

Abstract
In dystrophic mice, a model of merosin-deficient congenital muscular dystrophy, laminin-2 mutations produce peripheral nerve dysmyelination and render Schwann cells unable to sort bundles of axons. The laminin receptor and the mechanism through which dysmyelination and impaired sorting occur are unknown. We describe mice in which Schwann cell–specific disruption of β1 integrin, a component of laminin receptors, causes a severe neuropathy with impaired radial sorting of axons. β1-null Schwann cells populate nerves, proliferate, and survive normally, but do not extend or maintain normal processes around axons. Interestingly, some Schwann cells surpass this problem to form normal myelin, possibly due to the presence of other laminin receptors such as dystroglycan and α6β4 integrin. These data suggest that β1 integrin links laminin in the basal lamina to the cytoskeleton in order for Schwann cells to ensheath axons, and alteration of this linkage contributes to the peripheral neuropathy of congenital muscular dystrophy.
Keywords: axo–glial interactions; Cre/loxP; congenital muscular dystrophy; laminin; peripheral nerve
Introduction
Myelination in the peripheral nervous system is accomplished by Schwann cells. In embryonic nerves, Schwann cell precursors originate from neural crest cells and migrate along growing neurites. Neurites and Schwann cells contact and influence each other, reciprocally determining survival and differentiation (for review see Jessen and Mirsky, 1999; Witt and Brady, 2000). Before birth, Schwann cells send cytoplasmic processes into bundles of axons, and progressively sort individual axons so that a 1:1 relationship is established (promyelinating Schwann cells) in a process termed radial sorting (Webster, 1984). Axons that are destined to remain unmyelinated are ensheathed in groups by nonmyelinating Schwann cells. Assignment of one or the other Schwann cell–axon relationship is largely complete within the first few days after birth in the rodent (Webster, 1984). Both types of Schwann cells deposit and organize a basal lamina, but only promyelinating Schwann cells proceed to wrap concentric extensions of their membrane around axons, to produce myelin.
Bunge and Bunge (1983) proposed that Schwann cells resemble epithelia: they separate neurons from mesenchymal tissues by forming a basal lamina, and polarize their surfaces into an ab-axonal surface (similar to the basal surface of epithelia) contacting the basal lamina and an ad-axonal surface (similar to the apical surface of epithelia) contacting the axon. Subsequent studies showed that Schwann cells require the formation of an organized basal lamina (for review see Bunge, 1993), or at least laminin deposition (Podratz et al., 2001), to properly ensheath and myelinate dorsal root ganglion neurons (DRG)* in coculture. In support of this, the dystrophic (dy/dy and dy 2J/dy 2J) mice and merosin-deficient congenital muscular dystrophy (CMD) patients harbor mutations in the gene encoding the α2 laminin chain (dy 2J/dy 2J and CMD) that result in absent or defective laminin-2 (Xu et al., 1994; Helbling-Leclerc et al., 1995; Sunada et al., 1995) and cause muscular dystrophy (Michelson et al., 1955). Both the mouse and human disorders include a dysmyelinating peripheral neuropathy (Bradley and Jenkison, 1973). The most striking abnormality in dystrophic mice is present in spinal roots and cranial nerves, and to a lesser extent in peripheral nerves, where bundles of naked axons are not invested by any Schwann cell processes (Bradley and Jenkison, 1973). Basal laminae are discontinuous both in roots and peripheral nerves (Madrid et al., 1975). Despite extensive studies (Perkins et al., 1981), the mechanism for these abnormalities has not been elucidated. In particular, which laminin receptors determine the ability of Schwann cells to ensheath or myelinate axons is unclear.
Myelin-forming Schwann cells synthesize abundantly the laminin receptors α6β4, α6β1 integrins and dystroglycan, and minor amounts of α2β1 integrin (for review see Previtali et al., 2001). β1 integrin can dimerize with several α subunits, forming receptors for many extracellular matrix components in addition to laminins (for review see Previtali et al., 2001). β1 integrins, including α6β1, are detectable in the Schwann cell lineage from its first appearance (Bronnerfraser et al., 1992) and may play various roles in peripheral nerve development and myelination (Previtali et al., 2001). For example, antibodies to β1 integrin block myelination in neuronal-Schwann cell cocultures (Fernandez-Valle et al., 1994), although β1 is also present on axons (Tomaselli et al., 1993); the effect may be non-Schwann cell autonomous. Genetic studies of the role for β1 integrin in nerve development have been impossible, since targeted inactivation of β1 in mouse causes early embryonic death (Fassler and Meyer, 1995; Stephens et al., 1995).
Here, we report inactivation of β1 integrin specifically in Schwann cells using the Cre-loxP system, and show that β1 integrins are crucially important for Schwann cell–axon interactions. In the absence of β1 integrin, Schwann cells can populate nerves, proliferate, and survive normally, but cannot segregate axons, and leave fetal nerve–like bundles of axons in nerves of adult mice. Many Schwann cells form a basal lamina and initiate process extension and ensheathment of axons, but are unable to maintain extension and retract their processes. Intriguingly, these nerves closely resemble the roots and proximal nerves of dystrophic mice. Finally, we show that the few Schwann cells that achieve a 1:1 relationship with an axon can myelinate in the complete absence of β1 integrin. These data suggest a role for the cytoskeleton in the pathogenesis of neuropathy associated with CMD.
Results
Generation of mice lacking β1 integrin in Schwann cells
To inactivate the Itgβ1 gene only in Schwann cells, we crossed mice homozygous for β1 “floxed” (the first coding exon was flanked by two loxP sites [β1F/F]) with mice heterozygous for the β1 null allele and hemizygous for a P0Cre transgene (β1+/−//P0Cre) (Fig. 1 A). β1F/F and β1+/− mice appear normal (Stephens et al., 1995; Graus-Porta et al., 2001). Our P0Cre transgene, mP0TOT(Cre), is based on the whole P0 glycoprotein gene, and activates Cre-mediated recombination specifically in Schwann cells between embryonic days (E)13.5 and 14.5 (Feltri et al., 1999a). β1F/−//P0Cre (mutant) mice were generated in accord with Mendelian ratios, and were thus viable.
Figure 1.
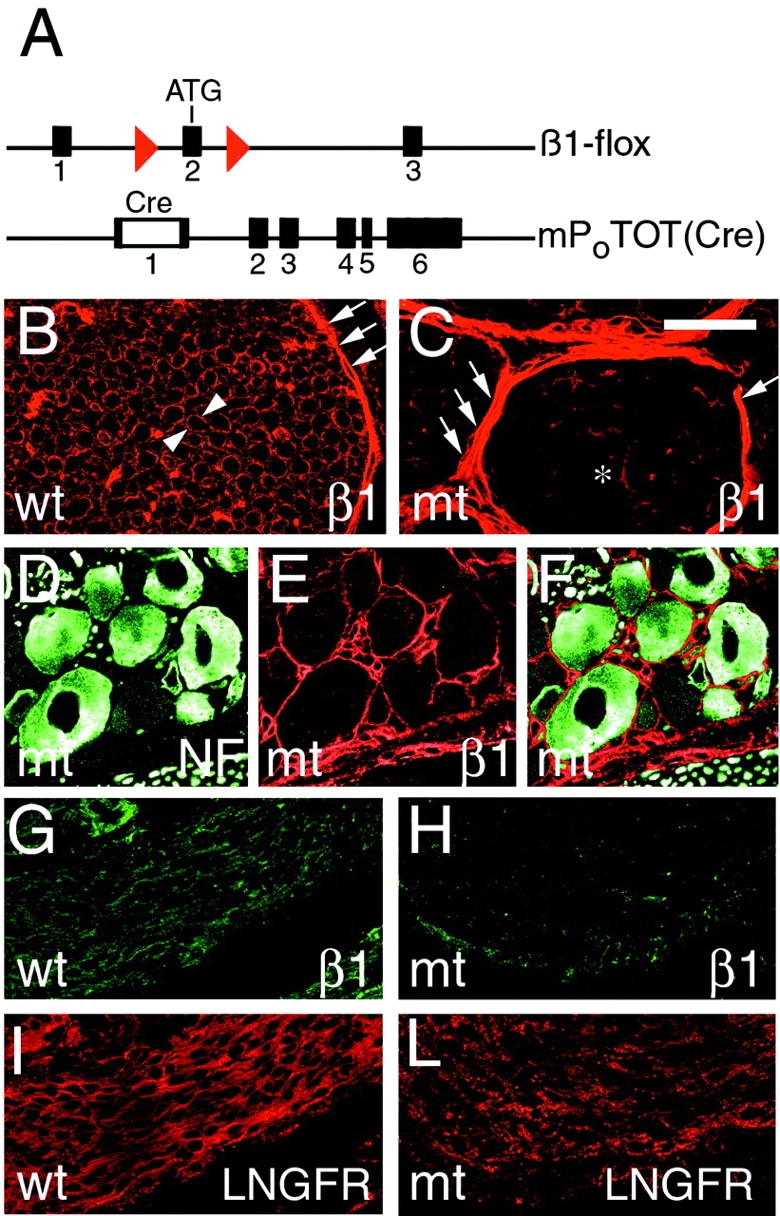
Disruption of β1 integrin specifically in Schwann cells in peripheral nerves. A schematic representation of the “floxed” Itgβ1 allele shows that two loxP sites (red) flank exon 2, containing the ATG start site of translation and the P0Cre-mP0TOT(Cre) transgene, containing the Cre gene inserted into exon 1 of the Mpz gene. Transverse sections of control (B; wt) or mutant (C; mt) sciatic nerves at P28 show that β1 is normally detected in the endoneurium at the outer surface of each myelinating Schwann cell (arrowheads) and in the perineurium (arrows), whereas in mutant nerves, β1 is absent in Schwann cells (asterisk), but is preserved in the perineurium (arrows). Low level endoneurial staining for β1 is associated with vessels and perineurial cells abnormally present in the endoneurium (C; see text). (D–F) Double staining of DRG for neurofilament (D; green) and β1 integrin (E; red) shows that β1 expression in mutant sensory neurons is preserved; (F) merge. Longitudinal sections of control (G and I) and mutant (H and L) nerves at E17.5 are double stained for β1 (G and H; green) and LNGFR (I and L; red). Note that the β1 integrin staining in Schwann cells is markedly reduced in mutant nerves by E17.5. Bar: (B and C) 30 μm; (D and F) 50 μm; (G and I) 60 μm; (H and L) 90 μm.
P0Cre disrupts β1 integrin specifically in Schwann cells
To verify the extent of β1 recombination in peripheral nerves, transverse sections of sciatic nerves from mutant and control mice were stained for β1 integrin at postnatal day (P)28. In control nerves, β1 integrin was detected in the endoneurium on the outer surface of Schwann cells and in blood vessels, and in the perineurium (Fig. 1 B). In contrast, in mutant nerves, β1 integrin was nearly absent in the endoneurium, whereas it remained easily detected in the perineurium (Fig. 1 C). Endoneurial β1 staining was accounted for by blood vessels and by perineurial cells not normally present within the endoneurium (see below). Thus, β1 integrin is absent in most, if not all, Schwann cells in nerves of adult mutant mice (see also below).
Schwann cells originate from multipotent neural crest precursors that also give rise to other components of peripheral nerve, DRG neurons, which contribute axons to peripheral nerves (Anderson, 1997). P0 mRNA is expressed at low levels in some neural crest precursors (Lee et al., 1997). Consistent with this, another P0Cre transgene activates recombination as early as E9.5 in the neural crest, producing somatic genomic alterations inherited by both Schwann cells and DRG neurons (Yamauchi et al., 1999). To determine if our P0Cre transgene targeted recombination only to Schwann cells, we stained DRG of adult mutant mice for β1 integrin and neurofilament, a neuronal marker. Fig. 1, D–F, shows that DRG neurons from mutant mice coexpress neurofilaments and β1 integrin. In addition, when our P0Cre mouse was crossed with a discontinuous lacZ “tester” mouse (Akagi et al., 1997), we never detected lacZ expression in DRG neurons (unpublished data). Therefore, DRG neurons and perineurial cells of mutant mice still express β1 integrin, whereas Schwann cells do not; any phenotype observed in nerves of mutant mice is Schwann cell autonomous.
Mice lacking β1 integrin in Schwann cells develop a severe dysmyelinating neuropathy
Mutant mice manifested progressive muscular weakness, appearing in the first 2 wk of life and evolving to hind limb paralysis by 3–5 mo of life (Fig. 2, C and D). Tremor, wide-based gait, and muscular atrophy were already evident by P28 (Fig. 2 B). This phenotype was variably severe, even among littermates. To quantify the neuromuscular deficit, we performed a rotarod behavioral test. 2–3-mo-old mutant and control mice were placed on a horizontal rod rotating at increasing speed, and the time that they remained on the bar was measured. Mutant mice remained on the rod for significantly less time than control mice (Fig. 2 E).
Figure 2.
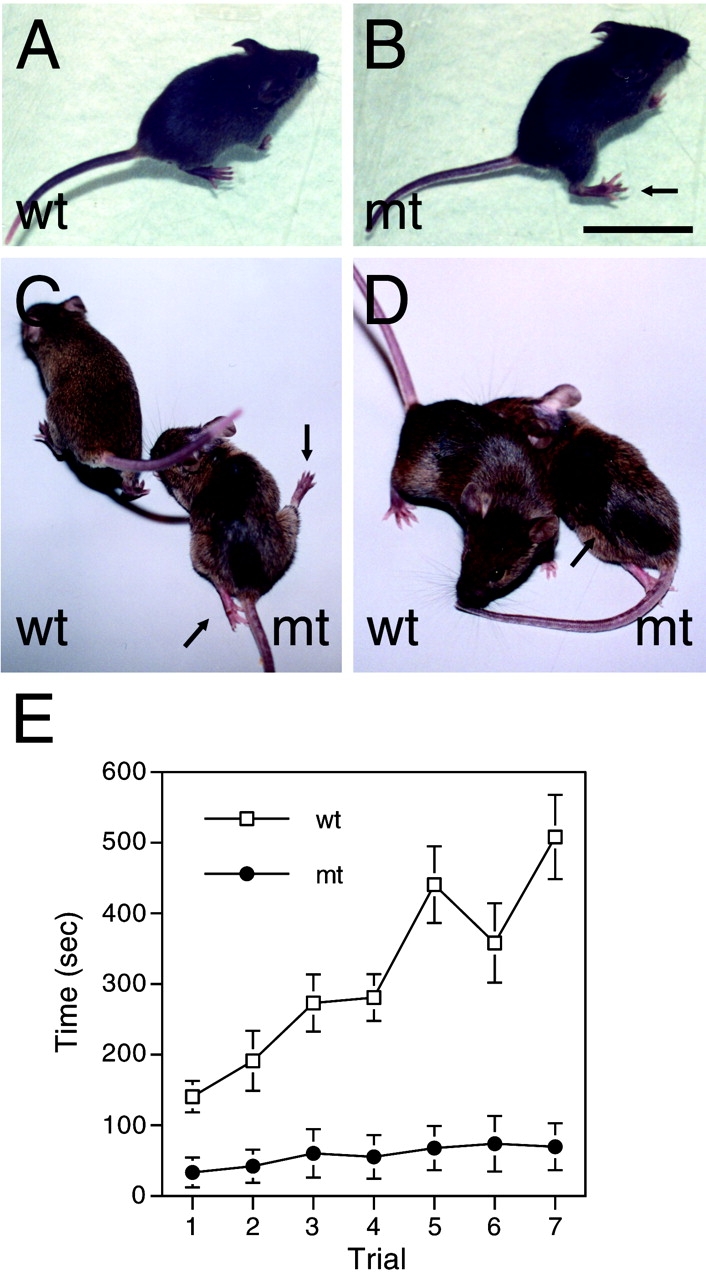
Mutant mice manifest severe motor impairment. (A–D) Mutant (mt) and control (wt) mice at P28 (A and B) or P90 (C and D) were observed sitting on an inclined surface (A and B) or walking on a flat surface (C and D). Mutant mice at P28 sit with their hind limbs displaced laterally (arrow in B) and walk with a wider base, swinging their hind limb laterally (unpublished data). By P90, many mutant mice manifest nearly paralyzed hind limbs (arrows in C) and more obvious muscle atrophy (arrow in D). (E) Rotarod test was performed with 2–3-mo-old mutant and control mice. In a series of seven consecutive trials the time (seconds; mean ± SEM) for which animals remained on a rod rotating at increasing speed is plotted. Hold time is significantly reduced in mutant mice (n = 10) as compared with control mice (n = 14). 0.01 < P < 0.001 in the first trial, and P < 0.001 in subsequent trials, by two-tailed Student t test. Bar: (A–C) 3 cm; (D) 2.7 cm.
We next examined the morphology of mutant and control sciatic nerves at P28 by analysis of transverse semithin sections or by electron microscopy. By this age every large caliber axon in a normal nerve has been myelinated (Fig. 3 H). In contrast, sciatic nerves from mutant mice show few myelinated axons (Fig. 3 G). Several axons had achieved an appropriate 1:1 relationship with a Schwann cell, but were not myelinated. Instead, most axons were tightly packed in large bundles, and not ensheathed by Schwann cells. These bundles of axons were surrounded by a ring of cells. Ultrastructural analysis revealed that some of these encircling cells lacked a basal lamina, and were presumably endoneurial fibroblasts. However, most frequently these cells displayed a basal lamina, caveolae, tight junctions, and lipid inclusions, identifying them as peurineurial-like cells (Fig. 4, A, B, and G). In addition, they stained for both β1 integrin (Fig. 1 C) and for occludin (unpublished data), a perineurial cell marker (Furuse et al., 1993; Parmantier et al., 1999), confirming that significant numbers of perineurial-like cells are found within the endoneurium of mutant, but not normal nerves. Sciatic nerves of older mice (up to 6 mo of age) appeared similar in semithin sections (unpublished data). Whether axonal degeneration and fiber loss occurs in distal nerves of older animals remains to be determined.
Figure 3.
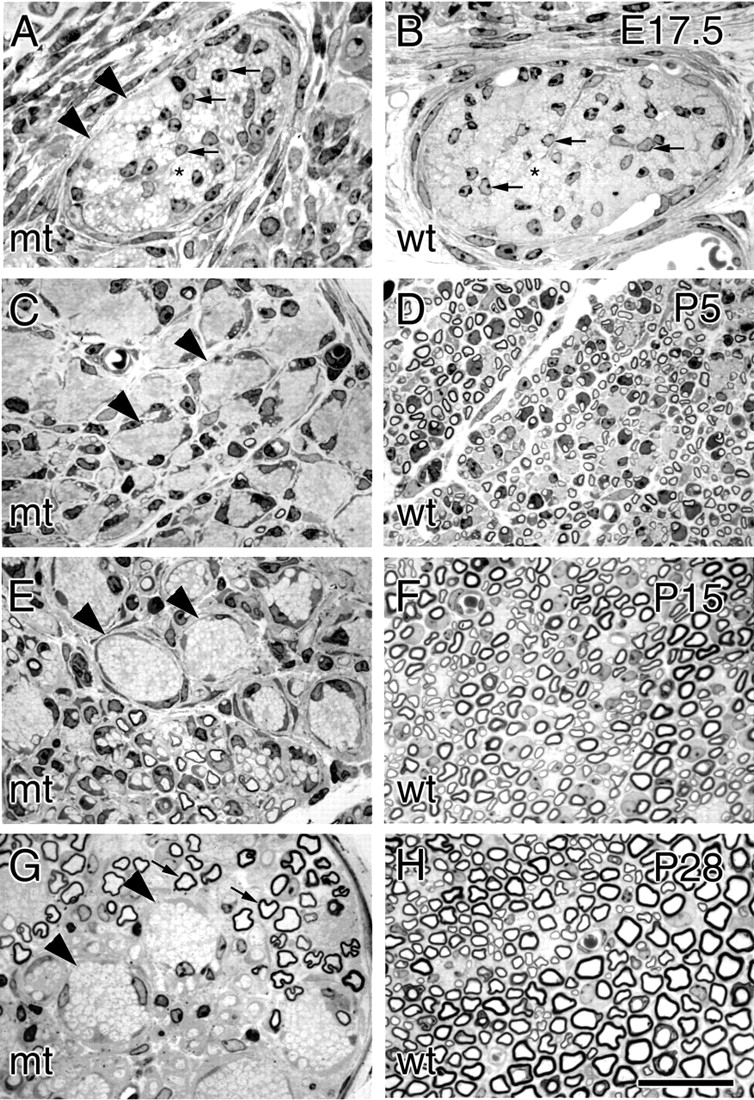
Mutant mice develop a severe dysmyelinating neuropathy. Transverse semithin sections of comparable nerves from mutant (mt; A, C, E, and G) and control (wt; B, D, F, and H) mice at E17.5 (A and B), P5 (C and D), P15 (E and F), and P28 (G and H). At P28, myelination is complete in normal nerves (H), whereas mutant nerves show large bundles of unsorted axons (G, arrowheads) and few myelinated fibers (arrows). At E17.5 (A and B) both control and mutant nerves contain groups of yet unsorted axons (asterisks) and numerous Schwann cells between them (arrows); groups of tightly packed axons begin to be distinguishable in mutant nerves (A, arrowheads), but not in controls (B). In P5 control nerves (D), most large caliber axons have reached the proper 1:1 relationship with a Schwann cell, and many thin myelin sheaths appear. In mutant nerves (C), in contrast, large groups of unsegregated axons are present (arrowheads), and only rare, very thin, myelin sheaths have formed. Unsorted groups of axons become progressively more obvious during nerve maturation (E and G, arrowheads), and are often grouped sectorially in the nerve. More thin myelin sheaths appear with delay in mutant nerves at P15 (E) and become thicker at P28 (G). Bar: (A and B) 27 μm; (C–H) 29 μm.
Figure 4.

Ultrastructural analysis of mutant nerves reveals perturbed relationships with axons. Transverse sections of mutant sciatic nerves at P28 (A and B) reveal bundles containing unsorted, mixed caliber axons (asterisks) that are not invested by Schwann cell cytoplasm (“naked”). Several Schwann cells have attained the proper 1:1 relationship with an axon and occasionally are forming a myelin sheath (A). At high magnification (C), the periodicity of compact myelin appears normal. Axonal bundles are always surrounded by a layer of perineurial cells, abnormally present in the endoneurium (arrowheads). Some of the perineurial cells contain lipid inclusions (B) and at high magnification (inset in B–G) display a basal lamina (arrowhead in G), tight junctions (arrow in G), and caveolae (arrows in insets indicated by asterisks in G; magnified 1.6×). In D–F several axons are shown surrounded by a loose, undulated basal lamina (arrowheads), which in some cases contain Schwann cell processes variably ensheathing axons (arrows), whereas in C, a myelinated fiber has a tightly apposed basal lamina (arrowheads). Transverse sections of control (H) and mutant (I) nerves at P1 show differences in radial sorting and cytoplasmic processes. In control nerves (H), most axons >1 μm are either segregated at the periphery of a bundle (black asterisks), in groups of 1–3 (arrows), or clearly in a 1:1 relationship with a Schwann cell. In contrast, in mutant nerves (I), most large axons are either unsegregated from bundles of smaller axons (black asterisks), or in groups of greater than three (thick arrows). Whereas control Schwann cells (H) send thin processes around axons, mutant Schwann cells (I) send abnormally shaped, thick cytoplasmic processes (white asterisks) in every direction (thin arrows). Bars: (A) 4.4 μm; (B) 3.7 μm; (C and G) 1.2 μm; (D and E) 1.6 μm; (F) 1 μm; (H and I) 7.6 μm.
β1-null Schwann cells variably retract their processes from ensheathed axons
The bundles of naked axons, not ensheathed by Schwann cell cytoplasm, were particularly interesting, because they are the hallmark feature of spinal roots in dystrophic mice. Typically they were more numerous in one or two fascicles of the nerve. By ultrastructural analysis (Fig. 4, A and B) we identified occasional Schwann cells at the margin or within bundles, in various stages of axonal ensheathment or myelination. Other fascicles contained only Schwann cells engaged in normal axonal relationships—Schwann cells in a 1:1 relationship with large diameter axons and normal nonmyelin–forming Schwann cells ensheathing multiple small axons (Fig. 4 B and unpublished data). Many Schwann cells had no myelin, but others formed myelin sheaths of various thickness (Fig. 4, A and B), and of normal periodicity (Fig. 4 C). Thus, at least some Schwann cells could bypass the impairment of sorting axons and differentiate normally toward myelin-forming or nonmyelin-forming phenotypes.
β1 integrin adheres to extracellular matrix components, such as laminin, in the basal lamina (Previtali et al., 2001). Mutant nerves contained some basal laminae that were continuous and adherent to the Schwann cell surface (Fig. 4 C). However, many Schwann cells produced a basal lamina that was either discontinous, or continuous but detached at points from the cell surface. Strikingly, many axons were surrounded by an empty, undulated basal lamina. Inside, we found Schwann cell cytoplasmic processes ranging from partially ensheathing, to only in tangential contact with an axon, to completely absent (examples are shown in Fig. 4, D–F), suggesting that a Schwann cell had originally ensheathed a single axon and then retracted its process. These data indicate that, in the absence of β1 integrin, many Schwann cells are unable to maintain normal contact with their basal lamina and consequently normal interactions with axons. We imagined that bundles of naked axons could result from both arrest in the process of radial sorting and retraction of ensheathing processes.
β1-null Schwann cells fail to ensheath and segregate axons in developing nerves
To further address the formation of bundles of naked axons, we examined developing nerves from mutant animals. Axonal segregation and sorting is normally largely complete by the first few days after birth (Fig. 3 D). In mutant nerves, bundles of tightly apposed axons were already distinguishable at E17.5 (Fig. 3 A) and became progressively more obvious with nerve maturation (Fig. 3, C, E, and G). By immunohistochemistry, we confirmed that β1 integrin was mostly absent from Schwann cells by E17.5 (Fig. 1 H and see below).
We next examined the ultrastructure of nerves at E17.5 and P1, when sorting and segregation normally produce promyelinating Schwann cells. At E17.5, some mutant Schwann cells had advanced cytoplasmic processes around axons, but these processes appeared redundant and disorganized as compared with controls (unpublished data). In control nerves at P1, most axons larger than 1 μm in diameter had already been segregated to the periphery of axonal bundles, or were already ensheathed by single Schwann cells (Fig. 4 H). Occasionally, we identified Schwann cells containing two or at most three large caliber axons. Small caliber axons, not destined to be myelinated, remained within bundles. Schwann cell processes were thin, with little cytoplasmic content, and uniformly distributed around axons or axon bundles.
In contrast, mutant nerves at P1 contained Schwann cells that had ensheathed groups of up to 8–9 large caliber axons, and bundles of unsorted, mixed caliber axons (Fig. 4 I). Strikingly, Schwann cell processes contained a larger volume of electron-dense cytoplasm and expanded in many directions, even away from axons. All processes were surrounded by a basal lamina; there was as yet no evidence of retraction of cytoplasmic processes. Thus, both impaired initial interactions with axons, as well as subsequent retraction of processes, may contribute to bundle formation.
Mutant Schwann cells populate nerves, proliferate, and die normally
One alternate explanation for bundles of naked axons was that the number of β1-null Schwann cells were insufficient to ensheath axons, due to a failure in their migration, proliferation, or survival. β1 integrins are involved in these processes in other cells (for review see De Arcangelis and Georges-Labouesse, 2000). Furthermore, as a source of mitogenic and survival factors (e.g., neuregulins), axons are a major determinant of Schwann cell number (Meyer and Birchmeier, 1995; Riethmacher et al., 1997; Wolpowitz et al., 2000). Thus, impaired relationships between mutant Schwann cells and axons could also reduce survival or proliferation.
Schwann cells in mutant nerves had migrated distally because comparable numbers of cells were visible within distal nerves of mutant and control mice at E15.5, E17.5, and P1 by DAPI and neurofilament staining in nerves of whole fetuses (Fig. 5), including in superficial nerves adjacent to the skin (unpublished data), or by semithin section analysis in nerves distal to the knee (Fig. 3 A and unpublished data). This is not surprising, as Schwann cell precursors populate the length of nerves before recombination by P0Cre at E13.5–E14.5. To determine if there was a failure in Schwann cell proliferation, we measured the percentage of nuclei incorporating BrdU in nerves of E15.5 and E17.5 mice. We found no significant difference in BrdU incorporation in mutant and control nerves (Fig. 5 and Table I), including at E17.5, the peak of Schwann cell proliferation (Stewart et al., 1993). To determine if β1-negative Schwann cells died by apoptosis more often than control Schwann cells, we measured the fraction of nuclei stained by terminal deoxynucleotidyl transferase–mediated dUTP-biotin nick-end labelling (TUNEL) in mutant and control nerves at E15.5, E17.5, and P5. The percentages were not significantly different in mutant and control nerves (Fig. 5 and Table I). The percentages in control nerves are in agreement with those previously reported in rat (Grinspan et al., 1996; Syroid et al., 1996) and mouse (Syroid et al., 2000). Thus, we found no major difference in migration, proliferation, and apoptosis of mutant Schwann cells. It should be noted that β1 class integrins probably play a role before E13.5–E14.5 in the processes of migration, proliferation, and survival in Schwann cell precursors (Lefcort et al., 1992; Milner et al., 1997; Haack and Hynes, 2001). These data further support that axons within empty or partially empty basal laminae are produced by process retraction, not death of Schwann cells, at least until P5.
Figure 5.
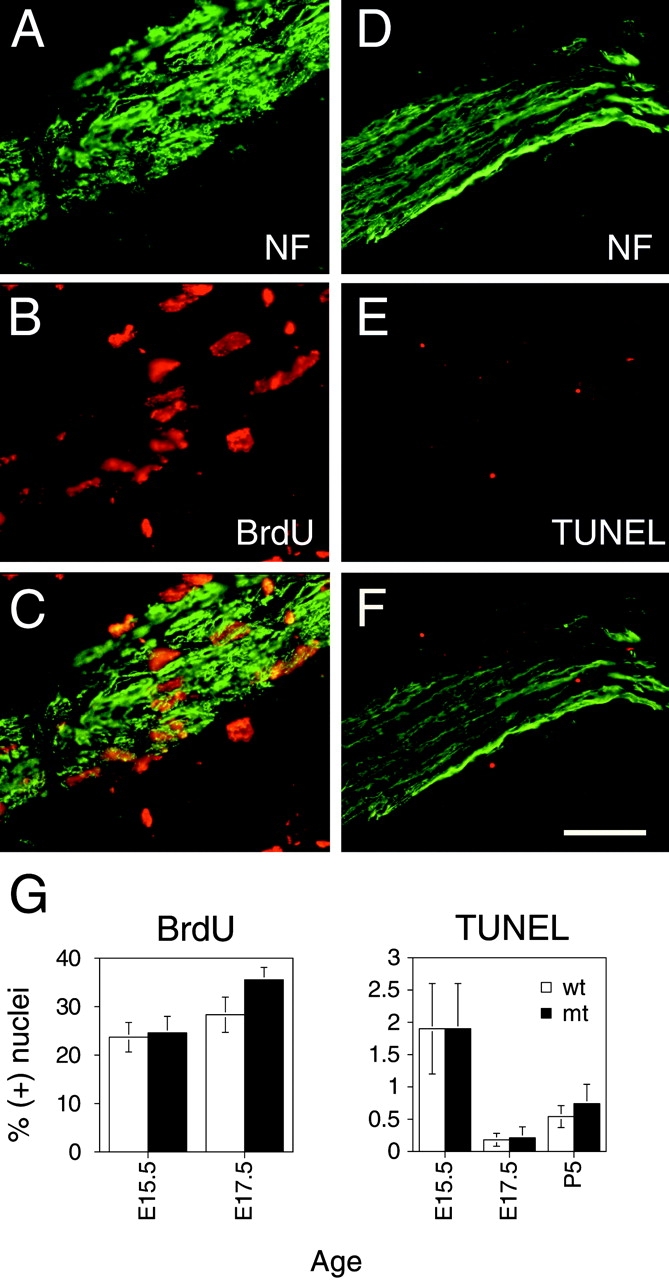
β1-null Schwann cells proliferate and survive normally. (A–C) Nuclei of mutant Schwann cells associated with longitudinal sections of nerves at E17.5 that have been stained for neurofilament (A, green) and BrdU (B, red) after a 1-h pulse; C, merge. (D–F) Apoptotic nuclei associated with mutant nerves at E17.5 that have been stained for neurofilament (NF) (D, green) were identified by TUNEL staining (E, red); F, merge. (G) The percentages (mean ± SEM) of BrdU- or TUNEL-positive nuclei in mutant (mt) and control (wt) nerves. See also Table I. By a paired, two-tailed t test the percentages in mutant and controls were not significantly different. Bar, (A–F) 60 μm.
Table I. Measurements of proliferation and apoptosis in mutant and control nerves.
| BrdU incorporating nuclei | TUNEL-positive cells | |||
|---|---|---|---|---|
| Age | mt | wt | mt | wt |
| % | % | % | % | |
| E15.5 | 24.6 ± 3.4(10) | 23.7 ± 3.0 (11) | 1.95 ± 0.67 (32) | 1.96 ± 0.75 (12) |
| E17.5 | 35.5 ± 2.5(6) | 28.3 ± 3.6 (7) | 0.21 ± 0.17 (24) | 0.18 ± 0.10 (23) |
| P5 | 0.74 ± 0.18 (14) | 0.54 ± 0.12 (11) | ||
Percentages (mean ± SEM) of nuclei incorporating BrdU and of TUNEL-positive cells in nerves of mutant (mt) and control (wt) mice. Number of experiments is in parentheses. By a paired, two-tailed t test, the percentages in mutant and control animals were not significantly different.
Schwann cells can synthesize myelin in the absence of β1 integrin
We identified ultrastructurally normal myelin sheaths in mutant sciatic nerves at P28 (see above). These findings were surprising, because previous antibody-blocking experiments in Schwann cell–DRG cocultures suggested that β1 integrin was necessary for myelination (Fernandez-Valle et al., 1994). One possible explanation was that some Schwann cells escaped Cre-mediated recombination and expressed β1 integrin. To test this, we stained nerves and spinal roots from adult mutant and control mice for β1 integrin and myelin basic protein (MBP), a myelin marker. In control nerves, each myelin sheath appeared as an MBP-positive ring (Fig. 6 A, green), encircled by a thin β1-positive ring at the abaxonal surface of the Schwann cell (Fig. 6 B, red). In contrast, in mutant nerves, the few MBP-positive myelin sheaths were not encircled by β1-positive rings (Fig. 6, E and F). This finding was more striking in nerve roots, where the majority of Schwann cells had achieved a 1:1 relationship with large axons and formed myelin. Here, none of the multiple MBP-positive myelin sheaths were surrounded by a β1-positive ring (Fig. 6, C and D).
Figure 6.

Synthesis of other laminin receptors and myelin in β1-null Schwann cells. Transverse sections of sciatic nerves from control (wt) (A and B) and mutant (mt) (E and F) mice at P28 were double stained for MBP (A and E; green) and β1 integrin (B and F; red). In the control nerve, every MBP-positive myelin sheath is encircled by a β1-positive Schwann cell cytoplasm, whereas in the mutant nerve, the MBP-positive myelin sheaths are not. Staining of serial sections from the same mutant nerve for dystroglycan (DG; G), β4 integrin (H), and α6 integrin (I) detects multiple myelinated fibers in the same field (next to an asterisk as a point of reference) positive for α6, β4 integrin, and dystroglycan. Transverse sections of spinal roots of mutant mice at P28 double stained for MBP (C) and β1 integrin (D) reveal numerous MBP-positive fibers with no associated β1 integrin. Bar: (A and B) 20 μm; (C and D) 40 μm; (E–I) 70 μm.
Alternatively, the ablation of β1 could have been delayed long enough in some Schwann cells to permit myelination to proceed before β1 disappeared. This could result from either delayed recombination by Cre or from a long half-life of β1 integrin. To test these possibilities, we stained nerves at E15.5, E17.5, and P5 for β1, low affinity nerve growth factor receptor (LNGFR) (or S-100 at P5), a marker of premyelinating Schwann cells, and occludin, a marker of perineurial cells and blood vessels (Furuse et al., 1993; Parmantier et al., 1999). At E17.5 or later, we detected minimal β1 staining in the endoneurium of mutant nerves that colocalized with occludin (unpublished data) but not with LNGFR, showing that the cells expressing β1 are perineurial cells or vessels. In sharp contrast, the cytoplasm of the majority of cells in the endoneurium of control nerves stained for β1 and LNGFR, but not occludin (Fig. 1, G–L, and unpublished data). Thus, in mutant nerves, premyelinating Schwann cells had already lost β1 by E17.5. This observation fits well with the finding that our P0Cre recombines a “tester” lacZ transgene along the length of peripheral nerves by E15.5 (unpublished data). The sum of the above data clearly shows that Schwann cells can synthesize myelin in the absence of β1 integrin. Moreover, it suggests that those Schwann cells that can form appropriate relationships with axons, and subsequently synthesize myelin, exploit other factors in the process.
In keeping with this idea, it was possible that myelination in mutant nerves depended on laminin receptors that substituted for β1 function. Serial sections from the nerve in Fig. 6 were stained for other laminin receptors not containing β1, and known to be expressed in Schwann cells: dystroglycan and α6β4 integrin (Einheber et al., 1993; Feltri et al., 1994; Yamada et al., 1994). β1-null, myelinated fibers contained dystroglycan, β4, and α6 integrins (Fig. 6, G–I).
Myelination is delayed in the absence of β1 integrin, despite the perinatal appearance of other laminin receptors
To further explore whether these laminin receptors compensate for the absence of β1, we compared the onset of myelination with that of dystroglycan and β4 integrin synthesis in developing mutant nerves. Myelination began later than normal in mutant nerves. Numerous thin myelin sheaths were visible in normal nerves at P5 (Fig. 3 D), whereas virtually no myelin was present at this age in mutant nerves (Fig. 3 C). Several myelin sheaths, thinner than those of littermate controls, were first evident at P15 (Fig. 3, E and F). At P28, thick myelin sheaths of normal periodicity were seen in both mutant and control nerves (Fig. 3, G and H). Dystroglycan expression immediately precedes myelination in normal nerves around birth (unpublished data). Thus, we stained nerves at P1 for dystroglycan and MBP. Dystroglycan was expressed in many fibers of normal nerves at this age, a subset of which had initiated MBP synthesis and myelination (Fig. 7, A–C). In mutant nerves, dystroglycan was also found in many fibers, but no MBP was detected (Fig. 7, D–F), consistent with delayed myelination. Similarly, β4 integrin appeared simultaneously in control and mutant nerves at P1 (unpublished data) in advance of significant myelin formation in mutant nerves (Fig. 3). Thus, dystroglycan and α6β4 integrin may compensate for β1 integrins in myelination, but they are not limiting. Other factors must intervene to permit the (delayed) onset of myelination in β1-ablated Schwann cells.
Figure 7.

Myelination is delayed despite normal onset of dystroglycan synthesis in mutant nerves. Longitudinal sections of control (wt) (A–C) and mutant (mt) (D–F) nerves at P1 were double stained for dystroglycan (A and D; red) and MBP (B and E; green). In all nerves, dystroglycan is detected in multiple fibers (A, C, D, and F), but only in control nerves do a subset of these fibers stain for MBP (compare B to E). Bar: (A–F) 60 μm.
Discussion
We describe mice in which β1 integrin was ablated specifically in Schwann cells in the late embryo using the Cre-loxP system. The lack of β1 integrin causes a severe dysmyelinating neuropathy due to the inability of Schwann cells to form and maintain proper interactions with axons. β1 integrin is not required at this time for Schwann cell migration, proliferation, and survival. Rather, ultrastructural changes in mutant nerves suggest that β1 is necessary to reorganize the cytoskeleton as Schwann cells ensheath axons and acquire the promyelinating phenotype. Interestingly, some Schwann cells can bypass the impaired axonal interaction even in the absence of β1 integrin and synthesize normal myelin, albeit with a delay. This data shows that Schwann cells require β1 integrins in order to segregate axons, and attain the proper 1:1 relationship with an axon, which is a prerequisite for myelination.
P0Cre directs Schwann cell–specific inactivation of β1
Using the transgenic vector mP0TOTA (Feltri et al., 1999b), we produced transgenic mice in which P0Cre directs recombination specifically in Schwann cells of peripheral nerves beginning at E13.5–E14.5, as revealed by lacZ “tester” mice (Feltri et al., 1999a, and unpublished data). Similarly, in P0Cre//β1F/− mice, β1 integrin was ablated in virtually all Schwann cells, whereas it remained in perineurial cells and DRG sensory neurons and their axons. This is particularly relevant because DRG neurons and Schwann cells derive from common neural crest precursors, some of which express P0 mRNA (Lee et al., 1997). The above data show that the observed phenotype is Schwann cell autonomous. Of note, this is the only transgenic mouse in which the expression of Cre has been targeted to Schwann cells and recombination in DRG neurons has been excluded experimentally (Akagi et al., 1997; Voiculescu et al., 2000). Instead, another P0Cre mouse has been reported to target recombination to both Schwann cells and DRG neurons (Yamauchi et al., 1999).
β1 integrin is required for Schwann cells to interact properly with axons
Process extension, axonal ensheathment, and spiral wrapping allow Schwann cells to form myelin. Many lines of evidence suggest that these changes in cell shape are mediated by the cytoskeleton and its linkage to the extracellular matrix (Bunge and Bunge, 1983; Fernandez-Valle et al., 1997). In mutant nerves, we observed several abnormalities that indicate an alteration of the cytoskeleton or its linkage. First, at the time of axonal ensheathment and sorting (between E17.5 and P1), Schwann cell processes are hypertrophic and are expanded in random directions. Second, associated with these morphological abnormalities, Schwann cells aberrantly ensheath groups of large diameter axons, or are unable to segregate them from small axons. Third, strong indirect evidence suggests that some Schwann cells initially ensheath axons, but subsequently retract their processes: at P28, many axons are surrounded by a basal lamina, either “empty” or containing Schwann cell processes that incompletely cover the axon. This indicates that Schwann cells once ensheathed the axon, synthesized the basal lamina, and then either died or retracted their processes. Since the amount of apoptosis during matching of Schwann cell and axon numbers is not significantly increased in mutant nerves, we conclude that Schwann cells must be unable to maintain process extension.
All of these characteristics (cell shape, formation, and maintenance of cytoplasmic processes) require dynamic alterations of the cytoskeleton. β1-containing integrins transmit signals that cause rearrangement of the actin-based cytoskeleton (Schoenwaelder and Burridge, 1999). In Schwann cells, β1 integrin associates with focal adhesion kinase, fyn kinase, paxillin, and F-actin (Chen et al., 2000). Moreover, Rho family GTP-ases that normally function in regulating actin polymerization are expressed and active in Schwann cells (Terashima et al., 2001), and actin depolymerization alters Schwann cell differentiation (Fernandez-Valle et al., 1997). It is therefore likely that ablation of β1 integrin results in abnormal cytoskeletal organization, causing failure of axonal ensheathment. Indeed, the lack of β1 integrin perturbs cell shapes in other cell types (Stephens et al., 1993; Fassler et al., 1995).
β1 integrin and myelination
Two reports have shown that β1-blocking antibodies prevent myelination in cocultures of Schwann cells and DRG neurons (Fernandez-Valle et al., 1994; Podratz et al., 2001). Thus, it was surprising to find that β1-negative Schwann cells can form normal myelin. Of note, Fernandez-Valle et al. (1994) have shown that high doses of β1-blocking antibodies indeed impair the ability of Schwann cells to sort axons in the DRG cocultures, consistent with our data. It is possible that higher amounts of antibodies are required to mimic the β1-ablated phenotype, with impaired sorting and ensheathment of axons, whereas lower doses of antibodies perturb axonal relationships subtly, and secondarily block myelination.
Instead, we propose that β1 integrin is most important for Schwann cells to reach the promyelinating stage, Schwann cells in a 1:1 relationship with an axon (Zorick et al., 1996; Arroyo et al., 1998), although a few Schwann cells can bypass the arrest and reach this stage with a few days delay. Once the promyelinating stage is reached, Schwann cells can eventually myelinate. Other laminin receptors, such as α6β4 integrin and dystroglycan, probably compensate or are redundant for the function of β1 integrins.
Despite the normal timing of appearance of α6β4 integrin and dystroglycan in mutant nerves, myelination is delayed. Many large caliber axons have achieved 1:1 relationships with mutant Schwann cells by P5 (Fig. 3), but significant myelination awaits P15. Thus, something else, not axonal relationship and not known laminin receptors, is limiting. Either new factors compensate but appear with delay, or Schwann cells need time to correct discoordinated expression or function of existing factors.
Basal lamina and β1 integrin
Basal lamina is first organized around “families” of Schwann cells that encircle bundles of axons before radial sorting (Webster, 1984) at the time that β1 is disappearing from mutant nerves. In fact, we show that genetic alterations of either laminin (dystrophic mice) or β1 integrin produce similar abnormalities in basal lamina and radial sorting. Thus, a functional, and probably physical, interaction between laminin and β1 integrin is required for normal radial sorting leading to the promyelinating Schwann cell phenotype. In support of this, the morphology of basal lamina in β1-null promyelinating Schwann cells is altered: basal lamina are no longer adherent to the surface of many Schwann cells and are sometimes discontinuous. This could represent lack of adhesion at the Schwann cell surface in the absence of β1, or a lack of appropriate basal lamina organization by β1 integrin.
Implications for the pathogenesis of CMD
β1-ablated nerves strikingly resemble the spinal roots and proximal nerves of dystrophic mice deficient in laminin-2. The Schwann cell basal lamina is discontinuous, or altered, and radial sorting is impaired, producing bundles of naked, mixed caliber axons. Finally, the ensheathing defect appears progressively in developing nerves just as it does in newborn dystrophic roots (Jaros and Bradley, 1978). It is interesting that the topography of impaired sorting differs between the two; it is most severe in the spinal roots of dystrophic mice and in the nerves of β1-ablated mice. We cannot eliminate the possibility that loss of a β1 function unrelated to laminin contributes to the phenotype of our mice. The β1 integrin subunit can interact with multiple α subunits, forming dimeric receptors with differing ligand specificities, many of which are present in Schwann cells (laminin: α1β1, α2β1, α3β1, and α6β1; fibronectin: α4β1 and α5β1; collagen: α1β1, α2β1, and α3β1) (Lefcort et al., 1992; Milner et al., 1997; Previtali et al., 2001). However, another appealing explanation is that the topographical difference in severity results from compensation for laminins that does not parallel that for β1 integrins in time or place in developing nerves and roots. Nonetheless, the striking morphological similarity between the two mutants, and the fact that laminin-2 defects appear before birth when the only laminin receptors present in nerve contain β1, indicates that β1 integrins are functionally downstream of laminin-2 in a pathway that regulates Schwann cell–axon interactions.
In conclusion, this study answers several longstanding and important questions. First, laminin 2 mutations have their effect through loss of β1 integrin function, rendering Schwann cells unable to segregate and ensheath axons perinatally. Second, the mechanism in dystrophic mice is very likely to involve uncoupling of the basal lamina from the cytoskeleton, such that the Schwann cell cannot reorganize its cytoskeleton to change shape appropriately. Third, β1 integrin is not directly required to form myelin, but to achieve the proper 1:1 promyelinating Schwann cell–axon relationship. Since laminin mutations are involved in CMD, and peripheral nerve dysfunction is an important contributor to the CMD phenotype (Kuang et al., 1998), understanding the molecular basis of how β1 signals to the cytoskeleton (with which α integrin partner and by which transduction pathways) will provide important clues to the pathogenesis of dysmyelination in CMD.
Materials and methods
Transgenic mice
β1-deficient mice and β1-floxed mice have been described (Stephens et al., 1995; Graus-Porta et al., 2001). mP0TOTA(Cre) (hereafter P0Cre) has been described previously (Feltri et al., 1999a). The mP0TOTA monomer contains a complete mouse P0 gene with 6 kilobases of promoter, in which the ATG start of translation has been mutated and substituted with Cre (Fig. 1). The P0Cre line used in this paper, Tg98.1 (TgN[MpzCre]26Mes), was maintained by backcrosses to FVB/N mice. Mice heterozygous for the β1-null allele (β1+/−), in a 129Sv/B6 mixed background were crossed with P0Cre mice to obtain P0Cre//β1+/− mice. These were then crossed with mice homozygous for β1-floxed (β1F/F), also in a 129Sv/B6 mixed background, to obtain P0Cre//β1F/− (hereafter mutant) or β1F/−, β1F/+, or P0Cre// β1F/+ (hereafter wild-type) mice. β1F/− were used as controls in most experiments, but occasionally β1F/+ or P0Cre//β1F/+ were added as controls; all three genotypes were associated with normal phenotypes. P0Cre//β1+/− and β1F/+ parents were maintained by backcrosses with C57BL6. Most progeny in this study resulted from parents that were N2–N5 generations congenic for C57BL6. Transgenic progeny were identified by Southern blot and PCR analysis of genomic DNA prepared from tail or other tissues samples as described previously (Stephens et al., 1995; Feltri et al., 1999a; Graus-Porta et al., 2001).
Behavioral analysis
Posture and gait of mutant and control animals from 1 to 6 mo of age were observed on a smooth plastic surface inclined at 30° relative to horizontal.
For Rotarod analysis, groups of 2.5–3 mo old mutant and control littermates were placed on a round metal bar rotating first at 4 rotations per minute (rpm) and then accelerating at 7.2 rpm2 (Ugo Basile). The animals were allowed to stay on the rod for a maximum of 700 s and the time of hold on the rotating rod was measured in subsequent trials (4 trials in the first 2 d and 1 trial on each of 3 consecutive days).
Morphological analysis
Mutant and control littermates were killed at the ages indicated, semithin and ultrathin morphological analyses of nerves were conducted as described previously (Wrabetz et al., 2000).
Immunohistochemistry
Complete fetuses, or the spinal column, the posterior limbs, or the sciatic nerve for post-natal animals, were collected in PBS and either directly, or after overnight fixation in 4% paraformaldehyde in 0.1M PBS at 4°C, cryoprotected in 20% sucrose, embedded in OCT (Miles), and snap-frozen in liquid nitrogen. Indirect immunofluorescence was performed on 8-μm thick cryosections fixed in cold acetone for 2 min, rinsed twice in PBS, and blocked with either 10% normal goat serum (Dako) or 20% calf or horse serum, 1% bovine serum albumin, 0.1% Triton X-100, and 0.05% sodium azide. The following antibodies were used: polyclonal antibody (pAb) anti–β-dystroglycan (AP83; a gift of K. Campbell, Howard Hughes Medical Institute, Iowa City, IA); mAb anti–α6 integrin (GoH3, a gift of A. Sonnenberg, The Netherlands Cancer Institute, Amsterdam); pAb anti-β1 integrin (a gift of K. Rubin, Biomedical Center, Uppsala, Sweden); mAb anti-β1 integrin MB1.2, pAb anti-200-kD neurofilament, and pAb anti-LNGFR (Chemicon International); pAb anti-β4 integrin (a gift of V. Quaranta, The Scripps Research Institute, La Jolla, CA); mAb anti-β4 integrin (gift of S.J. Kennel, National Laboratory, Oak Ridge, TN); mAb anti-MBP and mAb anti-neurofilament TA51 (gifts of V. Lee, University of Pennsylvania, Philadelphia, PA); mAb anti-160-kD neurofilament (NN18) and mAb anti-BrdU (Roche); pAb anti-occludin (Zymed Laboratories); pAb anti S-100 (Sigma-Aldrich). Secondary antibodies included: FITC- or TRITC-conjugated goat anti–mouse, –rat, or –rabbit IgG (Southern Biotechnology Associates, Inc.). To reduce background staining produced with mouse mAbs in mouse tissue, we used in some instances the mouse-to-mouse Dako ARK-kit (Dako). Sections were examined with a confocal (MRC 1024; Bio-Rad Laboratories) or fluorescence microscope (Olympus AX-70).
BrdU incorporation assay
BrdU incorporation was performed according to Stewart et al. (1993), with few modifications. Briefly, β1+/−//P0Cre females were mated with β1F/F mice, and 15 or 17 d after detection of a plug they were injected intraperitoneally with 100 μg BrdU/g body weight. 1 h later, fetuses were dissected; the upper body was used to determine genotype and the lower body was processed for immunohistochemistry, with no fixation (as above). Cryosections from at least 2 to 3 each of mutant or control fetuses were fixed in cold methanol, treated with 2 N HCl for 15 min at 37°C, and neutralized with 0.1 M sodium borate pH 8.5 for 10 min. Slides were then incubated with mAb antineurofilament, to identify nerves, and mAb anti-BrdU (see above). After staining with secondary antibodies, nuclei were counterstained with DAPI. Only cigar-shaped nuclei associated with nerves were counted, and the fraction of BrdU-positive nuclei was determined. At least 300 nuclei at E15.5 and 900 nuclei at E17.5 were examined.
TUNEL assay
TUNEL assay was performed as described by Grinspan et al. (1996), with few modifications. Briefly, E15.5 and E17.5 fetuses or P5 sciatic nerves were dissected, fixed in 4% paraformaldehyde in PBS for 1 h, then cryopreserved for immunohistochemistry as above. At least three fetuses and two P5 animals for each of the mutant or control genotypes were studied. Cryosections were treated with acetone, stained with antibodies, and then processed for TUNEL. P5 nerves were stained with anti-β1 integrin or anti-S100 antibodies (unpublished data), and fetuses were stained with antineurofilament antibodies to identify nerves (Fig. 5). TUNEL staining was then performed exactly as described (Grinspan et al., 1996). Nuclei were counterstained by DAPI. For quantification, only DAPI-positive nuclei associated with nerves were counted and the fraction of TUNEL-positive nuclei was determined. At least 1,000 nuclei at E15.5, 3,500 at E17.5, and 10,000 nuclei at P5 were examined.
Image analysis
Micrographs were digitalized using an AGFA Arcus 2 scanner, and figures were prepared using Adobe Photoshop® version 5.0.
Acknowledgments
We thank M. Fasolini, C. Ferri, and H. Peickert for excellent technical assistance, C. Damsky for β1-null mice, J. Grinspan for advice on TUNEL staining, E. Rugarli for the use of the Rotarod, and R. Mirsky for helpful discussion.
This work was supported by grants from Telethon D93 to L. Feltri and 1177 to L. Wrabetz), Progetto Finalizzato Ministero della Sanita (to L. Feltri and L. Wrabetz), Giovanni Armenise-Harvard Foundation (to L. Feltri and L. Wrabetz), Multiple Sclerosis Society of Great Britain (to L. Wrabetz), Italian Multiple Sclerosis Society (to L. Wrabetz), National Institutes of Health (to L. Feltri and L. Wrabetz NS-41319; A. Messing NS-23375), Novartis Research Foundation and the Swiss Foundation for Research on Neuromuscular Disease (to U. Mueller), and US PHS grant NS19090 (L.F. Reichardt). L.F. Reichardt is an investigator of the Howard Hughes Medical Institute.
Footnotes
Abbreviations used in this paper: CMD, congenital muscular dystrophy; DRG, dorsal root ganglion; E, embryonic day; LNGFR, low affinity nerve growth factor receptor; MBP, myelin basic protein; P, postnatal day; TUNEL, terminal deoxynucleotidyl transferase–mediated dUTP-biotin nick-end labelling.
References
- Akagi, K., V. Sandig, M. Vooijs, M. Van der Valk, M. Giovannini, M. Strauss, and A. Berns. 1997. Cre-mediated somatic site-specific recombination in mice. Nucleic Acids Res. 25:1766–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, D.J. 1997. Cellular and molecular biology of neural crest cell lineage determination. Trends Genet. 13:276–280. [DOI] [PubMed] [Google Scholar]
- Arroyo, E.J., J.R. Bermingham, Jr., M.G. Rosenfeld, and S.S. Scherer. 1998. Promyelinating Schwann cells express Tst-1/SCIP/Oct-6. J. Neurosci. 18:7891–7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, W.G., and M. Jenkison. 1973. Abnormalities of peripheral nerves in murine muscular dystrophy. J. Neurol. Sci. 18:227–247. [DOI] [PubMed] [Google Scholar]
- Bronnerfraser, M., M. Artinger, J. Muschler, and A.F. Horwitz. 1992. Developmentally regulated expression of α6 integrin in avian embryos. Development. 115:197–211. [DOI] [PubMed] [Google Scholar]
- Bunge, M.B. 1993. Schwann cell regulation of extracellular matrix biosynthesis and assembly. Peripheral Neuropathy. P.J. Dyck, P.K. Thomas, J. Griffin, P.A. Low, and J.F. Poduslos, editors. W.B. Saunders, Philadelphia. 299–316.
- Bunge, R.P., and M.B. Bunge. 1983. Interrelationship between Schwann cell function and extracellular matrix production. Trends Neurosci. 6:499–505. [Google Scholar]
- Chen, L.M., D. Bailey, and C. Fernandez-Valle. 2000. Association of β 1 integrin with focal adhesion kinase and paxillin in differentiating Schwann cells. J. Neurosci. 20:3776–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Arcangelis, A., and E. Georges-Labouesse. 2000. Integrin and ECM functions: roles in vertebrate development. Trends Genet. 16:389–395. [DOI] [PubMed] [Google Scholar]
- Einheber, S., T. Milner, F. Giancotti, and J. Salzer. 1993. Axonal regulation of Schwann cell integrin expression suggests a role for α6 ω4 in myelination. J. Cell Biol. 123:1223–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassler, R., and M. Meyer. 1995. Consequences of lack of β1 integrin gene expression in mice. Genes Dev. 9:1896–1908. [DOI] [PubMed] [Google Scholar]
- Fassler, R., M. Pfaff, J. Murphy, A.A. Noegel, S. Johansson, R. Timpl, and R. Albrecht. 1995. Lack of β1 integrin gene in embryonic stem cells affects morphology, adhesion, and migration but not integration into the inner cell mass of blastocysts. J. Cell Biol. 128:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri, M.L., S.S. Scherer, R. Nemni, J. Kamholz, H. Vogelbacker, M.O. Scott, N. Canal, V. Quaranta, and L. Wrabetz. 1994. β4 integrin expression in myelinating Schwann cells is polarized, developmentally regulated and axonally dependent. Development. 120:1287–1301. [DOI] [PubMed] [Google Scholar]
- Feltri, M.L., M. D'Antonio, S. Previtali, M. Fasolini, A. Messing, and L. Wrabetz. 1999. a. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann. NY Acad. Sci. 883:116–123. [PubMed] [Google Scholar]
- Feltri, M.L., M. D'Antonio, A. Quattrini, R. Numerato, M. Arona, S. Previtali, S.Y. Chiu, A. Messing, and L. Wrabetz. 1999. b. A novel P0 glycoprotein transgene activates expression of lacZ in myelin-forming Schwann cells. Eur. J. Neurosci. 11:1577–1586. [DOI] [PubMed] [Google Scholar]
- Fernandez-Valle, C., D. Gorman, A.M. Gomez, and M.B. Bunge. 1997. Actin plays a role in both changes in cell shape and gene-expression associated with Schwann cell myelination. J. Neurosci. 17:241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Valle, C., L. Gwynn, P.M. Wood, S. Carbonetto, and M.B. Bunge. 1994. Anti-β1 integrin antibody inhibits Schwann cell myelination. J. Neurobiol. 25:1207–1126. [DOI] [PubMed] [Google Scholar]
- Furuse, M., T. Hirase, M. Itoh, A. Nagafuchi, S. Yonemura, and S. Tsukita. 1993. Occludin: a novel integral membrane protein localizing at tight junctions. J. Cell Biol. 123:1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta, D., S. Blaess, M. Senften, A. Littlewood-Evans, C. Damsky, Z. Huang, P. Orban, R. Klein, J.C. Schittny, and U. Mueller. 2001. β1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 31:357–359. [DOI] [PubMed] [Google Scholar]
- Grinspan, J.B., M.A. Marchionni, M. Reeves, M. Coulaloglou, and S.S. Scherer. 1996. Axonal interactions regulate Schwann cell apoptosis in developing peripheral nerve: neuregulin receptors and the role of neuregulins. J. Neurosci. 16:6107–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack, H., and R.O. Hynes. 2001. Integrin receptors are required for cell survival and proliferation during development of the peripheral glial lineage. Dev. Biol. 233:38–55. [DOI] [PubMed] [Google Scholar]
- Helbling-Leclerc, A., X. Zhang, H. Topaloglu, C. Cruaud, F. Tesson, J. Weissenbach, F.M. Tome, K. Schwartz, M. Fardeau, K. Tryggvason, et al. 1995. Mutations in the laminin α2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet. 11:216–218. [DOI] [PubMed] [Google Scholar]
- Jaros, E., and W.G. Bradley. 1978. Development of the amyelinated lesion in the ventral root of the dystrophic mouse. Ultrastructural, quantitative and autoradiographic study. J. Neurol. Sci. 36:317–339. [DOI] [PubMed] [Google Scholar]
- Jessen, K.R., and R. Mirsky. 1999. Schwann cells and their precursors emerge as major regulators of nerve development. Trends Neurosci. 22:402–410. [DOI] [PubMed] [Google Scholar]
- Kuang, W., H. Xu, P.H. Vachon, L. Liu, F. Loechel, U.M. Wewer, and E. Engvall. 1998. Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J. Clin. Invest. 102:844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M.-J., A. Brennan, A. Blanchard, G. Zoidl, Z. Dong, A. Tabernero, C. Zoidl, M.A.R. Dent, K.R. Jessen, and R. Mirsky. 1997. Po is constitutively expressed in the rat neural crest and embryonic nerves and is negatively and positively regulated by axons to generate non-myelin-forming and myelin-forming Schwann cells, respectively. Mol. Cell Neurosci. 8:336–350. [DOI] [PubMed] [Google Scholar]
- Lefcort, F., K. Venstrom, J.A. McDonald, and L.F. Reichardt. 1992. Regulation of expression of fibronectin and its receptor, α5 β1, during development and regeneration of peripheral nerve. Development. 116:767–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid, R.E., E. Jaros, M.J. Cullen, and W.G. Bradley. 1975. Genetically determined defect of Schwann cell basement membrane in dystrophic mouse. Nature. 257:319–321. [DOI] [PubMed] [Google Scholar]
- Meyer, D., and C. Birchmeier. 1995. Multiple essential functions of neuregulin in development. Nature. 378:386–390. [DOI] [PubMed] [Google Scholar]
- Michelson, A., E. Russell, and P. Harman. 1955. Dystrophia muscolaris: a hereditary primary myopathy in the house mouse. Proc. Nat. Acad. Sci. USA. 41:1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner, R., M. Wilby, S. Nishimura, K. Boylen, G. Edwards, J. Fawcett, C. Streuli, R. Pytela, and C. French-Constant. 1997. Division of labor of Schwann cell integrins during migration on peripheral nerve extracellular matrix ligands. Dev. Biol. 185:215–228. [DOI] [PubMed] [Google Scholar]
- Parmantier, E., B. Lynn, D. Lawson, M. Turmaine, S.S. Namini, L. Chakrabarti, A.P. McMahon, K.R. Jessen, and R. Mirsky. 1999. Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron. 23:713–724. [DOI] [PubMed] [Google Scholar]
- Perkins, C.S., G.M. Bray, and A.J. Aguayo. 1981. Ongoing block of Schwann cell differentiation and deployment in dystrophic mouse spinal roots. Brain Res. 227:213–220. [DOI] [PubMed] [Google Scholar]
- Podratz, J.L., E. Rodriguez, and A.J. Windebank. 2001. Role of the extracellular matrix in myelination of peripheral nerve. Glia. 35:35–40. [DOI] [PubMed] [Google Scholar]
- Previtali, S.C., M.L. Feltri, J.J. Archelos, A. Quattrini, L. Wrabetz, and H. Hartung. 2001. Role of integrins in the peripheral nervous system. Prog. Neurobiol. 64:35–49. [DOI] [PubMed] [Google Scholar]
- Riethmacher, D., E. Sonnenberg-Riethmacher, V. Brinkmann, T. Yamaai, G.R. Lewin, and C. Birchmeier. 1997. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 389:725–730. [DOI] [PubMed] [Google Scholar]
- Schoenwaelder, S.M., and K. Burridge. 1999. Bidirectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 11:274–286. [DOI] [PubMed] [Google Scholar]
- Stephens, L.E., J.E. Sonne, M.L. Fitzgerald, and C.H. Damsky. 1993. Targeted deletion of β1 integrins in F9 embryonal carcinoma cells affects morphological differentiation but not tissue-specific gene expression. J. Cell Biol. 123:1607–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, L.E., A.E. Sutherland, I.V. Klimanskaya, A. Andrieux, J. Meneses, R.A. Pedersen, and C.H. Damsky. 1995. Deletion of β1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 9:1883–1895. [DOI] [PubMed] [Google Scholar]
- Stewart, H.J., L. Morgan, K.R. Jessen, and R. Mirsky. 1993. Changes in DNA synthesis rate in the Schwann cell lineage in vivo are correlated with the precursor—Schwann cell transition and myelination. Eur. J. Neurosci. 5:1136–1144. [DOI] [PubMed] [Google Scholar]
- Sunada, Y., S.M. Bernier, A. Utani, Y. Yamada, and K.P. Campbell. 1995. Identification of a novel mutant transcript of laminin α2 chain gene responsible for muscular dystrophy and dysmyelination in dy2J mice. Hum. Mol. Genet. 4:1055–1061. [DOI] [PubMed] [Google Scholar]
- Syroid, D.E., P.J. Maycox, M. Soilu-Hanninen, S. Petratos, T. Bucci, P. Burrola, S. Murray, S. Cheema, K.F. Lee, G. Lemke, and T.J. Kilpatrick. 2000. Induction of postnatal Schwann cell death by the low-affinity neurotrophin receptor in vitro and after axotomy. J. Neurosci. 20:5741–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syroid, D.E., P.R. Maycox, P.G. Burrola, N. Liu, D. Wen, K.F. Lee, G. Lemke, and T.J. Kilpatrick. 1996. Cell death in the Schwann cell lineage and its regulation by neuregulin. Proc. Natl. Acad. Sci. USA. 93:9229–9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima, T., H. Yasuda, M. Terada, S. Kogawa, K. Maeda, M. Haneda, A. Kashiwagi, and R. Kikkawa. 2001. Expression of Rho-family GTPases (Rac, cdc42, RhoA) and their association with p-21 activated kinase in adult rat peripheral nerve. J. Neurochem. 77:986–993. [DOI] [PubMed] [Google Scholar]
- Tomaselli, K.J., P. Doherty, C.J. Emmett, C.H. Damsky, F.S. Walsh, and L.F. Reichardt. 1993. Expression of β1 integrins in sensory neurons of the dorsal root ganglion and their functions in neurite outgrowth on two laminin isoforms. J. Neurosci. 13:4880–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voiculescu, O., P. Charnay, and S. Schneider-Maunoury. 2000. Expression pattern of a Krox-20/Cre knock-in allele in the developing hindbrain, bones, and peripheral nervous system. Genesis. 26:123–126. [DOI] [PubMed] [Google Scholar]
- Wolpowitz, D., T.B. Mason, P. Dietrich, M. Mendelsohn, D.A. Talmage, and L.W. Role. 2000. Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron. 25:79–91. [DOI] [PubMed] [Google Scholar]
- Webster, H. 1984. Development of peripheral nerve fibers. Peripheral Neuropathy. P.T. Dyck, E.H. Lambert, and R. Bunges, editors. W.B. Saunders, Philadelphia. 329–359.
- Witt, A., and S.T. Brady. 2000. Unwrapping new layers of complexity in axon/glial relationships. Glia. 29:112–117. [DOI] [PubMed] [Google Scholar]
- Wrabetz, L., M.L. Feltri, A. Quattrini, D. Imperiale, S. Previtali, M. D'Antonio, R. Martini, X. Yin, B.D. Trapp, L. Zhou, S.Y. Chiu, and A. Messing. 2000. P(0) glycoprotein overexpression causes congenital hypomyelination of peripheral nerves. J. Cell Biol. 148:1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H., X.R. Wu, U.M. Wewer, and E. Engvall. 1994. Murine muscular dystrophy caused by a mutation in the laminin α2 (Lama2) gene. Nat. Genet. 8:297–302. [DOI] [PubMed] [Google Scholar]
- Yamada, H., T. Shimizu, T. Tanaka, K.P. Campbell, and K. Matsumura. 1994. Dystroglycan is a binding protein of laminin and merosin in peripheral nerve. FEBS Lett. 352:49–53. [DOI] [PubMed] [Google Scholar]
- Yamauchi, Y., K. Abe, A. Mantani, Y. Hitoshi, M. Suzuki, F. Osuzu, S. Kuratani, and K. Yamamura. 1999. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev. Biol. 212:191–203. [DOI] [PubMed] [Google Scholar]
- Zorick, T.S., D.E. Syroid, E. Arroyo, S.S. Scherer, and G. Lemke. 1996. The transcription factors SCIP and Krox-20 mark distinct stages and cell fates in Schwann cell differentiation. Mol. Cell. Neurosci. 8:129–145. [DOI] [PubMed] [Google Scholar]