

Abstract
p120-catenin stabilizes epithelial cadherin (E-cadherin) in SW48 cells, but the mechanism has not been established. Here, we show that p120 acts at the cell surface to control cadherin turnover, thereby regulating cadherin levels. p120 knockdown by siRNA expression resulted in dose-dependent elimination of epithelial, placental, neuronal, and vascular endothelial cadherins, and complete loss of cell–cell adhesion. ARVCF and δ-catenin were functionally redundant, suggesting that proper cadherin-dependent adhesion requires the presence of at least one p120 family member. The data reveal a core function of p120 in cadherin complexes, and strongly predict a dose-dependent loss of E-cadherin in tumors that partially or completely down-regulate p120.
Keywords: tumor suppressor; p120-catenin; cell adhesion; tumor progression; metastasis
Introduction
p120-catenin (p120) is the prototypic and most abundant member of an Arm-domain protein subfamily that includes ARVCF, δ-catenin, and p0071 (for review see Anastasiadis and Reynolds, 2000). p120 was originally described as a substrate for Src- and receptor tyrosine kinases (Reynolds et al., 1989, 1992), and later was identified as a catenin (Reynolds et al., 1994; Shibamoto et al., 1995), one of several cofactors that interact with the cadherin tail and modulate cadherin function (for review see Anastasiadis and Reynolds, 2000). The classical catenins, α- and β-catenin, bridge the cadherin cytoplasmic domain to the underlying actin cytoskeleton. p120 is required to stabilize epithelial cadherin (E-cadherin) in SW48 cells (Ireton et al., 2002), and may also regulate cadherin–cytoskeletal connections indirectly through functional interactions with Rho GTPases (Anastasiadis et al., 2000; Noren et al., 2000; Grosheva et al., 2001; Magie et al., 2002; for review see Anastasiadis and Reynolds, 2001), but the underlying mechanisms have not been established.
E-cadherin is the main cell–cell adhesion molecule in epithelial tissues and is regarded as a master organizer of the epithelial phenotype (Takeichi, 1995). Direct mutation of the E-cadherin gene in gastric and lobular breast carcinomas indicates a classical tumor suppressor role in some tumors (Oda et al., 1994; Berx et al., 1995). In late-stage carcinomas of all types, E-cadherin down-regulation occurs frequently via epigenetic mechanisms (Comijn et al., 2001; Matsumura et al., 2001) and is closely correlated with the transition to metastasis (Frixen et al., 1991; Vleminckx et al., 1991; Birchmeier and Behrens, 1994; Perl et al., 1998). Together, these data establish E-cadherin as a tumor and/or metastasis suppressor, depending on the mechanism and timing of E-cadherin down-regulation (for review see Yap, 1998; Nollet et al., 1999).
In the event of E-cadherin down-regulation, α- and β-catenins are rapidly degraded (Nagafuchi et al., 1991) via an adenomatous polyposis coli–dependent mechanism (Polakis, 2000) that ultimately targets β-catenin for destruction by the proteosome (for review see Kikuchi, 2000). In contrast, p120 is stable in the absence of cadherins and becomes stranded in the cytoplasm (Thoreson et al., 2000). Ourselves and others have postulated that cytoplasmic p120 actively drives the metastatic phenotype in cadherin-deficient cells through inappropriate activation/suppression of various Rho-GTPases such as Rac1 and RhoA (Anastasiadis et al., 2000; Noren et al., 2000; Anastasiadis and Reynolds, 2001; Grosheva et al., 2001). These data suggest a metastasis promoter role for p120 when mislocalized through prior loss of E-cadherin.
Down-regulation of p120 occurs frequently in colon, prostate, breast, lung, and other carcinoma types (for review see Thoreson and Reynolds, 2002), but the consequences are unknown. Paradoxically, it is rare to see p120 down-regulation in established tumor cell lines. The lone exception is the SW48 colon carcinoma cell line, where genetic alterations result in extremely low levels of a mutated p120 that lacks the carboxy terminus (Ireton et al., 2002). Restoring normal levels of full-length p120 expression in these poorly organized cells stabilized E-cadherin and caused a striking rescue of epithelial morphology. Thus, in SW48 cells at least, p120 appears to be essential for E-cadherin stability and function (Ireton et al., 2002). On the other hand, recent reports in Drosophila (Myster et al., 2003; Pacquelet et al., 2003) and Caenorhabditis elegans (Pettitt et al., 2003) indicate that p120 is not essential, and that its absence causes only minor defects that are not fully apparent unless complemented by weak alleles of E-cadherin or α-catenin.
Here, to clarify the role of p120 in mammalian cells, we have knocked down p120 with siRNA in cells expressing epithelial (E-), placental (P-), neuronal (N-), and vascular endothelial (VE-) cadherins. We report that each of these cadherins, as well as α- and β-catenins, were rapidly degraded in the absence of p120, resulting in loss of cell–cell adhesion. The effect was clearly dose dependent, indicating that p120 expression levels may directly determine cadherin levels. Degradation of p120-uncoupled cadherin occurred after its arrival at the surface, indicating that p120 regulates cadherin turnover at the level of internalization or recycling. p120 homologues ARVCF and δ-catenin could substitute for p120, so at least one family member is likely required to maintain adhesion. Thus, cadherin complexes are rapidly turned over and degraded in mammalian cells in the absence of direct interaction with p120 or a p120 family member. These observations establish a core function for p120 in the cadherin complex and have additional implications in support of a role for p120 in tumor suppression.
Results
p120 loss leads to loss of the cadherin complex
To directly address the general consequences of p120 deficiency, we stably expressed p120-specific siRNA using the pRetroSuper (pRS) retrovirus to knockdown p120 in mammalian cell lines (Fig. 1). Human and murine p120 siRNAs (h siRNA and m siRNA, respectively) were generated against homologous human and murine sequences that differ by three mismatches at the nucleotide level (Fig. 1 a). Pilot experiments revealed that the h siRNA strongly knocked down p120 levels in human cells, but not murine cells, and vice versa. E-cadherin levels were also severely reduced by p120 knockdown in several different epithelial cell lines. These data indicate that the stabilizing effect of p120 is not limited to SW48 cells, but represents a mechanism that is likely common to all E-cadherin–expressing cells.
Figure 1.

p120 knockdown eliminates the E-cadherin complex and abolishes adhesion. (a) Human and murine p120 siRNAs (h siRNA and m siRNA, respectively) were generated against homologous human and murine sequences that contain three mismatches at the nucleotide level (asterisks). (b) Schematic depicting a novel method for in vitro p120 knock-down and knock-up. Human p120 was knocked down using the retroviral vector pRS to express human-specific p120 siRNA, and stable cell lines were selected. p120 was then reexpressed (knock-up) by infecting the knock-down cell line with an LZRS retrovirus containing murine p120 cDNA. (c) Wild-type A431 cells (lane 1) were infected with virus carrying the control m siRNA (lane 2) or h siRNA (lane 3), and stable cell lines were isolated. p120 expression was restored (knock-up) by infecting h siRNA–expressing cells with retrovirus containing murine p120 (lane 4). The indicated cadherin complex proteins were analyzed by Western blotting whole cell lysates. E-cadherin, β-catenin, and α-catenin levels were substantially reduced in p120 knockdown cells, and restoring p120 reversed the effect. (d) p120 (i and vii), E-cadherin (ii and viii), β-catenin (iii and ix), α-catenin (iv and x), tubulin (v and xi), and vinculin (vi and xii) were localized by immunofluorescence in stable A431 cell lines expressing the control m siRNA (i–vi) or h siRNA (vii–xii). Cells were plated sparsely to allow colonies to emerge from single cells. Note that p120 knockdown cells lack cadherin complexes and have lost cell–cell adhesion. The cadherin complex is selectively targeted because the levels of tubulin and vinculin are unaffected.
By intentionally targeting the above siRNA oligos to human and murine sequences that differed by several nucleotides, it was relatively straightforward to efficiently “knock down” p120 with the human-directed siRNA (pRS-h siRNA) and subsequently “knock up” p120 by infection with pLZRS-mp120, a retrovirus containing the murine p120 cDNA (Fig. 1 b). Restoring p120 levels by expressing murine p120 reversed the effects of the h siRNA and restored adhesion (Fig. 1, b and c). It is worth noting that this method is generally applicable to any protein. If a homologous gene is not available, a knock-up construct can be generated by making silent mutations in the region targeted by the siRNA. The method is a simple in vitro equivalent of transgenic knock-out and knock-in technology, and essentially solves the common dilemma associated with expressing mutant proteins in cells that already contain high levels of an endogenous counterpart. To our knowledge, this is the first example of this broadly applicable method.
To examine the effects of p120 knockdown in detail, we isolated stable clones of A431 cells expressing p120-specific siRNA and characterized them by Western blotting (Fig. 1 c) and by immunofluorescence (Fig. 1 d). p120 was nearly eliminated by h siRNA (Fig. 1 c, lane 3), but not by m siRNA (Fig. 1 C, lane 2), and p120 loss induced near complete loss of E-cadherin. Levels of α- and β-catenin were also severely reduced, as expected from the fact that these catenins are stabilized via interaction with cadherins. Thus, p120 loss essentially eliminated the entire cadherin complex. Levels of vinculin, which concentrate at focal adhesions in these cells, were unaffected, as were levels of tubulin.
Analysis of the p120 knockdown cells by immunofluorescence revealed near complete loss of junctional E-cadherin, loss of α- and β-catenins, and loss of cell–cell adhesion (Fig. 1 d). It is noteworthy that other adhesion systems (e.g., desmosomes) cannot compensate for loss of the core components of the adherens junction. These observations reveal that p120 is essential for adhesion and suggest a core function for p120 in regulating cadherin turnover.
The requirement for p120 is common to other cadherins
To determine whether the consequence of p120 knockdown pertains only to E-cadherin, we repeated the experiments described in Fig. 1 on cells expressing E-, P-, VE-, and N-cadherins (Fig. 2). A431 (human cervical carcinoma), human umbilical aortic vascular endothelial (HUAEC), and C2C12 (murine myoblast) cells were selected because they express E- and P-cadherin, VE-cadherin, and N-cadherin, respectively. Interestingly, the levels of each of these cadherins were substantially reduced by p120 knockdown (Fig. 2, lanes 3, 6, and 9). Note that because C2C12 cells are murine, the constructs are reversed relative to the human lines; m siRNA is the knockdown construct and the h siRNA is the control. The knockdown levels in these experiments are not quite as striking as in the clonal cell lines represented in Fig. 1 because they are polyclonal cell lines, and therefore represent the average siRNA expression and knockdown from multiple integration events. Nonetheless, these data indicate clearly that the mechanism of stabilization by p120 is common to a wide variety of cadherins, probably all cadherins that bind p120.
Figure 2.

The p120-associated destruction mechanism is common to multiple cadherins. A431 (human cervical carcinoma), HUAEC, and C2C12 (mouse myocyte) cells express E- and P-cadherin, VE-cadherin, and N-cadherin, respectively. Each cell line was infected with either human- or murine-specific p120 siRNA retrovirus to generate polyclonal knockdown cell lines, and levels of p120 or E-, P-, VE-, and N-cadherins were assayed by Western blotting of whole-cell lysates. Tubulin levels were used as a loading control. p120 knockdown reduced expression of all these cadherins, indicating that its function is common to most (if not all) p120-associated cadherins. Note that the effects of the h and m siRNAs used for knockdown and control in the human cell lines are reversed in the murine cell line C2C12.
p120 levels directly determine cadherin levels
To more accurately quantify the relationship between p120 and cadherin expression, we infected A431 cells with the p120 siRNA virus and analyzed individual cell clones by coimmunofluorescence for p120 and E-cadherin (unpublished data). We also performed the reverse experiment (knock-up) by introducing murine p120 into the h siRNA-expressing A431 cells (Fig. 3). In all cases, there was a striking correlation between the levels of p120 and E-cadherin, which was also reflected by the extent of cell–cell adhesion. In the absence of p120, there was essentially no E-cadherin present (Fig. 3, i and ii). By contrast, intermediate levels of p120 caused intermediate levels of E-cadherin and partial restoration of epithelial morphology (Fig. 3, iii and iv). When murine p120 was expressed at higher than normal levels, E-cadherin levels were correspondingly elevated and exceeded the wild-type levels observed in the parental cell lines (Fig. 3, v and vi; see also Fig. 1 c). Panels v and vi are overexposed because the common exposure time for the entire panel was chosen to allow better visualization of the low and intermediate p120 levels.
Figure 3.

p120 levels act as a set point mechanism for determining cadherin levels. (a) Assay of relationship between p120 and E-cadherin levels by immunofluorescent staining. A431 cells expressing p120 siRNA were infected with the murine p120 retrovirus and plated sparsely so that individual clones could emerge that expressed widely varying amounts of murine p120. Cells were costained by immunofluorescence to examine the p120–E-cadherin relationship and its affect on cell–cell adhesion. p120 loss (i) caused complete loss of E-cadherin (ii) and the cells were nonadhesive. Intermediate levels of p120 expression (panel iii) permitted intermediate levels of E-cadherin (panel iv), and cell–cell adhesion was partially restored. Higher than normal levels of p120 (panel v) strongly induced E-cadherin (vi) and cell–cell adhesion was robust. These experiments reveal a direct relationship between p120 and E-cadherin levels, and the extent of cell–cell adhesion is directly affected. (b) Quantitative assessment of relationship between p120 and E-cadherin levels. A polyclonal population of cells expressing p120 siRNA was generated by retroviral infection. Individual clones within the population express different levels of p120 depending on integration events that affect the efficiency of the siRNA expression. Using E-cadherin antibodies (HECD-1), the cells were separated by FACS® into pools expressing progressively lower levels of E-cadherin. Cell lysates from the samples were split and then Western blotted with anti-p120 (mAb pp120) or anti-E-cadherin (C-20820).
We also quantified the relationship between p120 and E-cadherin expression by FACS® analysis of a population of p120 siRNA-infected cells with mAb-HECD1, which recognizes the extracellular domain of human E-cadherin (Fig. 3 b). The cells were sorted into pools with progressively decreasing levels of surface E-cadherin. Cell lysates were generated from each pool, divided in half, and then Western blotted for E-cadherin and p120 (Fig. 3 b). As in the immunofluorescent assays, the levels of p120 closely paralleled the levels of E-cadherin.
Together, these data show that E-cadherin levels faithfully reflect the level of p120 expression in individual cells, and show that the levels of E-cadherin can be experimentally titrated by increasing or decreasing the levels of p120.
p120 family members can functionally substitute for p120
In most epithelial cell lines, p120 is abundant and its close relatives such as ARVCF and δ-catenin are poorly expressed or absent. Although p120 knockdown was sufficient to nearly eliminate E-cadherin in several epithelial cell lines tested, the effect was incomplete in cells such as the colon carcinoma cell line HCT116. An obvious explanation is that p120 family members might partially or completely substitute for p120, depending on their relative abundance. Indeed, HCT116 cells are unusual in that they express moderate levels of ARVCF (unpublished data). To determine whether other p120 relatives can also regulate E-cadherin turnover, we transiently expressed GFP-labeled ARCVF or δ-catenin in A431 cells that lack p120 as a result of siRNA knockdown (Fig. 4). As a negative control, we also tested plakophilin-3, a more distant p120 relative that binds desmosomal (but not classical) cadherins. A431 cells expressing p120 siRNA alone were almost completely E-cadherin negative (Fig. 4, i and ii), as described earlier in this paper, and were not affected by GFP expression (Fig. 4 i, fluorescent cells). ARVCF (Fig. 4, iii) and δ-catenin (Fig. 4, v) localized to adherens junctions and efficiently rescued adhesion by restoring normal E-cadherin levels (Fig. 4, iv and vi). In contrast, myc-tagged plakophilin-3 (Fig. 4 vii, stained cells) did not affect cadherin levels (Fig. 4 viii) and failed to restore cell–cell contacts. Thus, there is a clear redundant role among close family members with regard to cadherin stabilization, and the occasional significant presence of a p120 family member (e.g., ARVCF in colon HCT116 cells) is likely to account for the fact that E-cadherin loss does not perfectly parallel p120 loss in some cell lines.
Figure 4.
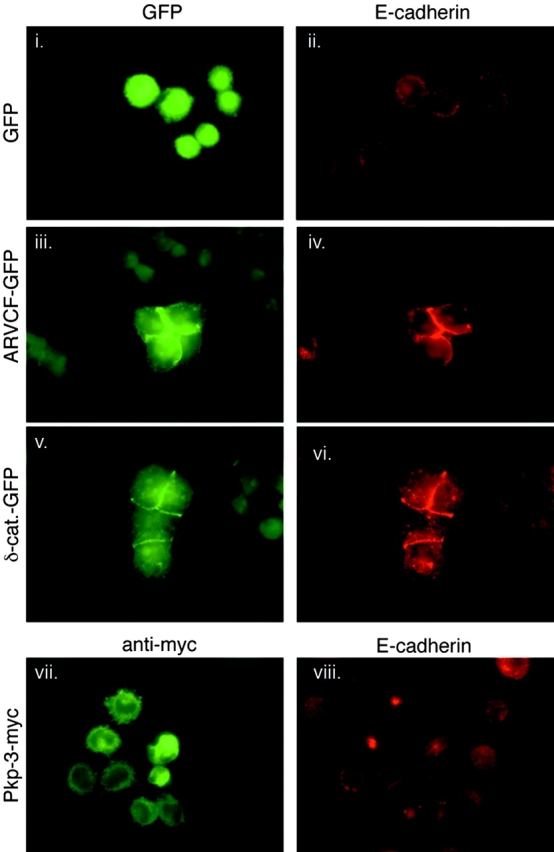
Redundant roles for p120 family members ARVCF and δ-catenin. A431 cells stably expressing human p120 siRNA were transiently transfected with ARVCF-GFP, δ-catenin–GFP, or myc-tagged plakophilin-3 (Pkp-3-myc). 24 h after transfection, cells were plated sparsely and individual colonies grew for 2 d. Levels of the transfected proteins and E-cadherin were then analyzed by immunofluorescence. GFP expression alone (i, eluminated cells) did not affect E-cadherin levels (ii). Both ARVCF-GFP (iii) and δ-catenin–GFP (v) substantially increased levels of E-cadherin (iv and vi) and rescued cell–cell adhesion. In contrast, plakophilin-3 (vii), a p120-related protein that does not bind classical cadherins, but had no effect on E-cadherin levels (viii) or cell–cell adhesion.
Mechanism of E-cadherin loss
p120 reportedly is the first of the catenins to bind newly synthesized N-cadherin, and coprecipitates with the nascent precursor form of N-cadherin (Wahl et al., 2003). Because of the extraordinary efficiency of E-cadherin destruction after p120 knockdown, we first considered the possibility that p120 binding was necessary to stabilize E-cadherin during or after protein translation and before arrival at the cell surface. To examine E-cadherin synthesis in the absence of p120, we labeled the p120 knockdown A431 cells with [35S]methionine and performed pulse-chase experiments (Fig. 5 a). Interestingly, the rate of E-cadherin synthesis was unaffected by the absence of p120 (Fig. 5 a, compare top panels). Moreover, the processing and turnover of both the precursor and mature forms of E-cadherin were identical for at least 1 h after the pulse, after which the cadherin degradation curves diverged rapidly. Degradation of α- and β-catenins paralleled the loss of E-cadherin, as expected from the fact that these catenins are stabilized by cadherin binding.
Figure 5.

p120 regulates E-cadherin turnover at the cell membrane. (a) E-cadherin synthesis and processing in p120 knockdown cells. E-cadherin turnover was examined by pulse-chase analysis of parental and h p120 siRNA-expressing A431 cells. α- and β-Catenin processing from the same experiment are shown below. Chase times are indicated across the top. At chase time 0 (15 min after initiation of the pulse labeling), E-cadherin synthesis was identical in the presence and absence of p120 (a, compare E-cadherin bands). The processing of the pro- (pro-E-cad) and mature (E-cad.) forms were identical for at least 1 h. Soon thereafter, E-cadherin degradation was significantly accelerated in the absence of p120. (b) Analysis of total E-cadherin surface levels in parental and p120 knockdown A431 cells. The parental and p120 knockdown A431 cells were biotinylated for 20 min at 4°C to label surface cadherins. To specifically measure the surface levels, E-cadherin was first immunoprecipitated directly with E-cadherin mAb HECD-1. The sample was eluted with 0.5% SDS and then reprecipitated with streptavidin-coated beads to isolate the surface-labeled pool. E-cadherin levels at the surface in p120 knockdown cells (lane 1) are at least 100-fold diminished relative to the parental cells (lane 2). The result in lane 2 shows that the surface E-cadherin can be efficiently labeled (and detected) by this method. (c) Tracking the arrival of newly synthesized E-cadherin to the cell surface. The methods in a and b were combined to determine whether newly synthesized E-cadherin could transit to the cell surface in the absence of p120. The results in c were quantified by densitometry and represented graphically in d. Parental and p120 knockdown (h siRNA) cells were labeled with [35S]methionine for 15 min, chased at 37°C for the times indicated across top, and placed on ice (4°C) to suspend trafficking. Cell surface proteins were immediately biotinylated at 4°C for 20 min as in b. Surface E-cadherin was then isolated as in b, and the nascent [35S]methionine E-cadherin pool was visualized by SDS-PAGE and radiography. Nascent E-cadherin appeared at the surface at 30 min and peaked at 1 h. The absence of p120 had no effect on this result. Therefore, p120 is not required for E-cadherin synthesis or trafficking, but is essential to regulate E-cadherin turnover soon after its arrival at the cell surface.
The fact that the newly synthesized cadherin behaved identically in the presence and absence of p120 for 1 h, and until after the precursor form disappeared, suggests that cadherin degradation occurred after arrival at the cell surface. The result was initially surprising because examination of total surface levels of E-cadherin in the p120 knockdown cells (h siRNA) and parental cell lines (Fig. 5 b) showed that although surface E-cadherin could be efficiently isolated by biotin labeling and streptavidin pulldown (e.g., Fig. 5 b, lane 2), it was ∼100-fold less abundant in the p120-deficient cells (Fig. 5 b, compare lane 1 with lane 2).
To definitively address this issue, we combined the pulse-chase and biotin surface-labeling strategies in order to selectively examine the fate of the nascent E-cadherin molecules with respect to their arrival at the cell surface (Fig. 5 c). The pulse labeling was conducted as in Fig. 5 a, except that surface E-cadherin was subsequently biotin labeled (as in Fig. 5 b) at each time point after the pulse chase. The surface-labeled cadherins were then isolated by streptavidin pulldown, and nascent cadherins were visualized by SDS-PAGE and autoradiography. The data show that the rate of nascent E-cadherin arrival at the cell surface is almost identical in the presence and absence of p120 (Fig. 5, c and d; compare parental and siRNA cell lines). The appearance and removal of E-cadherin from the cell surface (Fig. 5 c; E-cadherin + streptavidin immunoprecipitations) are quantified by densitometry and displayed graphically in Fig. 5 d. Note that peak levels of nascent (35S-labeled) E-cadherin at the cell surface occurred at 1 h, and by 4 h, the nascent cadherin was either moving off the surface or getting degraded. The timing is consistent with the 4-h time point in Fig. 5 a, which marks the first interval where degradation of the unbound cadherin sharply accelerates. Clearly, E-cadherin transits normally to the surface in the absence of p120, but is then rapidly turned over.
To identify the mechanism of degradation, we treated p120 knockdown cells with over 30 agents known to inhibit factors that have been reported to affect cadherin stability and turnover. Examples include inhibitors of presenilin-1, caspases, metalloproteinases, and calpain. Cells were incubated for 24 h with predetermined amounts of the various inhibitors, and then analyzed by immunofluorescence (unpublished data) or Western blotting for changes in levels of E-cadherin (Fig. 6). Although the majority of the inhibitors had no affect, several proteosome inhibitors (i.e., PS341, lactacystin, and MG132) significantly blocked E-cadherin degradation (Fig. 6; lactacystin, lanes 1 and 2; PS341, lanes 3 and 4). The reduced amount of E-cadherin at the higher PS341 dose (Fig. 6, compare lane 3 with lane 4) reflects toxicity of this compound. Of the two commonly used lysosomal inhibitors we tried, ammonium chloride (Fig. 6, lane 5; NH4Cl) had no effect, but chloroquine (Fig. 6, lane 6; Chl) blocked E-cadherin degradation almost as effectively as the proteosome inhibitors. In both the PS341- and chloroquine-treated cells, cytoplasmic pools of E-cadherin increased, but the increased levels were not reflected by increased adhesion or higher surface cadherin levels. Thus, these inhibitors appear to block cadherin degradation, but do not affect internalization. The data suggest that when newly synthesized E-cadherin arrives at the cell surface, p120 is required to prevent the immediate targeting of unbound E-cadherin for degradation by the proteosome and/or lysosome. We conclude that p120 regulates cadherin turnover by controlling either internalization, or possibly an immediately subsequent decision whereby internalized cadherins are sorted into recycling or degradation pathways.
Figure 6.

Mechanism of E-cadherin degradation. The effects of various inhibitors known to influence cadherin stability were assayed in the stable p120 knockdown A431 cells. E-cadherin levels from samples treated for 24 h (lanes 1–9) were monitored by Western blotting whole-cell lysates and were compared with normal E-cadherin levels in the parental cell line (lane 10). Inhibitor concentrations were as follows: lactacystin 10 μM (lane 1), 3.3 μM (lane 2); PS341 100 nM (lane 3), 33 nM (lane 4), ammonium chloride 5 mM (lane 5), Chloroquine 33 μM (lane 6), and IETD-CHO 10 nM (lane 7). DMSO is the control condition (lane 8). The proteosome inhibitors lactacystin and PS341 increased E-cadherin levels (compare lanes 1–4 to lanes 8 and 9). The lower cadherin levels after 100 nM PS341 (lane 3) relative to the 33-nM treatment (lane 4) is due to toxicity at the higher concentration. Of the lysosomal inhibitors, chloroquine (lane 6), but not ammonium chloride (lane 5), increased E-cadherin levels. A caspase 8 inhibitor (lane 7) that has been shown to inhibit E-cadherin degradation in myeloma cells had no effect.
Discussion
Here, we provide evidence that the core function of p120 in cadherin complexes is to regulate cadherin turnover. Previously, we showed that the stabilizing effect of p120 on E-cadherin in a p120-deficient SW48 cell line involved a post-transcriptional mechanism and required direct p120–E-cadherin interaction (Ireton et al., 2002). However, it was not clear whether this phenomenon was generally applicable beyond SW48 cells, nor could we determine the underlying mechanism. Here, using siRNA and/or p120 reconstitution, we show that E-cadherin levels depend absolutely on p120 expression. Importantly, this set point mechanism is common to other (probably all) p120-binding cadherins because p120 knockdown also induced significant down-regulation of P-, VE-, and N-cadherins. The timing and location of p120 action argue strongly that p120 regulates adhesion via controlling cadherin turnover at the cell surface. These observations have crucial implications for roles of p120 in cadherin function and cancer.
We believe that the only exception to the requirement for p120 occurs in cells that express p120 family members such as ARVCF or δ-catenin. This qualifier is based in part on cell lines such as HCT116 where the observed reduction in E-cadherin levels after p120 siRNA expression did not perfectly parallel the extent of p120 loss. Indeed, although ARVCF is typically difficult to detect in many epithelial cell lines, it is expressed at moderate levels in HCT116 cells (unpublished data). Our data show that ARVCF and δ-catenin efficiently compensate for p120 loss when ectopically expressed in A431 cell lines expressing p120 siRNA. Despite significant structural and sequence similarity, plakophilin-3 had no effect, presumably because it does not bind classical cadherins. These data strongly imply that surface cadherin stability is invariably dependent on the binding of either p120 or a closely related family member, and the presence of variable levels of p120 family members likely accounts for the discrepancy in cell lines where p120 knockdown does not cause a corresponding loss of resident classical cadherins.
The fact that p120 availability limits cadherin levels has several crucial implications. For example, overexpression of dominant-negative cadherins frequently down-regulates expression of endogenous cadherins (Kintner, 1992; Fujimori and Takeichi, 1993; Zhu and Watt, 1996), but the mechanism is unknown. Our data strongly suggest that a key action of dominant-negative cadherins is the sequestering of endogenous p120, thereby driving the turnover and degradation of endogenous cadherins. In addition, cadherin levels in cells may ultimately be controlled by factors that regulate p120 levels, and competition for interaction with p120 is likely to be physiologically relevant in cells that express more than one cadherin.
In theory, the absence of cadherins in p120-deficient cells indicates either a failure to normally synthesize cadherins or an efficient means of eliminating them when p120 is not present. However, our pulse-chase data indicate that p120 is not required for normal synthesis or transit of cadherin to the cell surface. Instead, p120 absence dramatically accelerates cadherin degradation after its arrival at the surface, indicating a role in regulating cadherin turnover at the membrane (modeled in Fig. 7). Our data do not precisely distinguish the point at which p120 acts to prevent degradation. The simplest explanation is that p120 limits degradation by regulating internalization. Only cadherin-bound p120 is phosphorylated (Thoreson et al., 2000), and p120 phosphorylation is the most likely means of regulating p120–cadherin affinity and/or p120 activity in the complex. We cannot rule out the less likely possibility that once internalized, p120 might control the next step, which targets the endocytosed cadherin for either degradation or recycling back to the surface. Regardless, it is likely that the ultimate destruction of the cadherin in p120-deficient A431 cells resides mainly in the proteosome, and to some extent in the lysosome.
Figure 7.

Model for p120 function in regulating cadherin turnover. The low affinity of p120 for cadherins, as judged by coimmunoprecipitation experiments, probably reflects the ability of p120 to rapidly alternate between cadherin-bound and -unbound states. (1) Our data suggest that the rate of cadherin turnover is controlled by cell surface events that transiently increase or decrease p120 affinity for cadherins. Thus, cadherin complexes exist in a dynamic equilibrium between p120-bound and -unbound states, which in turn may be regulated by p120 phosphorylation (not depicted). (2) Unbound cadherin is targeted for internalization, possibly via a Hakai-like ubiquitination mechanism (see Discussion). (3) We cannot yet rule out an alternative pathway where p120 binding is irrelevant for internalization, but mediates a sorting decision that recycles internalized cadherin back to the membrane. (4) Regardless of the exact decision point, unbound cadherin is targeted for degradation by the proteosome and/or lysosome. Considerable evidence indicates that signaling events at the cell surface modulate phosphorylation of the cadherin-bound pool of p120. The simplest interpretation of these observations is that p120 phosphorylation regulates its steady-state affinity for cadherins, which in turn regulates adhesion by controlling the rate of cadherin turnover. Note that α- and β-catenin are passive players in this model. They likely participate in clustering and certainly mediate the cytoskeletal interaction (not depicted), but their role may be secondary to regulating surface cadherin levels, which is almost completely determined by p120.
Under normal circumstances, cadherin turnover is constitutive and endocytosis is a crucial mechanism for down-regulating cadherin adhesiveness (Le et al., 1999, 2002; Xiao et al., 2003). Previously, we postulated that p120 acts as a switch, inducing the assembly or disassembly of cadherin complexes through transient signaling events (probably tyrosine and serine phosphorylation), which in turn might regulate cadherin clustering. Our new data strongly favor a mechanism whereby dynamic assembly and disassembly of cadherin complexes is driven primarily by regulation of cadherin turnover rather than physical clustering (Fig. 7). A plausible explanation is that the rate of cadherin turnover is dictated by events at the cell surface that transiently increase or decrease p120 affinity for cadherins. The off state favors internalization/degradation, whereas the on state favors retention/recycling. The low affinity of p120 for cadherins, as judged by coimmunoprecipitation experiments (Thoreson et al., 2000), probably reflects the ability of p120 to rapidly alternate between cadherin-bound and -unbound states. It is worth noting that α- and β-catenins are largely passive players in this model. Because their stability is controlled by cadherin binding, their fate is ultimately tied to cadherin levels, which are clearly controlled by p120. Of course, turnover and clustering are not mutually exclusive mechanisms, but our current data suggest that turnover may take precedence.
Recent experiments in C. elegans and Drosophila indicate that p120 is not essential in these organisms. Indeed, both worms (Pettitt et al., 2003) and flies (Myster et al., 2003) are viable when p120 is removed, and p120-uncoupled E-cadherin can substitute effectively for wild-type E-cadherin in flies (Pacquelet et al., 2003). In contrast, the murine p120 knockout is embryonic lethal (unpublished data). Additionally, our current data show clearly that p120 is essential in mammalian cells. It is possible that mammalian p120 has evolved both additional family members and increased complexity to accommodate the developmental demands of higher organisms.
An unanswered question is the exact targeting mechanism for internalization and/or degradation of cadherins not associated with p120. Because direct binding of p120 to E-cadherin is required, it is possible that p120 binding blocks the interaction of an unknown binding partner (or event) that targets E-cadherin for degradation. Candidates include presenilin-1 (Baki et al., 2001; Marambaud et al., 2002) and Hakai (Fujita et al., 2002), which are reported to compete with p120 for binding the cadherin juxtamembrane domain. Presenilin-1 binding promotes proteolytic degradation of E-cadherin (Baki et al., 2001; Marambaud et al., 2002), whereas Hakai is a ubiquitin ligase that binds tyrosine-phosphorylated E-cadherin, leading to its ubiquitination and destruction (Fujita et al., 2002). Several tyrosine kinase receptors are turned over via a similar mechanism involving the oncogene and ubiquitin ligase Cbl, which binds tyrosine-phosphorylated residues via its classical SH2 domain (for review see Hicke, 1999). However, we were unable to block E-cadherin destruction in the p120 siRNA cell lines with either presenilin or tyrosine kinase inhibitors (unpublished data). Moreover, the mechanism we describe is common to several cadherins, whereas the Hakai mechanism appears specific for E-cadherin. Nonetheless, our data favor a model where an E-cadherin–targeting event is triggered by the absence or transient off-loading of p120.
Finally, several lines of evidence suggest that this new role for p120 in regulating cadherin turnover may be important in cancer. In cell lines, E-cadherin loss leaves p120 stranded in the cytoplasm, but has little effect on p120 levels. It is well established that E-cadherin loss occurs frequently by mutation (Berx et al., 1998) and by epigenetic mechanisms (Comijn et al., 2001; Matsumura et al., 2001) that probably do not involve p120. In contrast, p120 loss clearly represents a different scenario that directly induces loss of E-cadherin, and thus ultimately, the entire cadherin complex. It follows that p120 loss may precede cadherin loss in the reported subset of tumors that have been shown to lack both proteins (for review see Thoreson and Reynolds, 2002). Accumulating evidence suggests that p120 down-regulation occurs frequently in colon, prostate, lung, bladder, breast, and several other malignancies (for review see Thoreson and Reynolds, 2002). p120 is both mutated and underexpressed in the colon carcinoma cell line SW48, and indeed, E-cadherin is indeed strongly down-regulated in these cells, providing the first physiologically relevant example of this phenomena in a carcinoma cell line. However, no other p120-deficient cell lines have been described, and physical alterations in the p120 gene locus have not been associated with malignancy. Together, these observations suggest that p120 down-regulation in tumors occurs by an epigenetic mechanism that has yet to be identified, and raise the possibility that like E-cadherin, p120 acts as a tumor suppressor.
In conclusion, we show that p120 levels determine steady-state levels of functional cadherins by regulating cadherin turnover at the cell surface. This is likely the core function of p120 in the cadherin complex and suggests that cadherin adhesiveness is modulated, in part, by signaling events that dynamically influence p120–cadherin affinity. In addition, p120 is clearly at the top of the cadherin food chain in terms of who controls the overall fate of the complex. Together with reports of p120 down-regulation in a wide range of epithelial tumors, these data suggest a role for p120 as a tumor suppressor.
Materials and methods
Cell culture, infections, and transfections
HUAECs (CC-2535; Cambrex) were thawed at passage one. They were grown in endothelial basal medium (CC-3121; Cambrex) supplemented with EGM SingleQuots® supplements and growth factors (CC-4133; Cambrex). Just before use, HUAEC culture dishes were treated with 0.2% gelatin (Sigma-Aldrich, G1393) in PBS for 20 min at 37°C. Culture conditions for Phoenix cells have been described previously (Ireton et al., 2002), and all other cell lines were cultured as described elsewhere (Anastasiadis et al., 2000). For siRNA expression, cells were infected with pRS and selected with 3 to 5 μg/ml puromycin. As indicated, some cells were infected again with LZRS–mp120–neomycin and selected with 600 μg/ml neomycin. pRS and LZRS retroviruses were produced in the Phoenix cell packaging line as described previously (Ireton et al., 2002). Clonal A431 cell lines were subcloned by limiting dilution. p120 expression was assessed by immunofluorescence and Western blotting. Transient transfections were performed with LipofectAMINE™ 2000 (Invitrogen) according to the manufacturer's instructions.
Immunofluorescence and FACS®
Cells were plated sparsely on glass coverslips and incubated for 2 d before immunofluorescent labeling. Cells were washed once with PBS, then fixed in 3% PFA for 30 min. Fixed cells were washed with PBS/10 mM glycine twice and permeabilized in 0.2% Triton X-100/PBS for 5 min. Cells were again washed in PBS/10 mM glycine and blocked in 3% milk/PBS before staining. Primary antibodies mAb pp120 (Transduction Laboratories), anti-β-catenin C-2206 (Sigma-Aldrich), anti-α-catenin C-2081 (Sigma-Aldrich), anti-E-cadherin C-20820 (Transduction Laboratories), and HECD-1 (a gift from Masatoshi Takeichi, Kyoto University, Kyoto, Japan) were used as described previously (Ireton et al., 2002). Other primary antibodies were used as follows: anti-tubulin (DM1a; Sigma-Aldrich) 1:1000, anti-vinculin (hvin-1; Sigma-Aldrich) 1:400, anti-myc (mAb 9E10) 1μg/ml, and SHE78–7 anti E-cadherin (Zymed Laboratories) 1μg/ml. Secondary antibodies goat anti–mouse IgG1 and IgG2a conjugated either to Alexa® 594 or 488 were used at 1.7 μg/ml. Cells were mounted in ProLong Antifade (Molecular Probes, Inc.) according to the manufacturer's instructions and were visualized on a microscope (Axioplan 2; Carl Zeiss MicroImaging, Inc.) with Immersol 518F oil (Carl Zeiss MicroImaging, Inc.) using a 63× Plan Apochromat 1.4 aperture objective lens (Carl Zeiss MicroImaging, Inc.). Pictures were acquired using a camera (Orca-ER; Hamamatsu) and Openlab v3.1.4 software (Improvision).
To isolate pools of cells expressing different levels of E-cadherin, a p120 siRNA-infected A431 cell population was sorted by FACS® as follows: cells were dissociated with GIBCO BRL cell dissociation buffer (enzyme free, PBS based) at 37°C for 45 min. Single-cell suspensions were enhanced by repeated pipetting, washed in PBS containing 1% serum, and then labeled with E-cadherin mAb HECD1 (10 μg for 5 × 106 cells in 1 ml), followed by washing and then additional labeling with the secondary antibody Alexa® 488–conjugated goat anti–mouse IgG (1/1,000 dilution in 1 ml; Molecular Probes, Inc.). After washing, cells were labeled with 7AAD (Molecular Probes, Inc.) to discriminate dead cells, and subjected to FACS® using a FACStar PLUS™ cell sorter (Becton Dickinson). All procedures were performed at 4°C to prevent E-cadherin endocytosis. Four gates were set based on preliminary experiments designed to separate cells into four categories of cells expressing high to low levels of E-cadherin. The resulting pools were expanded and then analyzed by Western blotting for p120 and E-cadherin levels.
Pulse chase, biotinylation, and cell surface trafficking
Pulse-chase experiments were performed exactly as described previously (Ireton et al., 2002). Biotinylation and the rate of cell surface trafficking were also performed exactly as described previously (Bonifacino et al., 2003). In brief, cells were plated at 5 × 105 cells per 60-mm dish for 36 h before pulse chase. Cells were 35S-labeled for 15 min before chase. At the end of the chase, cell surface proteins were labeled with 1 mg/ml EZ-Link sulfo-NHS-SS-biotin (Pierce Chemical Co.) at 4°C for 30 min. E-cadherin was immunoprecipitated from NP-40 cell lysates, and surface cadherin was detected by dividing the E-cadherin immunoprecipitations in half, eluting E-cadherin from the beads with 0.5% SDS, reconstituting elutions in 50 mM Tris-HCl (pH 7.5), and pulling down biotinylated E-cadherin with 10 μl per sample of packed streptavidin-coated agarose beads (Sigma-Aldrich) for 1 h at 4°C. Samples were washed three times with 50 mM Tris-HCl (pH 7.5), and protein was eluted with 2× Laemmli sample buffer and analyzed by SDS-PAGE and autoradiography as described previously (Ireton et al., 2002). Quantification was performed by densitometry using Image Gage software (Fujifilm Inc.). Arbitrary densitometry units were plotted with GraphPad Prism (GraphPad Software, Inc.) and adjusted for background. Biotinylation of total surface cadherin was performed as described above, but without the pulse-chase labeling.
Constructs
LZRS–mp120–Neo has been described in detail previously (Ireton et al., 2002). The pRS vector was a gift from Reuven Agami (The Netherlands Cancer Institute, Amsterdam, Netherlands). pRS human p120 siRNA and pRS m siRNA were generated according to Brummelkamp et al. (2002). In brief, a 64-bp linker was inserted into pRS using the BamHI and HindIII sites. Oligos for the linker contained p120-specific sense and corresponding antisense sequences, flanking a 6-base hairpin, and were PAGE purified by Integrated DNA Technologies. pEGFP-C1 δ-catenin (Lu et al., 1999) was a gift from Qun Lu (East Carolina University, Greenville, NC). pEGFP-C2 ARVCF C11 (Waibler et al., 2001) was a gift from Anna Starzinski-Powitz (Johann Wolfgang Goethe-Universität, Frankfurt, Germany). pEGFP-C1 (CLONTECH Laboratories, Inc.) was used as a negative control in transfection experiments.
Western blotting
Western blotting procedures were conducted as described by Mariner et al. (2001). In brief, cells were grown to confluence and lysed with either NP-40 or RIPA buffer. Protein concentrations in lysates were obtained by copper reduction/bicinchoninic acid (BCA) assay (Pierce Chemical Co.) according to the manufacturer's instructions. Primary antibodies were used as follows: mAb pp120 (0.1 μg/ml), anti-E-cadherin mAbs C-20820 (1/2,500) and HECD-1 (0.1 μg/ml), anti-β-catenin pAb C-2206 (1/5,000; Sigma-Aldrich), and anti-α-catenin pAb C-2081 (1/5,000; Sigma-Aldrich). Secondary antibodies were peroxidase-conjugated donkey anti–mouse IgG (1/10,000; Jackson ImmunoResearch Laboratories) and mouse anti–rabbit IgG (1/10,000; Jackson ImmunoResearch Laboratories). Anti-tubulin (DM1a; Sigma-Aldrich) and anti-vinculin (hvin-1; Sigma-Aldrich) were used at 1:1,000 and 1:400, respectively.
Inhibitors
Cells were plated at 5 × 105 cells per 60-mm dish for 36 h before treatment with inhibitors. Inhibitors were added to standard growth media at the following concentrations: 33 nM PS341 (Millenium Pharmaceuticals), 3.3 μM lactacystin (Calbiochem), 33 μM chloroquine (Sigma-Aldrich), 5 mM ammonium chloride (Sigma-Aldrich), and 10 nM IETD-CHO (Calbiochem). Cells were treated with inhibitors for 24 h before lysis in NP-40 buffer and analyzed by Western blotting with E-cadherin mAb HECD-1.
Acknowledgments
Special thanks to Reuven Agami for pRetroSuper, Masatoshi Takeichi for HECD-1, Qun Lu for δ-catenin, Anna Starzinski-Powitz for ARVCF, and Frans van Roy (Ghent University, Ghent, Belgium) for plakophilin-3.
Albert Reynolds was supported in part by National Institutes of Health (grant CA55724), GI SPORE (grant 1P50 CA95103), and by the Ingram-Vanderbilt Cancer Center through the Cancer Center support (grant CA69485).
M.A. Davis and R.C. Ireton contributed equally to this paper.
Abbreviations used in this paper: E-cadherin, epithelial cadherin; h siRNA, human small interfering RNA; HUAEC, human umbilical aortic endothelial cells; m siRNA, murine small interfering RNA; N-cadherin, neuronal cadherin; p120, p120-catenin; P-cadherin, placental cadherin; pRS, pRetroSuper; siRNA, small interfering RNA; VE-cadherin, vascular endothelial cadherin.
References
- Anastasiadis, P.Z., and A.B. Reynolds. 2000. The p120 catenin family: complex roles in adhesion, signaling and cancer. J. Cell Sci. 113:1319–1334. [DOI] [PubMed] [Google Scholar]
- Anastasiadis, P.Z., and A.B. Reynolds. 2001. Regulation of Rho GTPases by p120-catenin. Curr. Opin. Cell Biol. 13:604–610. [DOI] [PubMed] [Google Scholar]
- Anastasiadis, P.Z., S.Y. Moon, M.A. Thoreson, D.J. Mariner, H.C. Crawford, Y. Zheng, and A.B. Reynolds. 2000. Inhibition of RhoA by p120 catenin. Nat. Cell Biol. 2:637–644. [DOI] [PubMed] [Google Scholar]
- Baki, L., P. Marambaud, S. Efthimiopoulos, A. Georgakopoulos, P. Wen, W. Cui, J. Shioi, E. Koo, M. Ozawa, V.L. Friedrich, Jr., and N.K. Robakis. 2001. Presenilin-1 binds cytoplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates stability and function of the cadherin/catenin adhesion complex. Proc. Natl. Acad. Sci. USA. 98:2381–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berx, G., A.M. Cleton-Jansen, F. Nollet, W.J. de Leeuw, M. van de Vijver, C. Cornelisse, and F. van Roy. 1995. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 14:6107–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berx, G., K.F. Becker, H. Hofler, and F. van Roy. 1998. Mutations of the human E-cadherin (CDH1) gene. Hum. Mutat. 12:226–237. [DOI] [PubMed] [Google Scholar]
- Birchmeier, W., and J. Behrens. 1994. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim. Biophys. Acta. 1198:11–26. [DOI] [PubMed] [Google Scholar]
- Bonifacino, J.S., M. Dasso, J.B. Harford, J. Lippincott-Schwartz, and K.M. Yamada. 2003. Protein trafficking. Current Protocols in Cell Biology. K.S. Morgan, editor. John Wiley & Sons, Inc., New York. 15.4.
- Brummelkamp, T.R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science. 296:550–553. [DOI] [PubMed] [Google Scholar]
- Comijn, J., G. Berx, P. Vermassen, K. Verschueren, L. van Grunsven, E. Bruyneel, M. Mareel, D. Huylebroeck, and F. van Roy. 2001. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell. 7:1267–1278. [DOI] [PubMed] [Google Scholar]
- Frixen, U.H., J. Behrens, M. Sachs, G. Eberle, B. Voss, A. Warda, D. Lochner, and W. Birchmeier. 1991. E-cadherin-mediated cell–cell adhesion prevents invasiveness of human carcinoma cells. J. Cell Biol. 113:173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimori, T., and M. Takeichi. 1993. Disruption of epithelial cell-cell adhesion by exogenous expression of a mutated nonfunctional N-cadherin. Mol. Biol. Cell. 4:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita, Y., G. Krause, M. Scheffner, D. Zechner, H.E. Leddy, J. Behrens, T. Sommer, and W. Birchmeier. 2002. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 4:222–231. [DOI] [PubMed] [Google Scholar]
- Grosheva, I., M. Shtutman, M. Elbaum, and A.D. Bershadsky. 2001. p120 catenin affects cell motility via modulation of activity of Rho - family GTPases: a link between cell-cell contact formation and regulation of cell locomotion. J. Cell Sci. 114:695–707. [DOI] [PubMed] [Google Scholar]
- Hicke, L. 1999. Gettin' down with ubiquitin: turning off cell-surface receptors, transporters and channels. Trends Cell Biol. 9:107–112. [DOI] [PubMed] [Google Scholar]
- Ireton, R.C., M.A. Davis, J. van Hengel, D.J. Mariner, K. Barnes, M.A. Thoreson, P.Z. Anastasiadis, L. Matrisian, L.M. Bundy, L. Sealy, et al. 2002. A novel role for p120 catenin in E-cadherin function. J. Cell Biol. 159:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi, A. 2000. Regulation of beta-catenin signaling in the Wnt pathway. Biochem. Biophys. Res. Commun. 268:243–248. [DOI] [PubMed] [Google Scholar]
- Kintner, C. 1992. Regulation of embryonic cell adhesion by the cadherin cytoplasmic domain. Cell. 69:225–236. [DOI] [PubMed] [Google Scholar]
- Le, T.L., A.S. Yap, and J.L. Stow. 1999. Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J. Cell Biol. 146:219–232. [PMC free article] [PubMed] [Google Scholar]
- Le, T.L., S.R. Joseph, A.S. Yap, and J.L. Stow. 2002. Protein kinase C regulates endocytosis and recycling of E-cadherin. Am. J. Physiol. Cell Physiol. 283:C489–C499. [DOI] [PubMed] [Google Scholar]
- Lu, Q., M. Paredes, M. Medina, J. Zhou, R. Cavallo, M. Peifer, L. Orecchio, and K.S. Kosik. 1999. δ-catenin, an adhesive junction–associated protein which promotes cell scattering. J. Cell Biol. 144:519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magie, C.R., D. Pinto-Santini, and S.M. Parkhurst. 2002. Rho1 interacts with p120ctn and alpha-catenin, and regulates cadherin-based adherens junction components in Drosophila. Development. 129:3771–3782. [DOI] [PubMed] [Google Scholar]
- Marambaud, P., J. Shioi, G. Serban, A. Georgakopoulos, S. Sarner, V. Nagy, L. Baki, P. Wen, S. Efthimiopoulos, Z. Shao, et al. 2002. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 21:1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariner, D.J., P. Anastasiadis, H. Keilhack, F.D. Böhmer, J. Wang, and A.B. Reynolds. 2001. Identification of Src phosphorylation sites in the catenin p120ctn. J. Biol. Chem. 276:28006–28013. [DOI] [PubMed] [Google Scholar]
- Matsumura, T., R. Makino, and K. Mitamura. 2001. Frequent down-regulation of E-cadherin by genetic and epigenetic changes in the malignant progression of hepatocellular carcinomas. Clin. Cancer Res. 7:594–599. [PubMed] [Google Scholar]
- Myster, S.H., R. Cavallo, C.T. Anderson, D.T. Fox, and M. Peifer. 2003. Drosophila p120catenin plays a supporting role in cell adhesion but is not an essential adherens junction component. J. Cell Biol. 160:433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagafuchi, A., M. Takeichi, and S. Tsukita. 1991. The 102 kd cadherin-associated protein: similarity to vinculin and posttranscriptional regulation of expression. Cell. 65:849–857. [DOI] [PubMed] [Google Scholar]
- Nollet, F., G. Berx, and F. van Roy. 1999. The role of the E-cadherin/catenin adhesion complex in the development and progression of cancer. Mol. Cell Biol. Res. Commun. 2:77–85. [DOI] [PubMed] [Google Scholar]
- Noren, N.K., B.P. Liu, K. Burridge, and B. Kreft. 2000. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J. Cell Biol. 150:567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda, T., Y. Kanai, T. Oyama, K. Yoshiura, Y. Shimoyama, W. Birchmeier, T. Sugimura, and S. Hirohashi. 1994. E-cadherin gene mutations in human gastric carcinoma cell lines. Proc. Natl. Acad. Sci. USA. 91:1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacquelet, A., L. Lin, and P. Rorth. 2003. Binding site for p120/delta-catenin is not required for Drosophila E-cadherin function in vivo. J. Cell Biol. 160:313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl, A.K., P. Wilgenbus, U. Dahl, H. Semb, and G. Christofori. 1998. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 392:190–193. [DOI] [PubMed] [Google Scholar]
- Pettitt, J., E.A. Cox, I.D. Broadbent, A. Flett, and J. Hardin. 2003. The Caenorhabditis elegans p120 catenin homologue, JAC-1, modulates cadherin-catenin function during epidermal morphogenesis. J. Cell Biol. 162:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis, P. 2000. Wnt signaling and cancer. Genes Dev. 14:1837–1851. [PubMed] [Google Scholar]
- Reynolds, A.B., D.J. Roesel, S.B. Kanner, and J.T. Parsons. 1989. Transformation-specific tyrosine phosphorylation of a novel cellular protein in chicken cells expressing oncogenic variants of the avian cellular src gene. Mol. Cell. Biol. 9:629–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, A.B., L. Herbert, J.L. Cleveland, S.T. Berg, and J.R. Gaut. 1992. p120, a novel substrate of protein tyrosine kinase receptors and of p60v-src, is related to cadherin-binding factors beta-catenin, plakoglobin and armadillo. Oncogene. 7:2439–2445. [PubMed] [Google Scholar]
- Reynolds, A.B., J. Daniel, P.D. McCrea, M.J. Wheelock, J. Wu, and Z. Zhang. 1994. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol. Cell. Biol. 14:8333–8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibamoto, S., M. Hayakawa, K. Takeuchi, T. Hori, K. Miyazawa, N. Kitamura, K.R. Johnson, M.J. Wheelock, N. Matsuyoshi, M. Takeichi, et al. 1995. Association of p120, a tyrosine kinase substrate, with E- cadherin/catenin complexes. J. Cell Biol. 128:949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi, M. 1995. Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 7:619–627. [DOI] [PubMed] [Google Scholar]
- Thoreson, M.A., P.Z. Anastasiadis, J.M. Daniel, R.C. Ireton, M.J. Wheelock, K.R. Johnson, D.K. Hummingbird, and A.B. Reynolds. 2000. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J. Cell Biol. 148:189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreson, M.A., and A.B. Reynolds. 2002. Altered expression of the catenin p120 in human cancer: implications for tumor progression. Differentiation. 70:583–589. [DOI] [PubMed] [Google Scholar]
- Vleminckx, K., L. Vakaet, Jr., M. Mareel, W. Fiers, and F. van Roy. 1991. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 66:107–119. [DOI] [PubMed] [Google Scholar]
- Wahl, J.K., III, Y.J. Kim, J.M. Cullen, K.R. Johnson, and M.J. Wheelock. 2003. N-cadherin-catenin complexes form prior to cleavage of the proregion and transport to the plasma membrane. J. Biol. Chem. 278:17269–17276. [DOI] [PubMed] [Google Scholar]
- Waibler, Z., A. Schafer, and A. Starzinski-Powitz. 2001. mARVCF cellular localisation and binding to cadherins is influenced by the cellular context but not by alternative splicing. J. Cell Sci. 114:3873–3884. [DOI] [PubMed] [Google Scholar]
- Xiao, K., D.F. Allison, M.D. Kottke, S. Summers, G.P. Sorescu, V. Faundez, and A.P. Kowalczyk. 2003. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J. Biol. Chem. 278:19199–19208. [DOI] [PubMed] [Google Scholar]
- Yap, A.S. 1998. The morphogenetic role of cadherin cell adhesion molecules in human cancer: a thematic review. Cancer Invest. 16:252–261. [DOI] [PubMed] [Google Scholar]
- Zhu, A.J., and F.M. Watt. 1996. Expression of a dominant negative cadherin mutant inhibits proliferation and stimulates terminal differentiation of human epidermal keratinocytes. J. Cell Sci. 109:3013–3023. [DOI] [PubMed] [Google Scholar]