the adjacent zinc finger regions, together covering amino acids 1–325, as the Golgi compartment binding domain of PKCμ and demonstrate that intrinsic PKCμ kinase activity is not required for association with Golgi membranes.
Activation loop phosphorylation of PKCμ requires localization at the Golgi compartment
The data described above show the importance of the PKCμ NH2-terminal region for Golgi complex localization. As kinase-dead mutants of PKCμ-GFP remain associated with Golgi region (Fig. 3 A) and other intracellular membranes (Liljedahl et al., 2001), and complete inhibition of kinase activity of wild-type PKCμ-GFP by H89 did not result in a relocation to the cytosol (unpublished data), it appears that autophosphorylation is not required for membrane recruitment of PKCμ. However, as upstream kinases appear to be involved in PKCμ activation, it was necessary to analyze in detail individual phosphorylation sites in PKCμ with respect to their role in Golgi localization and activation of the kinase.
To correlate localization with the phosphorylation state, the various PKCμ-GFP constructs used in this study were expressed in HEK293 cells and monitored for expression level as well as for in vivo PKCμ phosphorylation using PKCμ phosphosite–specific antibodies. As expected, constitutive PKCμ kinase activity was detected by pSer910-specific antibodies (Fig. 4 A, middle). As a negative control, PKCμK612W-GFP was included. No autophosphorylation was detectable with pSer910 antibodies. The pSer738/742 antibody detected the PKCμK612W-GFP mutant, pointing to PKCμ kinase independent, constitutive phosphorylation of this site by an upstream kinase.
Figure 4.

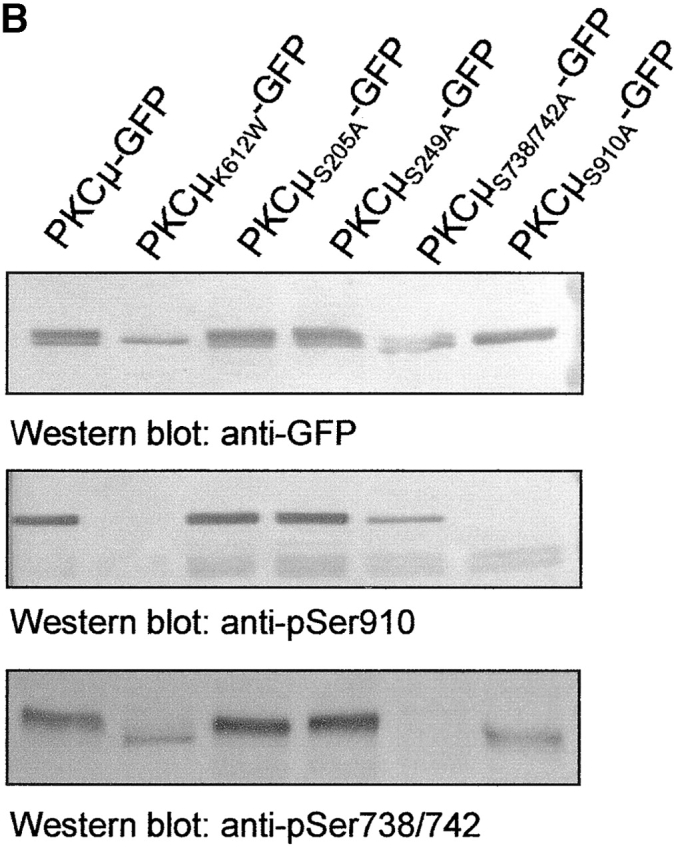
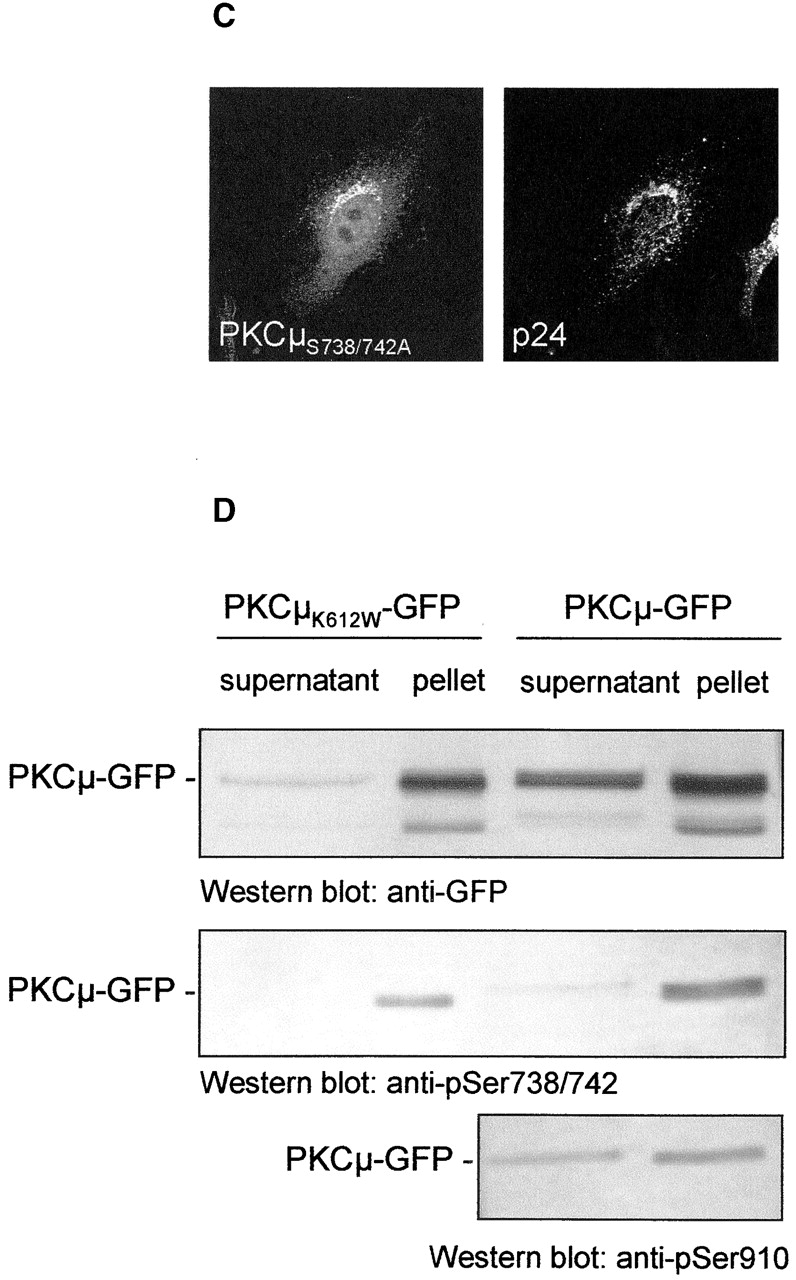
Localization of PKCμ-GFP at the Golgi compartment is required for phosphorylation of serines 738/742. (A) Differential phosphorylation of PKCμ-GFP deletion mutants. HEK293 cells were transfected with the indicated plasmids. Expression of the fusion proteins was monitored by Western blot analysis using an anti-GFP antibody. PKCμ-GFP phosphorylation was measured by phospho-specific antibodies recognizing phosphorylated Ser738/742 and Ser910. (B) Characterization of PKCμ-GFP phosphorylation mutants. (C) PKCμS738/742A-GFP colocalizes with the Golgi compartment–specific marker p24. (D) PKCμ-GFP with phosphorylated activation loop is exclusively recovered in the organelle fraction. HEK293 cells were transfected with PKCμ-GFP or PKCμK612W-GFP and separated into soluble proteins from organelles structures sedimenting at 100,000 g. Western blot analysis was performed by anti-GFP or phosphorylation-specific antibodies.
Deletion mutants of the PH domain, the acidic region, or deletions of either the first, second, or both cysteine-rich regions showed phosphorylation of Ser910. As the NH2-terminal deletion mutants are cytosolic, while the former two are Golgi bound (Fig. 3), Ser910 autophosphorylation appears localization independent. In contrast, only the PKCμΔPH-GFP and the PKCμΔAD-GFP mutant exerted significant phosphorylation at Ser738/742 (Fig. 4 A, bottom), whereas deletion mutants localized in the cytosol or in the nucleus show only weak phosphorylation at Ser738/742.
Five phosphorylation sites in PKCμ/PKD have been described recently (Vertommen et al., 2000). As well as three phosphorylation sites in the COOH-terminal region, two phosphorylation sites at Ser205 (equivalent with Ser203 in PKD) and Ser249 (Ser255 in PKD) were reported. The NH2-terminal phosphorylation sites are likely to contribute to PKCμ activation and/or regulation of PKCμ.
To further determine phosphorylation-dependent influence on Golgi complex localization of PKCμ, all predicted phosphorylation sites (Ser910, Ser738/742, Ser249, Ser205) were mutated to alanine and characterized for activation loop and COOH terminal phosphorylation. As shown in Fig. 4 B by Western blot analysis using phosphoserine-specific antibodies, mutations of NH2-terminal serine residues (S205A; S249A) did not influence phosphorylation sites on Ser738/742 or Ser910. Mutants of either Ser738/742 or Ser910 did effect detection by the respective antibodies, but did not influence other phosphorylation sites. Mutants were further analyzed for intracellular colocalization with p24. As shown for the Ser738/742Ala double mutation, Golgi complex localization (Fig. 4 C) was not affected, indicating that phosphorylation of these activation loop sites is not a prerequisite for Golgi complex localization, but instead suggests that activation loop phosphorylation requires Golgi complex localization of PKCμ. All other phosphorylation site mutants analyzed showed similar localization as wild-type PKCμ-GFP (unpublished data).
In addition, intracellular distribution of PKCμ-GFP and PKCμK612W-GFP was analyzed by biochemical methods. As shown in Fig. 4 D, after separation of soluble proteins from organelles and structures phosphorylation of PKCμ in the activation loop was exclusively recovered in the organelle fraction, whereas PKCμ was recovered in both fractions (Fig. 4 D). Phosphorylation of Ser910 was not affected by intracellular localization of PKCμ, as cytosolic and particular fractions contain approximately equal amounts of this phosphorylated species of PKCμ.
Golgi region–localized PKCμ is recruited from the cytosolic pool and is independent of activation loop phosphorylation. As shown by FRAP experiments (Fig. 5), cytosolic PKCμ-GFP and PKCμS738/742A-GFP rapidly translocate to the Golgi region. Upon bleaching of Golgi region–localized PKCμ-GFP and PKCμS738/742A-GFP within the circled area (Fig. 5 A, right), specific GFP fluorescence disappears leaving only the cytosolic and vesicular pool of PKCμ within the cell (Fig. 5 A, middle). Within a 15-min period, cytosolic PKCμ-GFP and PKCμS738/742A-GFP are rapidly recruited to the Golgi region (Fig. 5 A, right). As illustrated in Fig. 5 B by the reverse experiment, i.e., bleaching of cytosolic PKCμ-GFP and PKCμS738/742A-GFP, respectively, a decay of Golgi region–specific PKCμ-GFP and PKCμS738/742A-GFP staining was found (Fig. 5 B). Interestingly, in addition to an assumed cytosolic redistribution, which cannot be readily detected because of dilution of the fluorescence signal, we observed a redistribution of PKCμ-GFP, in particulate structures out of the defined region (Fig. 5 B, enlargements). Of note, no difference between wild-type and activation loop mutant PKCμ-GFP was observed. These data clearly indicate a translocation of cytosolic PKCμ to the Golgi region independent of its activation loop phosphorylation and point to a constitutive attachment of PKCμ to Golgi membranes.
Figure 5.
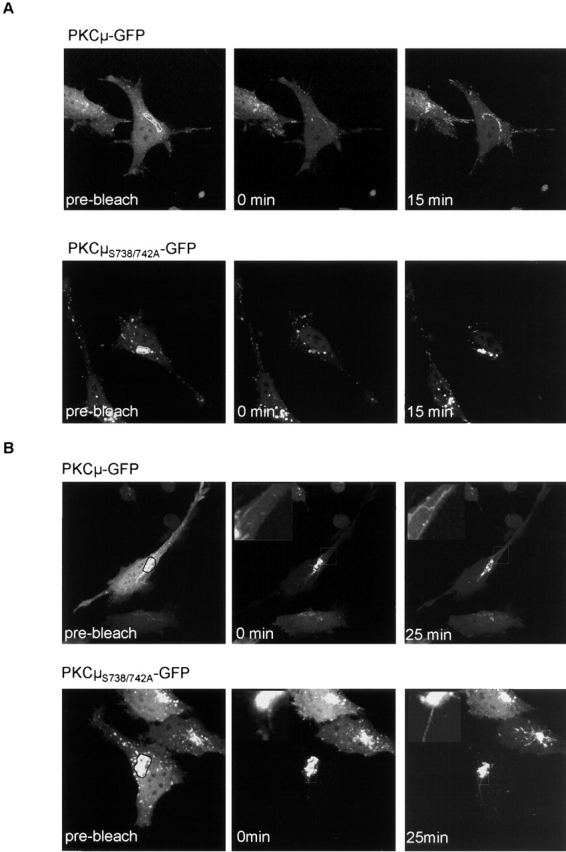
Constitutive recruitment of PKCμS738/742A-GFP to the Golgi compartment. (A) The Golgi pool of PKCμ-GFP recovers rapidly after photobleach independent of activation loop phosphorylation. The outlined area in the prebleach image (left) was photobleached. Pictures were taken after the indicated times shown in the middle and right panels. (B) Constitutive association of PKCμ-GFP with the Golgi compartment and membrane structures. Fluorescence outside of the marked region indicated in the prebleach image was eliminated by photobleaching. Note that the fluorescence intensity of PKCμ-GFP at the Golgi region is saturated in all of the images to allow visualization of less bright structures. Cells were preincubated with cycloheximide (20 μg/ml) for 2 h.
NH2-terminal phosphorylation is a consequence of activation loop phosphorylation of PKCμ at the Golgi compartment
The above studies already suggested a multistep process of PKCμ activation with auto- and transphosphorylation events for the COOH-terminal–located phosphorylation sites. To decipher the sequence of phosphorylation events leading to activation and regulation of PKCμ, we established an in vitro transphosphorylation assay using several NH2-terminal PKCμ domains expressed as GFP fusion proteins as substrates for PKCμ-GFP. As shown in Fig. 6 A, the PKCμ1–325-GFP domain could be efficiently phosphorylated by PKCμ-GFP, whereas the PKCμ1–86-GFP domain, as well as the PKCμPH-GFP domain, were not phosphorylated by PKCμ-GFP. According to published data, the phosphorylation site was predicted to be Ser205 within the 14-3-3 binding site (Hausser et al., 1999) or Ser249 predicted to be phosphorylated by an upstream kinase (Vertommen et al., 2000).
To further analyze whether the above-described NH2-terminal homologous transphosphorylation occurs in intact cells, PKCμ1–325 was coexpressed with wild-type or mutated PKCμ-GFP and analyzed by shift assays indicative of potential phosphorylation within this domain. As shown by Western blot analysis (Fig. 6 B), coexpression of PKCμ1–325 together with PKCμ-GFP led to the appearance of two bands at the expected size of the fragment. The slower migrating band of PKCμ1–325 represents the phosphorylated protein which is evident from coexpression of PKCμ1–325 with kinase-dead PKCμK612W-GFP, where only the faster migrating band appeared (Fig. 6 B, top). Conversely, coexpression of constitutively active PKCμΔPH-GFP led to the exclusive appearance of the slower migrating band, indicating strong transphosphorylation of the NH2-terminal fragment. Interestingly, coexpression of PKCμΔCRD-GFP did not result in phosphorylation of PKCμ1–325. As shown above, this mutant lacks the Golgi localization domain and is therefore not phosphorylated at the activation loop Ser738/742. Accordingly, these findings suggest a stepwise activation by phosphorylation of Ser910 and Ser738/742 followed by NH2-terminal phosphorylation of PKCμ.
To confirm this sequential phosphorylation process, mutations in known phosphorylation sites (S205A, S249A, S738/S742A, S910A) were introduced in PKCμ-GFP, expressed, and analyzed by kinase assay for auto/trans- and substrate phosphorylation. Immunoprecipitates of PKCμS738/ 742A-GFP did not show detectable aldolase- or PKCμ1–325-GFP phosphorylation, whereas in the case of all other mutants, auto- and substrate phosphorylation was not affected (Fig. 6 C). These data indicate that activation loop phosphorylation on Ser738/742 is essential for transphosphorylation of NH2-terminal residues.
Discussion
In this study, we analyzed the structural basis for Golgi compartment localization of PKCμ in epithelial cells. Using a set of deletion mutants we can show by confocal microscopy that NH2-terminal residues covering amino acids 1–325 constitute the Golgi compartment localization domain. Moreover, we show that phosphorylation of PKCμ is not required for binding to Golgi membranes but rather that phosphorylation of Ser738/742 requires Golgi localization. Our data further suggest a sequence of events in which transphosphorylation of NH2-terminal epitopes occurs subsequent to activation loop phosphorylation, whereas autophosphorylation at Ser910 is independent of localization and of phosphorylation of the activation loop. The findings presented in this study are illustrated in a model shown in Fig. 7.
Figure 7.

Model of recruitment to and activation of PKCμ at the Golgi compartment.
PKCμ is comprised of several structural domains which are putatively able to mediate membrane interactions, such as a hydrophobic NH2 terminus and two cysteine-rich zinc finger regions, highly conserved among PKC members and shown to be involved in Golgi compartment localization of PKCε (Lehel et al., 1995), as well as a PH domain considered to mediate membrane association of proteins via binding to phosphatidylinositol phosphate (Harlan et al., 1994). Biochemical studies have recently shown that the hydrophobic region of PKD does not function as a genuine transmembrane domain (Jamora et al., 1999), which is underlined by our studies showing that the expression of the human homologous fragment does not, on its own, localize to membranes. However, the analysis of the various NH2-terminal deletion mutants of PKCμ provide direct evidence that this region is, in concert with both zinc fingers involved in Golgi compartment localization of PKCμ, whereas the PH domain, unexpected from its functional relevance for PKCμ activation at the Golgi complex, is not involved. The functional importance of the NH2-terminal region is further stressed by Golgi complex localization of overexpressed PKCμ1–325-GFP, resulting in a similar appearance of vesicular structures as expression of kinase-dead PKCμ (Fig. 3). This points to a dominant negative effect of this mutant by competition with endogenous PKCμ for binding to Golgi membranes and thus negatively affecting structure and potential functions in Golgi complex (Liljedahl et al., 2001).
The simultaneous requirement of the three subdomains within the NH2-terminal regulatory region for PKCμ association with Golgi membranes points to the need for multiple interactions. In addition to potential hydrophobic interactions via the NH2 terminus and lipid messenger binding to the zinc finger regions, protein–protein interactions of this PKCμ domain with integral or associated Golgi membrane proteins are likely to be involved. Although these Golgi membrane interaction partners of PKCμ have to be identified in further studies, the NH2-terminal region is already known to serve as a binding domain for regulatory proteins. For example, 14-3-3 proteins can bind to PKCμ and negatively regulate its kinase activity (Hausser et al., 1999). Other proteins, such as the tyrosine kinase Btk and lipid PI4- and PI4-5 kinases, were also shown to be associated with PKCμ via the NH2-terminal region (Nishikawa et al., 1998; Johannes et al., 1999). As the PI4-5 kinase does not associate with kinase-dead PKCμ, a role of phosphorylation-triggering association with this target protein was predicted (Nishikawa et al., 1998). From the studies presented here, for PKCμ binding to the Golgi region, an essential role of phosphorylation is ruled out, as evident e.g., from Golgi membrane localization of kinase-dead, kinase domain–deficient, and activation loop–deficient PKCμ. Accordingly, a role of PI4-5 kinase in serving as a Golgi region receptor of PKCμ appears very unlikely.
The PKCμ PH domain does not contribute to the localization at Golgi membranes. As deletion resulted in constitutive kinase activity, these data support a specific regulatory function of this domain (Iglesias and Rozengurt, 1998; Hausser et al., 2001) (Fig. 2). Of note, the PH domain has been shown to mediate the interaction with PKCη, which is thought to play a role in PKCμ activation (Waldron et al., 1999). The participation of the PH domain of the murine PKCμ homologue, PKD, in function at the Golgi region during G-protein signaling events has been demonstrated previously (Jamora et al., 1999). Our data clearly indicate that the PKCμ PH domain serves a regulatory function, probably by coupling to upstream pathways and, in contrast to classical PH domains, does not mediate membrane localization.
Our data also shed light on the sequence of events leading to activation of PKCμ. We provide evidence that activation of PKCμ is a complex process involving auto- and transphosphorylation events at Ser910 and Ser738/742, respectively, followed by phosphorylation of NH2-terminal residues. The role of the NH2-terminal phosphorylation is currently unclear. As it is performed through a homologous transphosphorylation event by activated PKCμ (Fig. 6) its function might be in the generation of phosphoepitopes mediating the binding of regulatory proteins such as 14-3-3 (Hausser et al., 1999) or of potential substrates such as PI kinases (Nishikawa et al., 1998). Within the domain between amino acids 200–250 a clustering of potential phosphorylation sites are located (12xSer, 4xThr). Therefore, it presently cannot be excluded that, dependent on the cellular context, different residues might be phosphorylated and thus may differentially influence activity of PKCμ.
As cellularly expressed kinase-dead PKCμ is phosphorylated on Ser738/742, these sides can be considered as transphosphorylation sites for an upstream kinase. This reasoning is supported by H89 inhibition of PKCμ kinase, demonstrating selective inhibition of phosphorylation of Ser910 and not of Ser738/742 (unpublished data). Therefore, our data point to an H89-insensitive upstream kinase. According to published data and our own observations, PKCμ is activated by upstream PKCs (Zugaza et al., 1996). PKCη and also PKCε were recently implicated in PKD activation (Waldron et al., 1999). PKCε has been located at the Golgi compartment and a role in Golgi region–specific functions was suggested previously (Lehel et al., 1995). The data presented here are in accordance with a participation of PKCε in Golgi region functions via activation of PKCμ. In support of this, Golgi region localization domain mutants did not show phosphorylation on Ser738/742 (Fig. 4 A). On the other hand, activation loop mutants, similarly to wild-type PKCμ, were localized at the Golgi region (Fig. 4 C). This reemphasizes a phosphorylation-independent localization of PKCμ at the Golgi region and suggests PKCε as a candidate for an upstream kinase for activation loop phosphorylation of PKCμ at the Golgi compartment.
Materials and methods
Plasmid constructs and cell lines
cDNA constructs containing wild-type and various mutant PKCμ sequences in the pCDNA3 mammalian expression vector have been described previously (PKCμK612W, PKCμΔ1–78, PKCμΔ1–340, PKCμΔAD, and PKCμΔPH) (Johannes et al., 1998, 1999). Deletion of the CI motif, amino acids H147–C196 (PKCμΔCI); the CII motif, amino acids H271–C320 (PKCμΔCII); and the combination of both motifs (PKCμΔCRD) were generated by an overlap PCR using Taq-polymerase (MBI Fermentas). Site-specific mutations within PKCμ-GFP resulting in single amino acids substitutions (S205A, S249A, S738/742A, S910A) were performed by a PCR approach using the QuickChange site-directed mutagenesis system (Stratagene) according to the manufacturer's instructions. The integrity of the PCR-amplified plasmids were verified by sequencing. Fig. 1 shows a scheme of the different mutants used in this study. The GFP-tagged wild-type and mutant PKCμ expression plasmids were obtained by subcloning the respective PKCμ coding sequence into the EcoRI-BamHI sites of the polylinker of the pEGFP-N1 vector from CLONTECH Laboratories, Inc. HeLa and HEK293 (American Type Culture Collection) were cultured in RPMI medium supplemented with 5% FCS.
Antibodies and reagents
Antibodies directed against phosphoSer916 and phosphoSer744/748 of PKD were purchased from NEB/Cell Signaling. p24-specific antibodies were provided by F. Wieland (University of Heidelberg, Heidelberg, Germany). Anti-GFP antibodies were obtained from Roche Diagnostics. Anti-p230 and anti-GM130 antibodies were purchased from Transduction Laboratories. Anti-PKCμ rabbit antibody was obtained from Santa Cruz Biotechnology, Inc. Secondary alkaline phosphatase conjugated goat anti-mouse IgG and goat anti-rabbit IgG antibodies were purchased from Dianova or Sigma-Aldrich. The Alexa 546–conjugated goat anti–rabbit and anti–mouse antibodies were purchased from Molecular Probes. Protease- and phosphatase inhibitors were from Biomol.
HEK293 and HeLa cell transfections
HEK293 and HeLa cells were maintained at 37°C in a 5% CO2 atmosphere in RPMI medium supplemented with 5% FCS. The day before transfection, HEK293 cells were seeded at 3 × 105 cells per well in a 6-well plate (for in vitro kinase assays and Western blot). HeLa cells were seeded at 5 × 104 cells on glass coverslips (for immunofluorescence microscopy). DNA transfections (2 μg plasmid DNA per 3 × 105 cells and 1 μg plasmid DNA per 5 × 104 cells) were performed using Superfect reagent (QIAGEN) according to the manufacturer's instructions. In brief, appropriate DNA amounts were mixed with the Superfect reagent, incubated at room temperature for 10 min in order to allow the complex to form, and then directly added to the culture medium. 2–3 h later, cells were transferred to fresh RPMI supplemented medium and incubated for further 40 h at 37°C.
Immunoprecipitation and in vitro kinase assays
HeLa and HEK293 cells transiently expressing the indicated PKCμ-GFP mutants were lysed at 4°C in lysis buffer (20 mM Tris/HCl, pH 7.4, 1% Triton X-100, 150 mM NaCl, 5 mM MgCl2, 1 mM NaF, 1 mM sodium orthovanadate, 10 μg/ml leupeptin, 0.5 mM PMSF). After 30 min cell lysis, the lysates were centrifuged (10,000 g, 15 min, 4°C), the supernatant was collected, and immunoprecipitation of GFP fusion proteins was performed with 400 ng of anti-GFP antibody. After a 1.5-h incubation at 4°C, 30 μl of protein G sepharose was added and the mixture was incubated at 4°C for 1 h. The sepharose pellet was then washed two times in lysis buffer and once in kinase buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 2 mM DTT) and PKCμ activity (as measured by auto- and substrate phosphorylation) was determined by incubating immunocomplexes with 10 μl of kinase buffer containing 2 μCi [γ-32P]-ATP with or without 5 μg aldolase at 37°C for 15 min. Reactions were terminated by the addition of 5× SDS-PAGE sample buffer and analyzed by SDS-PAGE, Western blotting, and autoradiography. Autoradiographs were analyzed by quantitative phosphoimage analysis (Molecular Dynamics).
Western blot analysis
For Western blot analysis, transfected HEK293 cells were treated as described in the figure legends before being lysed in 200 μl lysis buffer followed by boiling with 5× SDS-PAGE sample buffer. Equal amounts of protein were loaded on a 12.5% SDS-PAGE. Upon fractionation, proteins were transferred to a nitrocellulose membrane (Schleicher & Schuell). Membranes were blocked, followed by incubation either with a monoclonal antibody against GFP (1:1,000), a mouse antiserum raised against the NH2-terminal region of PKCμ (1:1,000), or the rabbit antibodies phosphoSer744/748 and phosphoSer916 (both 1:500). Membranes were incubated with alkaline phosphatase–conjugated anti–mouse IgG or anti–rabbit IgG antibodies (1:5,000). Immunoblots were developed according to standard procedures.
For separation of soluble proteins from organelles 4 × 106 HEK293 cells were transfected with 20 μg of pEGFP-N1-PKCμ or pEGFP-N1-PKCμK612W and 100 μl Superfect reagent (QIAGEN) according to the manufacturer's instructions. 40 h after transfection, cells were harvested and resuspended in 500 μl lysis buffer without Triton X-100. Homogenization was done by applying 20 strokes with a “very tight fitting” 5-ml Dounce homogenizator (Braun). To remove cellular debris, the cellular extract was centrifuged at 1000 g followed by centrifugation of the supernatant for 1 h at 100 000 g (TLA 100; Beckman Coulter). Soluble proteins were recovered in the supernatant, whereas organelles and structures were recovered in the pellet. The pellet was resuspended in lysis buffer. For Western blot analysis equal amounts of protein were loaded onto a 12.5% SDS-PAGE.
Confocal immunofluorescence analysis
HeLa cells grown on glass coverslips and expressing the indicated GFP-tagged PKCμ mutants were washed once in PBS and fixed in 3.5% paraformaldehyde (pH 7.4) for 20 min at 37°C. Fixed cells were blocked and permeabilized in 5% normal goat serum and 0.05% Tween-20 for 30 min at room temperature. Coverslips were then incubated for 2 h at room temperature with the p24 rabbit antibody (1:200) or the p230 mouse antibody (1:200). Coverslips were washed three times in PBS and incubated with an anti–rabbit or an anti–mouse IgG Alexa 546–labeled antibody (1:500) for 1.5 h at room temperature. Cells were washed three times in PBS and mounted in Fluormount G (Dianova). Images were acquired using a confocal laser scanning microscope (TCS SP2; Leica) equipped with a 63×/1.4 HCX PlanAPO oil immersion objective. GFP was excited with an argon laser (488-nm line), whereas Alexa 546 was excited with a helium-neon laser (543-nm line). Each image represents a two-dimensional parallel projection of sections in the Z-series taken at 0.5–1-μm intervals across the depth of the cell.
Selective photobleaching was performed on the Leica TCS SP2 using 80 consecutive scans with a 488-nm laser line at full power. Live cells were held at 37°C and 5% CO2 atmosphere.
Acknowledgments
This work was supported by the Sonderforschungsbereich 495/B5 and by grant number 03121805 from the Bundesministerium für Bildung und Forschung (BMBF).
Footnotes
Abbreviations used in this paper: GFP, green fluorescent protein; PH, pleckstrin homology.
References
- Black, J.D. 2000. Protein kinase C-mediated regulation of the cell cycle. Front Biosci. 5:D406–D423. [DOI] [PubMed] [Google Scholar]
- Falasca, M., S.K. Logan, V.P. Lehto, G. Baccante, M.A. Lemmon, and J. Schlessinger. 1998. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain- mediated membrane targeting. EMBO J. 17:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, T.J., M. Hyvonen, A. Musacchio, M. Saraste, and E. Birney. 1994. PH domain: the first anniversary. Trends Biochem Sci. 19:349–353. [DOI] [PubMed] [Google Scholar]
- Gommel, D., L. Orci, E.M. Emig, M.J. Hannah, M. Ravazzola, W. Nickel, J.B. Helms, F.T. Wieland, and K. Sohn. 1999. p24 and p23, the major transmembrane proteins of COPI-coated transport vesicles, form hetero-oligomeric complexes and cycle between the organelles of the early secretory pathway. FEBS Lett. 447:179–185. [DOI] [PubMed] [Google Scholar]
- Gschwendt, M., F.J. Johannes, W. Kittstein, and F. Marks. 1997. Regulation of protein kinase Cmu by basic peptides and heparin. Putative role of an acidic domain in the activation of the kinase. J. Biol. Chem. 272:20742–20746. [DOI] [PubMed] [Google Scholar]
- Harlan, J.E., P.J. Hajduk, H.S. Yoon, and S.W. Fesik. 1994. Pleckstrin homology domains bind to phosphatidylinositol-4,5-bisphosphate. Nature. 371:168–170. [DOI] [PubMed] [Google Scholar]
- Hausser, A., P. Storz, S. Hubner, I. Braendlin, M. Martinez-Moya, G. Link, and F.J. Johannes. 2001. Protein kinase C mu selectively activates the mitogen-activated protein kinase (MAPK) p42 pathway. FEBS Lett. 492:39–44. [DOI] [PubMed] [Google Scholar]
- Hausser, A., P. Storz, G. Link, H. Stoll, Y.C. Liu, A. Altman, K. Pfizenmaier, and F.J. Johannes. 1999. Protein kinase C mu is negatively regulated by 14-3-3 signal transduction proteins. J. Biol. Chem. 274:9258–9264. [DOI] [PubMed] [Google Scholar]
- Hayashi, A., N. Seki, A. Hattori, S. Kozuma, and T. Saito. 1999. PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim. Biophys. Acta. 1450:99–106. [DOI] [PubMed] [Google Scholar]
- Iglesias, T., and E. Rozengurt. 1998. Protein kinase D activation by mutations within its pleckstrin homology domain. J. Biol. Chem. 273:410–416. [DOI] [PubMed] [Google Scholar]
- Jamora, C., N. Yamanouye, J. Van Lint, J. Laudenslager, J.R. Vandenheede, D.J. Faulkner, and V. Malhotra. 1999. Gβγ-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell. 98:59–68. [DOI] [PubMed] [Google Scholar]
- Johannes, F.J., A. Hausser, P. Storz, L. Truckenmuller, G. Link, T. Kawakami, and K. Pfizenmaier. 1999. Bruton's tyrosine kinase (Btk) associates with protein kinase C mu. FEBS Lett. 461:68–72. [DOI] [PubMed] [Google Scholar]
- Johannes, F.J., J. Horn, G. Link, E. Haas, K. Siemienski, H. Wajant, and K. Pfizenmaier. 1998. Protein kinase Cmu downregulation of tumor-necrosis-factor-induced apoptosis correlates with enhanced expression of nuclear-factor-κB-dependent protective genes. Eur. J. Biochem. 257:47–54. [DOI] [PubMed] [Google Scholar]
- Johannes, F.J., J. Prestle, S. Eis, P. Oberhagemann, and K. Pfizenmaier. 1994. PKCu is a novel, atypical member of the protein kinase C family. J. Biol. Chem. 269:6140–6148. [PubMed] [Google Scholar]
- Kjer-Nielsen, L., C. van Vliet, R. Erlich, B.H. Toh, and P.A. Gleeson. 1999. The Golgi-targeting sequence of the peripheral membrane protein p230. J. Cell Sci. 112:1645–1654. [DOI] [PubMed] [Google Scholar]
- Lehel, C., Z. Olah, G. Jakab, and W.B. Anderson. 1995. Protein kinase C ε is localized to the Golgi via its zinc-finger domain and modulates Golgi function. Proc. Natl. Acad. Sci. USA. 92:1406–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljedahl, M., Y. Maeda, A. Colanzi, I. Ayala, J. Van Lint, and V. Malhotra. 2001. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell. 104:409–420. [DOI] [PubMed] [Google Scholar]
- Matthews, S.A., T. Iglesias, E. Rozengurt, and D. Cantrell. 2000. a. Spatial and temporal regulation of protein kinase D (PKD). EMBO J. 19:2935–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, S.A., E. Rozengurt, and D. Cantrell. 2000. b. Protein kinase D. A selective target for antigen receptors and a downstream target for protein kinase C in lymphocytes. J. Exp Med. 191:2075–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa, K., A. Toker, K. Wong, P.A. Marignani, F.J. Johannes, and L.C. Cantley. 1998. Association of protein kinase Cμ with type II phosphatidylinositol 4-kinase and type I phosphatidylinositol-4-phosphate 5-kinase. J. Biol. Chem. 273:23126–23133. [DOI] [PubMed] [Google Scholar]
- Prestle, J., K. Pfizenmaier, J. Brenner, and F.J. Johannes. 1996. Protein kinase C μ is located at the Golgi compartment. J. Cell Biol. 134:1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennecke, J., F.J. Johannes, K.H. Richter, W. Kittstein, F. Marks, and M. Gschwendt. 1996. Immunological demonstration of protein kinase C mu in murine tissues and various cell lines. Differential recognition of phosphorylated forms and lack of down-regulation upon 12-O-tetradecanoylphorphol-13-acetate treatment of cells. Eur. J. Biochem. 242:428–432. [DOI] [PubMed] [Google Scholar]
- Sidorenko, S.P., C.L. Law, S.J. Klaus, K.A. Chandran, M. Takata, T. Kurosaki, and E.A. Clark. 1996. Protein kinase C μ (PKC μ) associates with the B cell antigen receptor complex and regulates lymphocyte signaling. Immunity. 5:353–363. [DOI] [PubMed] [Google Scholar]
- Sturany, S., J. Van Lint, F. Muller, M. Wilda, H. Hameister, M. Hocker, A. Brey, U. Gern, J. Vandenheede, T. Gress, et al. 2001. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J. Biol. Chem. 276:3310–3318. [DOI] [PubMed] [Google Scholar]
- Toker, A. 1998. Signaling through protein kinase C. Front Biosci. 3:D1134–D1147. [DOI] [PubMed] [Google Scholar]
- Valverde, A.M., J. Sinnett-Smith, J. Van Lint, and E. Rozengurt. 1994. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc. Natl. Acad. Sci. USA. 91:8572–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertommen, D., M. Rider, Y. Ni, E. Waelkens, W. Merlevede, J.R. Vandenheede, and J. Van Lint. 2000. Regulation of protein kinase D by multisite phosphorylation. Identification of phosphorylation sites by mass spectrometry and characterization by site-directed mutagenesis. J. Biol. Chem. 275:19567–19576. [DOI] [PubMed] [Google Scholar]
- Waldron, R.T., T. Iglesias, and E. Rozengurt. 1999. The pleckstrin homology domain of protein kinase D interacts preferentially with the eta isoform of protein kinase C. J. Biol. Chem. 274:9224–9230. [DOI] [PubMed] [Google Scholar]
- Zugaza, J.L., J. Sinnett-Smith, J. Van Lint, and E. Rozengurt. 1996. Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. EMBO J. 15:6220–6230. [PMC free article] [PubMed] [Google Scholar]