

Abstract
The localization of β-actin mRNA to the leading lamellae of chicken fibroblasts and neurite growth cones of developing neurons requires a 54-nt localization signal (the zipcode) within the 3′ untranslated region. In this study we have identified and isolated five proteins binding to the zipcode. One of these we previously identified as zipcode binding protein (ZBP)1, a 4-KH domain protein. A second is now investigated in detail: a 92-kD protein, ZBP2, that is especially abundant in extracts from embryonic brain. We show that ZBP2 is a homologue of the human hnRNP protein, KSRP, that appears to mediate pre-mRNA splicing. However, ZBP2 has a 47–amino acid (aa) sequence not present in KSRP. Various portions of ZBP2 fused to GFP indicate that the protein most likely shuttles between the nucleus and the cytoplasm, and that the 47-aa insert promotes the nuclear localization. Expression of a truncated ZBP2 inhibits the localization of β-actin mRNA in both fibroblast and neurons. These data suggest that ZBP2, although predominantly a nuclear protein, has a role in the cytoplasmic localization of β-actin mRNA.
Keywords: RNA localization; RNA binding proteins; nuclear-cytoplasmic trafficking; RNA splicing; KH domain proteins
Introduction
In diverse cell types, asymmetric localization of specific mRNAs generates cell polarity by controlling the translation sites and restricting the target proteins to the appropriate subcellular compartments (Bassell et al., 1999; Bassell and Singer, 1997, 2001; Jansen, 2001). During oogenesis in Drosophila and Xenopus, polarized localization of maternal mRNAs and consequent protein synthesis establish embryonic patterning (Bashirullah et al., 1998; Deshler et al., 1998; Ephrussi et al., 1991). In yeast Saccharomyces cerevisiae, mating-type switching is regulated by targeting ASH1 mRNA to the bud tip, where localized expression of Ash1p represses the HO endonuclease in the daughter cell (Amon 1996; Long et al., 1997; Takizawa et al., 1997). Asymmetrical segregation of mRNAs in subcellular locations is also observed in somatic cells, for example, MBP mRNA in oligodendrocytes (Ainger et al., 1993, 1997), MAP2 and tau mRNA in neurons (Kleiman et al., 1990; Litman et al., 1993; Behar et al., 1995), β-actin mRNA in fibroblasts (Kislauskis et al., 1993), and neurons (Bassell et al., 1998; Zhang et al., 1999).
The localization of β-actin mRNA in motile fibroblasts and growth cones of developing neurons provides good models by which to understand the molecular mechanism whereby specific mRNAs are transported and targeted to precise cytoplasmic environments, hence promoting cellular asymmetry. Spatially restricted synthesis of actin proteins results from targeting of β-actin mRNA at the leading edge of chicken embryonic fibroblasts (CEFs)* where actin polymerization drives cell motility (Kislauskis et al., 1994). Fibroblasts with localized β-actin mRNA migrate significantly further than those with nonlocalized β-actin mRNA (Kislauskis et al., 1997). In cultured rat and chicken developing neurons, the sorting of β-actin mRNA to neurite growth cones has also been observed (Bassell et al., 1998; Zhang, et al., 1999), and mRNA localization is necessary for enrichment of β-actin protein and forward movement of growth cones (Zhang et al., 2001). These data suggest that neurons and fibroblasts may share a similar mechanism for sorting of β-actin mRNA.
We have previously reported a cis-acting element, the zipcode, which is necessary and sufficient for asymmetric segregation of β-actin mRNA in fibroblasts. Deletion or mutation of the zipcode delocalized a reporter mRNA, and antisense treatment of the zipcode affected the regional synthesis of β-actin protein and, as a consequence, the cell motility (Kislauskis et al., 1994, 1997; Shestakova et al., 2001; Zhang et al., 2001). A cytoplasmic trans-acting factor, zipcode binding protein(ZBP)1, has been characterized (Ross et al., 1997); it bound to the zipcode of β-actin mRNA, but did not bind to a mutated zipcode incapable of asymmetrically localizing a reporter. Recently, the Xenopus homologue of ZBP1 has been identified by virtue of its binding to a localization element in Vg1 mRNA, an RNA that becomes localized to the vegetal pole of oocytes (Deshler et al., 1998; Havin et al., 1998). This implies that a common machinery may exist for targeting different mRNAs in diverse cell types.
It is likely that ZBP1 is a member of the locasome, a complex of proteins specialized for localization (Bertrand et al., 1998). Because β-actin mRNA is also localized in neurons, we searched for the complex in brain extracts. In this work we report a second protein that binds to the zipcode and is highly enriched in brain. We show that ZBP2 is a homologue of human hnRNP protein, KSRP, that regulates premRNA splicing (Min et al., 1997). Interestingly, ZBP2, like KSRP, is predominately a nuclear protein. The in vitro and in vivo data suggest that ZBP2 also has a small fraction present in the cytoplasm and may spend a short time in the cytoplasm, and in this way may contribute to the subcellular localization of β-actin mRNA.
Results
Identification of zipcode binding proteins
To identify proteins that bound to the zipcode of β-actin mRNA, RNA mobility shift and UV crosslinking approaches were used (Ross et al., 1997). Radiolabeled zipcode transcripts were used for RNA mobility shift assays. Three distinct RNA–protein complexes were formed when the labeled zipcode probe was incubated with brain or fibroblast extracts (Fig. 1 A, lanes 1–4; complexes are indicated by arrows). The intensity of shifted complexes was proportional to the amount of brain extract used (lanes 1–3). The fast-migrating complex (F) was seen in both brain and fibroblast extracts (lanes 3 and 4, respectively). However, the two slower migrating complexes (M and S) were more evident in brain than in fibroblast extracts (lanes 3 and 4). The specificity of the complexes was determined by competition assays in which 100-fold excess of unlabeled zipcode competed with the formation of the complexes (lane 5), whereas 100-fold excess of unlabeled RNA transcribed from pGEM 3Z vector did not interfere with the complex formation (unpublished data).
Figure 1.

Characterization of proteins binding to the zipcode in chicken embryo brain and fibroblast extracts. (A). Complexes formed from brain and fibroblasts. [32P]-labeled transcripts encoding the 54 nts zipcode of the 3′ UTR of chicken β-actin mRNA were incubated with aliquots of chicken embryo brain and fibroblast protein extracts, followed by incubation with RNase T1 (50 U/ml) and heparin (5 mg/ml). RNA–protein complexes were resolved in 4% polyacrylamide native gels that were then dried and exposed to Kodak X-Ray film. (Lanes 1, 2, and 3) [32P]-labeled transcript incubated with 1-, 2-, and 5-μl brain extracts, respectively. (Lane 4) [32P]-labeled transcript incubated with 5 μl fibroblast extract. (Lane 5) [32P]-labeled transcript incubated with 5-μl brain extract in the presence of 100× excess molar amounts of unlabeled 3′ UTR of actin mRNA transcripts. The arrows point to the slow (S), medium (M), and fast-migrating (F) RNA–protein complexes. (B) Specificity of the complexes for the zipcode. (Lane 1) Zipcode incubated with brain extract. (Lane 2) Mutant zipcode incubated with brain extract. (C) UV crosslinking assay. [32P]-labeled transcripts were incubated with brain extract, treated with RNase T1, and exposed to UV light for 3 min at a distance of 1 cm. Each of the RNA–protein complexes (F, M, and S) was identified and excised from a 4% polyacrylamide native gel, soaked in SDS buffer, and resolved in 10% SDS-PAGE. (Left three lanes) Excised RNA–protein complexes F, M, and S that were not UV crosslinked. (Right three lanes) Excised RNA–protein complexes F, M, and S that were crosslinked. The arrows indicate the protein mobilities with labeled RNA bound.
Because a mutated zipcode failed to localize the reporter mRNA (Ross et al., 1997), we reasoned that the mutant zipcode would not form an RNA–protein complex. To test this, we generated radiolabled mutant zipcode in which the two ACACCC motifs were replaced by GTGTGT, and we performed RNA mobility shift assays with brain extracts (Fig. 1 B). In contrast to the wild-type zipcode (lane 1), the mutant zipcode failed to form RNA–protein complexes (Fig. 1 B, lane 2). This indicated that proteins in the brain extract did not bind to the mutant zipcode, and demonstrated that a mutation that impairs mRNA localization can be correlated with impaired binding of the RNA–binding protein. To distinguish the protein components that bind to the zipcode, we used UV to crosslink RNA–protein complexes formed by mixing the radiolabeled zipcode with brain extracts, and analyzed the complexes by SDS-PAGE. All three RNA–protein complexes were not detectable without crosslinking (Fig. 1 C, no UV-X). When the RNA–protein complexes were crosslinked and stabilized, specific bands were visualized (UV-X). Complex F contained a crosslinked protein with an estimated molecular mass of ∼68 kD, the same molecular mass as ZBP1 (Ross et al., 1997). Complexes M and S (the brain-enriched bands) contained a crosslinked protein with an estimated molecular mass of ∼92 kD. The migration difference of complexes M and S in native gel, and the identity in molecular mass on SDS-PAGE, suggested that complex S could be a dimer of complex M. From these data we conclude that the 68- and 92-kD proteins bind to the zipcode, the β-actin mRNA localization signal. The 68-kD band is likely ZBP1, but the other ZBP is preferentially expressed in brain. The 92-kD protein was named ZBP2. The fact that they segregate into separate complexes indicates that ZBP1 and ZBP2 do not bind simultaneously to the zipcode.
Purification of ZBPs
To identify the brain-enriched protein, we purified ZBPs by using RNA affinity selection techniques. We passed brain or cultured fibroblast extracts over an affinity chromatography column containing zipcode RNA. After extensive washes, the proteins retained on the column were eluted with increasing salt concentration steps and analyzed by SDS-PAGE with silver staining (Fig. 2 A). Although the starting extract and flow-through fraction were heterogeneous (Fig. 2 A, lane 1), after a series of washes (lanes 2–5), specific proteins were eluted with increasing salt conditions (Fig. 2 A, lanes 7–9). The protein fraction eluted with 1 and 2 M KCl contained distinct protein bands (Fig. 2 A, lanes 8 and 9, arrows); three proteins were ∼68 kD and one was 92 kD. The estimated molecular masses were in close agreement with that of the UV-crosslinked RNA–protein complexes identified by SDS-PAGE (Fig. 1 B). A lower molecular mass protein (53 kD) had also been detected previously (Ross et al., 1997).
Figure 2.

Analysis of the proteins fractionated by RNA affinity chromatography. (A) SDS-PAGE analysis. Aliquots (30 μl) of proteins in each purification step eluted from the RNA affinity chromatography were resolved in 10% SDS-PAGE. (Lane 1) Flow-through fraction. (Lanes 2–6) Proteins in five successive washing steps, respectively. (Lanes 7–9) Proteins in 0.5, 1.0, and 2.0 KCl elution fractions, respectively. Protein molecular mass is marked at right position. The small arrows denote the five proteins that have been microsequenced. (B) Gel shift assay using the affinity selected proteins. Aliquots (5 μl) of each as in A (fraction) were incubated with 1 × 105 CPM of [32P]-labeled zipcode transcript and the complexes formed were resolved in 4% native gels. (C) Starting material. The arrows represent the RNA–protein complexes identified in Fig. 1 A.
To determine whether proteins eluted from the RNA affinity column contained RNA binding activity, we performed the band mobility shift assay by incubating the radiolabeled zipcode with proteins from each of the purification steps (Fig. 2 B). In comparison with the starting material (Fig. 2 B, lane C), enhanced RNA binding activity was detected in 1- and 2-M KCl fractions (lanes 7 and 8). The RNA binding activity was not found in the flow-through (Fig. 2 B, lane 2), wash steps (Fig. 2 B, lanes 3–6), and 0.5-M KCl fraction (Fig. 2 B, lane 6), indicating that the RNA binding proteins were enriched in the 1- and 2-M KCl fractions after RNA affinity purification. Surprisingly, although the 1- and 2-M KCl fractions contained 92- and 68-kD proteins, after incubation with radiolabeled zipcode, only complexes M and S were formed (Fig. 2 B, lanes 8 and 9). The relative absence of complex F in the band shift assay suggested either that the 68-kD protein lost its RNA binding activity in the high salt elution condition, or that the binding of the 68-kD protein to the zipcode needs additional proteins not present or active in the high salt fraction.
Microsequencing of affinity purified proteins
Proteins in the 2-M KCl fraction of RNA affinity column (Fig. 2 A, lane 9) were concentrated with a centricon-30 filter, resolved in 12% SDS-PAGE, and visualized by Coomassie blue staining. Four protein bands of ∼92, 70, 65, and 45 kD were eluted from the gel and microsequenced. Six peptides of the purified 92-kD protein, which we named ZBP2, were obtained after microsequencing. A database search with the peptide sequences of ZBP2 revealed that the six peptides matched a human nuclear protein, KSRP, that has been previously identified as a regulatory splicing factor (Min et al., 1997). The other three proteins were also identified by their peptide sequences. The 70-kD protein was a homologue of a human protein, FBP, which was known as a transcription factor for the c-myc gene (Duncan et al., 1994). The 65-kD protein was an unknown protein. We did not sequence the 68-kD protein, as we assumed that this protein was ZBP1 (Ross et al., 1997). The 45-kD protein was identified as ssDBF, a single-strand DNA binding factor in chicken (Smidt et al., 1995), and was homologous to a human protein, ABBP, a type A/B hnRNP protein that plays a role in mRNA editing (Lau et al., 1997).
Isolation of chicken cDNA for ZBP2
The cDNA-encoding ZBP2 was obtained by screening three chicken cDNA libraries and rapid amplification of cDNA ends (RACE)-PCR amplification of the 5′ terminus. The ZBP2 and the human KSRP share ∼81% identity in the nucleic acid sequence and >86% identity in amino acid (aa) sequence (Fig. 3), indicating that ZBP2/KSRP is a highly conserved protein. As with human KSRP and ZBP1, ZBP2 also contains four hnRNP type K homology RNA binding (KH) domains and a COOH-terminal region enriched in glutamines, with four repeats of an AWEEYYK motif and an NH2-terminal proline-glycine rich domain (Fig. 3). However, chicken ZBP2 contains a 47-aa segment before the first KH domain not found in KSRP, and significant sequence differences in the 5′ end of the mRNA.
Figure 3.

Complete sequence of ZBP2 compared with human KSRP. Proteins in the 2.0 KCl elution fraction of the RNA affinity column as shown in Fig. 2 were concentrated with a contricon-30 filter (Amicon) and electrophoresed in 10% SDS-PAGE. The proteins visualized by Coomassie blue staining were excised for microsequencing. The underlined sequences show the five microsequenced peptides of 16, 13, 13, 7, and 24 aa of purified ZBP2 (92 kD). The red regions indicate the four highly conserved K homology domains of the hnRNP. Note the proline, glycine-rich NH2-terminal, and glutamine-rich COOH-terminal domains of ZBP2. The identity of ZBP2 and KSRP is 86%. ZBP2 is available at GenBank/EMBL/DDBJ/accession no. AF461020.
ZBP2 is associated with the zipcode in chicken brain and fibroblasts
To facilitate our studies on the role of ZBP2 for mRNA localization, we generated antibodies by injecting gel-purified ZBP2 into rats. The specificity of ZBP2 antibodies was determined by Western blotting analysis (Fig. 4 A), in which the rat antibodies against ZBP2 specifically recognized ZBP2 in both fibroblast and brain extracts, as well as in affinity-purified protein fractions (Fig. 4 A, lanes 1–3).
Figure 4.

Characterization of antibodies to ZBP2. Antibodies against ZBP2 were made by injecting SDS-PAGE gel purified ZBP2 into rats. (A) Western blotting to ZBP2. (Lane 1) Crude brain extract. (Lane 2) Crude fibroblast extract. (Lane 3) 1.0 M KCl affinity- purified protein fraction of brain extract. The arrow indicates ZBP2. (B) Antiserum against ZBP2 or preimmune serum was incubated with brain extract for 2 h followed by incubation with agarose-protein G beads for 2 h at 4°C. The supernatants, after centrifugation at 1,500 rpm for 10 min, were analyzed by motility shift assay for their ability to form an RNA–protein complex. (Lane 1) Control brain extract. (Lane 2) Brain extract incubated with rat preimmune serum. (Lane 3) Brain extract incubated with anti-ZBP2 serum. Arrows denote the RNA–protein complexes identified in Fig. 1 A. (C) The same samples as in B were used for Western blotting. (Lane 1) Control brain extract. (Lane 2) Brain extract incubated with rat preimmune serum. (Lane 3) brain extract immunodepleted using anti-ZBP2 serum. Arrow indicates ZBP2.
To ensure that ZBP2 in brain extracts was the protein that formed complexes M and S with the zipcode, we immunodepleted ZBP2 from brain extracts and performed RNA mobility shift assays to detect RNA–protein complexes (Fig. 4 B). When the supernatants from an immunoprecipitation with ZBP2 antibodies were incubated with the zipcode, the specific RNA–protein complexes M and S were greatly reduced (Fig. 4 B, lane 3). The immunoprecipitation with anti-ZBP2 antibodies appeared to be specific, as complex F was only slightly reduced (Fig. 4 B, lane 3). In contrast, when the RNA mobility shift assay was performed with the supernatants after immunoprecipitation by a control rat preimmune serum, no reduction in the intensity of the expected RNA–protein complexes was found (Fig. 4 B, lane 2). This confirmed that the formation of complexes M and S was dependent on the presence of ZBP2. The formation of complex F was likely dependent on ZBP1, which is not recognized by the ZBP2 antibodies. To verify that ZBP2 was immunodepleted from extracts with anti-ZBP2 antibodies, the same supernatants used in the above mobility shift assay were analyzed by Western blotting (Fig. 4 C). The result showed that in contrast to control and rat preimmune serum–treated supernatants (Fig. 4 C, lanes 1 and 2), ZBP2 had been removed by the anti-ZBP2 antibodies (Fig. 4 C, lane 3).
Developmental regulation of ZBP2
To determine the expression pattern of ZBP2 in developing neurons, protein extracts were prepared from different developmental stages of chicken brain and analyzed for ZBP2 levels by Western blotting (Fig. 5). The highest expression of ZBP2 was seen in 6-d embryos (Fig. 5, 6 d), the level of expression was reduced to 30% before hatching and remained stable thereafter (Fig. 5). In contrast, the amount of β-actin protein is nearly constant in the same time points. This is consistent with the events during neural embryogenesis, when axons and dendrites are at maximal growth, and also coincides with the expression of ZBP1 (unpublished data).
Figure 5.

Embryonic expression of ZBP2. Proteins were extracted from chicken embryo brains at various stages and run on 10% SDS-PAGE before Western blotting using ZBP2 and β-actin antibodies. The relative ratios of ZBP2 per actin intensity were calculated for the stages indicated (in days).
Figure 6.

Distribution of β-actin mRNA and ZBP2 in CEFs and neurons. CEFs (A) and neurons (B) were cultured and fixed as described in Materials and methods. The cells were in situ hybridized to β-actin mRNA (Cy3, red) and by immunofluorescence to ZBP2 (FITC, green). Overlap is indicated in the right of A and in B. Image in B is a superimposed single, deconvolved optical sections. Arrowheads show the colocalization of ZBP2 and β-actin mRNA in neuronal processes and growth cone. Bars, 10 μm.
Cellular localization of ZBP2
To investigate the subcellular localization of ZBP2 and whether ZBP2 was associated with β-actin mRNA in vivo in fibroblasts and neurons, we performed a combined assay, using in situ hybridization with fluorescent-labeled oligo nucleotide probes for β-actin mRNA (red; Fig. 6, A, left panel, and B), and with antibodies for ZBP2 (green; Fig. 6, A, middle panel, and B). ZBP2 is predominantly present in cell nuclei. However, occasionally a significant amount of the ZBP2 was detected by immunofluorescence in the leading edge of CEFs and the growth cone of chicken developing neurons. We superimposed their images (Fig. 6, A, right, and B) where ZBP2 and β-actin mRNA were overlapped (yellow) in the leading edge of CEFs (Fig. 6 A, right) and the growth cone of neurons (Fig. 6 B, arrows). This provided evidence that ZBP2 and β-actin mRNA were spatially associated in vivo in the cells examined. Although the protein and RNA are generally distributed in the same region of the neurites, occasionally they are overlapping. Both the β-actin mRNA and the ZBP2 show a punctate distribution, particularly evident in neurons, consistent with observations of localizing particles (Ainger et al., 1993; Zhang et al., 2001).
ZBP2 and β-actin mRNA are physically associated
To determine whether the ZBP2 and β-actin mRNA could be physically associated, we employed immunoprecipitation followed by RT-PCR assays to determine whether the immunoprecipitated pellet with anti-ZBP2 antibodies contained β-actin mRNA (Fig. 7). RNA was isolated from the immunoprecipitated pellet from brain extract using either anti-ZBP2 antibodies or rat preimmune serum, and then subjected to RT-PCR. A 398-bp fragment of the β-actin mRNA 3′ untranslated region (UTR) was chosen for PCR amplification. We used a plasmid without β-actin cDNA as a negative control (Fig. 7, lane 1), and a plasmid with β-actin cDNA as a positive control (Fig. 7, lane 6). A fragment of the 3′ UTR of β-actin mRNA was amplified from the immunoprecipitations with ZBP2 antiserum or antibodies (Fig. 7, lanes 3 and 5). The immunoprecipitations with preimmune rat serum or IgG did not demonstrate any PCR-amplified DNA fragments (Fig. 7, lanes 2 and 4). This experiment indicated that β-actin mRNA and ZBP2 were most likely physically associated with each other in chicken brain extracts.
Figure 7.
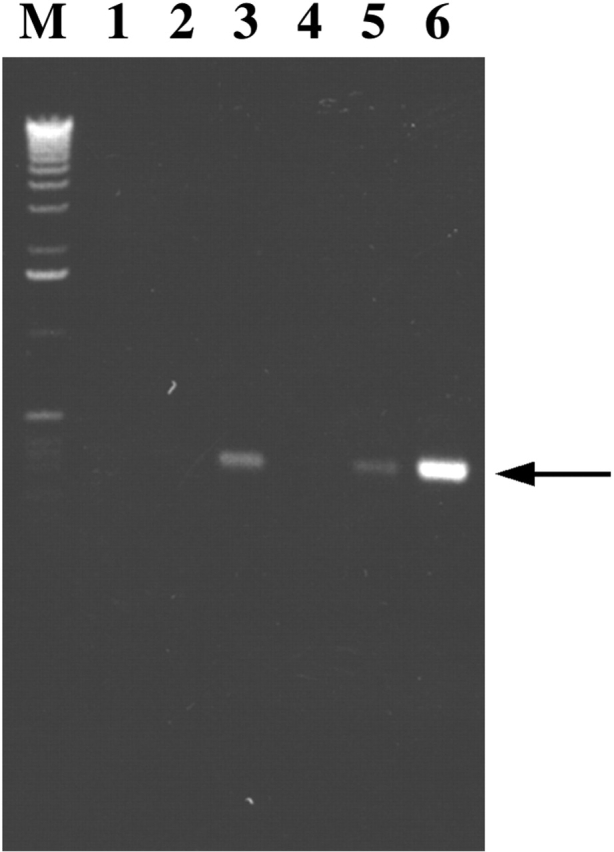
β-actin mRNA was coprecipitated with ZBP2. ZBP2 antibodies were used to immunoprecipitate a CEF extract. Primers to β-actin mRNA were used in an RT-PCK assay to detect the presence of β-actin mRNA in the pellets. (Lane 1) Negative PCR control no primers. (Lane 2) Preimmune rat serum. (Lane 3) ZBP2 antiserum. (Lane 4) Purified rat normal IgG. (Lane 5) affinity-purified ZBP2 IgG. (Lane 6) Positive PCR control using full-length cDNA of β-actin mRNA as template. M is the DNA molecular marker. The arrow denotes the PCR amplified DNA fragment of the 3′ UTR of β-actin mRNA.
Portions of ZBP2 can determine nuclear and cytoplasmic distribution
To determine whether ZBP2 shuttles between the nucleus and cytoplasm, we fused it to enhanced green fluorescence protein (EGFP) and analyzed the compartmentalization of the fluorescent protein in both fibroblasts and neurons. The various portions of ZBP2 that were fused to GFP included the 47-aa segment (red), the central four KH domains (blue), and the COOH-terminal repeats (gray) (Fig. 8 A). Different domains of ZBP2 control the nuclear or cytoplasmic compartmentalization of the EGFP fused to it (numbers indicate the percentage of cells with cytoplasmic fluorescence in fibroblasts). The COOH-terminal (Fig. 8 B) domain and four KH domains (Fig. 8 D) were distributed throughout the cell, whereas when the 47 aa were added to the 4-KH domain, which we call the central domain, the localization was almost entirely nuclear (Fig. 8 C). This suggested that the 47 aa determined nuclear distribution. This was confirmed by fusing the 47-aa segment to EGFP, which then became exclusively nuclear (Fig. 8 E). Inspection of the 47-aa segment revealed several arginine residues suggestive of a nuclear localization signal (NLS). When the full-length ZBP2 was fused to GFP, with or without the 47-aa segment (Fig. 8, F and G), the nuclear fluorescence was decreased but not eliminated in the construct without the 47 aa, and the cytoplasmic fluorescence increased, indicating that weak NLSs most likely existed elsewhere in the protein. The distribution in fibroblasts for these last two constructs was quantitated in neurons, and yielded the same percentages of cells with cytoplasmic fluorescence. Unlike ZBP2, human KSRP was entirely nuclear when fused to GFP and expressed in CEF (unpublished data).
Figure 8.
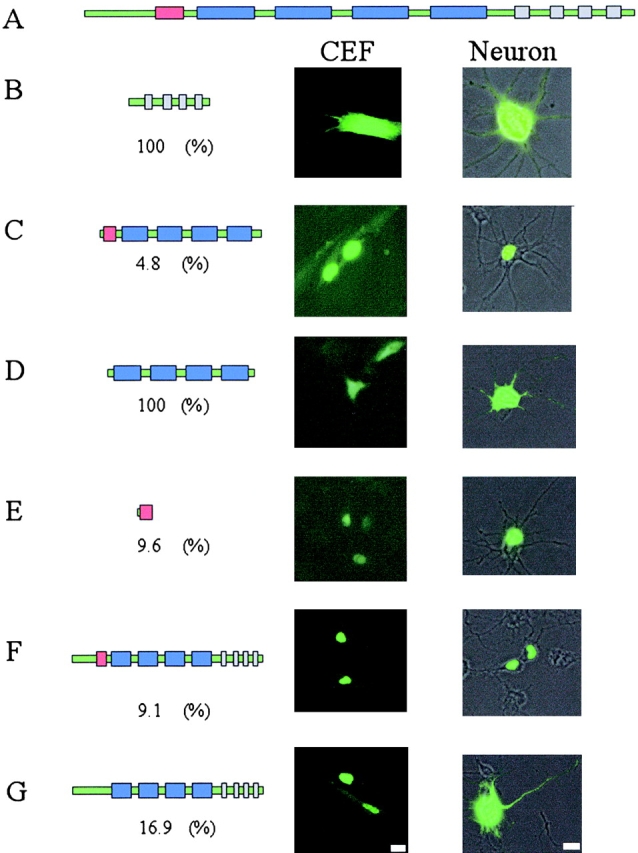
Nuclear versus cytoplasmic distribution of ZBP2 in fibroblasts and neurons. The gene structure of ZBP2 is demonstrated in A. Red bar, 47-aa insert; blue bars, KH domains; gray bars, COOH-terminal repeat of AWEEYYK motif. The various constructs of ZBP2; the full-length with or without the 47-aa insert (F and G), the 47-aa insert (E), the four KH domains (D), the central domain containing the four KH domains, and the 47-aa insert (C), or the COOH terminus of the protein (B) were fused to GFP and the cellular distribution characterized. The constructs are detailed in the left panels with cellular distributions in the right panels. The percent of cells with cytoplasmic signal was characterized for each construct in the number under each construct. Bars, 10 μm.
Overexpression of ZBP2 central domain partially disrupts β-actin mRNA localization in CEFs and neurons
A truncate of the ZBP2, the central domain, which contained ∼400 aa including the 47 aa with four KH domains (Fig. 8 C), was transfected into CEFs. We postulated that the KH domains would compete for RNA binding with the full-length ZBP2, analogous to ZBP1 (unpublished data), and hence act as a dominant negative for β-actin mRNA localization. The EGFP signal in fibroblasts transfected with the truncate was mainly present in the nucleus. Localization of the β-actin mRNA was visualized by fluorescence in situ hybridization. The percentage of cells with localized β-actin mRNA decreased roughly by half in the transfected CEFs (Fig. 9, top) in contrast to the cells transfected with EGFP or EGFP-KSRP as a control (the human ZBP2 homologue). The intensity of cytoplasmic signal of β-actin mRNA did not change significantly, indicating that the nuclear export of β-actin mRNA was not inhibited by the dominant negative construct (unpublished data). In transfected developing neurons, this truncation also caused a 35% decrease of β-actin mRNA signal in processes or growth cones compared with the control experiment (Fig. 9, middle). This suggested that the overexpression of the truncated, but still nuclear, ZBP2 affected the β-actin mRNA localization. To demonstrate that the dominant negative effect of ZBP2 truncate is specific, we performed cotransfections of both the full-length ZBP2 and central domain into CEFs. The delocalization of β-actin mRNA in transfected cells was partially rescued by using the wild-type ZBP2 (Fig. 9, bottom).
Figure 9.



A ZBP2 dominant negative construct suppresses β-actin mRNA localization. (Top) The percentage of transfected CEFs wth localized β-actin mRNA. CEFs were transfected for 6 h followed by fluorescence in situ hybridization. Transfected cells were scored for β-actin mRNA localization. GFP, vector pEGFPC1; GFP-CD, GFP fused with central domain; GFP-KSRP, GFP-human KSRP; Mock, no plasmid. The bars show percentage of transfected cells with localized β-actin mRNA in each experiment. At least 50 transfected cells were counted in each coverslip, two experiments each. (Middle) The β-actin mRNA signal in neuronal processes. Experiments were parallel to A except that the COOH terminus of ZBP2 (GFP-CT) was used (Fig. 8, construct B). β-actin mRNA signal was measured and then converted into fluorescence units. (Bottom) The percentage of cotransfected CEFs with localized β-actin mRNA. CEFs were transfected either with single construct (wild-type ZBP2 or the dominant negative central domain construct) or both constructs of different ratios (dominant negative vs. wild-type 1:1 or 1:4) for 6 h. After FISH, transfected cells were scored as in Fig. 9 A.
Discussion
We have purified and identified five proteins from embryonic brain extracts that bind to the zipcode sequence responsible for localizing β-actin mRNA in fibroblasts and neurons. One of these has been previously identified from cultured fibroblast extracts as ZBP1 (Ross et al., 1997). In this work we have characterized ZBP2, a ZBP highly enriched in chicken brain tissue. The binding specificity of this protein to the zipcode, its intracellular colocalization with β-actin mRNA, and the dominant negative effect of a truncation of this protein suggests a role in localization of β-actin mRNA. Consistent with its homology to the human splicing factor, KSRP, the protein is predominantly nuclear in its intracellular distribution and the possibility exists that it exerts its effect there with subsequent consequences for the cytoplasmic compartmentalization. The other proteins identified in this complex have homology to the transcription factor, FBP1 (Duncan et al., 1994), a single-stranded DNA binding protein, ssDBF (Smidt et al., 1995) also homologous to Drosophila Squid, known to localize ftz (Lall et al., 1999), and an A/B hnRNP associated with mRNA editing (Lau et al., 1997). All of these are likewise nuclear in location and function, and some may shuttle between the nucleus and cytoplasm and could play a concerted role in vetting the RNA before, during, or after export.
The amino acid sequence predicted from ZBP2 cDNA demonstrates that ZBP2 is a chicken homologue of human KSRP, a regulatory splicing factor (Min et al., 1997). Like ZBP1 (Ross et al., 1997), KSRP/ZBP2 contains four KH domains, a glutamine-rich COOH-terminal domain, and a proline, glycine-rich NH2-terminal domain that may provide flexibility for the protein to fold properly. In addition, ZBP2 is particularly abundant in cell nuclei where it could also be involved in splicing of many pre-mRNAs, possibly including β-actin, as well as their localization.
Our results provide evidence that ZBP2 interacts with the zipcode in vitro and participates in β-actin mRNA localization in vivo. First, the recognition of the zipcode by ZBP2 is specific: it does not bind to nonzipcode RNA. This was demonstrated using RNA competition and immunodepletion assays wherein the formation of the RNA–protein complexes was abolished. That the mutant zipcode failed to bind ZBP2 in vitro is consistent with the previous study showing that mutated zipcode delocalized β-actin mRNA in vivo (Ross et al., 1997). Second, we verified the physical association of ZBP2 with the zipcode using UV to covalently crosslink it to the zipcode, and by immunoprecipitation where the β-actin mRNA copurified with it in brain extracts. Third, immunocytochemistry and in situ hybridization showed ZBP2 present infrequently in the cytoplasm, where it colocalized with β-actin mRNA at the leading edge of fibroblasts and growth cones of developing neurons. Moreover, EGFP–ZBP2 transfected into fibroblasts, although also infrequently cytoplasmic, showed a similar colocalization pattern to the endogenous ZBP2 and β-actin mRNA, suggesting that the movement of ZBP2 toward the cell's leading edge was associated with localization of the mRNA. Importantly, the expression of a truncated construct of ZBP2 containing the central KH domains and the nuclear localizer 47 aa interfered with endogenous β-actin mRNA localization.
ZBP2, a predominantly nuclear protein, is shown here to play a role in cytoplasmic transport of β-actin mRNA by binding to the localization signal. Of interest, ZBP2 has similarities to ZBP1, a predominantly cytoplasmic protein, which also is involved in β-actin mRNA localization in fibroblasts and neurons (Zhang et al., 2001). However, unlike ZBP2, ZBP1 has a nuclear export signal (Ross et al., 1997), which may explain its predominate cytoplasmic localization. Although both ZBP1 and ZBP2 are in different cellular compartments, they share many common features. Both ZBP1 and ZBP2 recognize the wild-type zipcode, but not the mutant zipcode in vitro, and colocalize with β-actin mRNA in vivo (Ross et al., 1997; Zhang et al., 2001). Therefore, the two hnRNP proteins, one cytoplasmic and the other nuclear, are involved in localization of the same mRNA. We propose that the process for β-actin mRNA localization may initiate in nuclei where ZBP2 may interact with the zipcode, perhaps with other proteins, such as ssDBF and FBP. Upon nuclear export of the RNA, ZBP1 may take over the process of cytoplasmic localization. The fact that the proteins form different complexes with the zipcode suggests that they bind sequentially rather than simultaneously.
Increasing evidence demonstrates that specific interactions between RNA localization elements and cellular factors play an essential role in cytoplasmic sorting of mRNAs to their destinations. Strikingly, although mRNA localization is a cytoplasmic event, proteins that shuttle between the nucleus and cytoplasm participate in this pathway. A number of other nuclear proteins participating in cytoplasmic mRNA localization have been documented in recent studies (Hoek et al., 1998; Cote et al., 1999; Lall et al., 1999; Long et al., 2001). VgRBP60, a hnRNP I type protein in Xenopus, binds to the VM1 localization motif of Vg1 mRNA in vitro and colocalizes with Vg1 mRNA in vivo (Cote et al., 1999). Lall et al. (1999) reported that sqd, a Drosophila hnRNP protein is required for ftz mRNA localization in embryos. In yeast, an exclusively nuclear protein (does not shuttle), Loc1p, binds to the 3′ UTR zipcode of ASH 1 mRNA and is required for its efficient cytoplasmic localization to the bud tip (Long et al., 2001). Therefore, our finding that ZBP2, an hnRNP protein with known nuclear location, was involved in cytoplasmic localization of β-actin mRNA was consistent with hnRNPs being part of a common localization mechanism. It is possible that hnRNPs including ZBP2/KSRP are required for packaging the RNA in nucleus in a way that marks it for a localization pathway in the cytoplasm.
The fact that ZBP2 is only found in the cytoplasm of 5–10% of the cells strongly suggests that it rapidly shuttles, spending a short time there. This is the opposite of ZBP1, which spends only a short time in the nucleus (unpublished data) and is predominantly cytoplasmic. The 47-aa segment has a strong effect on ZBP2 nuclear retention, just as the nuclear export signal in ZBP1 has a strong effect on its cytoplasmic presence (unpublished data). When removed from ZBP2, the 47-aa segment increases the presence in the cytoplasm. KSRP, which does not have this 47-aa segment, also does not appear in the cytoplasm, at appreciable levels. Possibly KSRP is an alternatively spliced variant of ZBP2, but we have not found evidence for this in chicken, which does not appear to have KSRP. Like KSRP, ZBP2 is a member of a KH domain–containing family of neuronally expressed proteins which include KSRP, ZBP1, FMRP (Fridell et al., 1996), and NOVA (Polydorides et al., 2000), all involved in some aspects of the nuclear regulation of RNA.
Microsequencing of the other proteins copurified with ZBP2/KSRP by RNA affinity selection showed that the 70-kD protein is also a KH domain containing protein, a homologue of FBP, a human transcription factor, that binds to single-stranded DNA and activates the transcription of the c-myc gene (Duncan et al., 1994). It is possible that FBP was copurified with ZBP2 because it has been shown that FBP associates with KSRP in human nuclear extracts (Min et al., 1997). However, this does not eliminate the possibility that FBP may have a function in cytoplasmic mRNA distribution. The 45-kD protein, which was copurified with ZBP2, is ssDBF, a nuclear hnRNP A/B type protein that binds to the regulatory site of apo VLDL II gene (Smidt et al., 1995), and a human homologue of ssBDF is involved in Apo mRNA editing (Lau et al., 1997). ssDBF shares ∼74% identity with MBP mRNA binding protein (Hoek et al., 1998), and 41% identity with sqd, the ftz binding protein in Drosophila (Lall et al., 1999), both of which are also hnRNA A/B proteins. Whereas both MBP mRNA binding protein and sqd appear to perform an essential role for localization of their respective mRNAs, ssDBF involvement in cytoplasmic β-actin mRNA segregation remains to be determined.
These proteins could be part of a complex that we have termed the locasome (Bertrand et al., 1998). This structure most likely contains proteins unique to both the nucleus and the cytoplasm, in the first case marking the RNA for localization, and in the second case directing the peripheral location to the leading edge of the fibroblast or the growth cone of the developing neuron.
Materials and methods
Preparation of chicken brain and fibroblast extracts
Brain cytoplasmic extract was prepared from 12-d-old chick embryos. Whole brains were removed and washed three times with buffer A (20 mM Tris Cl, 3 mM MgCl2, 40 mM KCl and 1 mM DTT, 0.7 μg/ml leupeptin, 1 μg/ml aprotinin, 0.7 μg/ml pepstantin and 1 mM PMSF). 1 ml of buffer A was added per gram of wet tissue, and the mixture was homogenized in a Teflon glass homogenizer. Homogenates were centrifuged for 10 min at 5,000 g, and the supernatant was centrifuged for 2 h at 28,000 rpm in a Beckman SW-40.1 rotor. The high-speed supernatant was either used immediately or stored in aliquots at –70°C. The protein concentration of the brain extract was ∼10–20 μg/μl. To prepare CEF extract, fibroblasts were isolated from breast muscle tissue of 11-d-old chick embryos. Cells were grown to ∼95% confluency (Kislauskis et al., 1994), and then scraped and washed in cold buffer A and pelleted by centrifugation (Ross et al., 1997). The cells were resuspended in buffer A and homogenized in a Dounce homogenizer. Homogenates were centrifuged as described above. The protein concentration of prepared extract was 5–10 μg/μl.
Neuronal cultures
Primary cultures of embryonic chick forebrain neurons were generated as described previously (Zhang et al., 1999). Briefly, forebrains were dissected from 8-d chick embryos, trypsinized, dissociated, and plated on poly-l-lysine– (0.2 mg/ml, 16 h) and laminin- (0.02 mg/ml, 12 min) coated coverslips in MEM with 10% FBS for 2 h. Cells were inverted onto a monolayer of chick astrocytes in N2-conditioned medium with serum (0.2% FBS) and cultured for 4 d at 37°C in 5% CO2.
In vitro RNA transcription
The pCZIP was constructed by inserting the 54 nucleotide zipcode of chick β-actin mRNA into a pSP64-Poly(A) vector (Promega) at Hind III and Ava I sites. To construct the pCZIPm plasmid, the following pair of complementary oligos were synthesized, annealed, and inserted between Hind III and Ava I sites of pSP64 Poly(A) vector: Sense: 5′ AGCTTACCGGACT-GTTACCATGTGTGTGTGTGCTGTGATGAAACAAAACCCATAAATGC 3′ and antisense: 5′ CCGGGCATTTATGGGTTTTGTTTCATCCAGCACACACACACATGGTAACAGTCCGGTA 3′.
For gel mobility shift assays, [32P]-labeled RNA was generated by SP6 RNA polymerase directed in vitro transcription from Ava I linearized pCZIP DNA. The transcribed RNA was gel purified by 6%. For RNA affinity purification, polyadenylated transcripts were synthesized in vitro with SP6 polymerase (MEGAScript kit; Ambion) from EcoRI-linearized pCZIP constructs. Trace amounts of [32P]-CTP were added to allow detection and quantitation of transcribed RNA. The transcribed RNA contained the 54 nts of pCZIP RNA and a poly(A) tail of 30 nts.
Gel mobility shift assay and UV crosslinking
Briefly, 105 CPM of the [32P]-labeled RNA probe was incubated at room temprature with 5 μl of brain or fibroblast protein extract for 20 min in a 20-μl binding solution containing 20 mM Hepes, pH 7.4, 50 mM KCl, 3 mM MgCl2, 2 mM DTT, and 5% glycerol. Unbound RNAs were degraded by a 10-min incubation with 1 U of RNase T1, and nonspecific RNA–protein interactions were minimized by incubation with 5 mg/ml heparin for 10 min. The RNA–protein complexes formed were separated in a 4% native gel and visualized by autoradiography. To establish the specificity of RNA-protein interactions, competition assays were performed by preincubating the protein extract with unlabeled RNA competitors.
UV crosslinking of RNA–protein complexes was performed by irradiating the reactions on ice in a UV Chamber (GS gene linker; Bio-Rad Laboratories) with 254-nm, 8-W UV bulbs for 10 min and resolved by 4% native gel. The UV crosslinked RNA–protein complexes, detected by autoradiography, were cut from the gel, mixed with SDS loading buffer and incubated with 10 U RNase T1 for 30 min at room temperature. The gel slices were loaded on a 10% SDS-polyacrylamide gel and electrophoresed to distinguish the RNA–protein complexes.
Affinity purification of proteins that bind to RNA
2 ml of poly(U) agarose beads (type 6; Amersham Pharmacia Biotech) were suspended in RNA binding buffer (25 mM Tris HCl, pH 7.4, 100 mM KCl) and packed into a 10-ml column. About 1 mg in vitro synthesized poly(A) zipcode RNA was added to the column and cycled four times. The efficiency of RNA bound to poly(U) agarose beads was monitored by measuring the amount of [32P]-labeled RNA present in the RNA preparation. After binding, the beads were equilibrated with the extract buffer, mixed with 40 ml of brain protein extracts or 20 ml fibroblast extracts containing 50 U/ml RNasin (Promega), and incubated for 1 h at room temperature with gentle shaking. To lower nonspecific protein binding, yeast tRNA and heparin were added to 50 μg/ml and 5 mg/ml to binding buffer, respectively. The beads were then centrifuged for 2 min at 1,000 g, resuspended in binding buffer, and repacked into a 10-ml column. The column was extensively washed in 5 × 20 ml of binding buffer as follows: (1) binding buffer; (2) binding buffer + 40 μg/ml yeast RNA; (3) binding buffer + 5 mg/ml heparin; (4) binding buffer + 0.1% Triton X-100; and (5) binding buffer only. Proteins retained on the RNA affinity column were step eluted with 20 mM Hepes buffer, pH 7.4, containing 0.5, 1, and 2 M KCl. The eluted proteins were analyzed by SDS-PAGE and band shift assay.
Production of rat anti-ZBP2 antibodies
The 2-M KCl elution fraction from zipcode affinity column was concentrated with centricon-30 filter and eletrophoresed in 10% SDS-PAGE. After staining with Coomassie blue, the expected protein bands were cut from the gel, crushed, mixed with adjuvant, and injected into rats (Covance, Inc.) for antibody production. The titer of the antiserum was tested by immunoblot.
Immunoblotting
Aliquots of proteins were resolved in 10% SDS-PAGE and transferred onto Zeta membranes by a semidry transferring blotter (Bio-Rad Laboratories). The membranes were blotted overnight with PBS containing 5% nonfat milk at 4°C and then incubated with antiserum against ZBP2 (1:2,000) in PBS containing 1% BSA for 2 h at room temperature. After washing three times with PBS/0.3% Tween-20, the membranes were incubated with horseradish peroxidase conjugated goat anti–rat antibodies (1:8,000), ZBP2 was detected with the ECL system (Amersham Pharmacia Biotech) or with DAB/H2O2.
Immunoprecipitation and β-actin mRNA detection
10 μg purified antibodies or 5 μl of antiserum against ZBP2 were incubated with 100 μl of brain protein extract for 1 h at 4°C with gentle agitation. 30 μl of protein G–coupled agarose beads (Pierce Chemical Co.) were added to the mixture and incubated for another 1 h at 4°C. After centrifugation for 5 min at 1,500 g, the supernatant was taken for immunoblotting and RNA band shift assays. For analysis of ZBP2, the pelleted agarose beads were extensively washed with PBS and the proteins bound to the beads were eluted with 100 μl of ImmunoPure IgG Elution Buffer (Pierce Chemical Co.). For detecting β-actin mRNA, the pelleted agarose beads were washed three times with DEPC-PBS, resuspended in 100 μl DEPC water and boiled for 10 min. Total RNA was extracted from the supernatant using TRIzol RNA isolation reagent (GIBCO BRL). RT-PCR for β-actin mRNA was performed for 20 cycles with 1 μl of RNA as template using ReadyToGo RT-PCR-beads (Amersham Pharmacia Biotech). The following primers were used for amplification: 5′ CTGACACCTTCACCATTCCAG 3′ and 5′ ATTGCTGACAGGATGCAGAAG 3′. The expected PCR product is a 398-bp DNA fragment of β-actin mRNA in 3′ UTR at the positions of 1021–1419.
Isolation and cloning of ZBP2 cDNA
Two primers, zbp2–1 and zbp2–2, were designed and synthesized according to the two peptide sequences (ZBP2 amino acids 621–627 and 685–709, respectively) and human KSRP cDNA sequence (zbp2–1 GCTTGGGAGGAGTACTACAA, zbp2–2 AGGACCTGGGGTCTGTCCGTA): A 200-bp DNA fragment was amplified from a chicken embryo brain library (CLONTECH Laboratories, Inc.) and verified by sequencing. This fragment was used for screening the chicken embryo brain library. Two cDNA clones were isolated from the library screening. After sequence analysis, it was evident that both clones contained a 500-bp COOH-terminal coding sequence followed by 150 bp of 3′ UTR. 5′ RACE PCR was applied to obtain an NH2-terminal fragment of ZBP2 (CLONTECH Laboratories, Inc.) RACE 4 primers were used in the 5′ RACE experiment (RACE6–1: CACGCGGCGTTGGGGTCGGCTGC, RACE7: TTATAGGCGGCCCA-CGCGGCGTTGG, RACEF1: GAACGCGTCCTTCCGGATCC, RACEF2: CCCAATTTTTGCCGCTATCTG). The sequences of DNA fragments amplified by RACE were determined by automated DNA sequence analyses. The full-length gene was spliced together by PCR and confirmed by sequencing.
In situ hybridization and immunocytochemistry
In situ hybridization was performed on fibroblasts as described by Kislauskis et al. (1993), and on neurons as described by Zhang et al. (1999). 9-d-old CEFs were cultured in MEM supplemented with 10% FCS. After growing on coverslips for 2 d, the cells were fixed in 4% paraformaldehyde in PBS for 10 min and permeabilized with 0.5% Triton X-100 for another 10 min at room temperature. Distribution of ZBP2 in CEFs was visualized by incubation of rat antiserum against ZBP2 (1:800) followed by Cy3 or FITC conjugated rabbit anti–rat antibodies (1:500; Jackson ImmunoResearch Lab). Neurons were cultured in N2 conditioned media with 2% fetal bovine serum (FBS) for 2 or 4 d and fixed in 4% paraformaldehyde in PBS for 15 min at room temperature. After permeabilization with 0.5% Triton X-100, the cells were incubated with rat antiserum against ZBP2 (1:750) followed by Cy3-labeled rabbit anti–rat antibodies (1:750). For in situ hybridization and immunochemistry, cells were first processed for hybridization of β-actin mRNA, washed with PBS, and immunostained as previously described (Zhang et al., 1999). Cells were viewed with an Olympus B×60 with 60× Plan Apo 1.4NA objective. Images were acquired with a cooled CCD camera, operated by Espirit imaging software.
GFP fusion protein constructs
Human KSRP cDNA (the homologue of chicken ZBP2), a gift from D. Black (University of California at Los Angeles, Los Angeles, CA), was subcloned to pEGFP C1 vector (CLONTECH Laboratories, Inc.) at the EcoRI and Hind III sites. (pGFP-KSRP). All other EGFP fusion constructs are either full-length or truncated chicken ZBP2. The central domain fragment containing aa sequence 103–650, including the 47-aa sequence unique to ZBP2 and 4-KH domains, was amplified by PCR and cloned into pEGFP-C1 vector at EcoRI and Xbal sites to generate plasmid pGFP-CD. The 4-KH domains of ZBP2 were cloned into pEGFP-C1 vector at SalI and XbaI site to produce construct pGFP-KH. The 47-aa unique sequence was amplified by PCR and introduced into pEGFPC1 at EcoRI Hind III (site pGFP-IN). The COOH-terminal domain, including from amino acids 650 to the stop codon, were excised from the plasmid pBS-LS2 (isolated from library screening) using EcoRI and XhoI sites and cloned into pEGFPC1 vector, generating pGFP-CT. Full-length ZBP2 was cloned into pEGFPC2 (CLONTECH Laboratories, Inc.) at EcoRI and Hind III sites to generate pGFP-FULL. The 47-aa unique sequence was deleted from full-length ZBP2 by two-step splicing PCR, and was cloned into pEGFPC2 vector by EcoRI and Hind III sites to generate pGFP-Δ47. All constructs mentioned above were verified by DNA sequencing.
Transfection and imaging of transfected cells
CEFs were transfected for 4–5 h with GFP–ZBP2 constructs using Qiagen's Effectene Reagent according to manufacturer's protocol. Cultured neurons were transfected with DOTAP as described (Zhang et al., 1999). GFP imaging with or without FISH was performed (Zhang et al., 1999). For CEFs, the GFP and β-actin mRNA signals were visualized by fluorescence microscopy using an Olympus B×60 microscope with a 60× objective, n.a. 1.4. At least 50 transfected cells were counted per coverslip for the β-actin mRNA localization.
The EGFP/ZBP2 fusion expressed in neurons was identified using a fluorescence microscopy (Nikon Eclipse inverted microscope) equipped with 60× Plan-Neofluar objective, phase optics, 100 W mercury arc lamp and HiQ bandpass filters (ChromaTech). Fluorescence images were immediately acquired in a constant exposure time (1 s) with a cooled CCD camera that was run by IP Lab computer software. The perimeter of each axonal dendrites (first 30 μm from cell body) was traced using phase image. Region of interest was transferred to the fluorescence image in the same cell, and fluorescence intensity of β-actin mRNA was measured by IP Lab software. The fluorescence intensity of ROI within untransfected cells on the coverslip was measured as normal control. Over 20 cells in each group were analyzed.
Acknowledgments
We would like to thank Yuri Oleynikov for important advice, Shailesh Shenoy for microscopy and figure preparation assistance, Kim Farina for critical reading of the manuscript, and Dr. Doug Black for KSRP construct.
This work was supported by grants from the National Institutes of Health (AR41480 to R.H. Singer, and GM55599 to G.J. Bassell).
W. Gu and F. Pan contributed equally to this work.
Footnotes
Abbreviations used in this paper: aa, amino acid(s); CEF, chicken embryonic fibroblast; EGFP, enhanced green fluorescence protein; NLS, nuclear localization signal; RACE, rapid amplification of cDNA ends; UTR, untranslated region; ZBP, zipcode binding protein.
References
- Ainger, K., D. Avossa, F. Morgan, S.J. Hill, C. Barry, E. Barbarese, and J.H. Carson. 1993. Transport and localization of exogenous myelin basic protein mRNA microinjected into oligodendrocytes. J. Cell Biol. 123:431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainger, K., D. Avossa, A.S. Diana, C. Barry, E. Barbarese, and J.H. Carson. 1997. Transport and localization elements in myelin basic protein mRNA. J. Cell Biol. 138:1077–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon, A. 1996. Mother and daughter are ng fine asymmetric cell division in yeast. Cell. 84:651–654. [DOI] [PubMed] [Google Scholar]
- Bassell, G.J., and R.H. Singer. 1997. MRNA and cytoskeletal filaments. Curr. Opin. Cell Biol. 9:109–115. [DOI] [PubMed] [Google Scholar]
- Bassell, G.J., and R.H. Singer. 2001. Neuronal RNA localization and the cytoskeleton. In Results and Problems in Cell Differentiation. D. Richter, editor. Springer-Verlag, 34:41–56. [DOI] [PubMed] [Google Scholar]
- Bassell, G.J., H.L. Zhang, A.L. Byrd, A.M. Femino, R.H. Singer, K.L. Taneja, L.M. Lifshitz, I.M. Herman, and K.S. Kosik. 1998. Sorting of β-actin mRNA and protein to neurites and growth cones in culture. J. Neurosci. 18:251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell, G.J., Y. Oleynikov, and R.H. Singer. 1999. The travels of mRNAs through all cells large and small. FASEB J. 13:447–454. [DOI] [PubMed] [Google Scholar]
- Bashirullah, A., R.L. Cooperstock, and H.D. Lipshitz. 1998. RNA localization in development. Annu. Rev. Biochem. 67:335–394. [DOI] [PubMed] [Google Scholar]
- Behar, L., R. Marx, E. Sadot, J. Barg, and I. Ginzburg. 1995. cis-acting signals and trans-acting proteins are involved in tau mRNA targeting into neurites of differentiating neuronal cells. Int. J. Dev. Neurosci. 13:113–127. [DOI] [PubMed] [Google Scholar]
- Bertrand, B., C. Pascal, M. Schaefer, S.M. Shenoy, R.H. Singer, and R.M. Long. 1998. Localization of ASH1 mRNA particles in living yeast. Mol. Cell. 2:437–445. [DOI] [PubMed] [Google Scholar]
- Cote, C.A., D. Gautreau, J.M. Denegre, T. Kress, N.A. Terry, and K.L. Mowry. 1999. A Xenopus protein related to hnRNP I has a role in cytoplasmic RNA localization. Mol. Cell. 4:431–437. [DOI] [PubMed] [Google Scholar]
- Deshler, J.O., M.I. Highett, T. Abramson, and B.J. Schnapp. 1998. A highly conserved RNA-binding protein for cytoplasmic mRNA localization in vertebrates. Curr. Biol. 8:489–496. [DOI] [PubMed] [Google Scholar]
- Duncan, R., L. Bazar, G. Michelotti, T. Tomonaga, H. Krutzsch, M. Avigan, and D. Levens. 1994. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 8:465–480. [DOI] [PubMed] [Google Scholar]
- Ephrussi, A., L.K. Dickinson, and R. Lehmann. 1991. Oskar organizes the germ plasm and directs localization of the posterior determinant nanos. Cell. 66:37–50. [DOI] [PubMed] [Google Scholar]
- Fridell, R.A., R.E. Benson, J. Hua, H.P. Bogerd, and B.R. Cullen. 1996. A nuclear role for the fragile X mental retardation protein. EMBO J. 19:5408–5414. [PMC free article] [PubMed] [Google Scholar]
- Havin, L., A. Git, Z. Elisha, F. Oberman, K. Yaniv, S.P. Schwartz, N. Standart, and J.K. Yisraeli. 1998. RNA-binding protein conserved in both microtubule- and microfilament-based RNA localization. Genes Dev. 12:1593–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek, K.S., G.J. Kidd, J.H. Carson, and R. Smith. 1998. hnRNP A2 selectively binds the cytoplasmic transport sequence of myelin basic protein mRNA. Biochemistry. 37:7021–7029. [DOI] [PubMed] [Google Scholar]
- Jansen, R.P. 2001. mRNA localization: message on the move. Nat. Rev. Mol. Cell. Biol. 4:247–256. [DOI] [PubMed] [Google Scholar]
- Kislauskis, E.H., Z.F. Li, R.H. Singer, and K.L. Taneja. 1993. Isoform-specific 3′-untranslated sequences sort α-cardiac and β-cytoplasmic actin messenger RNAs to different cytoplasmic compartments. J. Cell Biol. 123:165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislauskis, E.H., X.-C. Zhu, and R.H. Singer. 1994. A sequence required for intracellular localization of β-actin messenger RNA also affects cell phenotype. J. Cell Biol. 127:441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislauskis, E.H., X.-C. Zhu, and R.H. Singer. 1997. β-actin messenger RNA localization and protein synthesis augment cell motility. J. Cell Biol. 136:1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman, R., G. Banker, and O. Steward. 1990. Differential subcellular localization of particular mRNAs in hippocampal neurons in culture. Neuron. 6:821–830. [DOI] [PubMed] [Google Scholar]
- Lall, S., H. Francis-Lang, A. Flament, A. Norvell, T. Schüpbach, and D. Ish-Horowicz. 1999. Squid hnRNA protein promotes apical cytoplasmic transport and localization of Drosophila pair-rule transcripts. Cell. 98:171–180. [DOI] [PubMed] [Google Scholar]
- Lau, P.P., H.-J. Zhu, M. Nakamuta, and L. Chan. 1997. Cloning of an apobec-1-binding protein that also interacts with apolipoprotein B mRNA and evidence for its involvement in RNA editing. J. Biol. Chem. 272:1452–1455. [DOI] [PubMed] [Google Scholar]
- Litman, P., J. Barg, L. Rindzoonski, and I. Ginzburg. 1993. Subcellular localization of tau mRNA in differentiating neuronal cell culture: Implications for neuronal polarity. Neuron. 10:627–638. [DOI] [PubMed] [Google Scholar]
- Long, R.M., R.H. Singer, X.H. Meng, I. Gonzales, K. Nasmyth, and R.-P. Jansen. 1997. Mating type switching in yeast controlled by asymmetric localization of ASH1 mRNA. Science. 277:383–387. [DOI] [PubMed] [Google Scholar]
- Long, R.M., W. Gu, X. Meng, G. Gonsalvez, R.H. Singer, and P. Chartrand. 2001. An exclusively nuclear RNA-binding protein affects asymmetric localization of ASH1 mRNA and Ash1p in yeast. J. Cell Biol. 153:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min, S., C.W. Turck, J.M. Nikolic, and D.L. Black. 1997. A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev. 11:1023–1036. [DOI] [PubMed] [Google Scholar]
- Ross, A.F., Y. Oleynikov, E.H. Kislausksi, K. Taneja, and R.H. Singer. 1997. Characterization of a β-actin mRNA zipcode-binding protein. Mol. Cell. Biol. 17:2158–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydorides, A.D., H.J. Okano, Y.Y. Yang, G. Stefani, and R.B. Darnell. 2000. A brain-enriched polypyrimidine tract-binding protein antagonizes the ability of Nova to regulate neuron-specific alternative splicing. Proc. Natl. Acad. Sci. USA. 12:6350–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestakova, E.A., R.H. Singer, and J. Condeelis. 2001. The physiological significance of β-actin mRNA localization in determining cell polarity and directional motility. Proc. Natl. Acad. Sci. USA. 98:7045–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smidt, M.P., B. Russchen, L. Snippe, J. Wijnholds, and A.B. Geert. 1995. Cloning and characterization of a nuclear, site specific ssDNA binding protein. Nucl. Acid Res. 23:2389–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa, P.A., A. Sil, J.R. Swedlow, I. Herskowitz, and R.D. Vale. 1997. Actin-dependent localization of an RNA encoding a cell-fate determinant in yeast. Nature. 389:90–93. [DOI] [PubMed] [Google Scholar]
- Zhang, H.L., R.H. Singer, and G.J. Bassell. 1999. Neurotrophin regulation of β-actin mRNA and protein localization within growth cones. J. Cell Biol. 147:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H.L., T. Eom, Y. Oleynikov, S.M. Shenoy, D.A. Liebelt, J.B. Dictenberg, R.H. Singer, and G.J. Bassell. 2001. Neurotrophin-induced transport of zipcode binding protein with β-actin mRNA increases β-actin levels and stimulates growth cone motility. Neuron. 31:261–275. [DOI] [PubMed] [Google Scholar]