

Abstract
Phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) on serine 51 is effected by specific stress-activated protein kinases. eIF2α phosphorylation inhibits translation initiation promoting a cytoprotective gene expression program known as the integrated stress response (ISR). Stress-induced activation of GADD34 feeds back negatively on this pathway by promoting eIF2α dephosphorylation, however, GADD34 mutant cells retain significant eIF2α-directed phosphatase activity. We used a somatic cell genetic approach to identify a gene encoding a novel regulatory subunit of a constitutively active holophosphatase complex that dephosphorylates eIF2α. RNAi of this gene, which we named constitutive repressor of eIF2α phosphorylation (CReP, or PPP1R15B), repressed the constitutive eIF2α-directed phosphatase activity and activated the ISR. CReP RNAi strongly protected mammalian cells against oxidative stress, peroxynitrite stress, and more modestly against accumulation of malfolded proteins in the endoplasmic reticulum. These findings suggest that therapeutic inhibition of eIF2α dephosphorylation by targeting the CReP-protein–phosphatase-1 complex may be used to access the salubrious qualities of the ISR.
Keywords: signal transduction; protein folding; pre-conditioning; somatic cell genetics; translation control
Introduction
Diverse stressful conditions are associated with phosphorylation of the α subunit of eukaryotic translation initiation factor 2 (eIF2α) on serine 51. This signaling event, which is conserved from yeast to mammals, negatively regulates the guanine nucleotide exchange factor, eIF2B and inhibits the recycling of eIF2 to its active GTP bound form. As a consequence, the initiation event in mRNA translation is inhibited (Hinnebusch, 2000), however, the impact on translation is not the same for all mRNAs. Although global protein synthesis is inhibited, the translation of specific mRNAs, such as those encoding the yeast transcription factor Gcn4p or the mammalian transcription factor ATF4 are induced under conditions of modest eIF2α phosphorylation (Hinnebusch, 1997; Harding et al., 2000a). Thus, in mammalian cells eIF2α phosphorylation emerges as an important event in stress signaling that impacts on gene expression at both the translational and transcriptional levels (Scheuner et al., 2001; Harding et al., 2003).
Four eIF2α kinases responsive to distinct stress signals have been discovered to date and experimental manipulation of their activity has provided insight into the physiological significance of regulated eIF2α phosphorylation in vertebrates. PKR (protein kinase repressor) is activated by double-stranded RNA produced during viral infection and plays an important role in translational inhibition and apoptosis of virally infected cells (Kaufman, 2000). But other eIF2α kinases protect cells against the impact of their cognate activating stress. GCN2, which responds to uncharged tRNAs, adapts cells to amino acid starvation (Natarajan et al., 2001; Zhang et al., 2002b). The heme-repressed kinase, HRI, protects developing erythroid precursors against the proteotoxic stress of free globin chains in iron-deficient vertebrates (Han et al., 2001). Whereas the ER localized eIF2α kinase PERK (PKR-like ER kinase), which is activated when ER chaperones are saturated by an excess of client proteins, protects cells against ER stress (Harding et al., 1999, 2000b, 2001a; Zhang et al., 2002a).
Mutant cells in which serine 51 of eIF2α has been replaced by alanine reveal that much of the protective effect of the aforementioned kinases is attributed to signaling through eIF2α phosphorylation (Scheuner et al., 2001). The diversity of the upstream signals that activate eIF2α kinases has led us to propose that the downstream response, coordinated by the phosphorylation of eIF2α on serine 51 be termed an integrated stress response (ISR), because it integrates signaling in multiple stress pathways (Harding et al., 2002; Ron, 2002). Gene expression profiling showed that eIF2α phosphorylation induces a gene expression program with a special role in promoting resistance to oxidative stress, which commonly accompanies conditions that activate eIF2α kinases (Harding et al., 2003). It has been further demonstrated that genetic manipulations that reduce eIF2 activity, and thereby mimic the effect of eIF2α phosphorylation, also promote resistance to oxidative stress (Tan et al., 2001b). This last paper even suggested that eIF2α phosphorylation and activation of its downstream ISR might promote preemptive resistance to oxidative stress in otherwise normal cells.
Specific phosphatase complexes can counteract phosphorylation of eIF2α on serine 51. The first such complex to be identified consists of a viral regulatory subunit encoded by the herpes simplex virus γ134.5 gene and a cellular catalytic subunit, protein phosphatase-1 (PP1c). By dephosphorylating eIF2α, γ134.5 enables the virus to escape the inhibitory effect of PKR activation (He et al., 1997, 1998). GADD34 is a stress-induced cellular homologue of γ134.5, which recruits PP1c to specifically dephosphorylate eIF2α (He et al., 1996; Connor et al., 2001; Novoa et al., 2001). GADD34 is not detected in unstressed cells, but stressful conditions associated with eIF2α phosphorylation promote GADD34 gene expression (Novoa et al., 2001; Ma and Hendershot, 2003). Under severely stressful conditions, cells lacking GADD34 accumulate high levels of phosphorylated eIF2α, and the resulting sustained inhibition of protein synthesis interferes with stress-induced gene expression programs (Kojima et al., 2003; Novoa et al., 2003). Thus, GADD34 is part of a negative feedback loop, promoting translational recovery during the later phases of diverse stress responses.
In unstressed GADD34 mutant cells, eIF2α phosphorylation was indistinguishable from that of wild-type cells, suggesting that GADD34 does not regulate basal levels of eIF2α phosphorylation. Here, we report on identification of a regulatory subunit of a constitutive eIF2α phosphatase complex that regulates basal levels of eIF2α phosphorylation. Our observations suggest that this novel complex may serve as a target for therapeutic inhibition to activate the ISR and elicit a stress-resistant state in cultured cells.
Results
GADD34-independent eIF2α dephosphorylation
GADD34 protein first becomes detectable 2–3 h after onset of an ER stress response (Fig. 1; Novoa et al., 2001, 2003). To determine if cells have GADD34-independent mechanisms for terminating signaling by phosphorylated eIF2α, we exploited the fact that ER stress in cells exposed to the reducing substance DTT is rapidly reversible (Bertolotti et al., 2000). A brief 30-min pulse of DTT resulted in rapid activation of PERK and phosphorylation of eIF2α on serine 51. After DTT washout, PERK was rapidly restored to its inactive, higher mobility state. The level of phosphorylated eIF2α also diminished after DTT washout. The decline in phosphorylated eIF2α occurred before any detectable accumulation of GADD34 protein (Fig. 1 A). Moreover, levels of phosphorylated eIF2α declined with similar kinetics after DTT washout in wild-type and mutant mouse fibroblasts lacking GADD34-mediated phosphatase activity (Fig. 1 A). Similar observations were made in mouse embryonic stem cells (Fig. 1 B), indicating that GADD34-independent mechanism(s) for terminating signaling by phosphorylated eIF2α were present in diverse cell types.
Figure 1.

Reversal of eIF2α phosphorylation in the absence of GADD34. (A) Immunoblots of eIF2α phosphorylated on serine 51, detected with an epitope-specific primary antiserum, eIF2α (P), total eIF2α(T), PERK (which detects both the unphosphorylated, inactive form of the kinase PERK° and activated, phosphorylated form PERK(P)), and GADD34 on lysates prepared from mouse embryonic fibroblasts with the indicated GADD34 genotypes. The cells were treated for 30 min with 1 mM dithiothreitol (DTT) and placed in DTT-free media for the indicated period of time (wash). Cells were also treated for 4 h with the ER stress-inducing drug thapsigargin (400 nM) serving as positive control for GADD34 induction. (B) Similar experiment to A performed in mouse embryonic stem cells.
We hypothesized that overexpression of genes active in this GADD34-independent pathway for terminating signaling by phosphorylated eIF2α might inhibit signaling in the ISR. Therefore, we used a modified version of a genetic screen previously used to isolate genetic suppressor elements of the signaling pathway by which ER stress culminates in induction of the downstream ISR target gene CHOP (Gudkov and Roninson, 1997; Novoa et al., 2001). It had previously been shown that activation of CHOP during ER stress is promoted by PERK phosphorylation of eIF2α on serine 51, translationally activating ATF4 (Harding et al., 2000a; Novoa et al., 2001; Scheuner et al., 2001), a transcription factor that binds to and activates the CHOP promoter (Fawcett et al., 1999; Harding et al., 2000a; Ma et al., 2002). Therefore, the activity of a CHOP::GFP transcriptional fusion gene serves as a faithful reporter for the transcriptional aspects of the ISR (Novoa et al., 2001). We transduced a reporter-containing CHO cell line with a cDNA library made in a retroviral vector and used FACS® to select cells that had abnormally low levels of CHOP::GFP expression after treatment with tunicamycin, a drug that causes ER stress and normally activates the ISR.
We found that successive cycles of FACsorting of GFP-dull cells selected for reduced reporter gene activity independent of retroviral transduction. To circumvent this background, we “rescued” the integrated, replication defective, retroviruses from pools of CHO cells with reduced CHOP::GFP expression by transient transfection of GAG, ENV, and POL, and infected parental CHOP::GFP cells with this rescued pool of recombinant retroviruses enriched in genetic suppressors of the ISR. Three rounds of enrichment for pools of recombinant retroviruses that suppressed CHOP::GFP activation by tunicamycin, yielded clonal populations of transduced cells; the retroviral inserts of which were sequenced. Most recombinant retroviruses identified by this method encoded the COOH terminus of GADD34, as predicted (Novoa et al., 2001); however one clone, named CD, contained an insert from a novel gene.
Constitutive repressor of eIF2α phosphorylation (CReP)
Transduction of the CD retrovirus markedly attenuated CHOP::GFP activation by tunicamycin and arsenite, an agent that activates the ISR independently of ER stress (Fig. 2 A). The inhibitory effect of the CD retrovirus extended to the endogenous CHOP gene (Fig. 2 B) and correlated with a profound defect in eIF2α phosphorylation (Fig. 2 C).
Figure 2.

Identification of a retroviral clone encoding CReP. (A) FACS®-derived fluorescent profiles of untreated (UT), tunicamycin (Tm, 2 μg/ml, 8 h), and sodium arsenite (As, 50 μM, 8 h)–treated parental CHOP::GFP cells and CHOP::GFP cells transduced with the CD retrovirus. (B) Immunoblot of endogenous CHOP and total eIF2α from cells as in A. (C) Immunoblot of eIF2α phosphorylated on serine 51 and total eIF2α from cells as in A.
Sequencing of the CD insert revealed it to consist of the 3′ coding region of a putative hamster homologue of a gene, PPP1R15B, for which full-length coding region mouse and human cDNAs had been isolated in the various genome sequencing projects. At its COOH terminus the encoded protein contained a motif predicted to bind PP1c. The insert of retrovirus CD corresponded to amino acids 314–688 of the predicted mouse protein (GenBank/EMBL/DDBL accession no. NP_598580.1) and amino acids 320–705 of its predicted human homologue (GenBank/EMBL/DDBL accession no. NP_116222.1). Furthermore, whereas this protein had little overall similarity to any known protein in the database, alignment of the COOH termini with that of GADD34 revealed strong conservation of residues known to be involved in PP1c binding by the latter (Fig. 3 A). For reasons that will become apparent below, we named the novel protein CReP.
Figure 3.

CReP is a regulatory subunit of an eIF2α holophosphatase complex with similarity to GADD34 in its COOH-terminal, PP1c–binding region. (A) Alignment of the predicted amino acid sequence of the COOH termini of mouse (mu) and human (hu) CReP and GADD34. The asterisk indicates the phenylalanine residue conserved in all PP1 regulatory subunits that is required for interaction with PP1c. (B) Immunoblot of FLAG-epitope tagged CReP COOH-terminal fragment encoded by the CD retrovirus and full-length CReP immunoprecipitated from lysates of transfected 293T cells (top). Immunoblot of endogenous PP1c in the immunoprecipitates (bottom). (C) Autoradiogram of an in vitro dephosphorylation assay of 32P-radiolabeled eIF2α on serine 51. The labeled protein was incubated with immune complexes purified from untreated or tunicamycin-treated (Tm) HT22 cells by means of preimmune sera (PI) or antisera directed to CReP or GADD34. Incubation times were 0, 10, and 20 min (lanes 1, 2, and 3, respectively). The bottom panels are immunoblots of the endogenous CReP and GADD34 from the immunoprecipitates used in the dephosphorylation assay. (D) Autoradiogram of an in vitro dephosphorylation assay of 32P-radiolabeled eIF2α on serine 51. The labeled protein solution was incubated with buffer alone (No lysate) or crude lysate from parental CHO cells or CHO cells stably transduced with the CReP-expressing CD retrovirus described in Fig. 2 (CReP). Reaction times were 10 and 20 min. The asterisk indicates GST-PERK, which is autophosphorylated.
Full-length mouse CReP and the peptide corresponding to the insert of the CD retrovirus were both tagged with a FLAG epitope and expressed in 293T cells by transient transfection. Immunopurification of these FLAG-tagged proteins revealed that they are associated in a stable complex with endogenous PP1c (Fig. 3 B). Antiserum raised against bacterial-expressed CReP immunopurified an endogenous activity from unstressed cell lysates that dephosphorylated phospho-serine 51 of eIF2α in vitro. By contrast, antiserum to GADD34 immunopurified an active eIF2α phosphatase only from ER-stressed cells (Fig. 3 C), which is consistent with the absence of GADD34 from unstressed cells (Fig. 3 C, bottom). The CReP-directed phosphatase activity exhibits specificity to eIF2α, as lysates of CReP overexpressing cells have increased phosphatase activity directed to eIF2α but not to other radiolabeled proteins present in the 32P-labeled eIF2α preparation (Fig. 3 D). These findings suggested that CReP encodes a regulatory subunit of a holophosphatase complex that dephosphorylates eIF2α, distinct from the stress-inducible GADD34. However, the existence of other substrates for CReP-PP1c complexes is not excluded.
Unlike GADD34, whose expression is tightly regulated by stress and whose basal level of expression is almost undetectable (Novoa et al., 2001, 2003), CReP is constitutively present in cells (Fig. 4 A). Pulse-chase labeling followed by immunoprecipitation showed that CReP is a relatively short-lived protein, whose t 1/2 in cells is ∼45 min (Fig. 4 B). Inhibition of protein synthesis with cycloheximide led to rapid disappearance of the CReP protein, as predicted by its short t 1/2. Interestingly, disappearance of CReP correlated with accumulation of phosphorylated eIF2α in the cycloheximide-treated cells (Fig. 4 C). This last observation is consistent with a role for CReP in maintaining low levels of eIF2α phosphorylation basally in unstressed cells.
Figure 4.
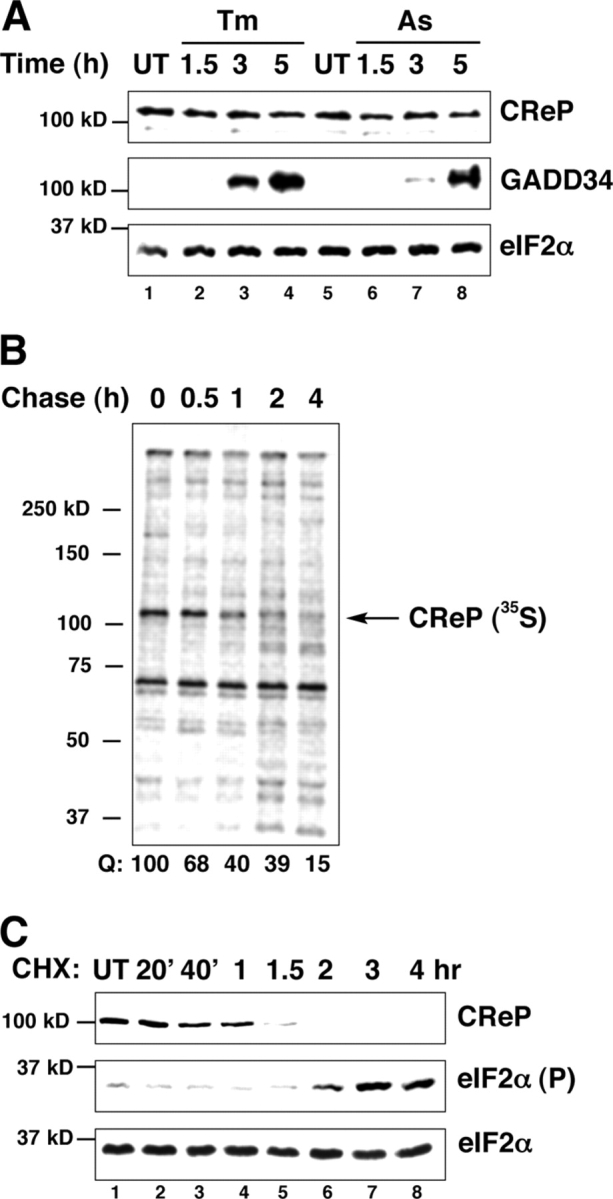
CReP is a constitutively expressed short-lived protein. (A) Immunoblot of endogenous CReP, GADD34, and total eIF2α in untreated (UT), and tunicamycin (Tm, 2 μg/ml)- and arsenite-treated cells (As, 50 μM). (B) Autoradiogram of pulse chase–labeled endogenous CReP immunoprecipitated with an anti-CReP immune serum. The label in the CReP band is quantified with the signal at the end of the pulse set arbitrarily at 100 (Q). (C) Immunoblot of endogenous CReP, phosphorylated eIF2α and total eIF2α in cells treated with the protein synthesis inhibitor cycloheximide (CHX, 50 μg/ml).
Blocking CReP expression activated the ISR and protected cells from oxidative, nitrosative, and ER stress
Because CReP is a short-lived protein whose levels depend on continuous translation of its mRNA, we attempted to interfere with its expression by transient RNA inhibition in cultured cells. A double-stranded RNA oligonucleotide directed to a region of the CReP mRNA that is conserved in all known mammalian species was transfected into the CHOP::GFP expressing CHO cells. This activated the transcriptional reporter gene, an observation consistent with activation of the eIF2α phosphorylation-dependent ISR. An identically sized oligonucleotide directed to human CD2 did not result in substantial activation of CHOP::GFP (Fig. 5 A). CReP RNAi activated CHOP::GFP only in fraction of the transfected cells, consistent with relatively low efficiency of the transient siRNA transfection into CHO cells (the latter was assessed by GFP RNAi of GFP-expressing CHO cells; unpublished data). Importantly, CReP RNAi failed to activate CHOP::GFP in cells overexpressing GADD34 (Fig. 5 A). The last observation is consistent with a functional redundancy in the activity of the two proteins.
Figure 5.

CReP RNAi activates CHOP::GFP, an ISR target gene. (A) FACS®-derived fluorescent profiles of nontransfected, CReP RNAi and CD2 RNAi transfected parental CHOP::GFP cells or CHOP::GFP cells stably overexpressing GADD34. The profile of nontransfected cells is shown in black. The light and dark tracings were obtained 16 and 28 h after RNAi transfection. (B) Immunoblots of eIF2α phosphorylated on serine 51, detected with an epitope-specific primary antiserum, eIF2α (P), total eIF2α(T), and PERK, which detects both the unphosphorylated, inactive form of the kinase (PERK°) and activated, phosphorylated form PERK(P) on lysates prepared from mouse ES cells with a CReP knockdown elicited by stable transduction of a CReP shRNA expressing plasmid or control scrambled shRNA plasmid. The cells were treated for 30 min with 1 mM dithiothreitol (DTT) and placed in DTT-free media for the indicated period of time (wash). (C) Immunoblot of endogenous CReP and PP1c in lysates of parental ES cells, CReP shRNA cells, and cells stably transfected with the control scrambled shRNA plasmid. The abundantly expressed TLS protein, detected with a specific antiserum, served as a loading control in this gel.
We could not reliably detect changes in levels of phosphorylated eIF2α in cells transiently transfected with the CReP siRNA oligonucleotide; perhaps because eIF2α phosphorylation is a transient event that can only be detected in cell populations responding to a highly synchronous perturbation. To further assess the role of CReP in regulating levels of phosphorylated eIF2α, we established embryonic stem (ES) cells in which CReP levels were stably reduced by an integrated transgene expressing a short hairpin RNA species corresponding to the aforementioned CReP sequence (CReP shRNA). As a control, we established cells expressing a short hairpin RNA species identical in base composition, but diverging from the CReP sequence (scrambled shRNA). Transient exposure of cells to DTT, followed by DTT washout, revealed the existence of a GADD34-independent mechanism for reversing eIF2α phosphorylation (Fig. 1, A and B). This procedure was applied to the CReP shRNA and control, scrambled shRNA cells. Peak levels of phosphorylated eIF2α were reproducibly higher in the CReP shRNA cells and their decline followed delayed kinetics, when compared with the control scrambled shRNA cells (Fig. 5, B and C). These observations support CReP's hypothesized role as a constitutive component of an eIF2α(P)-directed phosphatase. Cellular levels of PP1c were unaffected by CReP RNAi (Fig. 5 C), suggesting that the integrity of other PP1c-containing holophosphatase complexes was not compromised by this manipulation.
Interfering with eIF2α expression by antisense RNA had been noted to promote resistance to oxidative stress in HT22 cells (Tan et al., 2001b). This protective effect was specifically sought in HT22 cells because they are exquisitely sensitive to cell death caused by oxidative glutamate toxicity (Tan et al., 2001a). In these cells glutamate exposure promotes massive accumulation of reactive oxygen species and cell death that can be prevented by knockdown of eIF2α protein level (Tan et al., 2001b). Therefore, we examined the impact of CReP RNAi on the resistance of HT22 cells to glutamate and to other stressful conditions.
Immunoblot of HT22 cells transiently transfected with the CReP siRNA oligonucleotide revealed loss of CReP protein 24–36 h after transfection, with protein levels recovering to near normal levels by 48 h (Fig. 6 A). Total eIF2α, serving as a reference protein in the treated cell lysates, did not change with CReP RNAi and GFP RNAi (or CD2 RNAi) did not affect CReP protein levels (Fig. 6 A and not depicted). After a 7-h exposure to 5 or 10 mM glutamate, only 43 and 20%, respectively, of nontransfected HT22 cells survived. Mock transfection or transfection of GFP siRNA did not increase cell survival, however after transfection with CReP siRNA, >80% of the cells exposed to either dose of glutamate survived. The survival benefit of CReP RNAi was also noted in cells treated with H2O2, the nitric oxide donor SIN-1, and to a lesser extent in response to the ER stress-causing agent tunicamycin (Fig. 6 B).
Figure 6.

CReP RNAi promotes a stress-resistant state. (A) Immunoblot of endogenous CReP in lysates of nontransfected HT22 cells and cells transfected with a small interfering RNAi directed to CReP or to GFP. Lysates were harvested for immunoblot at the indicated time. (B) Survival, measured by MTT assay after exposure to glutamate (7 h at 5 mM, gray bar; or 10 mM, white bar), H2O2 (7 h at 1 mM, gray bar; 2 mM, white bar), SIN-1 (7 h at 1.5 mM, gray bar; 3 mM, white bar) and tunicamycin (12 h at 0.125 μg/ml, gray bar; 0.25 μg/ml, white bar followed by 24 h recovery). Cells were nontransfected, mock transfected with no siRNA, or transfected with siRNA to GFP (a control) or CReP. Shown are mean ± SEM of experiments performed in triplicates and reproduced three times. The MTT signal of the untreated, nontransfected cells is arbitrarily set at 100. (C) Dual channel FACscans of dichlorofluorescein fluorescence (DCF axis, reporting on endogenous peroxides) and propidium iodide fluorescence (P.I. axis, reporting on permeabilized, damaged cells) of untreated (boxes 1 and 4) and glutamate-treated (boxes 2, 3, 5, and 6) mock-transfected (boxes 1–3), and CReP RNAi-transfected (boxes 4–6) HT22 cells. The fraction of cells in each quadrant of the FACS® is indicated in Table I.
To determine if increased survival of the CReP RNAi cells correlated with decreased accumulation of reactive oxygen species, we incubated cells with dichlorodihydrofluorescein, which is oxidized by cellular reactive oxygen species to the fluorescent compound dichlorofluorescein (DCF). The cells were also stained with propidium iodide (PI), to which live cells are impermeable and which therefore marks dead cells. As the dual channel DCF/PI FACS® show, in mock transfected HT22 cells exposure to glutamate increased DCF fluorescence, which, as expected of a vital die, only stained the live, PI-negative cells. Glutamate treatment also markedly increased the fraction of PI-positive, permeabilized, dead cells (Fig. 6 C and Table I). DCF fluorescence was lower in glutamate-treated CReP RNAi cells and the number of PI-positive, dead cells was markedly lower (Fig. 6 C and Table I). These observations suggest that the state of resistance to glutamate oxidative stress induced by transient CReP RNAi is associated with reduced accumulation of reactive oxygen species.
Table I.
FACS ® of glutamate-treated cells
|
|
|
FACS® 1
|
FACS® 2
|
FACS® 3
|
FACS® 4
|
FACS® 5
|
FACS® 6
|
|---|---|---|---|---|---|---|---|
| % | % | % | % | % | % | ||
| Top Lt | PI+,DCF− | 1.49 | 14.65 | 48.36 | 3.01 | 4.32 | 17.97 |
| Top Rt | PI+,DCF+ | 0.11 | 6.07 | 5.38 | 0.58 | 3.35 | 5.69 |
| Bottom Lt | PI−,DCF− | 96.54 | 20.52 | 7.72 | 94.87 | 59.11 | 15.30 |
| Bottom Rt | PI−,DCF+ | 1.86 | 58.76 | 38.54 | 1.54 | 33.22 | 61.04 |
The fraction of cells in each of the four quadrants of the six FACscans shown in Fig. 6 C is tabulated above.
Discussion
Past studies of regulated translation initiation have focused on stress inducible eIF2α kinases, however recent experiments suggest that eIF2α dephosphorylation also contributes to regulating this process. One of the important targets of the gene expression program induced by eIF2α phosphorylation is GADD34, which encodes an inducible regulatory subunit of an eIF2α holophosphatase complex that forms a negative feedback loop to dephosphorylate eIF2α and promote translational recovery in the late phases of diverse stress responses (He et al., 1996; Novoa et al., 2001, 2003; Brush et al., 2003; Kojima et al., 2003; Ma and Hendershot, 2003). However, cells lacking GADD34-mediated eIF2α-directed phosphatase activity, nonetheless, are able to rapidly reverse eIF2α phosphorylation induced by transient activation of the upstream kinase PERK (Fig. 1). Furthermore, levels of phosphorylated eIF2α are not measurably elevated in unstressed GADD34 mutant cells (Fig. 1; Kojima et al., 2003; Novoa et al., 2003). Given the importance of eIF2α phosphorylation to translational and transcriptional responses in diverse stress conditions, the nature of the activity reversing eIF2α phosphorylation in GADD34 mutant cells emerges as a matter of considerable interest.
Here, we report on a novel protein identified as the product of a gene whose overexpression blocks the eIF2α phosphorylation-dependent ISR. The protein, which we named CReP, has sequence similarity to GADD34 in the region that binds PP1c. The ISR-suppressing retrovirus isolated in our screen encoded the COOH-terminal half of CReP, which binds PP1c and is sufficient to promote eIF2α dephosphorylation in vivo. A similar fragment of GADD34, which contains the region of similarity to CReP, also possesses eIF2α-directed phosphatase activity (He et al., 1996; Novoa et al., 2001; Brush et al., 2003). Like GADD34, CReP also forms a stable complex with PP1c that specifically dephosphorylates eIF2α. Both proteins have extended NH2-terminal portions of unknown function that are dispensable to eIF2α phosphatase activity of the overexpressed COOH-terminal portion.
CReP expression was not altered by stressful stimuli that promote eIF2α phosphorylation and the phosphatase activity of the immunoprecipitated endogenous CReP-containing complex was unaffected by ER stress (Fig. 3 C and not depicted). CReP overexpressing cells had lower levels of both basal and stress-inducible phosphorylated eIF2α, and markedly attenuated signaling in the ISR. Knockdown of CReP by RNAi led to reduced basal eIF2α-directed phosphatase activity and induced CHOP::GFP, a stable marker of the ISR. These findings point to CReP as a constitutively active counterpart to GADD34 that contributes to regulation of basal levels of eIF2α phosphorylation. We were unable to reliably detect induction of other ISR markers in CReP RNAi cells, but this may not be too surprising, as the effects of CReP RNAi on basal levels of eIF2α phosphorylation are likely to be transient and resisted by various homeostatic mechanisms. Furthermore, the ISR markers are for the most part short-lived proteins and the uptake of siRNA oligonucleotides may be rather asynchronic, rendering detection of an endogenous ISR marker difficult. Of note, CReP itself is a labile protein, and its disappearance from cells in which protein synthesis has been inhibited may contribute to elevated levels of eIF2α phosphorylation noted under these conditions.
The ISR, a gene expression program activated by eIF2α phosphorylation, has strong pro-survival benefits in certain circumstances. ISR target genes encode ER chaperones (Scheuner et al., 2001; Harding et al., 2003), amino acid transporters, enzymes involved in metabolism of thiol-containing amino acids to glutathione and proteins like HMOX1 and SQSTM1 with known antioxidant properties (Tan et al., 2001b; Harding et al., 2003). The significance of this gene activation program to cell survival is revealed by the phenotype of cells and animals lacking key components of the ISR (Harding et al., 2000b, 2003; Han et al., 2001; Scheuner et al., 2001; Zhang et al., 2002a), indicating that the ISR is required for survival of cells and tissues exposed to physiological levels of stress.
Recent work suggests that activation of the ISR can also promote additional stress resistance in otherwise wild-type cells, a phenomenon referred to as preconditioning. For example, Tan and colleagues have shown that either stable knockdown of eIF2α protein levels by expression of an antisense RNA (a manipulation presumed to phenocopy certain aspects of eIF2α phosphorylation on serine 51) or expression of a phospho-mimetic form of eIF2α (S51D), promote resistance to oxidative stress (Tan et al., 2001b). We have found that eIF2α phosphorylation by a genetically engineered, conditionally active, PERK kinase that was uncoupled from its cognate upstream signal, ER stress, promoted preemptive resistance to oxidative stress, peroxynitrite stress, and to a lesser degree ER stress in cultured HT22 cells (Lu et al., 2004). These experiments suggested that preemptive activation of the ISR might pharmacologically precondition cells to stress. However, modified PERK, expressed as a transgene, or knockdown of eIF2α levels, are not practical means of exploiting this phenomenon. Neither, for that matter, are manipulations that activate endogenous eIF2α kinases, as these likely expose cells to destructive stressful stimuli. However, inactivation of an endogenous phosphatase that controls basal levels of eIF2α phosphorylation might access the ISR, without promoting cell stress. Support for this idea is provided by the observation that CReP inactivation was markedly protective against subsequent application of oxidative, nitrosative, or ER stress.
Elevated levels of phosphorylated eIF2α have been linked to apoptosis, but this association seems particularly important in virally infected cells (Der et al., 1997; Clemens, 2001; Williams, 2001). In many other physiological contexts eIF2α phosphorylation is cytoprotective. One of the more dramatic examples of this association is the finding of markedly elevated levels of phosphorylated eIF2α during hibernation, a naturally stress-resistant state (Frerichs et al., 1998). The properties of CReP described here suggest that it might serve as a target for inhibiting eIF2α dephosphorylation effecting a transient pharmacologically induced stress-resistant state resembling hibernation. This could be applied as a therapeutic modality in circumstances where acute tissue injury by reactive oxygen species can be anticipated, with applications ranging from organ procurement for transplantation to vascular surgery.
Materials and methods
Cell culture, treatment, and survival assays
The HT22 subclone of HT-4 cells was a gift of P. Maher (Scripps Research Institute, La Jolla, CA), and was maintained at ∼50% confluence in regular media consisting of DME (Life Technologies), 10% FCS (Atlanta Biologicals), 1 × pen/strep, and 2 mM l-glutamine (Life Technologies) as described previously (Maher and Davis, 1996). CHO-K1 cells and 293T cells were obtained from American Type Culture Collection and maintained as described previously (Novoa et al., 2001). The CHO-K1 cell line stably expressing CHOP::GFP and FACScan™ analysis of the GFP signal in these cells was performed as described previously (Novoa et al., 2001). The W4 mouse embryonic stem cell line is a gift of A. Joyner (New York University School of Medicine). Tunicamycin, sodium arsenite, DTT, cycloheximide, sodium glutamate, H2O2, and SIN-1 (3-morpholinosydnonimine) were obtained from Sigma-Aldrich. Treatment of cells with 1 mM DTT to transiently induce PERK was performed as described previously (Bertolotti et al., 2000).
Cell survival assays were based on MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide; Sigma-Aldrich) cleavage. HT22 cells were plated at a density of 2.5–5.0 × 103 cells/96 well. Cells were exposed to the indicated concentrations of l-glutamic acid, tunicamycin, H2O2, or SIN-1 for the indicated period of time; at the end of which the cells were switched to media containing 0.5 mg/ml MTT for 4 h, and the reaction was stopped by the addition of an equal volume of stop buffer (50% isopropanol, 10% SDS, and 10 μM HCl). The formazan crystals were allowed to dissolve during a 16-h incubation at 37°C and the OD was read at 550 nm. Background correction values were subtracted from each sample. Experiments were performed in triplicate.
FACScan™ analysis to detect in vivo conversion of dichlorodihydrofluoroscein to DCF and cellular permeability to PI was performed after exposing the cells to the indicated concentration of glutamate. The media were removed to collect floating cells and these were pooled with the adherent cells trypsinized from the dishes. The cells were pelleted and resuspended in fresh media containing 10 μM 2'7'dichloro-dihydrofluoroscein diacetate (Molecular Probes) and incubated for 15 min at 37°C. Cells were pelleted, washed once in ice-cold PBS-2% FCS, and resuspended in PBS-2% FCS containing 1 μg/ml PI (Roche). After a 5-min incubation on ice, the cells were analyzed for DCF fluorescence (FL-1, green channel) and PI positivity (FL-3, red channel) by a FACScan™ (BD Biosciences). 10,000 ungated cells were counted.
Immunoblot, metabolic labeling immunoprecipitation, and in vitro dephosphorylation assays
Cell lysates and methods to detect total and phosphorylated eIF2α, endogenous GADD34, PERK, and CHOP have been described previously in detail (Harding et al., 2001b; Novoa et al., 2001, 2003). The antiserum to CReP was raised in rabbit against a His-tagged bacterially expressed protein corresponding to the 280 COOH-terminal residues of the murine protein. It was used in immunoblot at a dilution of 1:1,000. In immunoprecipitation reactions, 1 μl of the crude serum was bound to 10 μl of protein A–Sepharose resin and used to purify the endogenous CReP from whole cell lysates containing 280 μg of total protein. HT22 cells were metabolically labeled with 33 μCi/ml of TransLabel (ICN Biomedicals) for 30 min, followed by cold chase in complete, unlabeled media. The immunoprecipitates containing labeled CReP were washed three times in RIPA buffer before being resolved on a 8% SDS-PAGE gel.
Methods to radiolabel the eIF2α in reticulocyte lysates using recombinant bacterially expressed PERK have been described previously (Harding et al., 1999; Novoa et al., 2001). The radiolabeled eIF2α was incubated with endogenous CReP or GADD34 immunopurified from cell lysate (3 mg of total protein) with anti-CReP or anti-GADD34 antiserum (20 μl of antiserum were bound to 40 μl of protein A Sepharose) as described previously (Novoa et al., 2001). The labeled proteins resolved on a 10% SDS PAGE before exposure to autoradiography. Similar in vitro dephosphorylation assays were performed in crude detergent lysates of parental CHO cells and CD-transduced cells as described previously (Novoa et al., 2001).
Isolation of the CReP-encoding genetic suppressor element and construction of expression plasmids
The procedure for isolating recombinant retrovirus encoding genetic suppressor element that interfere with stress-induced activation of CHOP::GFP has been described previously (Gudkov and Roninson, 1997; Novoa et al., 2001) and was modified in this screen. In brief, a retrovirally expressed CHO cDNA library constructed in pBABEpuro was transduced in batches of 105 clones into a CHO line stably expressing CHOP::GFP. The cells were treated with 1.75 μg/ml tunicamycin for 3 h, followed by overnight recovery in the absence of tunicamycin and the 1% dullest cells were FACsorted. The pool of dull cells obtained by FACsorting was expanded and the resident replication defective retroviruses were rescued by transient transfection of plasmids encoding the helper functions: VSV-G protein (pseudo ENV), GAG, and POL, as described previously (Landau and Littman, 1992). This pool of retroviruses, enriched in genetic suppressor elements, was used to transduce the parental CHOP::GFP cells, and the process of selection of dull cells and viral rescue was repeated three times. After the third cycle of enrichment, sub-clones of transduced cells selected from those pools of retrovirus that effected a significant (>50%) reduction of GFP in 50% of the transduced cells were procured, examined by FACScan™ to confirm that they had impaired induction of CHOP::GFP, and the retroviral insert was amplified from the genomic DNA by PCR and sequenced. 19 of the 20 pools evaluated by this method encoded different COOH-terminal fragments of GADD34 (an observation consistent with the fact that the cDNA library was constructed from mRNA obtained from ER stressed cells). One pool, CD, harbored a retrovirus encoding the COOH terminus of CReP.
The CReP cDNA expression plasmids were constructed by ligating the cDNA coding region, amplified by RT-PCR from mouse mRNA (aa residues 24–698 for the “full length” and 314–698 for the “COOH terminus”), in frame with FLAG epitope tag in pFLAG-CMV2.
CReP RNAi
The double-stranded CReP siRNA oligonucleotide, 5′ AAGGGAUGGAUGCAGGUUCCA 3′, corresponds to a sequence conserved in mouse, hamster, and human CReP. The sequences used for the control RNAi experiments were: 5′ GGUGCAGUCUCCAAAGAGA 3′ (human CD2 gene) and 5′ GCAGCACGACUUCUUCAAG 3′ (GFP). The annealed double-stranded siRNA oligonucleotide (Dharmacon) was transfected following the manufacturer's instructions. Cells growing in 35-mm wells (50 × 103/well) were incubated with 1 μM double-stranded oligonucleotide in 200 μl serum-free media and 3 μl Oligofectamine™ (Invitrogen) for 4 h followed by addition of serum to 10% and further incubation until analysis.
Stable knockdown of CReP in ES cells was obtained by transfection of W4 ES cells with a pTU6.puro plasmid (a gift of X. Wang, Northwestern University Medical School, Chicago, IL) containing a CReP shRNA insert of the following sequence: 5′ tttGAACCTGCATCCATCCCTTGCAgaagcttgTGCGAGGGGTGGATGTAGGTTCtttttc 3′. The scrambled control shRNA sequence was 5′ tttGAACCT CCATGCATCCGTTCCAgaagcttgTGGGACGGGTGTATGGAGGTTCtttttc 3′. The position of the sequence substitutions in the predicted scrambled hairpin is underlined.
Acknowledgments
We thank Chris Arendt for assistance with the siRNA procedures.
This work was supported by National Institutes of Health grants ES08681 and DK47119. C. Jousse was supported in part by a postdoctoral fellowship from the Institut Francais de Nutrition. D. Ron is a Scholar of the Ellison Medical Foundation.
Abbreviations used in this paper: CReP, constitutive repressor of eIF2α phosphorylation; DCF, dichlorofluorescein; eIF2, eukaryotic translation initiation factor 2; ES, embryonic stem; ISR, integrated stress response; PI, propidium iodide; PP1c, protein phosphatase-1 catalytic subunit.
References
- Bertolotti, A., Y. Zhang, L. Hendershot, H. Harding, and D. Ron. 2000. Dynamic interaction of BiP and the ER stress transducers in the unfolded protein response. Nat. Cell Biol. 2:326–332. [DOI] [PubMed] [Google Scholar]
- Brush, M.H., D.C. Weiser, and S. Shenolikar. 2003. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 23:1292–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens, M.J. 2001. Initiation factor eIF2 alpha phosphorylation in stress responses and apoptosis. Prog. Mol. Subcell. Biol. 27:57–89. [DOI] [PubMed] [Google Scholar]
- Connor, J.H., D.C. Weiser, S. Li, J.M. Hallenbeck, and S. Shenolikar. 2001. Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol. Cell. Biol. 21:6841–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der, S.D., Y.L. Yang, C. Weissmann, and B.R. Williams. 1997. A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc. Natl. Acad. Sci. USA. 94:3279–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett, T.W., J.L. Martindale, K.Z. Guyton, T. Hai, and N.J. Holbrook. 1999. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 339:135–141. [PMC free article] [PubMed] [Google Scholar]
- Frerichs, K.U., C.B. Smith, M. Brenner, D.J. DeGracia, G.S. Krause, L. Marrone, T.E. Dever, and J.M. Hallenbeck. 1998. Suppression of protein synthesis in brain during hibernation involves inhibition of protein initiation and elongation. Proc. Natl. Acad. Sci. USA. 95:14511–14516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudkov, A.V., and I.B. Roninson. 1997. Isolation of genetic suppressor elements (GSEs) from random fragment cDNA libraries in retroviral vectors. Methods Mol. Biol. 69:221–240. [DOI] [PubMed] [Google Scholar]
- Han, A.P., C. Yu, L. Lu, Y. Fujiwara, C. Browne, G. Chin, M. Fleming, P. Leboulch, S.H. Orkin, and J.J. Chen. 2001. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 20:6909–6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, H.P., Y. Zhang, and D. Ron. 1999. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 397:271–274. [DOI] [PubMed] [Google Scholar]
- Harding, H.P., I. Novoa, Y. Zhang, H. Zeng, R.C. Wek, M. Schapira, and D. Ron. 2000. a. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 6:1099–1108. [DOI] [PubMed] [Google Scholar]
- Harding, H.P., Y. Zhang, A. Bertolotti, H. Zeng, and D. Ron. 2000. b. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell. 5:897–904. [DOI] [PubMed] [Google Scholar]
- Harding, H.P., H. Zeng, Y. Zhang, R. Jungreis, P. Chung, H. Plesken, D. Sabatini, and D. Ron. 2001. a. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in survival of secretory cells. Mol. Cell. 7:1153–1163. [DOI] [PubMed] [Google Scholar]
- Harding, H.P., I. Novoa, A. Bertolotti, H. Zeng, A. Zhang, F. Urano, C. Jousse, and D. Ron. 2001. b. Translational regulation in the cellular response to biosynthetic load on the endoplasmic reticulum. Cold Spring Harb. Symp. Quant. Biol. 66:499–508. [DOI] [PubMed] [Google Scholar]
- Harding, H.P., M. Calfon, F. Urano, I. Novoa, and D. Ron. 2002. Transcriptional and translational control in the mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 18:575–599. [DOI] [PubMed] [Google Scholar]
- Harding, H.P., Y. Zhang, H. Zeng, I. Novoa, P. Lu, M. Calfon, N. Sadri, C. Yun, B. Popko, R. Paules, et al. 2003. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 11:619–633. [DOI] [PubMed] [Google Scholar]
- He, B., J. Chou, D.A. Liebermann, B. Hoffman, and B. Roizman. 1996. The carboxyl terminus of the murine MyD116 gene substitutes for the corresponding domain of the gamma(1)34.5 gene of herpes simplex virus to preclude the premature shutoff of total protein synthesis in infected human cells. J. Virol. 70:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, B., M. Gross, and B. Roizman. 1997. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA. 94:843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, B., M. Gross, and B. Roizman. 1998. The gamma134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem. 273:20737–20743. [DOI] [PubMed] [Google Scholar]
- Hinnebusch, A.G. 1997. Translational regulation of yeast GCN4. A window on factors that control initiator-tRNA binding to the ribosome. J. Biol. Chem. 272:21661–21664. [DOI] [PubMed] [Google Scholar]
- Hinnebusch, A.G. 2000. Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes. Translational Control of Gene Expression. N. Sonenberg, J.W.B. Hershey, and M.B. Mathews, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 185–243.
- Kaufman, R.J. 2000. The double-stranded RNA-activated protein kinase PKR. Translational Control of Gene Expression. N. Sonenberg, J.W.B. Hershey, and M.B. Mathews, editors. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 503–527.
- Kojima, E., A. Takeuchi, M. Haneda, F. Yagi, T. Hasegawa, K.I. Yamaki, K. Takeda, S. Akira, K. Shimokata, and K.I. Isobe. 2003. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress-elucidation by GADD34-deficient mice. FASEB J. 17:1573–1575. [DOI] [PubMed] [Google Scholar]
- Landau, N., and D. Littman. 1992. Packaging system for rapid production of murine leukemia virus vectors with variable tropisim. J. Virol. 66:5110–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, P.D., C. Jousse, S.J. Marciniak, Y. Zhang, I. Novoa, D. Scheuner, R.J. Kaufman, D. Ron, and H.P. Harding. 2004. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y., J.W. Brewer, J.A. Diehl, and L.M. Hendershot. 2002. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 318:1351–1365. [DOI] [PubMed] [Google Scholar]
- Ma, Y., and L.M. Hendershot. 2003. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 278:34864–34873. [DOI] [PubMed] [Google Scholar]
- Maher, P., and J.B. Davis. 1996. The role of monoamine metabolism in oxidative glutamate toxicity. J. Neurosci. 16:6394–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan, K., M.R. Meyer, B.M. Jackson, D. Slade, C. Roberts, A.G. Hinnebusch, and M.J. Marton. 2001. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol. Cell. Biol. 21:4347–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa, I., H. Zeng, H. Harding, and D. Ron. 2001. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 153:1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa, I., Y. Zhang, H. Zeng, R. Jungreis, H.P. Harding, and D. Ron. 2003. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 22:1180–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron, D. 2002. Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 110:1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner, D., B. Song, E. McEwen, P. Gillespie, T. Saunders, S. Bonner-Weir, and R.J. Kaufman. 2001. Translational control is required for the unfolded protein response and in-vivo glucose homeostasis. Mol. Cell. 7:1165–1176. [DOI] [PubMed] [Google Scholar]
- Tan, S., D. Schubert, and P. Maher. 2001. a. Oxytosis: a novel form of programmed cell death. Curr. Top. Med. Chem. 1:497–506. [DOI] [PubMed] [Google Scholar]
- Tan, S., N. Somia, P. Maher, and D. Schubert. 2001. b. Regulation of antioxidant metabolism by translation initiation factor 2α. J. Cell Biol. 152:997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, B.R. 2001. Signal integration via PKR. Sci STKE. 2001:RE2. [DOI] [PubMed]
- Zhang, P., B. McGrath, S. Li, A. Frank, F. Zambito, J. Reinert, M. Gannon, K. Ma, K. McNaughton, and D.R. Cavener. 2002. a. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 22:3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, P., B.C. McGrath, J. Reinert, D.S. Olsen, L. Lei, S. Gill, S.A. Wek, K.M. Vattem, R.C. Wek, S.R. Kimball, et al. 2002. b. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol. Cell. Biol. 22:6681–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]