

Abstract
Cleavage of membrane-anchored heparin-binding EGF-like growth factor (proHB-EGF) via metalloprotease activation yields amino- and carboxy-terminal regions (HB-EGF and HB-EGF-C, respectively), with HB-EGF widely recognized as a key element of epidermal growth factor receptor transactivation in G protein–coupled receptor signaling. Here, we show a biological role of HB-EGF-C in cells. Subsequent to proteolytic cleavage of proHB-EGF, HB-EGF-C translocated from the plasma membrane into the nucleus. This translocation triggered nuclear export of the transcriptional repressor, promyelocytic leukemia zinc finger (PLZF), which we identify as an HB-EGF-C binding protein. Suppression of cyclin A and delayed entry of S-phase in cells expressing PLZF were reversed by the production of HB-EGF-C. These results indicate that released HB-EGF-C functions as an intracellular signal and coordinates cell cycle progression with HB-EGF.
Keywords: HB-EGF; ADAM12; shedding; PLZF; transcriptional repression
Introduction
Interreceptor cross-talk has received significant attention recently as an essential element in understanding the increasingly complex signaling networks identified within cells. Transactivation of the epidermal growth factor receptor (EGFR) has been shown to play a crucial role in the signaling by G protein–coupled receptors (GPCRs), cytokine receptors, receptor tyrosine kinases, and integrins to a variety of cellular responses (Hackel et al., 1999; Moghal and Sternberg, 1999). Transactivation of EGFR is mediated, at least in some cases, by the EGFR ligand heparin-binding EGF-like growth factor (HB-EGF), which is cleaved from its membrane-anchored form (proHB-EGF) in a process termed “ectodomain shedding” (Prenzel et al., 1999).
The proHB-EGF molecule is proteolytically cleaved by “a disintegrin and metalloprotease” (ADAM) 9, 12, 10, or 17 (Izumi et al., 1998; Asakura et al., 2002; Lemjabbar and Basbaum, 2002; Sunnarborg et al., 2002; Yan et al., 2002) to release a soluble form of HB-EGF. This cleavage can be stimulated by treating cells with various agents, including the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA), an activator of PKC (Goishi, et al., 1995). Recent analyses have shown that the processing of proHB-EGF by metalloproteases plays important roles in cutaneous wound healing (Tokumaru et al., 2000) and branching morphogenesis of the submandibular gland (Umeda et al., 2001). Furthermore, the cleavage of proHB-EGF is required for EGFR transactivation by GPCR signaling (Prenzel et al., 1999), which is involved in various biological processes such as cardiac hypertrophy (Asakura et al., 2002), cystic fibrosis (Lemjabbar and Basbaum, 2002), and the mitogenic effects of arachidonic acid metabolites (Chen et al., 2002; Cussac et al., 2002).
Although much has been learned about the functions of extracellular domains produced by ectodomain shedding, very little attention has been paid to the remnant cell-associated domains also created by the processing event. Here, we focus on a biological role played by the carboxy-terminal remnant (HB-EGF-C) produced in parallel with HB-EGF, and characterize it as a novel intracellular signaling molecule acquired posttranslationally. Using fluorescent protein–tagged proHB-EGF and an antibody recognizing the cytoplasmic region of proHB-EGF, we visualized the translocation of HB-EGF-C from the plasma membrane into the nucleus after ectodomain shedding of proHB-EGF. Yeast two-hybrid screening resulted in the cloning of an HB-EGF-C binding protein, promyelocytic leukemia zinc finger (PLZF), previously identified as a transcriptional repressor and a negative regulator of the cell cycle. The proteolytic release of HB-EGF-C via metalloprotease activation caused nuclear export of PLZF and reversal of cyclin A suppression and delayed entry of S-phase by PLZF in human fibrosarcoma HT1080 cells. Intracellular trafficking of endogenous HB-EGF-C into the nucleus and the subsequent nuclear export of PLZF after metalloprotease processing of proHB-EGF were also observed in human primary cultured keratinocytes. Thus, our present data provide new insights into the inter- and intracellular communication generated by proHB-EGF processing.
Results
Translocation of HB-EGF-C after TPA-inducible processing
To investigate the behavior of HB-EGF-C after metalloprotease processing of proHB-EGF, we constructed an expression vector encoding a protein in which YFP was fused to the carboxy terminus of proHB-EGF (HB-EGF–YFP). This vector was then transfected into human fibrosarcoma HT1080 cells. HB-EGF–YFP was localized at the plasma membrane of the transfected cells and its ectodomain was released by TPA-inducible proteolysis, as shown previously with wild-type proHB-EGF (unpublished data). In transfected cells treated with TPA for 60 min, fluorescent images revealed the translocation and accumulation of HB-EGF–YFP around the nucleus (Fig. 1, A and B). Time-lapse imaging showed that HB-EGF–YFP began to internalize and accumulate in the cytoplasm 15 min after TPA stimulation, and that a majority of the fluorescent protein had disappeared from the plasma membrane and was observed within the cytoplasm 30 min after TPA stimulation (Fig. 1 C).
Figure 1.
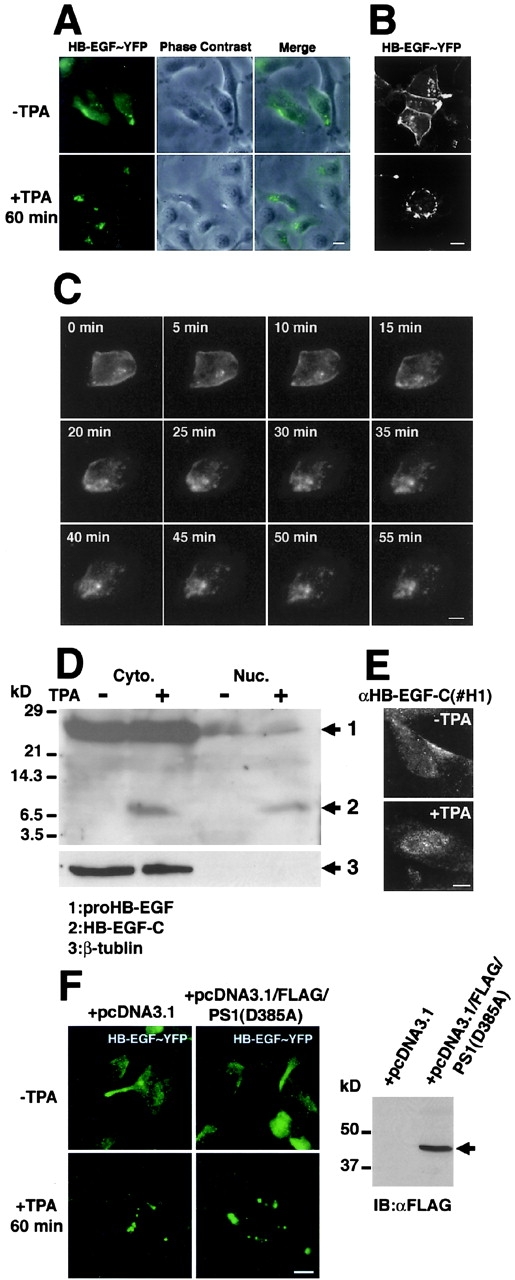
Translocation of HB-EGF-C after TPA-inducible processing. (A–C) HT1080 cells were transiently transfected with a plasmid encoding the fusion protein HB-EGF–YFP, in which YFP was fused to the carboxy terminus of proHB-EGF. After 24 h, the subcellular localization of the YFP sequence was visualized with fluorescent microscopy. (A) HB-EGF–YFP in unstimulated cells was observed at the plasma membrane. After stimulation with 100 nM TPA for 60 min, HB-EGF–YFP was absent from the plasma membrane, but detected in the cytoplasm and localized around the nucleus. (B) Localization of the fusion protein was confirmed by confocal microscopy. (C) Representative time-lapse images of HB-EGF–YFP transfected cells are shown every 5 min for up to 55 min after TPA treatment. Note that HB-EGF–YFP at the plasma membrane was translocated into the cytoplasm after TPA stimulation of the cells. (D and E) Accumulation of HB-EGF-C in the nucleus of HT1080 cells overexpressing proHB-EGF (HT1080/HB-EGF cells). (D) Cytoplasmic (Cyto.) and nuclear (Nuc.) fractions were prepared from HT1080/HB-EGF cells (2.0 × 107). After the incubation with or without TPA for 30 min, cells were lysed and immunoblotted using anti-HB-EGF-C antibody (#H1). In addition to a 25-kD band (proHB-EGF), the 6.7-kD band (HB-EGF-C) was detected in the cytoplasmic fraction when cells were treated with TPA. The 6.7-kD band was also detectable in the nuclear fraction after TPA stimulation. (E) Accumulation of HB-EGF-C after TPA treatment was also observed by immunofluo-rescence microscopy using the #H1 antibody. (F) Transient expression of the amino-terminally FLAG-tagged dominant-negative form of presenilin-1 (D385A) was not altered HB-EGF–YFP translocation after TPA treatment in HT1080 cells (left). Expression of the FLAG-tagged presenilin-1 mutant was confirmed by immunoblotting with anti-FLAG antibody (right, arrow). Bars, 10 μm.
Next, we examined the localization of HB-EGF-C by separating cytoplasmic and nuclear fractions from HT1080 cells overexpressing proHB-EGF (HT1080/HB-EGF cells). We used an antibody recognizing the cytoplasmic region of proHB-EGF (#H1) to detect proHB-EGF and HB-EGF-C. A large amount of proHB-EGF was accumulated in the cytoplasmic fraction, and the band of HB-EGF-C was not observed in cytoplasmic and nuclear fractions without the stimulation (Fig. 1 D). After TPA treatment for 30 min, the 6.7-kD band (the expected size of an HB-EGF-C with transmembrane and cytoplasmic domains of proHB-EGF) was detectable in both cytoplasmic and nuclear fractions of HB-EGF-C after TPA treatment (Fig. 1 E).
To test whether γ-secretase activity is involved with the proteolytic release of HB-EGF-C, we investigated the effect of a dominant-negative mutant of presenilin-1 (PS1 D385A), which reduces γ-secretase activity (Wolfe et al., 1999), on HB-EGF-C translocation after TPA treatment. Transient expression of amino-terminally FLAG-tagged dominant-negative PS1 in HT1080 cells was not altered the HB-EGF–YFP translocation induced by TPA treatment (Fig. 1 F).
Identification of PLZF as a proHB-EGF cytoplasmic domain–binding protein
The translocation of HB-EGF-C after proHB-EGF processing suggested that after proHB-EGF processing, HB-EGF-C could interact with cytoplasmic or nuclear proteins. To identify potential binding proteins of HB-EGF-C, we used yeast two-hybrid cloning and screened a human heart cDNA library using the cytoplasmic region of proHB-EGF (residues 185–208) as bait. Screening of 106 transformants yielded 16 positive clones. One of the clones (referred to as clone 3) encoded a carboxy-terminal sequence of PLZF protein (Fig. 2 A), a transcriptional repressor that is localized in the nucleus (Chen et al., 1993; Reid et al., 1995). Immunoprecipitation from COS cells expressing proHB-EGF and CFP-tagged PLZF (CFP-PLZF) revealed that CFP-PLZF was coimmunoprecipitated with the #H1 antibody once the cells were treated with TPA (Fig. 2 B).
Figure 2.

Identification of PLZF as an HB-EGF-C binding protein. (A) We used the yeast two-hybrid method and screened a human heart cDNA library with the cytoplasmic region of HB-EGF (HB-EGF-Cyto; residues 185–208) as bait. One of the positive clones (clone 3) was identified as encoding a portion of PLZF. (B) COS cells were transiently transfected with a proHB-EGF expression vector and a CFP-PLZF expression vector encoding a fusion protein in which CFP was fused to the amino terminus of PLZF, stimulated for 60 min with TPA, and lysed. Expression of CFP-PLZF was confirmed by immunoblotting of the whole lysate with anti-GFP antibody (lane 1). The lysates were immunoprecipitated with anti-HB-EGF-C antibody (#H1), and the precipitates were immunoblotted with anti-GFP antibody (lanes 2 and 3). Note that the interaction between PLZF and HB-EGF was observed when cells were stimulated with TPA. (C–F) GST pull-down assay. Cell lysates containing the FLAG-tagged PLZF derivatives were incubated with GST (1) or GST–HB-EGF-C (2) beads, and bound proteins were detected by immunoblotting using an anti-FLAG antibody (right). Expression of the derivatives was also confirmed by immunoblotting using an anti-FLAG antibody (left). (C) Schematic diagrams of FLAG-tagged PLZF derivatives. Nine zinc finger motifs in the PLZF protein are numbered from first to ninth. Binding properties of PLZF derivatives to GST–HB-EGF-C is summarized in the right lane of each structure. The binding properties are based on the estimation from the intensity of bands as compared with the control band and are indicated as ++ (>50%), + (50–10%), and − (<10%). (D) Full-length PLZF and the fragment containing nine zinc finger motifs (Zn Finger) were bound to GST–HB-EGF-C, but not to GST alone. (E) The fragments consisting of zinc finger motifs 4∼9 (Zn4∼9) and 5∼8 (Zn5∼8) were bound to GST–HB-EGF-C stronger than the fragment with zinc finger motifs 1∼6 (Zn1∼6). The fragment consisting of zinc finger motifs 6∼7 (Zn6∼7) was not efficient for the interaction. (F) Deletion mutant (ΔZn6∼7) showed that the region with Zn6∼7 was essential for HB-EGF-C–PLZF interaction.
To determine the region of PLZF that interacts with HB-EGF-C, we performed a GST pull-down assay. Various FLAG-tagged PLZF derivatives (Fig. 2 C) were incubated with glutathione Sepharose beads containing recombinant HB-EGF-C fused to GST (GST–HB-EGF-C). In agreement with the data obtained from the yeast study, the zinc finger region containing nine zinc finger motifs (the carboxy terminus of PLZF protein) interacted with GST–HB-EGF-C (Fig. 2 D). We further characterized the zinc finger region of PLZF required for interaction with HB-EGF-C. GST–HB-EGF-C pulled down zinc finger 5∼8 motifs (Zn5∼8) efficiently and zinc finger 1∼6 motifs (Zn1∼6) partially, but not the zinc finger 6∼7 motifs (Zn6∼7; Fig. 2 E). However, deletion of Zn6∼7 abrogated the binding of PLZF to GST–HB-EGF-C (Fig. 2 F). These data suggest that the Zn6∼7 region is essential and Zn5∼8 is sufficient for the PLZF-HB-EGF-C interaction.
Nuclear export of PLZF triggered by TPA-inducible ectodomain shedding of proHB-EGF
Next, we examined the subcellular localization of PLZF by using the expression vector encoding CFP-PLZF. This expression vector was transfected into four types of cell lines as follows: HT1080 cells; a stable transfectant of HT1080 expressing proHB-EGF (HT1080/HB-EGF); a stable double transfectant of HT1080 cells expressing proHB-EGF and a metalloprotease domain–deleted mutant of ADAM12 (HT1080/ΔMP-ADAM12/HB-EGF); and a stable transfectant of HT1080 expressing an uncleavable mutant (L148G; Hirata et al., 2001) of proHB-EGF (HT1080/HB-EGF-UC). Endogenous HB-EGF expression was very low in parental HT1080 cells (unpublished data).
CFP-PLZF was predominantly localized in the nucleus in HT1080 cells and in the three transfectants. TPA treatment did not alter the subcellular localization of CFP-PLZF in HT1080 cells. In contrast, TPA treatment for 60 min distributed CFP-PLZF in the entire cytoplasm of HT1080/HB-EGF cells (Fig. 3 A). A previous experiment had revealed that ADAM12 can mediate HB-EGF shedding, and that expression of a dominant-negative (metalloprotease domain–deleted mutant) form of ADAM12 inhibited proHB-EGF processing in HT1080 cells (Asakura et al., 2002). In HT1080/ΔMP-ADAM12/HB-EGF cells, the export of CFP-PLZF from the nucleus to the cytoplasm was not observed after TPA stimulation. Similarly, HT1080/HB-EGF-UC cells did not show the nuclear export of CFP-PLZF despite TPA treatment (Fig. 3 A). Quantitative analyses (see Materials and methods) verified that the number of cells with nuclear-localized CFP-PLZF was reduced by the TPA treatment in HT1080/HB-EGF cells, but not in parental HT1080 cells, HT1080/ΔMP-ADAM12/HB-EGF cells, or HT1080/HB-EGF-UC cells (Fig. 3 C).
Figure 3.

Processing of proHB-EGF by ADAM12 causes nuclear export of PLZF. Subcellular localization of CFP-PLZF fusion protein in HT1080 cells and its variants. The CFP-PLZF expression vector was transiently transfected into four types of cells: HT1080 cells (HT1080); proHB-EGF–overexpressing HT1080 cells (HT1080/HB-EGF); proHB-EGF– and metalloprotease domain–deleted mutant of ADAM12-overexpressing HT1080 cells (HT1080/ΔMP-ADAM12/HB-EGF); and uncleavable-type proHB-EGF–overexpressing HT1080 cells (HT1080/HB-EGF-UC). (A and C) 24 h after the transfection, CFP-PLZF was predominantly localized at the nucleus in these four cell types. In HT1080/HB-EGF cells (but not the other types of cells), CFP-PLZF was distributed in the entire cytoplasm after treatment of the cells with 100 nM TPA for 60 min (B and D). A metalloprotease inhibitor KB-R7785 (10 μM), but not an EGFR-neutralizing antibody (10 μg/ml), inhibited nuclear export of CFP-PLZF in HT1080/HB-EGF cells in response to treatment with TPA for 60 min. Note that nuclear export of CFP-PLZF was dependent on the processing of HB-EGF, but that EGFR signaling was not involved in this process. 10 ng/ml leptomycin B (LMB), a specific inhibitor of CRM1-dependent nuclear export, also inhibited nuclear export of CFP-PLZF after TPA treatment. (E and F) The expression vectors encoding CFP-PLZF (CFP-PLZF) or Zn6∼7 deletion mutant of CFP-PLZF (CFP-PLZFΔZn6∼7) were transiently transfected into HT1080/HB-EGF cells. After 24 h, changes in subcellular localization of CFP-tagged proteins after TPA treatment were examined. Note that the export of CFP-PLZFΔZn6∼7 was suppressed in HT1080/HB-EGF cells after TPA stimulation. Bars, 10 μm.
Proteolytic cleavage of proHB-EGF induced by TPA yields at least two fragments, HB-EGF and HB-EGF-C. Therefore, we next investigated whether EGFR activation by HB-EGF was involved in triggering the nuclear export of CFP-PLZF in HT1080/HB-EGF cells. HT1080/HB-EGF cells transfected with the plasmid encoding CFP-PLZF were preincubated with 10 μg/ml of EGFR-neutralizing antibody (an amount of antibody sufficient to inhibit tyrosine phosphorylation of EGFR in HT1080/HB-EGF cells after TPA addition; unpublished data). Nuclear export of CFP-PLZF by TPA stimulation was still observed in the presence of the antibody (Fig. 3, B and D). On the other hand, by pretreatment with 10 μM KB-R7785, a metalloprotease inhibitor that blocks proHB-EGF processing by ADAM12 (Asakura et al., 2002), the frequency of the nuclear export of CFP-PLZF after TPA stimulation of HT1080/HB-EGF cells was reduced (Fig. 3, B and D). The export of CFP-PLZF was also inhibited by preincubation of the cells with 10 ng/ml leptomycin B (LMB), a specific inhibitor of CRM1 (designated exportin 1)-dependent nuclear export (Kudo et al., 1999; Fig. 3, B and D). These results indicate that generation of HB-EGF-C by proHB-EGF processing is required for the nuclear export of PLZF.
The GST pull-down analysis indicated that deletion of Zn6∼7 of PLZF abrogated the binding to HB-EGF-C. Therefore, we investigated the nuclear export of CFP-PLZF with deletion of Zn6∼7 (CFPPLZFΔZn6∼7) in HT1080/HB-EGF cells. The frequency of the CFP-PLZFΔZn6∼7 export much decreased as compared with CFP-PLZF after TPA stimulation (Fig. 3, E and F). This result indicates that binding of PLZF to HB-EGF-C is essential for the nuclear export of PLZF.
Translocation and interaction of HB-EGF-C with PLZF precedes nuclear export of PLZF
Next, we performed the simultaneous visualization of proHB-EGF processing and PLZF transport. Processing of HB-EGF–YFP did not promote nuclear export of PLZF (unpublished data), possibly due to the fused YFP interfering with the interaction between HB-EGF-C and PLZF. Therefore, HB-EGF–YFP and CFP-PLZF expression vectors were cotransfected into HT1080/HB-EGF or HT1080/ΔMP-ADAM12/HB-EGF cells that stably expressed wild-type proHB-EGF. Images were collected every 15 min up to 90 min after TPA treatment. In HT1080/HB-EGF cells, internalization of HB-EGF–YFP was observed 30 min after the treatment, but nuclear export of CFP-PLZF did not occur until 45 min after TPA stimulation (Fig. 4 A). In HT1080/ΔMP-ADAM12/HB-EGF cells, subcellular localization of HB-EGF–YFP and CFP-PLZF were not changed despite TPA treatment (Fig. 4 B).
Figure 4.

Nuclear export of PLZF follows internalization of HB-EGF-C. (A and B) Time-lapse imaging of CFP-PLZF and HB-EGF–YFP in living cells. Cells cotransfected with CFP-PLZF and HB-EGF–YFP expression vectors were cultured for 24 h before observation, and then the culture medium was replaced with medium containing 100 nM TPA. Shown are representative serial images of transfected HT1080/HB-EGF (A) and HT1080/ΔMP-ADAM12/HB-EGF (B) cells taken every 15 min up to 90 min after TPA stimulation. Note that translocation of HB-EGF–YFP preceded nuclear export of CFP-PLZF in HT1080/HB-EGF cells. Changes in subcellular localization of the CFP and YFP fusion proteins were not observed after TPA treatment of HT1080/ΔMP-ADAM12/HB-EGF cells. (C) Time course–dependent changes in interaction of HB-EGF-C with CFP-PLZF. The HB-EGF-C–PLZF interaction was investigated by immunoprecipitation using an anti-HB-EGF-C antibody in HT1080/HB-EGF and HT1080/ΔMP-ADAM12/HB-EGF cells. The band of CFP-PLZF coimmunoprecipitated with HB-EGF-C (top) was evident at 15 min and the later after TPA treatment in HT1080/HB-EGF but not HT1080/ΔMP-ADAM12/HB-EGF cells, despite same expression of CFP-PLZF in cells (bottom). Bars, 10 μm.
Time course–dependent changes in interactions of HB-EGF-C with CFP-PLZF were investigated in these cells by immunoprecipitation experiments. Anti-HB-EGF-C antibody #H1 clearly coimmunoprecipitated CFP-PLZF at 15 min and the later with maximal binding at 30 and 75 min after TPA stimulation in HT1080/HB-EGF cells, but not in HT1080/ΔMP-ADAM12/HB-EGF cells (Fig. 4 C). These results, together with time-lapse images, indicate that internalized HB-EGF-C interacted with nuclear PLZF.
The proHB-EGF cytoplasmic region is required for PLZF transport
The data presented in Fig. 3 indicated that proteolytic release of HB-EGF-C by proHB-EGF processing could trigger the nuclear export of PLZF. Therefore, we designed three types of cytoplasmic deletion mutants of proHB-EGF (HBΔC10, HBΔC19, and HBΔC22; Fig. 5 A), constructed their expression vectors, and established stable transfectants of HT1080 cells with each of the vectors. All YFP-tagged deletion mutants of proHB-EGF were also localized at the plasma membrane and internalized into the cytoplasm by TPA treatment (unpublished data). HT1080/HBΔC10 cells (as well as HT1080/HB-EGF cells) showed TPA-responsible nuclear export of PLZF. However, in HT1080/HBΔC19 and HT1080/HBΔC22 cells, nuclear export of PLZF did not occur despite TPA stimulation (Fig. 5, B and C).
Figure 5.

Cytoplasmic region of proHB-EGF is required for PLZF export after TPA stimulation. (A) Structures of cytoplasmic deletion mutants of proHB-EGF. These mutants, HBΔC10, HBΔC19, and HBΔC22, were truncated by 10, 19, and 22 amino acids, respectively, from the carboxy terminus of proHB-EGF. (B and C) Subcellular localization of CFP-PLZF in HT1080 cells stably expressing proHB-EGF and its cytoplasmic deletion mutants. CFP-PLZF was predominantly localized in the nucleus in these stable transfectants. In HT1080/HB-EGF and HT1080/HBΔC10 cells, TPA treatment distributed CFP-PLZF to the entire cytoplasm. However, in HT1080/HBΔC19 and HT1080/HBΔC22 cells, the export of CFP-PLZF from nucleus was not observed after TPA stimulation. Bar, 10 μm.
Increased expression of cyclin A and promotion of S-phase progression associated with nuclear export of PLZF
It has been reported that PLZF is a transcriptional repressor of cyclin A and inhibits cell growth (Shaknovich et al., 1998; Yeyati et al., 1999). HT1080/HB-EGF cells transfected with an expression vector for FLAG-tagged PLZF (FLAG-PLZF) were cultured for 48 h in the medium containing 3% serum, and then the culture medium was replaced with fresh medium with and without 100 nM TPA. After 8 h, expression of both FLAG-PLZF and cyclin A was examined by immunofluorescence microscopy. The intensity of cyclin A immunofluorescence in the transfected cells expressing FLAG-PLZF (Fig. 6 A, arrows) was much less than that in untransfected cells (Fig. 6 A, arrowheads). TPA treatment distributed FLAG-PLZF into the entire cytoplasm and up-regulated the immunofluorescence intensity of cyclin A in the transfected cells (Fig. 6 A, arrows) to the same level as that in the untransfected cells (Fig. 6 A; arrowheads). The range of relative fluorescence intensity of cyclin A in TPA-treated cells was narrower than that in nontreated cells, and the reduced expression of cyclin A associated with nuclear-localized FLGA-PLZF was not observed (Fig. 6 B). However, in HT1080, HT1080/ΔMP-ADAM12/HB-EGF, and HT1080/HB-EGF-UC cells, FLAG-PLZF was localized in the nucleus and suppressed cyclin A expression in the absence or presence of TPA (unpublished data).
Figure 6.

Processing of proHB-EGF reverses PLZF-dependent cyclin A suppression and delayed entry of S-phase. (A and B) Expression analysis of FLAG-PLZF and cyclin A by immunofluorescence microscopy. An expression vector encoding FLAG-tagged PLZF (FLAG-PLZF) was transiently transfected into HT1080/HB-EGF cells, and the expression of FLAG-PLZF and cyclin A was detected with anti-FLAG and anti-cyclin A antibodies, respectively. (A) In transfected cells (arrows), FLAG-PLZF was localized at the nucleus and suppressed cyclin A expression in the absence of TPA treatment. In the presence of TPA, FLAG-PLZF was distributed into the entire cytoplasm, and the expression level of cyclin A in the transfected cells (arrows) was the same as that in the untransfected cells (arrowheads). (B) Relative fluorescence intensity of cyclin A and FLAG-PLZF was measured at the nucleus of the transfected cells. (C and D) Cell cycle analysis by DNA staining and cyclin A immunoblotting. HT1080 cells and its variants infected with PLZF or LacZ (control) expression adenoviruses were synchronized in G0/G1-phase by serum deprivation for 36 h and were restimulated into cell cycle with 10% serum. At the indicated times, cells were collected and analyzed for cell cycle phase and cyclin A expression. (C) Exogenous expression of PLZF by adenovirus induced the delay of S-phase entry after the serum stimulation in HT1080, but not in HT1080/HB-EGF cells. Cells were stained with propidium iodide and analyzed by FACS® to determine their DNA content and cell cycle phase at 0 and 8 h after addition of 10% serum. Cell number is plotted on the y axes, and DNA content is plotted on the x axes. (D) The delay of S-phase entry and suppression of cyclin A expression were reversed by proHB-EGF processing. The percentage of cells in S-phase is plotted versus time after serum stimulation. Whole-cell lysates from HT1080 and HT1080/HB-EGF cells (105) harvested at each point were immunoblotted with anti-cyclin A antibody. The expression of β-actin was examined as a control (not depicted). Bar, 10 μm.
The effect of PLZF on cell cycle was investigated in HT1080 and HT1080/HB-EGF cells. To control cell cycle progression and proHB-EGF processing simultaneously, we synchronized the cells infected with the PLZF or LacZ expression adenoviruses in G0/G1-phase by serum deprivation, and then stimulated the cells back into cycle by addition of 10% serum. proHB-EGF processing is induced by serum-containing medium without any particular stimuli (Goishi et al., 1995; Hirata et al., 2001), and a large amount of released HB-EGF was detected in the conditioned medium of HT1080/HB-EGF cells (unpublished data). After the serum stimulation, the cells were collected every 4 h up to 24 h and analyzed for DNA content and cyclin A expression by flow cytometry and immunoblotting, respectively. Entrance into S-phase was delayed in the PLZF-expressing HT1080 cells compared with the control LacZ-expressing HT1080 cells (Fig. 6, C and D). Consistent with the delay of S-phase entry, increased expression of cyclin A after the stimulation was suppressed in HT1080 cells expressing PLZF (Fig. 6 D). However, S-phase entry and cyclin A regulation were reversed in HT1080/HB-EGF cells expressing PLZF (Fig. 6, C and D).
Intracellular localization of HB-EGF-C in human keratinocyte cell line HaCaT and primary cultured cells
We examined human keratinocyte cell line HaCaT and primary cultured cells to determine the localization of endogenous HB-EGF-C. TPA treatment of HaCaT cells (Fig. 7 A) as well as primary cultured cells (unpublished data) resulted in the production of an ∼6.7-kD band, the expected size of an HB-EGF-C retaining both the transmembrane and cytoplasmic domains of proHB-EGF. HB-EGF-C was in the Golgi apparatus after 30 min of TPA treatment, as determined by its colocalization with the Golgi protein p115 (Nelson et al., 1998; Fig. 7 B). HB-EGF-C accumulated and partially colocalized with FLAG-tagged PLZF at the nucleus of HaCaT cells after 60 min of TPA treatment (Fig. 7 C).
Figure 7.

Characterization of endogenous HB-EGF-C in HaCaT cells. (A) Characterization of endogenous HB-EGF-C in untransfected HaCaT cells. 108 HaCaT cells that had been incubated in the presence or absence of TPA were lysed and immunoprecipitated with an HB-EGF-C antibody #H1, and the precipitates were immunoblotted with antibodies #H1 or #H6 (recognizing HB-EGF ectodomain). Both antibodies detected the 20–30-kD heterogenous bands of proHB-EGF. A 6.7-kD band was detected with the #H1 antibody, but not with #H6, and the density of this band was enhanced by TPA treatment. The apparent size of this band was consistent with the putative size of a form of HB-EGF-C containing the transmembrane and cytoplasmic domains of proHB-EGF. (B) Staining with the #H1 antibody showed that the cell surface HB-EGF–associated fluorescence was reduced once untransfected HaCaT cells were treated with TPA for 30 min. The perinuclear accumulation of HB-EGF-C, more evident after TPA treatment for 30 min, was colocalized with p115, a marker protein of the Golgi apparatus. (C) The plasmid encoding FLAG-PLZF was transiently transfected into HaCaT cells, and after 48 h, the localization of FLAG-PLZF and endogenous HB-EGF-C was visualized with immunofluorescent microscopy. HB-EGF-C accumulated and partially colocalized with FLAG-PLZF at the nucleus of HaCaT cells after 30 min of TPA treatment. Bars, 10 μm.
Gene expression of PLZF was detected by RT-PCR in human primary keratinocytes, but not in HT1080 or HaCaT cells (unpublished data). Immunostaining of endogenous HB-EGF-C, PLZF, and cyclin A in primary cultured keratinocytes revealed their complemental localization in nucleus, cytoplasm, and plasma membrane under different conditions (Fig. 8 A). Keratinocytes in culture medium supplemented with bovine brain extracts showed heterogeneous staining of HB-EGF-C, PLZF, and cyclin A. However, TPA treatment clearly localized HB-EGF-C in nuclei and PLZF in the cytoplasm (Fig. 8, A and B). On the other hand, KB-R7785 treatment mainly localized HB-EGF at the plasma membrane and PLZF in the nuclei despite the addition of TPA (Fig. 8 A). Cyclin A was detected in the nuclei of most cells in response to the nuclear export of PLZF (Fig. 8 A). Increased expression of cyclin A in cultured keratinocytes after proHB-EGF processing was also confirmed by immunoblotting (Fig. 8 C). Further, treatment with angiotensin II, which causes GPCR-mediated EGFR transactivation through proHB-EGF processing in keratinocytes (unpublished data), also triggered the localization of HB-EGF-C in nuclei and PLZF in the cytoplasm, which was canceled effectively by the addition of KB-R7785 (Fig. 8 D).
Figure 8.
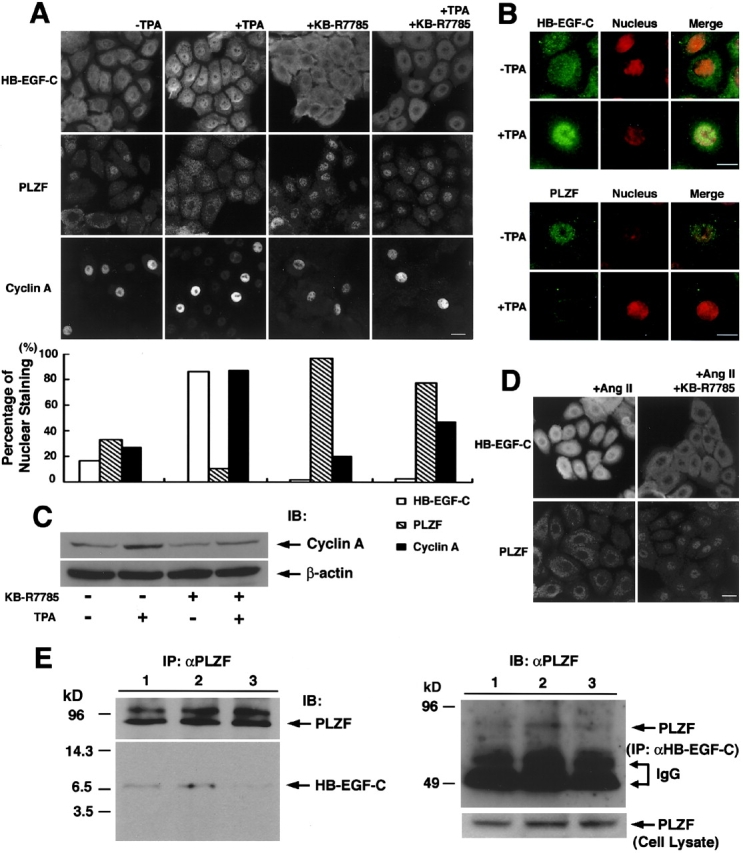
Interaction and traffic of endogenous HB-EGF-C and PLZF in human primary cultured keratinocytes. (A) Detection and localization of endogenous HB-EGF-C, PLZF, and cyclin A in primary human cultured keratinocytes by immunofluorescence microscopy. Keratinocytes were cultured in medium supplemented with bovine brain extracts and stained with antibodies against HB-EGF-C, PLZF, and cyclin A. Heterogeneous staining of HB-EGF-C, PLZF, and cyclin A was observed under the normal culture conditions. However, TPA treatment clearly localized HB-EGF-C in nuclei and PLZF in the cytoplasm, and up-regulated the expression of cyclin A in nuclei. KB-R7785 treatment resulted in HB-EGF-C staining at the plasma membrane and PLZF staining in nuclei despite the addition of TPA. (B) Nuclear accumulation of HB-EGF-C and export of PLZF after TPA stimulation. To facilitate the changes in subcellular localization of HB-EGF-C and PLZF after TPA treatment, we performed a nuclear staining using Hoechst 33258 in combination with immunostaining using anti-HB-EGF-C or PLZF. (C) Analysis of cyclin A expression in primary human cultured keratinocytes by immunoblotting. Consistent with immunostaining data, increased expression of cyclin A was observed in the keratinocytes treated with TPA. KB-R7785 suppressed cyclin A expression despite TPA stimulation. The expression of β-actin was examined as a control. (D) Staining of HB-EGF-C in nuclei and PLZF in the cytoplasm was also observed after angiotensin II treatment of the cells for 6 h, and this effect was reversed by the addition of KB-R7785 to the medium. (E) Interaction of HB-EGF-C and PLZF in human primary cultured keratinocytes. Cell lysates (107 cells) were immunoprecipitated with anti-PLZF antibody and immunoblotted with anti-PLZF (top left) and HB-EGF-C #H1 (bottom left) antibodies. Coimmunoprecipitated HB-EGF-C was slightly detectable in lysates from cells in normal culture conditions (lane 1), but the intensity of this band was increased by TPA treatment (lane 2). Addition of KB-R7785 (lane 3) resulted in the loss of the HB-EGF-C from the PLZF immunoprecipitates. Anti-HB-EGF-C antibody #H1 also coimmunoprecipitated PLZF (top right; each lane indicates the same condition as in left panel). PLZF was equally present in each cell lysate, as shown by Western blotting (bottom right). Bars, 10 μm.
Immunoprecipitation of PLZF in primary cultured keratinocytes resulted in the coprecipitation of HB-EGF-C, and the amount of the coprecipitated HB-EGF-C increased when the cells were incubated with TPA before lysis (Fig. 8 E). Thus, the interaction between PLZF and HB-EGF-C also occurs in primary cells. Similarly, anti-HB-EGF-C antibody #H1 also coimmunoprecipitated PLZF in the same cells (Fig. 8 E).
PLZF-HB-EGF-C interaction in vivo
We also examined the interaction of PLZF and HB-EGF-C in vivo using a TPA-treated mouse skin model of keratinocyte hyperplasia. TPA treatment of mouse skin tissue for 24 and 48 h produced keratinocyte hyperplasia as reported previously (Hawighorst et al., 2001; Fig. 9 A). Immunoprecipitation of PLZF in the homogenates of TPA-treated and control mouse skin tissues resulted in the coprecipitation of a 6.7-kD band recognized by anti-HB-EGF-C antibody #H1 in TPA-treated tissue homogenates alone (Fig. 9 B), suggesting that the PLZF-HB-EGF-C interaction occurs in vivo.
Figure 9.

Involvement of the HB-EGF-C–PLZF interaction in epidermal hyperplasia. In vivo interaction between HB-EGF-C and PLZF in TPA-treated mouse skin hyperplasia. (A) Mouse skin was treated with TPA, and keratinocyte hyperplasia was successively observed for 48 h by HE staining. (B) The homogenates of TPA-treated and control mouse skin tissues were immunoprecipitated with anti-PLZF antibody and immunoblotted with anti-PLZF (top) or HB-EGF-C #H1 (bottom) antibodies. A 6.7-kD band identified as HB-EGF-C was detected in TPA-treated tissue homogenates, but not in control skin tissues. Bar, 10 μm.
Discussion
The biological functions of the EGFR ligand HB-EGF, shed by proteolytic cleavage from the amino terminus of the transmembrane proHB-EGF, have been well studied. In contrast, roles of the carboxy-terminal fragment (HB-EGF-C) produced by proHB-EGF processing have not been previously investigated. Our present work has three major findings concerning HB-EGF-C. First, HB-EGF-C generated by ectodomain shedding of proHB-EGF is translocated from the plasma membrane to the nucleus. Second, the proteolytic release of HB-EGF-C results in nuclear export of the transcriptional repressor PLZF. Third, cell cycle regulation by nuclear PLZF is reversed by proHB-EGF processing.
Using the technology of fluorescent proteins, we first demonstrated that HB-EGF-C was internalized into the cytoplasm after ectodomain shedding. Immunoblotting of the nuclear fraction and immunostaining using the antibody recognizing the cytoplasmic region of proHB-EGF (#H1) revealed the accumulation of endogenous HB-EGF-C in the nucleus after the processing. The 6.7-kD band of HB-EGF-C and no effect of dominant-negative presenilin-1 on HB-EGF-C translocation indicate that HB-EGF-C has the proHB-EGF transmembrane domain and is not further processed by γ-secretase. The colocalization of HB-EGF-C with p115, a marker protein of the Golgi apparatus, indicates that it is internalized by vesicular trafficking. Although the detailed transport mechanism of HB-EGF-C containing transmembrane domain remains unclear, recent analyses of nuclear localization of receptor tyrosine kinases raise the possibility that the Golgi–ER pathway is a route to the nucleus of type I transmembrane molecules (Carpenter, 2003). As shown in Fig. 1, TPA treatment localized HB-EGF–YFP around the nucleus, but not in the nucleus. HB-EGF–YFP did not promote the nuclear export of PLZF after TPA treatment (unpublished data). Therefore, we speculate that the YFP moiety fused to the carboxy terminus of HB-EGF-C might aggregate and fail to enter the nucleus, which abrogates the HB-EGF-C–PLZF interaction.
Recently, it has been reported that neuregulin-1 is cleaved at the transmembrane domain and the released intracellular domain (Nrg-1-ICD) enters the nucleus to repress expression of several regulators of apoptosis (Bao et al., 2003). Bao et al. (2003) also mentioned that Nrg-1-ICD forms the complex with a second zinc finger–containing protein related to PLZF. Although the machinery of carboxy-terminal signaling of proHB-EGF and neuregulin-1 seems to be different, these results, together with our present data, suggest that the carboxy-terminal fragments of the EGF family precursors are functional molecules that control gene expression by regulating transcription factors in the nucleus.
It is apparent that nuclear accumulation of HB-EGF-C, nuclear export of PLZF, and interaction between HB-EGF-C and PLZF are mutually exclusive. Although the machinery of this signaling is still unclear, one possible mechanism suggested by time course–dependent changes in the interaction and localization of HB-EGF-C and PLZF is as follows: (1) HB-EGF-C is translocated into the nucleus after proHB-EGF processing; (2) HB-EGF-C associates with nuclear PLZF; and (3) PLZF is exported from the nucleus in a CRM1-dependent manner (Fig. 10 A).
Figure 10.

Schematic models of the roles of HB-EGF-C. (A) An intracellular signaling mediated by HB-EGF-C produced by proHB-EGF processing. (B) Coordination of cell cycle progression by shed HB-EGF and HB-EGF-C.
The cleavage of proHB-EGF is required for EGFR transactivation in response to GPCR signaling (Prenzel et al., 1999, Asakura et al., 2002, Chen et al., 2002; Cussac et al., 2002; Lemjabbar and Basbaum, 2002), suggesting that posttranslational processing of proHB-EGF is involved in a variety of biological processes. In the present report, we show that nuclear export of PLZF also results from angiotensin II stimulation in human primary cultured keratinocytes. This result suggests that proHB-EGF processing by the GPCR signaling cascade leads to the nuclear export of PLZF. PLZF is expressed in a large number of tissues, and perinatal up-regulation of this factor is observed in the kidney, liver, and heart (Cook et al., 1995; Reid et al., 1995). PLZF produces transcriptional repression through recruitment of a repressor complex that contains N-CoR, SMRT, Sin3a, and histone deacetylase (Hong et al., 1997; David et al., 1998; Grignani et al., 1998; Guidez et al., 1998; He et al., 1998; Lin et al., 1998). The study of PLZF knockout mice indicates that PLZF represses Hox gene expression though chromatin remodeling to regulate the patterning of limb and axial skeleton, and is involved in apoptosis and cell proliferation during limb development (Barna et al., 2000, 2002). Thus, these findings suggest potential roles for HB-EGF-C mediated nuclear export of PLZF and the subsequent effects on regulation of cell proliferation and Hox gene expression in various signaling cascades during the development and maintenance of adult tissues.
It has been reported that PLZF is a transcriptional repressor of cyclin A and suppresses cell growth by inhibiting entry or progression of S-phase in the cell cycle (Shaknovich et al., 1998; Yeyati et al., 1999). Gene expression control by transcriptional regulators occurs in the nucleus, and nuclear export of these factors results in loss of the regulation. The present data show that HB-EGF-C generated by ectodomain shedding of proHB-EGF causes nuclear export of PLZF, increases the expression of cyclin A, and promotes S-phase entry. Furthermore, the interaction of HB-EGF-C and PLZF occurs in the TPA-treated mouse skin model of keratinocyte hyperplasia. On the other hand, proHB-EGF processing also generates HB-EGF, a soluble ligand of EGFR. It is well known that HB-EGF activates EGFR signaling and promotes G1-phase progression in the cell cycle by regulating the expression of cyclin D via the Ras-MAPK signaling cascade (Hackel et al., 1999; Prober and Edgar, 2001). Therefore, our current paper suggests that proHB-EGF has two functional domains affecting mitogenic signaling. Posttranslational processing of proHB-EGF by metalloprotease activation produces intercellular and intracellular signaling molecules simultaneously (Fig. 10 A). The coordination of the resulting dual mitogenic signals may be important for cell cycle progression in various signaling cascades (Fig. 10 B).
Materials and methods
Plasmid construction
A plasmid for recombinant expression of YFP-tagged proHB-EGF was generated by subcloning the human HB-EGF cDNA with the stop codon deleted into pEYFP-N1 (CLONTECH Laboratories, Inc.). A plasmid for recombinant expression of CFP-tagged PLZF was generated by subcloning the human PLZF cDNA into pECFP-C1 (CLONTECH Laboratories, Inc.). DNA fragments encoding the deleted cytoplasmic region of proHB-EGF with or without stop codon were generated by PCR and substituted for the corresponding region in pEYFP-N1-HB-EGF. A plasmid encoding FLAG-tagged PLZF was generated by subcloning the FLAG sequence and the human PLZF cDNA into pcDNA3.1 (Invitrogen). pcDNA3.1/FLAG/PS1(D385A) was a gift from Dr. Okochi (Osaka University, Osaka, Japan). Plasmids for recombinant expression of FLAG-tagged PLZF derivatives were generated by subcloning the FLAG and various PLZF derivative sequences made by PCR into pME18S. A plasmid for recombinant expression of GST-fused HB-EGF-C was generated by subcloning the HB-EGF-C sequence made by PCR into pGEX6P-1 (Amersham Biosciences). All cDNA constructs were verified by DNA sequencing.
Yeast two-hybrid assay
The cytoplasmic domain of proHB-EGF (residues 185–208; Higashiyama et al., 1991) was used to screen a human heart cDNA library in the yeast two-hybrid assay, following the manufacturer's instructions for the Matchmaker™ two-hybrid assay system (CLONTECH Laboratories, Inc.). About 106 transformants were screened, and library plasmids from 16 positive clones were analyzed by transformation tests and DNA sequencing (Gietz et al., 1992). β-Galactosidase activity was measured by liquid and filter assays.
Cellular fractionation, immunoprecipitation, and immunoblotting
Cytoplasmic and nuclear fractions were prepared using the CellLytic NuClear extraction kit (Sigma-Aldrich). Immunoprecipitation and immunoblotting of cell lysates was performed as described previously (Goishi et al., 1995). Primary antibodies were used as follows: mouse monoclonal IgG antibodies to FLAG (Sigma-Aldrich), PLZF (Oncogene Research Products), and cyclin A (Neomarkers); rabbit pAbs to GFP (MBL International Corporation), and HB-EGF-C (#H1) and HB-EGF (#H6) (Miyagawa et al., 1995). Secondary antibodies were HRP-conjugated goat anti–mouse and anti–rat IgG (Promega).
GST pull-down assay
GST and GST–HB-EGF-C were expressed in and purified from the Escherichia coli BL21 strain according to standard protocol. After binding of GST and GST–HB-EGF-C to the glutathione Sepharose beads, cell lysates containing various FLAG-tagged PLZF derivatives were incubated with 20 μl of the beads for 2 h at 4°C. After being washed, bound proteins were analyzed by immunoblotting using anti-FLAG antibody (Sigma-Aldrich).
Cell lines and transfection
HT1080/HB-EGF and HT1080/ΔMP-ADAM12/HB-EGF cells were described previously (Asakura et al., 2002). For the establishment of HT1080/HB-EGF-UC or HT1080/HBΔC cells, the plasmids encoding uncleavable-type proHB-EGF, HBΔC mutants were introduced into HT1080 cells using LipofectAMINE™ 2000 (Life Technologies), and stably transfected clones were isolated. HT1080 cells and its transfectants were grown in MEM supplemented with nonessential amino acids (Life Technologies), 10% FBS, and antibiotics. COS and HaCaT cells were maintained in DME containing 10% FBS. The culture of primary human keratinocytes was as described previously (Hashimoto et al., 1994). All cells were cultured in a humidified 37°C/5% CO2 incubator.
For transient transfections, 4.0 × 105 cells were seeded per 35-mm cell culture dish (Corning), grown for 12 h in the respective medium, and then transfected with expression vectors using LipofectAMINE™ 2000 (Life Technologies).
Imaging of YFP or CFP fusion proteins
Transiently transfected cells were cultured for 24 h and then used for experiments. For treatment with the EGFR-neutralizing antibody (Upstate Biotechnology), KB-R7785 (Asakura et al., 2002), or leptomycin B (Sigma-Aldrich), the cells were incubated in serum-free medium with 10 μg/ml antibody for 2 h, 10 μM KB-R7785 for 30 min, or 10 ng/ml leptomycin B for 2 h, and then cultured in the same medium containing 100 nM TPA for 1 h. Subcellular localization of YFP or CFP fusion proteins was examined under an epifluorescence microscope (Eclipse TE300; Nikon) (Fig. 1, A and F, Fig. 3, and Fig. 5). Time-lapse observations were made with the same epifluorescence microscope with a stage incubator (Fig. 1 C and Fig. 4).
Quantitation of the fraction of cells with nuclear-localized CFP-PLZF
To quantitate the fraction of cells in a population that displayed predominantly nuclear localization of CFP-PLZF, fields of cells were scored using a completely blind manner. The cells expressing CFP-PLZF were categorized into two classes: those in which CFP-PLZF was predominantly localized in the nucleus (N), and those in which CFP-PLZF was distributed throughout the entire cytoplasm (C). The ratio of the number of cells with nuclear CFP-PLZF among total transfected cells (N/[N + C] × 100) was then calculated to generate the percentage of cells with nuclear-localized CFP-PLZF. This ratio was found to be in good agreement with the qualitative impression of microscopic observations. The values (means ± SD) were determined based on the results obtained in at least two independent transfections, and at least 200 independent cells expressing CFP-PLZF were examined in each experiment.
Immunofluorescence microscopy
Cells were fixed in 4% PFA in PBS at 4°C for 10 min and permeabilized for 10 min in 0.2% Triton X-100 in PBS. Cells were blocked with 1% BSA, and subsequently incubated at RT with primary and secondary antibodies. Primary antibodies were used as follows: mouse monoclonal IgG antibodies to FLAG (Sigma-Aldrich), PLZF (Oncogene Research Products), and cyclin A (Neomarkers); a rabbit pAb to HB-EGF-C (#H1), and a goat pAb to p115 (Santa Cruz Biotechnology, Inc.). Secondary antibodies were used as follows: FITC- and rhodamine-conjugated goat anti–mouse IgG, rhodamine-conjugated goat anti–rat IgG, FITC-conjugated goat anti–rabbit IgG (CHEMICON International), and Alexa® Fluor 568–conjugated donkey anti–goat IgG (Molecular Probes, Inc.). Some cells were also stained with Hoechst 33258 (Molecular Probes, Inc.). Stained cells were viewed with an epifluorescence microscope (Eclipse TE300; Nikon) (Fig. 1 E, Fig. 6, and Fig. 7) or a confocal microscope (model LSM 510; Carl Zeiss MicroImaging, Inc.) (Fig. 1 B and Fig. 8). The thickness of optical sections was 0.8 μm.
Adenovirus vector construction and infection
Adenovirus vectors carrying genes encoding PLZF and LacZ were prepared using the adenovirus expression vector kit (Takara Biomedicals). Purified, concentrated, and titer-checked viruses were infected to the cells at a multiplicity of infection of 50.
Cell cycle analysis
For DNA staining, cells were fixed in 70% ethanol for 2 h at 4°C and incubated with 0.25 mg/ml RNase for 1 h at 37°C. After being washed, cells were stained with 0.05 mg/ml propidium iodide. Data acquisition was performed with a FACScan™ (Becton Dickinson) flow cytometer. Cell cycle distribution was analyzed with ModFit software (Nippon Becton Dickinson).
TPA treatment of mouse skin
200 μl of 0.1 mM TPA, dissolved in acetone, was applied topically to the shaved back skin of 20-wk-old female C57/BL6 mice every 24 h. After 24 or 48 h, skin samples were harvested as an 8-mm punch biopsy and stored at −80°C until use. For immunoprecipitation, protein was extracted in 1,000 μl lysis buffer with protease inhibitors using a polytron homogenizer.
Acknowledgments
We thank Dr. Y. Shirakata for mouse experiments; Drs. K. Yamasaki, Y. Yahata, and K. Shiraishi for adenovirus vector construction, helpful comments, and discussion; Drs. H. Ueno, M. Matsubara, and K. Yoshino for providing KB-R7785; Dr. M. Okochi for providing PS1 plasmid; Dr. M. Tanaka for plasmid construction; Dr. K. Kameda for cell cycle analysis; and A. Ohno, K. Nakahira, and F. Toki for technical assistance. We are also grateful to Dr. J.A. Abraham for editing the manuscript.
This work is supported by Grants-in-aid for Scientific Research (no. 13670139, 13216057, and 15390097) to S. Higashiyama from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
A. Mammoto's present address is Department of Surgical Research, Children's Hospital, Boston, MA 02115.
Abbreviations used in this paper: ADAM, a disintegrin and metalloprotease; EGFR, epidermal growth factor receptor; GPCR, G protein–coupled receptor; HB-EGF, heparin-binding EGF-like growth factor; PLZF, promyelocytic leukemia zinc finger; proHB-EGF, membrane-anchored heparin-binding EGF-like growth factor; TPA, 12-O-tetradecanoylphorbol-13-acetate.
References
- Asakura, M., M. Kitakaze, S. Takashima, Y. Liao, F. Ishikura, T. Yoshinaka, H. Ohmoto, K. Node, K. Yoshino, H. Ishiguro, et al. 2002. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat. Med. 8:35–40. [DOI] [PubMed] [Google Scholar]
- Bao, J., D. Wolpowitz, L.W. Role, and D.A. Talmage. 2003. Back signaling by the Nrg-1 intracellular domain. J. Cell Biol. 161:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna, M., N. Hawe, L. Niswander, and P.P. Pandolfi. 2000. Plzf regulates limb and axial skeletal patterning. Nat. Genet. 25:166–172. [DOI] [PubMed] [Google Scholar]
- Barna, M., T. Merghoub, J.A. Costoya, D. Ruggero, M. Branford, A. Bergia, B. Samori, and P.P. Pandolfi. 2002. Plzf mediates transcriptional repression of HoxD gene expression through chromatin remodeling. Dev. Cell. 3:499–510. [DOI] [PubMed] [Google Scholar]
- Carpenter, G. 2003. Nuclear localization and possible functions of receptor tyrosine kinases. Curr. Opin. Cell Biol. 15:143–148. [DOI] [PubMed] [Google Scholar]
- Chen, J.K., J. Capdevila, and R.C. Harris. 2002. Heparin-binding EGF-like growth factor mediates the biological effects of P450 arachidonate epoxygenase metabolites in epithelial cells. Proc. Natl. Acad. Sci. USA. 99:6029–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z., N.J. Brand, A. Chen, S.J. Chen, J.H. Tong, Z.Y. Wang, S. Waxman, and A. Zelent. 1993. Fusion between a novel Krüppel-like zinc finger gene and the retinoic acid receptor-α locus due to a variant t(11;17) translocation associated with acute promyelocytic leukaemia. EMBO J. 12:1161–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, M., A. Gould, N. Brand, J. Davies, P. Strutt, R. Shaknovich, J. Licht, S. Waxman, Z. Chen, S. Gluecksohn-Waelsch, et al. 1995. Expression of the zinc-finger gene PLZF at rhombomere boundaries in the vertebrate hindbrain. Proc. Natl. Acad. Sci. USA. 92:2249–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cussac, D., S. Schaak, C. Denis, and H. Paris. 2002. α2B-adrenergic receptor activates MAPK via a pathway involving arachidonic acid metabolism, matrix metalloproteinases and EGF-R transactivation. J. Biol. Chem. 277:19882–19888. [DOI] [PubMed] [Google Scholar]
- David, G., L. Alland, S.H. Hong, C.W. Wong, R.A. DePinho, and A. Dejean. 1998. Histone deacetylase associated with mSin3A mediates repression by the acute promyelocytic leukemia-associated PLZF protein. Oncogene. 16:2549–2556. [DOI] [PubMed] [Google Scholar]
- Gietz, D., A.S. Jean, R.A. Woods, and R.H. Schiestl. 1992. Improved method for high efficiency transformation of intact yeast cell. Nucleic Acids Res. 20:1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goishi, K., S. Higashiyama, M. Klagsbrun, N. Nakano, T. Umata, M. Ishikawa, E. Mekada, and N. Taniguchi. 1995. Phorbol ester induces the rapid processing of cell surface heparin-binding EGF-like growth factor: conversion from juxtacrine to paracrine growth factor activity. Mol. Biol. Cell. 6:967–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grignani, F., S. De Matteis, C. Nervi, L. Tomassoni, V. Gelmetti, M. Cioce, M. Fanelli, M. Ruthardt, F.F. Ferrara, I. Zamir, et al. 1998. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 391:815–818. [DOI] [PubMed] [Google Scholar]
- Guidez, F., S. Ivins, J. Zhu, M. Soderstrom, S. Waxman, and A. Zelent. 1998. Reduced retinoic acid-sensitivities of nuclear receptor corepressor binding to PML- and PLZF-RARα underlie molecular pathogenesis and treatment of acute promyelocytic leukemia. Blood. 91:2634–2642. [PubMed] [Google Scholar]
- Hackel, P.O., E. Zwick, N. Prenzel, and A. Ullrich. 1999. Epidermal growth factors: critical mediators of multiple receptor pathways. Curr. Opin. Cell Biol. 11:184–189. [DOI] [PubMed] [Google Scholar]
- Hashimoto, K., S. Higashiyama, H. Asada, E. Hashimura, T. Kobayashi, K. Sudo, T. Nakagawa, D. Damm, K. Yoshikawa, and N. Taniguchi. 1994. Heparin-binding EGF-like growth factor is an autocrine growth factor for human keratinocytes. J. Biol. Chem. 269:20060–20066. [PubMed] [Google Scholar]
- Hawighorst, T., P. Velasco, M. Streit, Y.K. Hong, T.R. Kyriakides, L.F. Brown, P. Bornstein, and M. Detmar. 2001. Thrombospondin-2 plays a protective role in multistep carcinogenesis: a novel host anti-tumor defense mechanism. EMBO J. 20:2631–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, L.Z., F. Guidez, C. Tribioli, D. Peruzzi, M. Ruthardt, A. Zelent, and P.P. Pandolfi. 1998. Distinct interactions of PML-RARa and PLZF-RARa with transcriptional co-repressors determine differential responses to retinoic acid in APL. Nat. Genet. 18:126–135. [DOI] [PubMed] [Google Scholar]
- Higashiyama, S., J.A. Abraham, J. Miller, J.C. Fiddes, and M. Klagsbrun. 1991. A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science. 251:936–939. [DOI] [PubMed] [Google Scholar]
- Hirata, M., T. Umata, T. Takahashi, M. Ohnuma, Y. Miura, R. Iwamoto, and E. Mekada. 2001. Identification of serum factor inducing ectodomain shedding of proHB-EGF and studies of noncleavable mutants of proHB-EGF. Biochem. Biophys. Res. Commun. 283:915–922. [DOI] [PubMed] [Google Scholar]
- Hong, S.H., G. David, C.W. Wong, A. Dejean, and M.L. Privalsky. 1997. SMRT corepressor interacts with PLZF and with the PML-retinoic acid receptor-α (RARα) and PLZF-RARα oncoproteins associated with acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA. 94:9028–9033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi, Y., M. Hirata, H. Hasuwa, R. Iwamoto, T. Umata, K. Miyado, Y. Tamai, T. Kurisaki, A. Sehara-Fujisawa, S. Ohno, and E. Mekada. 1998. A metalloprotease-disintegrin, MDC9/meltrin-γ/ADAM9 and PKCδ are involved in TPA-induced ectodomain shedding of membrane-anchored heparin-binding EGF-like growth factor. EMBO J. 17:7260–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo, N., M. Matsumori, H. Taoka, D. Fujiwara, E. Schreiner, B. Wolff, M. Yoshida, and S. Horinouchi. 1999. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA. 96:9112–9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemjabbar, H., and C. Basbaum. 2002. Platelet-activating factor receptor and ADAM10 mediate responses to Staphylococcus aureus in epithelial cells. Nat. Med. 8:41–46. [DOI] [PubMed] [Google Scholar]
- Lin, R.J., L. Nagy, S. Inoue, W. Shao, W.H. Miller, Jr., and R.M. Evans. 1998. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 391:811–814. [DOI] [PubMed] [Google Scholar]
- Miyagawa, J., S. Higashiyama, S. Kawata, Y. Inui, S. Tamura, K. Yamamoto, M. Nishida, T. Nakamura, S. Yamashita, Y. Matsuzawa, and N. Taniguchi. 1995. Localization of heparin-binding EGF-like growth factor in the smooth muscle cells and macrophages of human atherosclerotic plaques. J. Clin. Invest. 95:404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghal, N., and P.W. Sternberg. 1999. Multiple positive and negative regulators of signaling by the EGF-receptor. Curr. Opin. Cell Biol. 11:190–198. [DOI] [PubMed] [Google Scholar]
- Nelson, D.S., C. Alvarez, Y.S. Gao, R. Garcia-Mata, E. Fialkowski, and E. Sztul. 1998. The membrane transport factor TAP/p115 cycles between the Golgi and earlier secretory compartments and contains distinct domains required for its localization and function. J. Cell Biol. 143:319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenzel, N., E. Zwick, H. Daub, M. Leserer, R. Abraham, C. Wallasch, and A. Ullrich. 1999. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 402:884–888. [DOI] [PubMed] [Google Scholar]
- Prober, D.A., and B.A. Edgar. 2001. Growth regulation by oncogenes—new insights from model organisms. Curr. Opin. Genet. Dev. 11:19–26. [DOI] [PubMed] [Google Scholar]
- Reid, A., A. Gould, N. Brand, M. Cook, P. Strutt, J. Li, J. Licht, S. Waxman, R. Krumlauf, and A. Zelent. 1995. Leukemia translocation Gene, PLZF, is expressed with a speckled nuclear pattern in early hematopoietic progenitors. Blood. 86:4544–4552. [PubMed] [Google Scholar]
- Shaknovich, R., P.L. Yeyati, S. Ivins, A. Melnick, C. Lempert, S. Waxman, A. Zelent, and J.D. Licht. 1998. The promyelocytic leukemia zinc finger protein affects myeloid cell growth, differentiation, and apoptosis. Mol. Cell. Biol. 18:5533–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunnarborg, S.W., C.L. Hinkle, M. Stevenson, W.E. Russell, C.S. Raska, J.J. Peschon, B.J. Castner, M.J. Gerhart, R.J. Paxton, R.A. Black, and D.C. Lee. 2002. Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J. Biol. Chem. 277:12838–12845. [DOI] [PubMed] [Google Scholar]
- Tokumaru, S., S. Higashiyama, T. Endo, T. Nakagawa, J. Miyagawa, K. Yamamori, Y. Hanakawa, H. Ohmoto, K. Yoshino, Y. Shirakata, et al. 2000. Ectodomain shedding of epidermal growth factor receptor ligands is required for keratinocyte migration in cutaneous wound healing. J. Cell Biol. 151:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda, Y., Y. Miyazaki, H. Shiinoki, S. Higashiyama, Y. Nakanishi, and Y. Hieda. 2001. Involvement of heparin-binding EGF-like growth factor and its processing by metalloproteinases in early epithelial morphogenesis of the submandibular gland. Dev. Biol. 237:202–211. [DOI] [PubMed] [Google Scholar]
- Wolfe, M.S., W. Xia, B.L. Ostaszewski, T.S. Diehl, W.T. Kimberly, and D.J. Selkoe. 1999. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature. 398:513–517. [DOI] [PubMed] [Google Scholar]
- Yeyati, P.L., R. Shaknovich, S. Boterashvili, J. Li, H.J. Ball, S. Waxman, K. Nason-Burchenal, E. Dmitrovsky, A. Zelent, and J.D. Licht. 1999. Leukemia translocation protein PLZF inhibits cell growth and expression of cyclin A. Oncogene. 18:925–934. [DOI] [PubMed] [Google Scholar]
- Yan, Y., K. Shirakabe, and Z. Werb. 2002. The metalloprotease Kuzbanian (ADAM10) mediates the transactivation of EGF receptor by G protein-coupled receptors. J. Cell Biol. 158:221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]