

Abstract
Breast epithelial cells differentiate into tubules when cultured in floating three-dimensional (3D) collagen gels, but not when the cells are cultured in the same collagen matrix that is attached to the culture dish. These observations suggest that the biophysical properties of collagenous matrices regulate epithelial differentiation, but the mechanism by which this occurs is unknown. Tubulogenesis required the contraction of floating collagen gels through Rho and ROCK-mediated contractility. ROCK-mediated contractility diminished Rho activity in a floating 3D collagen gel, and corresponded to a loss of FAK phosphorylated at Y397 localized to 3D matrix adhesions. Increasing the density of floating 3D collagen gels also disrupted tubulogenesis, promoted FAK phosphorylation, and sustained high Rho activity. These data demonstrate the novel finding that breast epithelial cells sense the rigidity or density of their environment via ROCK-mediated contractility and a subsequent down-regulation of Rho and FAK function, which is necessary for breast epithelial tubulogenesis to occur.
Keywords: contractility; FAK; Rho; ROCK; breast tubulogenesis
Introduction
The interaction between breast epithelial cells and the surrounding ECM is necessary for polarization, differentiation, and growth control. In vitro, breast epithelial cells differentiate into polarized duct-like tubules when cultured in a floating three-dimensional (3D) collagen matrix (Keely et al., 1995). Interestingly, we and others observe that this differentiation does not occur when the cells are cultured in the same 3D collagen matrix that is attached to the culture dish, making it more rigid (Parry et al., 1985). This implies that a biophysical component is involved in regulating breast epithelial tubulogenesis. This observation is notable because women with dense breast tissue, which is associated with a substantial increase in collagen deposition in the stroma, have a four- to sixfold increased risk of developing breast cancer (Boyd et al., 1998). However, the mechanism by which breast epithelial cells sense and respond to the mechanical properties of their surrounding environment is completely unknown.
In nonepithelial cells, relatively more is known about the response to biophysical signals. Because all cells contact the ECM through integrins, which link external environmental cues to the actin cytoskeleton, focal adhesions are poised to transduce responses to mechanical stimuli (Geiger and Bershadsky, 2002). Specifically, focal adhesion kinase (FAK), a key signaling component in focal adhesions, is emerging as a critical player in mechanosensing, as FAK-null fibroblasts cannot detect soft vs. rigid substrata (Wang et al., 2001). Phosphorylation of FAK at its autophosphorylation site, Y397, seems to be involved in this response because FAK is phosphorylated when mechanical strain is applied to smooth muscle and endothelial cells (Yano et al., 1996; Tang et al., 1999). Autophosphorylation of FAK at Y397 is also lost in fibroblasts cultured in cell-derived 3D matrices (Cukierman et al., 2001). Focal adhesion components in these cells were different from those found in characteristic focal adhesions observed on two-dimensional (2D) substrates, supporting the theories that focal adhesions may act as mechanosensors (Geiger and Bershadsky, 2002) and that focal adhesions formed under different mechanical environments have different composition and could therefore transmit different signals.
The small GTPase Rho is implicated in generating contractility necessary to drive focal adhesion and stress fiber formation (Ridley and Hall, 1992; Chrzanowska-Wodnicka and Burridge, 1996; Amano et al., 1997). Rho can be activated downstream of integrin engagement (Ren et al., 1999, 2000; Arthur et al., 2000). Because integrins are needed to maintain breast epithelial differentiation (Keely et al., 1995), we hypothesized that Rho may also be an important regulator of breast tubulogenesis. The purpose of this study was to investigate the mechanism by which breast cells sense and respond to differences in the mechanical properties of a 3D collagen matrix. Here, we show that in vitro breast epithelial tubulogenesis is accompanied by a decrease in focal adhesion formation and a loss of FAK phosphorylated at Y397 localized to focal adhesions. Although contractility through Rho and its effector Rho Kinase (ROCK) is needed to sense ECM rigidity, Rho activity must then be down-regulated, which is necessary for breast epithelial differentiation into tubules.
Results
The rigidity of the ECM regulates breast epithelial tubulogenesis
To investigate the effects of matrix rigidity on breast tubulogenesis, well-differentiated T47D breast carcinoma cells were cultured in 3D collagen gels (1.3 mg/ml collagen). Gels were detached from the sides and bottom of the dish, allowing them to float in medium (floating), or left attached to the dish (attached). Cells in floating 3D gels were able to form duct-like tubules (Fig. 1 A, c), whereas cells in attached 3D gels did not form tubules (Fig. 1 A, b); rather, the cells spread, and their morphology was similar to cells plated on a 2D collagen substratum (Fig. 1 A, a).
Figure 1.
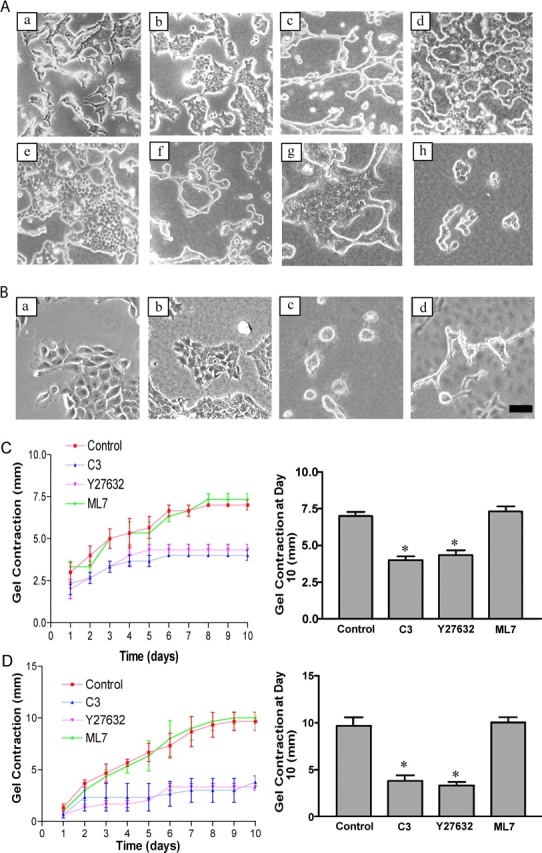
The rigidity of the collagen matrix regulates tubulogenesis through Rho- and ROCK-mediated contractility. (A) T47D breast epithelial cells were cultured on collagen-coated plastic (a), for 10 d in 3D collagen gels (1.3 mg/ml) that were left attached to the dish (b) or floated into the culture medium (c). Inhibition of Rho with C3 exoenzyme (10 μg/ml) (d) or ROCK with Y27632 (10 μM) (e) in floating 3D collagen gels blocked tubulogenesis, whereas inhibition of MLCK with ML7 (10 μM) did not alter tubulogenesis (f). Inhibition of actin–myosin contractility with BDM (20 mM) (g) or H-7 (300 μM) (h) also blocked tubulogenesis. (B) MCF10A cells do not differentiate when cultured on collagen-coated plastic (a) or in attached 3D gels (b), but do form acini in a floating collagen gel (c). Addition of HGF (50 ng/ml) promotes the formation of tubules in a floating gel (d). Bar: (A and B) 100 μm. (C) Inhibition of Rho with C3 (10 μg/ml) or ROCK with Y27632 (10 μM), but not MLCK with ML7 (10 μM), blocks contraction of floating collagen gels by T47D cells. The diameter of the gel was measured every day for 10 d, and graphed as total change in diameter in millimeters (left). The graph on the right shows the extent of gel contraction on the tenth day, expressed as total change in diameter. Data is expressed ±SEM for four separate experiments. Inhibition of Rho or ROCK significantly (*P < 0.05) decreased gel contraction. (D) Contraction of floating gels by MCF10A cells also shows significant (*P < 0.05) sensitivity to Rho and ROCK inhibition, but not MLCK inhibition.
This phenomenon was also observed in other breast cell lines. Noncancerous MCF10A breast epithelial cells do not form differentiated structures when plated on a 2D collagen substratum (Fig. 1 B, a) or when cultured in an attached 3D gel (Fig. 1 B, b). However, when cultured in a floating 3D collagen gel, MCF10A cells form differentiated acinar-like structures (Fig. 1 B, c). Addition of hepatocyte growth factor (HGF), a growth factor that induces epithelial branching (Montesano et al., 1991), promoted tubule formation in cells cultured in floating 3D gels (Fig. 1 B, d). In addition, primary mouse (unpublished data) and rat (Larsen, M., and C. Jefcoate, personal communication) mammary epithelial cells also differentiate only in floating 3D collagen matrices. These data suggest that breast epithelial differentiation in floating 3D collagen gels is not specific to particular breast cell lines. Moreover, these results suggest that the biophysical properties of the ECM regulate breast epithelial differentiation.
Breast epithelial cells require the α2β1 integrin for tubulogenesis (Keely et al., 1995) and use this integrin to exert force on collagen gels (Zutter et al., 1999). Therefore, we determined the ability of breast epithelial cells to contract floating collagen gels. T47D cells in floating 3D gels contracted the gels by several millimeters (Fig. 1 C, control). MCF10A cells (Fig. 1 D) and primary mouse mammary cells (unpublished data) also contracted floating 3D collagen gels, suggesting a correlation between cellular contractility and epithelial tubulogenesis.
ROCK-mediated contractility regulates breast tubulogenesis and collagen gel contraction
Cellular contraction is mediated by myosin light chain (MLC) phosphorylation, which regulates actin–myosin interactions by increasing myosin ATPase activity. MLC kinase (MLCK) can phosphorylate MLC on S19 and T18, leading to increased contractility (Ikebe and Hartshorne, 1985). In addition, the Rho effector ROCK promotes contractility by phosphorylating MLC on S19 and by inactivating MLC phosphatase (Amano et al., 1996; Kimura et al., 1996; Kureishi et al., 1997).
These pathways were inhibited in order to further investigate the relationship between cellular contractility and epithelial tubulogenesis. Inhibitors were dissolved in media and added to the gels the day after they were cast, when the gels were detached or left attached to the culture dish. Inhibition of Rho, with C3 exoenzyme, or ROCK, with Y27632, blocked the tubulogenesis of T47D breast cells in floating 3D collagen gels (Figs. 1 A, d and e). These inhibitors also blocked the contraction of the collagen gels by the cells (Fig. 1 C). Addition of C3 or Y27632 at day 6, when tubules have already formed, also disrupted tubulogenesis (unpublished data), suggesting that cells continue to require Rho-mediated contractility even after tubules have formed. In contrast, inhibition of MLCK with ML7 did not disrupt tubulogenesis or contraction of the gels (Fig. 1 A, f, and C). The contraction of collagen gels by MCF10A cells was also sensitive to inhibition of Rho and ROCK, but not MLCK (Fig. 1 D).
Since both ROCK and MLCK lead to MLC phosphorylation and contractility, it was of interest that we find minimal effect of MLCK inhibition on gel contraction and tubulogenesis. Differences between ROCK and MLCK have also been demonstrated in fibroblasts, where they differ in spatial localization and regulation (Totsukawa et al., 2000). In addition, although ROCK and MLCK both modulate MLC phosphorylation, they do so differently. Therefore, inhibition of either kinase may yield different results if differential phosphorylation of MLC on T18 by MLCK is functionally significant.
To confirm that contraction downstream of ROCK is required for tubulogenesis, the general inhibitors of actin–myosin contractility, BDM and H7, were added to the cells. Addition of BDM or H7 the day after the gels were poured blocked the cells from growing (unpublished data). However, when BDM and H7 were added at day 6, when tubules were already formed, both inhibitors disrupted the tubulogenesis of T47D cells (Figs. 1 A, g and h) and blocked further 3D gel contraction (unpublished data). This finding demonstrates that tubulogenesis functionally requires actin–myosin contractility.
Breast cells in a floating 3D collagen gel down-regulate Rho activity
Because tubulogenesis required Rho and ROCK (Fig. 1 A), the role of Rho in tubulogenesis was further examined. Surprisingly, stable expression of either constitutively activated Rho(63L) or dominant–negative Rho(19N) disrupted tubulogenesis of T47D breast cells in floating 3D gels (Figs. 2 A, b and c) compared with control cells (Fig. 2 A, a). In addition, the misregulation of Rho by the stable expression of constitutively activated exchange factors specific for Rho, Lsc (Fig. 2 A, d) or Lfc (unpublished data) (Glaven et al., 1996), kept Rho activity high (Fig. 2 C), and also disrupted tubulogenesis in cells cultured in floating 3D collagen gels. These findings suggest that proper regulation of Rho activity is a necessary aspect of breast tubulogenesis in a collagen gel.
Figure 2.

Rho activity is regulated by ECM rigidity. (A) Compared with control cells (a), stable expression of constitutively activated Rho (63L) (b) or dominant–negative Rho (19N) (c) disrupted tubulogenesis. Expression of truncated, constitutively activated Lsc, an exchange factor specific for Rho, also disrupted tubule formation (d). Bar, 100 μm. (B) ECM rigidity regulates Rho activity. T47D cells were stimulated in suspension with collagen fibers (3D) or on a collagen coated plate (30 μg/ml) (2D). At the times indicated, cells were lysed, and 30 μg RBD–GST was incubated with the cell lysates to pull down active Rho. Active and total Rho was detected by Western blotting. (C) Rho activity is significantly down-regulated in a floating 3D collagen gel by 1 h. Quantitation was performed on five individual experiments, and is shown in the bar graph on the right (*P < 0.05 vs. 2D). (D) Rho activity significantly (*P < 0.05) remains down-regulated in a floating, but not attached, collagen gel after 24 h. Quantitation was performed on four individual experiments.
To determine if Rho is regulated in response to the biophysical properties of the ECM, Rho activity was analyzed, making use of the Rho-binding domain (RBD) of the Rho effector, Rhotekin, fused to GST, to pull down Rho-GTP from cell lysates. T47D cells plated on a rigid 2D collagen substratum sustained high Rho activity during the 60-min assay, whereas cells incubated with 3D flexible collagen fibers down-regulated Rho activity by 60 min (Fig. 2 B), suggesting that the physical properties of the ECM can regulate Rho activity.
T47D cells were then cultured in 3D collagen gels for 1 or 24 h, and Rho activity was analyzed. In the absence of collagen stimulation, Rho activity was high (Fig. 2 C). Cells plated on a rigid 2D collagen substratum or in an attached 3D collagen gel sustained high Rho activity, whereas cells in a floating 3D collagen matrix significantly down-regulated Rho activity by 1 or 24 h (Figs. 2, C and D, respectively). This effect was specific for Rho, since tyrosine phosphorylation of several proteins was stimulated under these conditions (unpublished data). This indicates that the rigidity of the ECM can regulate Rho activity in cells cultured in attached vs. floating collagen gels.
ROCK-mediated contractility assists in the down-regulation of Rho activity, tubulogenesis, and collagen gel contraction
ROCK is an effector of Rho that was necessary for tubulogenesis (Fig. 1 A, e). Since Rho misregulation disrupted tubulogenesis and contraction of floating 3D collagen gels, we reasoned that misregulation of ROCK would also affect these processes. T47D cells stably expressing control vector, wild-type ROCK (WT), kinase-dead ROCK that is also defective in Rho binding, ROCK (KD-IA), or a deletion mutant that renders ROCK constitutively activated, ROCK (Δ3), were cultured in floating 3D collagen gels. Cells expressing ROCK (WT) formed tubules, although the tubules were more developed (Fig. 3 A, b) when compared with control cells (Fig. 3 A, a). The expression of ROCK (KD-IA) disrupted tubulogenesis; these cells formed large, abnormal structures (Fig. 3 A, d). Because we used the ROCK (KD-IA) construct that does not bind to Rho, the effect is not likely due to nonspecific sequestration of Rho by ROCK (KD-IA). Cells expressing ROCK (Δ3) had altered larger tubules that often had projections that attached the tubules to one another (Fig. 3 A, c).
Figure 3.
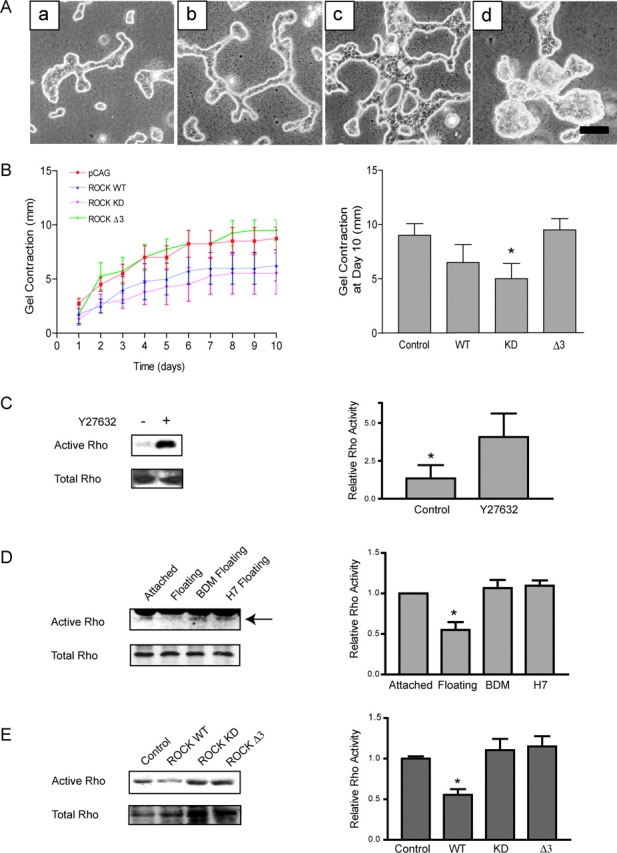
Proper ROCK regulation is required for tubulogenesis, collagen gel contraction, and Rho regulation. (A) Control cells (a) and cells expressing wild-type (WT) ROCK (b) form tubules in floating collagen gels. Misregulation of ROCK by the expression of constitutively activated ROCK (Δ3) (c) or kinase-dead ROCK (KD-IA) (d) alter tubulogenesis. Bar, 100 μm. (B) Expression of ROCK (KD-IA) decreases the contraction of floating 3D gels (*P = 0.066) compared with control cells. ROCK (WT) and (ROCK Δ3) do not significantly affect contraction (P = 0.253 and 0.750, respectively compared with control cells). (C) The presence of Y27632 (10 μM) for 1 h significantly (*P < 0.05) reduced the down-regulation of Rho activity, suggesting a negative feedback loop to Rho from its effector, ROCK. In the example shown, two noncontinuous lanes from the same gel have been moved next to one another to remove intervening lanes treated with other inhibitors. The quantitation shown (right) is a representation of four similar experiments. (D) Rho-GTP was determined in cells cultured in floating 3D collagen gels in the presence or absence of 20 mM BDM or 300 μM H7 for 60 min. Both inhibitors prevented the down-regulation of Rho activity. Quantitation was performed on eight individual experiments (bar graph; *P<0.05 vs. BDM and H7). (E) Expression of ROCK (WT) significantly decreases Rho activity in a floating 3D collagen gel, but misregulation of ROCK activity by expression of ROCK (Δ3) or (KD) increases Rho activity, suggesting proper regulation of ROCK is required to down-regulate Rho. The experiment was repeated five times and quantitation performed (bar graph; *P < 0.05 vs. control).
The ability of these cells to contract floating 3D gels was also examined. Compared with control cells, cells expressing ROCK (KD-IA) decreased contraction of the surrounding matrix (Fig. 3 B). These data support the hypothesis that ROCK mediates collagen gel contraction.
Finally, Rho activity was analyzed in the presence of the ROCK inhibitor, Y27632, to determine if ROCK-mediated contractility modulates Rho activity. Inhibition of ROCK significantly reduced the ability of the cells to down-regulate Rho in a floating 3D gel (Fig. 3 C). Furthermore, inhibition of contractility with BDM or H7 in cells in floating gels also kept Rho activity significantly high (Fig. 3 D), indicating that contractility downstream of ROCK helps down-regulate Rho activity. Because this suggests a ROCK-dependent feedback loop regulating Rho activity, Rho activity was assessed in the above cell lines. Cells expressing ROCK (WT) had significant decreased Rho activity compared with control cells (Fig. 3 E) when cultured in a floating 3D collagen gel for 1 h, again demonstrating that ROCK down-regulates Rho. Cells expressing ROCK (KD-IA) retained high Rho activity, confirming the effect of the pharmacological inhibitor. Cells expressing ROCK (Δ3) also retained high Rho activity, suggesting that, like Rho, ROCK is highly regulated and its misregulation will alter Rho activity, tubulogenesis, and cellular contraction.
ECM rigidity and cellular contractility regulate the localization of FAK phosphorylated at Y397 to focal adhesions
Because Rho is implicated in focal adhesion formation, and we find that Rho is regulated by ECM rigidity, the structure and composition of focal adhesions were analyzed in T47D cells cultured in attached and floating 3D collagen gels. Cells in 3D gels were immunostained for the focal adhesion components FAK and vinculin. Cells in floating 3D gels, which form tubules, localized vinculin to cell–cell junctions and to the periphery of cells, but notably not to adhesive structures (Fig. 4, insets). FAK immunostaining was diffuse in cells in floating collagen gels. This differed from cells in attached gels, in which FAK and vinculin localized to small punctate matrix adhesions located at extensions in the cell periphery (Fig. 4). Cells cultured in floating 3D gels had subcortical actin at sites of cell–cell junctions, whereas cells in attached 3D gels still formed small actin fibers localized at protruding cell peripheries (Fig. 4).
Figure 4.

The rigidity of the matrix regulates 3D matrix adhesion formation in breast epithelial cells. Components of focal adhesions were analyzed by immunostaining T47D cells cultured in collagen gels (1.3 mg/ml). Breast cells in attached, but not floating, 3D gels show FAK, vinculin, and p16 localized to small punctate 3D matrix adhesions (insets). Actin fibers are also localized to these structures (insets). Bar: 50 μm; (insets) 10 μm.
Cells in floating 3D gels did not form these extensions into the collagen matrix. Because protrusive activity is often associated with Arp2/3 activity (Svitkina and Borisy, 1999; Blanchoin et al., 2000), cells were also stained for p16, a component of the Arp2/3 complex. Cells in floating 3D gels had cytoplasmic p16 staining, which did not localize to adhesions at the cell–ECM interface (Fig. 4). However, cells in attached 3D gels showed punctate p16 staining at the cell periphery and enhanced staining at cell protrusions extending into the ECM (Fig. 4).
Because FAK phosphorylation at Y397 is regulated by the physical properties of the matrix (Cukierman et al., 2001), cells in attached or floating collagen gels were analyzed for FAK phosphorylated at Y397 (pY397). Breast cells cultured on 2D collagen-coated coverslips had prominent FAK pY397 at focal adhesions (Fig. 5 A, a). Cells in attached collagen gels also retained FAK pY397 localized to 3D matrix adhesions extending from the cells (Fig. 5 A, c). However, breast cells cultured in floating 3D collagen gels, a condition that promotes tubulogenesis (Fig. 1 A, c), lost the localization of FAK pY397 to matrix adhesions (Fig. 5 A, b), even though these cells did show diffuse cytoplasmic FAK staining (Fig. 4). Western blot analysis revealed that the levels of FAK pY397 were not significantly different in cells in attached vs. floating gels (Fig. 5 C), indicating that ECM rigidity is not regulating the phosphorylation of FAK at this site, but rather the localization of FAK pY397 to matrix adhesions.
Figure 5.
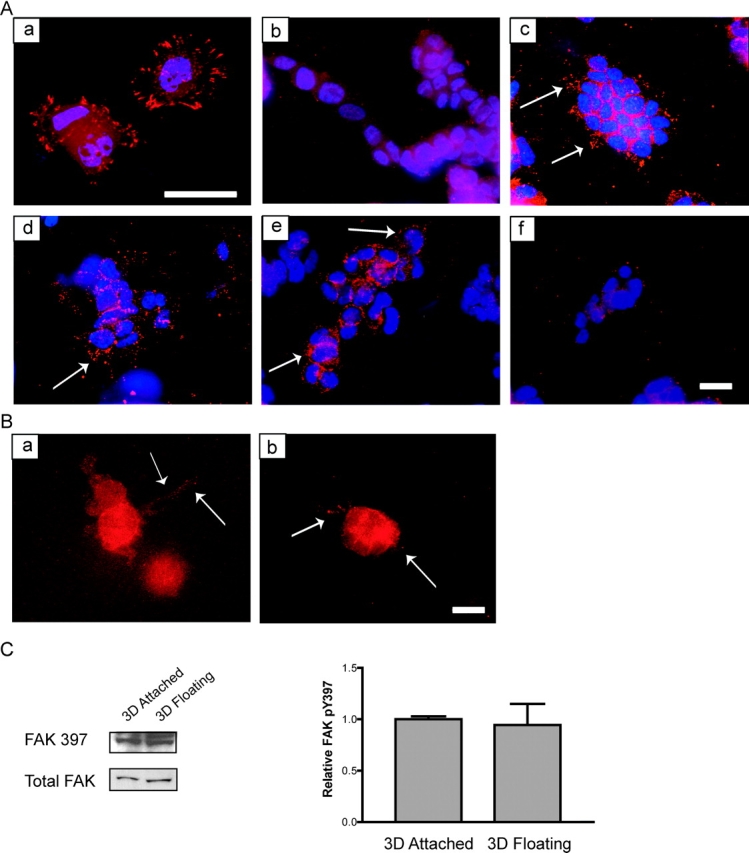
The rigidity of the matrix and cellular contractility regulates the localization of FAK phosphorylated at Y397 at 3D matrix adhesions. (A) FAK phosphorylated at Y397 is localized to focal adhesions when T47D cells are plated on 2D collagen-coated coverslips (a). In a floating 3D gel, FAK phosphorylation at Y397 is minimal (b). Cells cultured in an attached 3D collagen gel localized FAK pY397 to punctate adhesions (c). Cells in floating 3D gels treated with C3 (10 μg/ml) (d) or Y27632 (10 μM) (e) localized FAK pY397 to small 3D matrix adhesions (see arrows), whereas cells treated with ML7 (10 μM) lost this localization (f). FAK pY397 is shown in red, and nuclei are shown in blue. Bar: (a) 25 μm; (b–f) 50 μm. (B) Cells treated with BDM (20 mM) (a) or H7 (300 μM) (b) also localized FAK pY397 to punctuate matrix adhesions. Bar, 50 μm. (C) The localization, and not phosphorylation, of FAK Y397 is regulated by ECM rigidity. Western blot analysis of FAK pY397 in attached vs. floating collagen gels (1.3 mg/ml) show statistically similar (P = 0.8014) levels of phosphorylation and FAK expression. The quantitation represents four individual experiments (right).
FAK pY397 localization was determined in cells cultured in floating gels that had been treated with the Rho inhibitor, C3 exoenzyme, or the ROCK inhibitor, Y27632, both of which blocked contraction and tubulogenesis. These cells retained FAK pY397 localized to punctate matrix adhesions (Fig. 5 A, d and e, respectively). In contrast, treatment with the MLCK inhibitor, ML7, which did not block contraction or tubulogenesis, did not localize FAK pY397 to matrix adhesions (Fig. 5 A, f). Treatment with the more general contractility inhibitors, BDM (Fig. 5 B, a) or H7 (Fig. 5 B, b), localized FAK pY397 to matrix adhesions, further suggesting a role for contraction of the collagen matrix in down-regulation of Rho activity and matrix adhesions.
ECM rigidity regulates breast epithelial cell proliferation
The above results suggest that ROCK-mediated cellular contractility regulates the recruitment of FAK pY397 into matrix adhesions, and implicate this event in regulating breast differentiation into tubules. An important aspect of epithelial differentiation is controlled cell growth. It has been reported that FAK regulates cell cycle progression into S phase and that this function of FAK is dependent on phosphorylation on Y397. Because we see differences in the localization of FAK pY397 in attached vs. floating collagen gels, cell proliferation was examined under these conditions. Cells were cultured in attached or floating 3D gels for 7 d and then stained for Ki67, a marker of proliferation (Brown and Gatter, 2002). Cells cultured in an attached gel show a significant increase in proliferation when compared with cells in a floating gel (Fig. 6). This demonstrates an association between FAK activity and cell proliferation. Furthermore, these data suggest that increased ECM rigidity can increase epithelial proliferation, which will likely disrupt normal tubule structure.
Figure 6.

ECM rigidity regulates breast epithelial proliferation. T47D cells were cultured in floating and attached 3D collagen gels and allowed to grow for 7 d. The gels were then fixed and costained for Ki67, a marker of proliferation, and bisbenzimide, which will stain all nuclei regardless of cell cycle state. Cells in floating collagen gels have significantly (*P < 0.05) decreased proliferation compared with cells in attached gels. The graph is representative of three individual experiments.
Collagen density regulates tubulogenesis, cellular contraction, and FAK phosphorylation at Y397
In vivo, differences in the biophysical properties of the ECM are potentially important. In the breast, an increase in stromal density, which corresponds to an increase in collagen and fibronectin deposition and an increase in fibroblasts, is correlated with a significant increased risk of developing breast cancer (Boyd et al., 1998). We hypothesize that an increase in collagen concentration in vivo causes an increase in the rigidity of the ECM, which may promote tumor formation due to increased FAK and Rho activity, which will disrupt differentiation and promote cellular proliferation.
To test whether altering collagen density alone, in the absence of other ECM components and fibroblasts, is sufficient to alter breast cell behavior, T47D cells were cultured in gels of varying collagen concentrations. Collagen gels composed of higher concentrations of collagen were less translucent than gels made of lower concentrations of collagen, correlating with an increase in density (Demou and McIntire, 2002). Low concentrations of collagen, from 0.8 mg/ml up to 1.5 mg/ml, promoted tubulogenesis in floating 3D gels (Fig. 7 A, left panels). However, when the collagen concentration was increased over a narrow range, tubulogenesis was disrupted, even though these cells were in floating gels, which normally promote tubulogenesis (Fig. 7 A). This indicates that an increase in collagen concentration alone is sufficient to disrupt tubulogenesis. We do not think this effect is due to increased integrin ligand binding due to higher concentrations of collagen, since tubulogenesis and localization of FAK phosphorylated at Y397 were also altered when gels of identical collagen concentrations were compared in attached vs. floating gels (Figs. 1 A and 5 A). This supports the notion that the biophysical properties of the ECM, and not simply its concentration, are determinants for tubulogenesis and phosphorylation of FAK.
Figure 7.
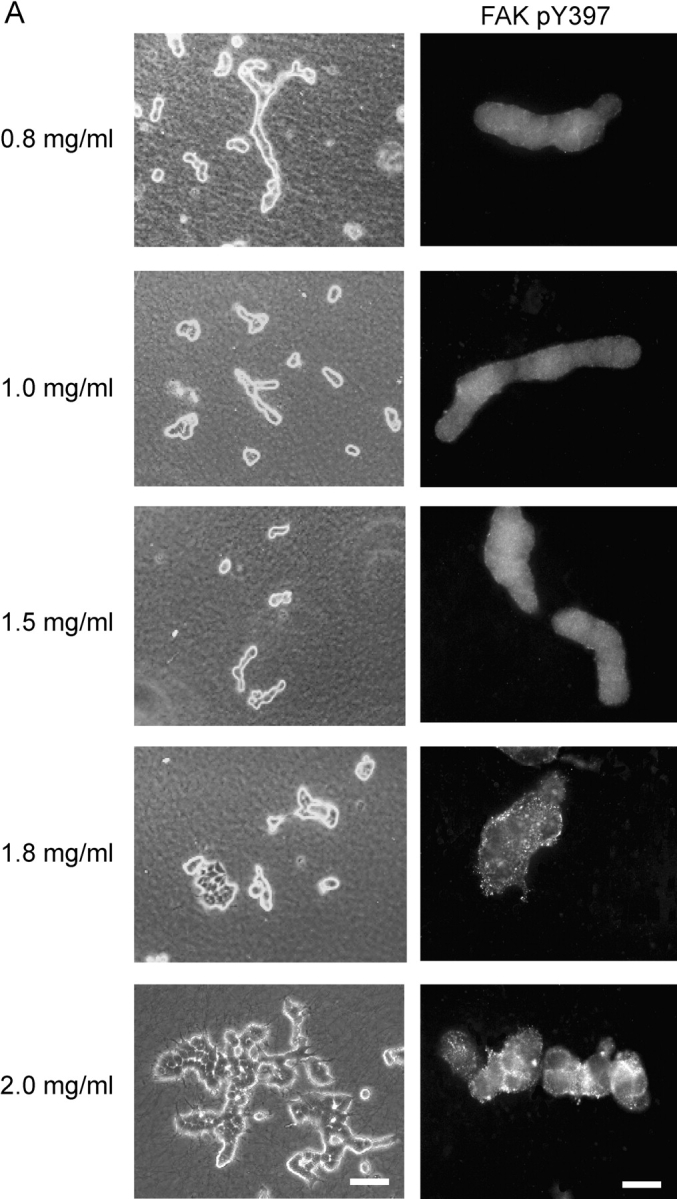
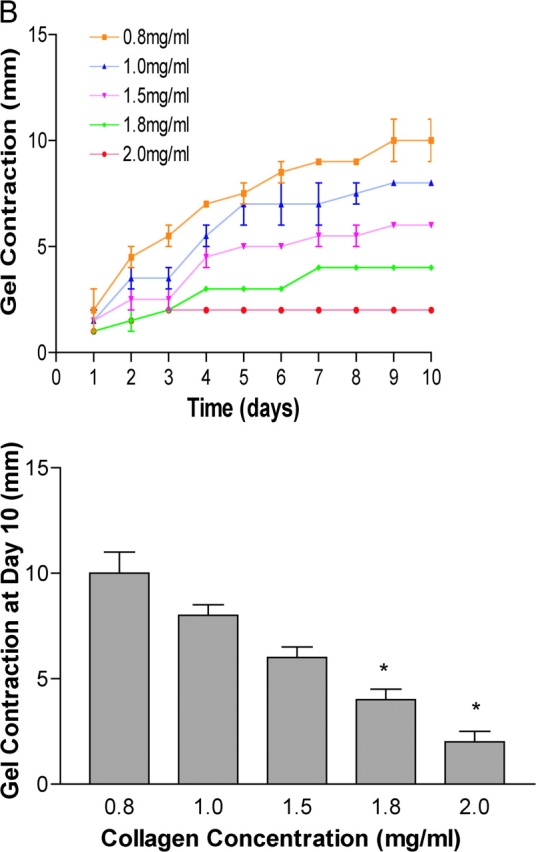
Collagen density regulates tubulogenesis, the localization of FAK phosphorylation at Y397, and cellular contraction. (A) T47D cells were cultured in floating collagen gels composed of increasing concentrations of collagen, as indicated. Collagen concentrations equal to or above 1.8 mg/ml disrupt tubulogenesis (left panels). Bar, 100 μm. Increasing collagen concentration also increased FAK pY397 localized to 3D matrix adhesions (right panels). Bar, 50 μm. (B) Increasingly dense collagen gels significantly (*P < 0.05) decrease the contraction of floating 3D collagen gels by T47D breast epithelial cells. The data are an average of three experiments.
Altering the collagen concentration also altered the ability of the cells to contract the gel. T47D breast cells cultured in gels composed of high concentrations of collagen showed a significant decrease in their ability to contract a floating 3D collagen gel (Fig. 7 B). The most straightforward explanation is that the increased density created by greater collagen concentrations makes the gel more rigid so that the cells cannot contract the gel, comparable to the situation cells encounter in an attached 3D collagen gel.
Phosphorylation of FAK at Y397 was also analyzed in these cells. Breast cells in 3D gels composed of lower concentrations of collagen (≤1.5 mg/ml), which formed tubules, had diffuse FAK pY397 throughout the cell (Fig. 7 A, right panels). However, cells cultured in gels composed of higher concentrations of collagen (≥1.8 mg/ml ), in which tubulogenesis was disrupted, localized FAK pY397 to matrix adhesions (Fig. 7 A).
Finally, we performed the Rho activity assay on cells cultured in floating collagen gels of high concentration (1.8 mg/ml). Cells in floating collagen gels composed of 1.3 mg/ml have decreased Rho activity compared with cells in attached gels (Fig. 2, C and D). However, cells cultured in high concentration collagen significantly sustained high Rho activity at 1 and 24 h (Fig. 2, C and D). Together, these data indicate that an increase in collagen density can promote a phenotype similar to an attached 3D collagen gel: the cells cannot contract the gel, Rho activity remains high, FAK pY397 is localized to matrix adhesions, and tubulogenesis is disrupted.
Discussion
It has long been appreciated that breast epithelial cells maintain differentiated structures and produce milk proteins unique to differentiated cells if they are cultured in a floating, but not an attached, 3D collagen gel (Emerman and Pitelka, 1977; Lee et al., 1984). Here, we set out to determine how breast cells sense and respond to the rigidity of their ECM, and how this regulates breast epithelial tubulogenesis.
We propose that breast cells use ROCK-mediated contractility as a sensor to detect the rigidity of the ECM (Fig. 8). In a flexible ECM (such as a floating 3D collagen gel), cellular contraction on the matrix is met with minimal resistance, which then feeds back to diminish Rho activity, focal adhesion formation, and the localization of FAK pY397 to matrix adhesions, resulting in breast tubulogenesis. In cells encountering a rigid or dense ECM, contractile forces do not contract the matrix, but rather result in tension generated within the cell. In this case, Rho activity remains high and matrix adhesions are formed that include FAK pY397. We hypothesize that formation of 3D matrix adhesions containing FAK pY397 fundamentally alters subsequent signaling events in rigid vs. flexible matrices, leading to an increase in cell proliferation and a disruption of breast epithelial tubulogenesis.
Figure 8.

Model for regulation of breast epithelial differentiation by the rigidity of 3D matrices. The α2β1 integrin binds to collagen, leading to Rho activation. Rho activates its effector, ROCK, to promote cellular contractility. If the cells are unable to contract their matrix; that is, if the matrix is rigid, attached, or dense, tension is generated within the cell. Rho activity remains high and promotes 3D matrix adhesion formation including FAK phosphorylated at Y397. This phosphorylation event links a rigid matrix to subsequent cell proliferation and the disruption of tubulogenesis. If the cells are able to contract their matrix (if the matrix is flexible, floating, or composed of low collagen density), ROCK down-regulates Rho activity. Matrix adhesions are changed and FAK phosphorylated at Y397 is not localized to adhesive structures, fundamentally altering subsequent signaling events and resulting in differentiation into tubules.
The regulation of Rho activity by ECM rigidity
Here, we report the novel observation that Rho activity is regulated by the biophysical properties of the ECM. Breast cells in flexible, floating 3D collagen gels down-regulate Rho activity, whereas cells in rigid, attached 3D collagen gels do not. Precise regulation of Rho activity is required for proper breast epithelial differentiation, as misregulation of Rho activity by expression of either constitutively active or dominant–negative Rho disrupts tubulogenesis (Fig. 2).
Surprisingly, the Rho effector, ROCK, mediates down-regulation of Rho in floating 3D matrices. This suggests the presence of a regulatory feedback loop that enhances the response of cells to matrix rigidity. The mechanism by which this occurs is currently unknown, but likely reflects changes in either Rho GEFs or GAPs in a manner regulated by contractility and the biophysical properties of the ECM. It is therefore interesting that down-regulation of Rho through p190RhoGAPB has been implicated in mammary gland development by regulating terminal end bud formation and ductal morphogenesis by controlling IGF signaling (Chakravarty et al., 2000, 2003). This suggests that proper formation of in vivo differentiated structures in the breast requires precise regulation of Rho activity, which is consistent with our in vitro observations.
There are important implications for these observations. Besides promoting focal adhesion and stress fiber formation, Rho has several other effectors that direct signaling events leading to cell proliferation, gene expression, cell motility, transformation, and invasion. Differentiated breast epithelium exists in a highly regulated state in which cellular proliferation and motility are diminished to maintain tissue organization. Therefore, our observation that matrix rigidity regulates Rho activity may clarify how matrix rigidity regulates breast epithelial differentiation. If Rho activity remained high to promote proliferation, motility, and focal adhesion formation, it is likely that breast cells would lose their differentiated organization, which is the case in attached gels.
ROCK-generated contractility is required for breast tubulogenesis in 3D floating gels
The Rho effector, ROCK, is implicated in generating actin–myosin contractility, and here we find that this contractility regulates breast tubulogenesis. Although ROCK has several potential effectors, our data suggests a role for ROCK effects on contractility per se, since tubulogenesis (Fig. 1 A) and the decrease in Rho activity (Fig. 3 D) could be inhibited by the actin–myosin inhibitors, BDM or H7. Indeed, gel contraction precedes tubulogenesis since contraction is observed within the first 24 h and is greatest through day 6, followed by tubulogenesis, which is first observed around days 5 or 6.
ROCK has also been shown to regulate tubulogenesis or gel contraction in other systems. Fibroblasts require both ROCK and FAK activity to contract 3D matrices composed of fibronectin and fibrin (Midwood and Schwarzbauer, 2002), which is consistent with our observations that ROCK is an important regulator of matrix contraction. In MDCK cells cultured in a 3D matrix, inhibition of ROCK disrupts the formation of cysts and causes a striking increase in the number and length of cellular protrusions (Yu et al., 2003). This is consistent with our data, where inhibition of ROCK also disrupts normal tubule formation. Together, these data implicate ROCK as an important regulator of epithelial contractility and tubulogenesis.
In contrast to these findings, ROCK has been implicated as a negative regulator of adherens junctions in MDCK cells growing as a monolayer (Sahai and Marshall, 2002). Because tubulogenesis requires adherens junction formation, these results predict that inhibiting ROCK would stabilize junctions and enhance tubule formation. However, since we observe the opposite, that inhibition of ROCK disrupts tubule formation, our results suggest a role for ROCK in tubulogenesis other than on adherens junctions. Alternatively, the difference may relate to the examination by Sahai and Marshall (2002) of junction formation in cell monolayers plated on a rigid 2D substratum rather than in a 3D matrix. ROCK-mediated contraction may pull adherens junctions apart in cells encountering opposing forces supplied by a rigid matrix, but may help to bring these same junctions together when cells are in a flexible matrix.
Focal adhesion formation and the localization of FAK phosphorylated at Y397 is regulated by ECM rigidity
Focal adhesions are emerging as mechanosensors that relay biophysical information to the cell through integrins. Several experiments demonstrate that external force exerted on integrins increases the strength and rigidity of the linkage between integrins and the actin cytoskeleton (Choquet et al., 1997; Pelham and Wang, 1997; Balaban et al., 2001; Riveline et al., 2001; Koo et al., 2002). Therefore, cells within a rigid or dense collagen matrix will encounter a restraining force predicted to increase integrin–cytoskeletal linkages (Choquet et al., 1997; Balaban et al., 2001; Riveline et al., 2001; Galbraith et al., 2002; Tan et al., 2003). In contrast, cells in a floating 3D gel will encounter an unrestrained matrix, with the prediction that integrin attachments to the cytoskeleton will not be strengthened. Consistent with these observations, we find that matrix adhesions are not seen in flexible 3D gels, but are observed in attached gels. Like breast epithelial cells, MDCK cells also down-regulate focal adhesions in a collagen gel, when compared with cells plated on collagen (Wang et al., 2003). Focal adhesion formation is significant because it will likely affect the signaling complexes formed, which will regulate subsequent cellular behavior.
The phosphorylation of FAK at Y397 in fibroblasts is regulated by the physical properties of the ECM (Cukierman et al., 2001). We now demonstrate that this occurs in epithelial cells, as cells in attached, but not floating, 3D collagen gels localize autophosphorylated FAK to matrix adhesions. This observation has many important implications, and suggests that cells in a floating 3D matrix do not receive the same biochemical signals as cells in an attached 3D matrix. Phosphorylation of FAK at Y397 is a key signaling event, linking FAK to cell proliferation, survival, and migration (Chen et al., 1996; Zhao et al., 1998; Han et al., 2000; for review see Schwartz and Assoian, 2001), all of which are regulated when breast cells differentiate. Since FAK autophosphorylation regulates cell cycle control (Zhao et al., 1998), this may explain why breast cells cultured in floating 3D gels have decreased proliferation compared with cells in attached gels (Fig. 6).
We find that inhibition of Rho, ROCK, or actin–myosin contractility promotes the localization of FAK pY397 to 3D matrix adhesions. It is surprising that we still see FAK phosphorylated at this site, since decreased Rho activity is normally associated with decreased FAK activity, whereas activation of Rho increases FAK phosphorylation (Kumagai et al., 1993; Clark et al., 1998). Given the many biochemical and morphological differences we see in breast cells cultured on 2D vs. 3D matrices, it is likely that Rho regulates FAK differently in a 3D matrix.
Collagen density and breast carcinoma
An increase in stromal density is a risk factor for breast carcinoma and involves an increase in the number of fibroblasts in the stroma, as well as an increase in collagen deposition (Guo et al., 2001). However, it is not known which of these factors contributes to the observed increased carcinoma risk associated with dense breast tissue. Our results demonstrate the novel finding that an increase in collagen density alone is sufficient to alter cellular behavior and disrupt tubulogenesis.
We also find that Rho activity and the localization of FAK pY397 to matrix adhesions is regulated by collagen density. This result implies that increased collagen density in the breast could enhance signaling through FAK, although this remains to be tested in an in vivo environment. Because phosphorylation at Y397 links FAK to downstream pathways leading to cell proliferation and migration, an intriguing possibility is that phosphorylation at this site contributes to the effects of breast stromal density on the development of breast carcinoma.
The mechanism by which cells sense and respond to the biophysical properties of their environment is physiologically important. Although emerging work places FAK and focal adhesions as key mechanosensors in fibroblasts, almost nothing is known about how epithelial cells interpret mechanical signals from the ECM. This study demonstrates the novel finding that ROCK-mediated contractility is essential for breast epithelial cells to sense the biophysical properties of their environment and to respond by down-regulating Rho and FAK activity and altering focal adhesions. The implications of this are significant, as cells that fail to do so, either due to a rigid matrix or increasing collagen density, cannot correctly differentiate into tubules.
Materials and methods
Collagen gel culture
T47D and MCF10A cells were obtained from American Type Culture Collection and maintained as described previously (Keely et al., 1995). T47D cells were stably transfected with Rho19N in pZIP, or pZIP vector alone, truncated Lfc and Lsc (a gift from Dr. Ian Whitehead, New Jersey Medical School, Newark, NJ) Rho63L, or pCAG-myc-ROCK KD-IA (a kinase-dead and non-Rho binding mutant), -ROCK Δ3 (truncated after aa 727, rendering ROCK constitutively active), -ROCK WT, or pCAG vector alone (a gift from Dr. Shuh Narumiya, Kyoto University, Kyoto, Japan) (Ishizaki et al., 1997), and expression of constructs verified by immunoblotting, using approaches described previously (Keely et al., 1997). Primary mouse mammary epithelial cells (MECs) were a gift from Dr. Caroline Alexander (University of Wisconsin, Madison, WI).
T47D and MCF10A breast epithelial cells were cultured in collagen type I gels at a final collagen concentration of 1.3 mg/ml as previously described (Keely et al., 1995). The day after the gels were poured, one set of gels was left attached to the dish (attached) and one set of gels was detached from the sides and bottom of the dish (floating). Media with or without inhibitors was added to the gels at this time, unless otherwise specified in the text. The gels were fed (with inhibitors, if they were used in the experiment) approximately every 5 d. For MCF10A cells, media with or without 50 ng/ml HGF (Calbiochem) was added to the gels at this time. C3 exoenzyme was purified in bacteria and used at 10 μg/ml. Y27632 (Welfide Corporation) was used at 10 μM, ML7 (Calbiochem) was used at 10 μM or 20 μM, 2,3-Butanedione-Monoxime (BDM) (Sigma-Aldrich) was used at 20 mM, and 1-(5-isoquinolinylsulfonyl)-2-methylpiperazine (H-7) (Sigma-Aldrich) was used at 300 μM. The cells were grown for 7–10 d, and phase contrast pictures were taken to assess morphology using a Nikon 35 mm camera attached to a TE300 Nikon inverted microscope.
For contraction studies, the diameter of the gels was measured and recorded every day for 10 d. Three to five experiments were performed and statistically analyzed using Prism GraphPad software. For collagen concentration studies, collagen gels were made composed of increasing concentrations of collagen (BD Bioscience). Morphology and FAK phosphorylation (below) were assessed after 7 d.
Rho activity assays
Subconfluent T47D cells were harvested in 0.5 mM EDTA in PBS and resuspended in 5 mg/ml fatty acid–free BSA (ICN Biomedicals) in RPMI, and equal numbers of cells were stimulated with collagen. For 3D flexible collagen, collagen was neutralized to form fibers, which were added to the cells in suspension. Rigid collagen was created by coating petri plates with 30 μg/ml collagen and allowing the cells to adhere to the collagen. For analysis of Rho activity in 3D collagen gels, the gels were made as described above containing ∼20 million cells. If the gel was to be incubated for 1 h, the gel was made with BSA in RPMI in order to determine the effect of collagen alone on the cells, and without serum stimulation. However, for 24 h time points, the gels were made with full medium, including serum, to prevent cell death.
The gels were allowed to polymerize for 1 h and then one gel released and another gel left attached. These gels were then incubated for 1 or 24 h at 37° and then the assay performed. Cells in gels were lysed in TBS buffer containing 0.1% SDS, 1% Triton X-100, protease inhibitor cocktail (Sigma-Aldrich), and sodium pervanadate. RBD-GST pull-down assays were performed as described (Arthur et al., 2000). Samples were run on a SDS-PAGE gel, transferred to a PVDF membrane, and the membrane was probed with anti-Rho (1:250; Santa Cruz Biotechnology, Inc.) followed by anti–mouse HRP (1:5000; Jackson ImmunoResearch Laboratories). Rho was detected using ECL substrate (Amersham Biosciences).
For inhibitor studies, cells were pretreated with 10 μM Y27632, 20 mM BDM, or 300 μM H7 for 15 min at 37°C before collagen stimulation, and then Rho-GTP was determined.
To quantitate results, the bands from four to eight individual experiments (pairs of active and total Rho) were quantitated using ImageQuant software (Molecular Dynamics). The quantitation was statistically analyzed using Prism GraphPad software.
Immunoblotting for FAK Y397
Lysates were prepared from cells cultured in 3D collagen gels for 1 h as described above. Relative levels of FAK phosphorylated at Y397 were determined as previously described (Kwong et al., 2003).
Immunofluorescence
Cells were grown in collagen gels for 7–10 d. The gels were fixed in 4% paraformaldehyde in PBS for 15 min at room temperature and washed for 10 min with PBS while shaking, and then 0.15 M glycine in PBS was added to the gels to quench the formaldehyde. After washing again with PBS for 10 min, 0.02% Triton X-100 in PBS was added for 10 min to permeabilize the cells. The gels were washed with PBS for 10 min and then blocked overnight at 4° with 1% fatty acid–free BSA, 1% donkey serum in PBS. The gels were then soaked in 100 μl PBS containing primary antibody diluted in PBS with 1% donkey serum for 30 min at room temperature or overnight at 4°C. Antibodies used are as follows: anti-FAK Y397 (1:100; Biosource International), anti-vinculin (1:400; Sigma-Aldrich), anti-FAK (1:200; Upstate Biotechnology), anti-Ki67 (1:80; a gift from Dr. Andreas Friedl, University of Wisconsin), and anti-p16 (1:400; a gift from Dr. William Bement, University of Wisconsin). After extensive washing in PBS, 100 μl anti–rabbit TRITC (1:100; Jackson ImmunoResearch Laboratories) or anti–mouse TRITC (1:100; Jackson ImmunoResearch Laboratories) with phalloidin-FITC (1:1000; Jackson ImmunoResearch Laboratories), Bisbenzimide (Sigma-Aldrich), and 1% donkey serum in PBS was added for 30 min at room temperature. The gels were washed again twice in PBS for 10 min and in water for 10 min, and mounted on a slide with 30 μl Prolong Antifade mounting medium (Molecular Probes). To ensure staining was specific, gels were also stained only for the secondary antibody, which showed no specific staining (unpublished data). For cell proliferation studies, 200 nuclei (cells) per experiment were counted along with the number of Ki67-positive cells. Statistical analysis was performed using Prism GraphPad software. Immunostaining was analyzed by epifluorescence using a TE300 Nikon inverted microscope equipped with a Photometrics CoolSnap fx CCD camera. Images were collected and deconvolution performed using Inovision software.
Acknowledgments
The authors are grateful to Dr. Shuh Narumiya for ROCK constructs, to Dr. Ian Whitehead for Lfc and Lsc, to Dr. Deane Mosher for providing C3 exoenzyme, to Dr. Caroline Alexander and Bob Liu for providing primary mouse mammary epithelial cells, to Dr. William Bement for providing the p16 antibody, to Dr. Andreas Friedl for providing the Ki67 antibody, to Drs. Keith Burridge and William Arthur for providing the RBD-GST construct and technical advice for RhoGTP assays, and to Drs. Caroline Alexander and Jeff Hardin for critically reading the manuscript.
This work was supported by grant RPG-00-339 from the American Cancer Society (to P.J. Keely).
Abbreviations used in this paper: 2D, two-dimensional; 3D, three-dimensional; FAK, focal adhesion kinase; FAK pY397, FAK phosphorylated on tyrosine 397; HGF, hepatocyte growth factor; MLC, myosin light chain; MLCK, MLC kinase; ROCK, Rho kinase.
References
- Amano, M., K. Chihara, K. Kimura, Y. Fukata, N. Nakamura, Y. Matsuura, and K. Kaibuchi. 1997. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science. 275:1308–1311. [DOI] [PubMed] [Google Scholar]
- Amano, M., M. Ito, K. Kimura, Y. Fukata, K. Chihara, T. Nakano, Y. Matsuura, and K. Kaibuchi. 1996. Phosphorylation and activation of myosin by Rho-associated kinase (Rho- kinase). J. Biol. Chem. 271:20246–20249. [DOI] [PubMed] [Google Scholar]
- Arthur, W.T., L.A. Petch, and K. Burridge. 2000. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr. Biol. 10:719–722. [DOI] [PubMed] [Google Scholar]
- Balaban, N.Q., U.S. Schwarz, D. Riveline, P. Goichberg, G. Tzur, I. Sabanay, D. Mahalu, S. Safran, A. Bershadsky, L. Addadi, and B. Geiger. 2001. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat. Cell Biol. 3:466–472. [DOI] [PubMed] [Google Scholar]
- Blanchoin, L., K.J. Amann, H.N. Higgs, J.B. Marchand, D.A. Kaiser, and T.D. Pollard. 2000. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature. 404:1007–1011. [DOI] [PubMed] [Google Scholar]
- Boyd, N.F., G.A. Lockwood, J.W. Byng, D.L. Tritchler, and M.J. Yaffe. 1998. Mammographic densities and breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 7:1133–1144. [PubMed] [Google Scholar]
- Brown, D., and K. Gatter. 2002. KI67 protein: the immaculate deception? Histopathology. 40:2–11. [DOI] [PubMed] [Google Scholar]
- Chakravarty, G., D. Roy, M. Gonzales, J. Gay, A. Contreras, and J.M. Rosen. 2000. P190-B, a Rho-GTPase-activating protein, is differentially expressed in terminal end buds and breast cancer. Cell Growth Differ. 11:343–354. [PubMed] [Google Scholar]
- Chakravarty, G., D. Hadsell, W. Buitrago, J. Settleman, and J. Rosen. 2003. p190-B RhoGAP regulates mammary ductal morphogenesis. Molec. Endocrin. 17:1054–1065. [DOI] [PubMed] [Google Scholar]
- Chen, H.C., P.A. Appeddu, H. Isoda, and J.L. Guan. 1996. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J. Biol. Chem. 271:26329–26334. [DOI] [PubMed] [Google Scholar]
- Choquet, D., D.P. Felsenfeld, and M.P. Sheetz. 1997. Extracellular matrix rigidity causes strengthening of integrin- cytoskeleton linkages. Cell. 88:39–48. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka, M., and K. Burridge. 1996. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133:1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, E.A., W.G. King, J.S. Brugge, M. Symons, and R.O. Hynes. 1998. Integrin-mediated signals regulated by members of the Rho family of GTPases. J. Cell Biol. 142:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman, E., R. Pankov, D.R. Stevens, and K.M. Yamada. 2001. Taking cell-matrix adhesions to the third dimension. Science. 294:1708–1712. [DOI] [PubMed] [Google Scholar]
- Demou, Z.N., and L.V. McIntire. 2002. Fully automated three-dimensional tracking of cancer cells in collagen gels: determination of motility phenotypes at the cellular level. Cancer Res. 62:5301–5307. [PubMed] [Google Scholar]
- Emerman, J.T., and D.R. Pitelka. 1977. Maintenance and induction of morphological differentiation in dissociated mammary epithelium on floating collagen membranes. In Vitro. 13:316–328. [DOI] [PubMed] [Google Scholar]
- Galbraith, C.G., K.M. Yamada, and M.P. Sheetz. 2002. The relationship between force and focal complex development. J. Cell Biol. 159:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger, B., and A. Bershadsky. 2002. Exploring the neighborhood: adhesion-coupled cell mechanosensors. Cell. 110:139–142. [DOI] [PubMed] [Google Scholar]
- Glaven, J.A., I.P. Whitehead, T. Nomanbhoy, R. Kay, and R.A. Cerione. 1996. Lfc and Lsc oncoproteins represent two new guanine nucleotide exchange factors for the Rho GTP-binding protein. J. Biol. Chem. 271:27374–27381. [DOI] [PubMed] [Google Scholar]
- Guo, Y.P., L.J. Martin, W. Hanna, D. Banerjee, N. Miller, E. Fishell, R. Khokha, and N.F. Boyd. 2001. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer Epidemiol. Biomarkers Prev. 10:243–248. [PubMed] [Google Scholar]
- Han, D.C., T.L. Shen, and J.L. Guan. 2000. Role of Grb7 targeting to focal contacts and its phosphorylation by focal adhesion kinase in regulation of cell migration. J. Biol. Chem. 275:28911–28917. [DOI] [PubMed] [Google Scholar]
- Ikebe, M., and D.J. Hartshorne. 1985. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J. Biol. Chem. 260:10027–10031. [PubMed] [Google Scholar]
- Ishizaki, T., M. Naito, K. Fujisawa, M. Maekawa, N. Watanabe, Y. Saito, and S. Narumiya. 1997. p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of Rho and induces focal adhesions. FEBS Lett. 404:118–124. [DOI] [PubMed] [Google Scholar]
- Keely, P., A. Fong, M. Zutter, and S. Santoro. 1995. Alteration of collagen-dependent adhesion, motility, and morphogenesis by the expression of antisense α2 integrin mRNA in mammary cells. J. Cell Sci. 108:595–607. [DOI] [PubMed] [Google Scholar]
- Keely, P.J., J.K. Westwick, I.P. Whitehead, C.J. Der, and L.V. Parise. 1997. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 390:632–636. [DOI] [PubMed] [Google Scholar]
- Kimura, K., M. Ito, M. Amano, K. Chihara, Y. Fukata, M. Nakafuku, B. Yamamori, J. Feng, T. Nakano, K. Okawa, et al. 1996. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 273:245–248. [DOI] [PubMed] [Google Scholar]
- Koo, L.Y., D.J. Irvine, A.M. Mayes, D.A. Lauffenburger, and L.G. Griffith. 2002. Co-regulation of cell adhesion by nanoscale RGD organization and mechanical stimulus. J. Cell Sci. 115:1423–1433. [DOI] [PubMed] [Google Scholar]
- Kumagai, N., N. Morii, K. Fujisawa, T. Yoshimasa, K. Nakao, and S. Narumiya. 1993. Lysophosphatidic acid induces tyrosine phosphorylation and activation of MAP-kinase and focal adhesion kinase in cultured Swiss 3T3 cells. FEBS Lett. 329:273–276. [DOI] [PubMed] [Google Scholar]
- Kureishi, Y., S. Kobayashi, M. Amano, K. Kimura, H. Kanaide, T. Nakano, K. Kaibuchi, and M. Ito. 1997. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J. Biol. Chem. 272:12257–12260. [DOI] [PubMed] [Google Scholar]
- Kwong, L., M. Wozniak, A. Collins, S. Wilson, and P. Keely. 2003. R-Ras promotes focal adhesion formation through focal adhesion kinase and p130Cas by a novel mechanism that differs from integrins. Mol. Cell. Biol. 23:933–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, E.Y., G. Parry, and M.J. Bissell. 1984. Modulation of secreted proteins of mouse mammary epithelial cells by the collagenous substrata. J. Cell Biol. 98:146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midwood, K.S., and J.E. Schwarzbauer. 2002. Tenascin-C modulates matrix contraction via focal adhesion kinase- and Rho-mediated signaling pathways. Mol. Biol. Cell. 13:3601–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesano, R., G. Schaller, and L. Orci. 1991. Induction of epithelial tubular morphogenesis in vitro by fibroblast- derived soluble factors. Cell. 66:697–711. [DOI] [PubMed] [Google Scholar]
- Parry, G., E.Y. Lee, D. Farson, M. Koval, and M.J. Bissell. 1985. Collagenous substrata regulate the nature and distribution of glycosaminoglycans produced by differentiated cultures of mouse mammary epithelial cells. Exp. Cell Res. 156:487–499. [DOI] [PubMed] [Google Scholar]
- Pelham, R.J., Jr., and Y. Wang. 1997. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA. 94:13661–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, X.D., W.B. Kiosses, and M.A. Schwartz. 1999. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, X.D., W.B. Kiosses, D.J. Sieg, C.A. Otey, D.D. Schlaepfer, and M.A. Schwartz. 2000. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 113:3673–3678. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J., and A. Hall. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 70:389–399. [DOI] [PubMed] [Google Scholar]
- Riveline, D., E. Zamir, N.Q. Balaban, U.S. Schwarz, T. Ishizaki, S. Narumiya, Z. Kam, B. Geiger, and A.D. Bershadsky. 2001. Focal contacts as mechanosensors. Externally applied local mechanical force induces growth of focal contacts by an mdia1-dependent and rock-independent mechanism. J. Cell Biol. 153:1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai, E., and C.J. Marshall. 2002. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat. Cell Biol. 4:408–415. [DOI] [PubMed] [Google Scholar]
- Schwartz, M.A., and R.K. Assoian. 2001. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J. Cell Sci. 114:2553–2560. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1999. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, J.L., J. Tien, D.M. Pirone, D.S. Gray, K. Bhadriraju, and C.S. Chen. 2003. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc. Natl. Acad. Sci. USA. 100:1484–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, D., D. Mehta, and S. Gunst. 1999. Mechanosensitive tyrosine phosphorylation of paxillin and focal adhesion kinase in tracheal smooth muscle. Am. J. Physiol. 276:C250–C258. [DOI] [PubMed] [Google Scholar]
- Totsukawa, G., Y. Yamakita, S. Yamashiro, D.J. Hartshorne, Y. Sasaki, and F. Matsumura. 2000. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 150:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H.B., M. Dembo, S.K. Hanks, and Y. Wang. 2001. Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc. Natl. Acad. Sci. USA. 98:11295–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., Y. Wang, C. Wang, J. Sung, W. Chiu, S. Lin, Y. Chang, and M. Tang. 2003. Rigidity of collagen fibrils controls collagen gel-induced down-regulation of focal adhesion complex proteins mediated by a2b1 integrin. J. Biol. Chem. 278:21886–21892. [DOI] [PubMed] [Google Scholar]
- Yano, Y., J. Geibel, and B. Sumpio. 1996. Tyrosine phosphorylation of pp125FAK and paxillin in aortic endothelial cells induced by mechanical strain. Am. J. Physiol. 271:C635–C649 [DOI] [PubMed] [Google Scholar]
- Yu, W., L.E. O'Brien, F. Wang, H. Bourne, K.E. Mostov, and M.M. Zegers. 2003. Hepatocyte growth factor switches orientation of polarity and mode of movement during morphogenesis of multicellular epithelial structures. Mol. Biol. Cell. 14:748–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J.H., H. Reiske, and J.L. Guan. 1998. Regulation of the cell cycle by focal adhesion kinase. J. Cell Biol. 143:1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zutter, M.M., S.A. Santoro, J.E. Wu, T. Wakatsuki, S.K. Dickeson, and E.L. Elson. 1999. Collagen receptor control of epithelial morphogenesis and cell cycle progression. Am. J. Pathol. 155:927–940. [DOI] [PMC free article] [PubMed] [Google Scholar]