

Abstract
Mmutations in paraplegin, a putative mitochondrial metallopeptidase of the AAA family, cause an autosomal recessive form of hereditary spastic paraplegia (HSP). Here, we analyze the function of paraplegin at the cellular level and characterize the phenotypic defects of HSP patients' cells lacking this protein. We demonstrate that paraplegin coassembles with a homologous protein, AFG3L2, in the mitochondrial inner membrane. These two proteins form a high molecular mass complex, which we show to be aberrant in HSP fibroblasts. The loss of this complex causes a reduced complex I activity in mitochondria and an increased sensitivity to oxidant stress, which can both be rescued by exogenous expression of wild-type paraplegin. Furthermore, complementation studies in yeast demonstrate functional conservation of the human paraplegin–AFG3L2 complex with the yeast m-AAA protease and assign proteolytic activity to this structure. These results shed new light on the molecular pathogenesis of HSP and functionally link AFG3L2 to this neurodegenerative disease.
Keywords: spasticity; mitochondria; respiratory complex; neurodegeneration; AAA protease
Introduction
Both clinical and genetic heterogeneity apply to hereditary spastic paraplegia (HSP), a disabling disorder affecting 1 in 10,000 people in the Western world (Polo et al., 1993). The main phenotypic trait common to all forms of spastic paraplegia is a progressive lower limb weakness, spasticity, and decreased vibratory sense. Complicated forms show additional neurological signs such as retinopathy, optic neuropathies, ataxia, dementia, and mental retardation (Harding, 1983). Neuropathological studies on pure HSP patients describe specific alterations of the cortical spinal tract due to axonal degeneration of motor and sensory neurons without cell body loss (Schwarz and Liu, 1956).
A more precise classification of HSP forms originates from the inheritance pattern, which may be autosomal dominant, autosomal recessive, or X-linked recessive. 20 different loci have been associated with diverse forms of HSP (Crosby and Proukakis, 2002; Reid, 2003); however, causative mutations have been found in eight genes only. The most recent gene associated with HSP is the neuronal specific kinesin (KIF5A) gene SPG10, a member of the kinesin superfamily of molecular motors that transport cargoes along microtubules in an ATP-dependent manner. Mutations in the cell adhesion module L1, L1CAM (SPG1), and in the proteolipid protein SPG2 cause X-linked forms of HSP characterized by defects in the development of the corticospinal tract and in axonal–glial interactions, respectively. Little is known about the four known autosomal HSP genes encoding atlastin (SPG3A), spartin (SPG20), spastin (SPG4), and paraplegin (SPG7), whereas HSP60 is a well-characterized shock response protein (SPG13). Atlastin is a novel GTPase that has sequence homology to members of the dynamin family of large GTPases, particularly guanylate binding protein-1 (Zhao et al., 2001). Spartin is responsible for Troyer Syndrome, an autosomal recessive HSP, and is homologous to proteins involved in endosomal morphology and protein trafficking of late endosomal component (Patel et al., 2002). Both spastin and paraplegin are AAA proteins, which are ATPases associated with different cellular activities (Casari et al., 1998; Hazan et al., 1999; Ogura and Wilkinson, 2001). Members of this superfamily of ATPases exert chaperone-like activities and mediate assembly and disassembly of macromolecular structures involved in different cellular processes. Although a still unclear role at the nuclear level exists (Charvin et al., 2003), spastin interacts with microtubules and seems to modulate microtubule dynamics, which is an essential attainment for maintenance of long axons (Errico et al., 2002).
Apart from these mutant proteins that associate the HSP pathogenetic cascade to intracellular and axonal trafficking, another pathogenetic mechanism points to the mitochondrion. In fact, both paraplegin and HSP60 are nuclear-encoded mitochondrial proteins (Casari and Rugarli, 2001; Hansen et al., 2002).
Paraplegin is closely related to ATP-dependent metalloproteases (Langer, 2000). By database scanning, two additional mitochondrial AAA proteases that are highly homologous to paraplegin have been identified, namely AFG3L2 and YME1L1 (Banfi et al., 1999; Coppola et al., 2000). Yeast orthologues of these proteins are membrane-bound peptidases, which ensure the quality control of mitochondrial inner membrane proteins (Leonhard et al., 1996).
Here, we analyze the function of paraplegin and its homologous protein, AFG3L2. They assemble in the mitochondrial inner membrane into a high molecular mass complex, which is aberrant in HSP patients. Impaired complex formation in HSP cells causes a decreased activity of respiratory complex I and increases the sensitivity to oxidative stress. Heterologous expression of paraplegin and AFG3L2 in yeast cells and complementation studies establish the complex as a proteolytically active structure involved in maintenance of respiratory competence. Together, these findings provide novel insights into the pathogenic mechanism of neurodegeneration in HSP patients.
Results
Paraplegin and AFG3L2 assemble in the inner mitochondrial membrane into a high molecular mass complex, which is aberrant in HSP patients
Paraplegin is homologous to mitochondrial AAA metalloproteases that have been exclusively studied in fungi. Two additional human members of the AAA metalloprotease family, AFG3L2 and YME1L1, localize to mitochondria (Banfi et al., 1999; Coppola et al., 2000). Although YME1L1 is orthologous to the yeast protein Yme1p and forms a homooligomeric structure able to rescue the Δyme1 mutant yeast phenotype (Shah et al., 2000), the function of paraplegin and AFG3L2 is not known.
The presence of two hydrophobic regions, which have the characteristics of transmembrane domains (Casari et al., 1998; Banfi et al., 1999), suggests that both paraplegin and AFG3L2 are integral membrane proteins. To test this prediction, we determined their submitochondrial localization. Upon selective disruption of the mitochondrial outer membrane by digitonin, both proteins are found exclusively in the mitoplast fraction (Fig. 1 A, lane 1) as well as in HSP60, which is a matrix marker (Fig. 1 A, lane 1). We also monitored the presence of porin, as an outer membrane marker, and cytochrome c, as outer membrane and intermembrane fractional markers, respectively. Porin, which resides on the outer membrane, but takes connections to the inner membrane, was found either in the outer membrane–intermembrane fraction (Fig. 1 A, lane 2) or in the mitoplast fraction as expected (Fig. 1 A, lane 1). Cytochrome c appears in the outer membrane/intermembrane fraction (Fig. 1 A, lane 2).
Figure 1.

Submitochondrial localization and structural organization of the paraplegin–AFG3L2 complex. (A) Submitochondrial localization of paraplegin and AFG3L2 in human primary fibroblasts. Disruption of the mitochondrial outer membrane by digitonin: pellet/mitoplast (lane 1) and supernatant fraction (lane 2); alkaline treatment of mitoplasts, isolating integral membrane proteins (lane 3) and soluble/peripheral membrane proteins (lane 4). (B) Coimmunoprecipitation of paraplegin and AFG3L2. HEK293 mitochondrial lysate was immunoprecipitated with AFG3L2 antibody (lane 1) and paraplegin antibody (lane 3). Precipitates were analyzed by SDS-PAGE and immunostained using paraplegin (lane 1) or AFG3L2 (lane 3) antibodies. Corresponding preimmune sera were used as controls for the specificity of the coimmunoprecipitation (lanes 2 and 4). (C) Gel filtration analysis of mitochondrial extracts from HEK293. Isolated mitochondria are solubilized and fractionated by Superose 6-gel chromatography. Fractions are analyzed by SDS-PAGE and immunostained by antibodies recognizing paraplegin or AFG3L2 (fractions are from two gels: top, fractions 18 and 19, and 20–33; bottom, fractions 18–26 and 27–33) . The peaks of elution for various proteins of known molecular mass are indicated by lines (900 kD, ferritin dimer; 450 kD, ferritin monomer; 232 kD, catalase). (D) Gel filtration analysis of mitochondrial extracts from control and HSP fibroblasts. Fractions are analyzed by SDS-PAGE and immunostained by AFG3L2 antibody (fractions 18–26 and 27–33 are from two gels).
Isolated mitoplasts were subjected to alkaline extraction and divided into supernatant (containing matrix and peripheral membrane proteins) and membrane fractions (containing integral proteins of the inner membrane) by ultracentrifugation. Paraplegin and AFG3L2 were recovered from the membrane fraction, indicating that both are integral proteins of the mitochondrial inner membrane (Fig. 1 A, lane 3).
To explore a potential physical interaction between paraplegin and AFG3L2, we performed coimmunoprecipitation studies in HEK293. Mitochondria were solubilized and supplemented with either AFG3L2 or paraplegin antibodies. Immunoblotting of the precipitates revealed that paraplegin was isolated with AFG3L2 antibody and that, accordingly, AFG3L2 coprecipitated with paraplegin antibody (Fig. 1 B, lanes 1 and 3). Preimmune sera failed to precipitate any protein. We conclude from these experiments that paraplegin assembles with AFG3L2.
To substantiate this conclusion, we performed gel filtration experiments. Mitochondria isolated from HEK293 cells or from primary human control fibroblasts were solubilized and fractionated. Eluate fractions were analyzed by immunoblotting using AFG3L2 and paraplegin antibodies. Both proteins, which show a similar molecular mass of ∼80 kD, eluted in a rather broad peak from the column with fractions corresponding to a molecular mass of ∼800–900 kD (Fig. 1, C and D). Notably, the elution pattern of AFG3L2 was altered in mitochondria from primary fibroblasts of HSP patients lacking paraplegin (see Materials and methods, Cell cultures). In fact, AFG3L2 was present in a complex with a reduced native molecular mass of ∼250 kD (Fig. 1 D).
These data are in agreement with the complex formation of paraplegin and AFG3L2, and they show that the paraplegin–AFG3L2 complex is aberrant in HSP cells lacking paraplegin. The latter also suggests the inability of AFG3L2 to homopolymerize or to assemble with other proteins to form the 900-kD complex.
HSP cells show increased susceptibility to reactive oxygen species (ROS)
Because mitochondria are sensitive targets for ROS, we examined the sensitivity of HSP patients' cells towards hydrogen peroxide (H2O2) challenge (1 h after 100 μM H2O2 treatment). To assess the activity of mitochondria, the membrane potential was monitored by staining control and HSP fibroblasts with the potentiometric probes rhodamine 123 and JC-1.
Upon rhodamine staining, mitochondria in HSP fibroblasts subjected to ROS challenge displayed an increased punctuate and diffuse pattern when compared with control cells (Fig. 2 A). By using JC-1, the hyperpolarized orange-staining mitochondria of HSP cells shift to greenish upon H2O2 treatment faster than mitochondria of control cells (Fig. 2 B). These results point to an increased sensitivity of HSP fibroblasts towards oxidative damage, which is accompanied by depolarization of mitochondria.
Figure 2.
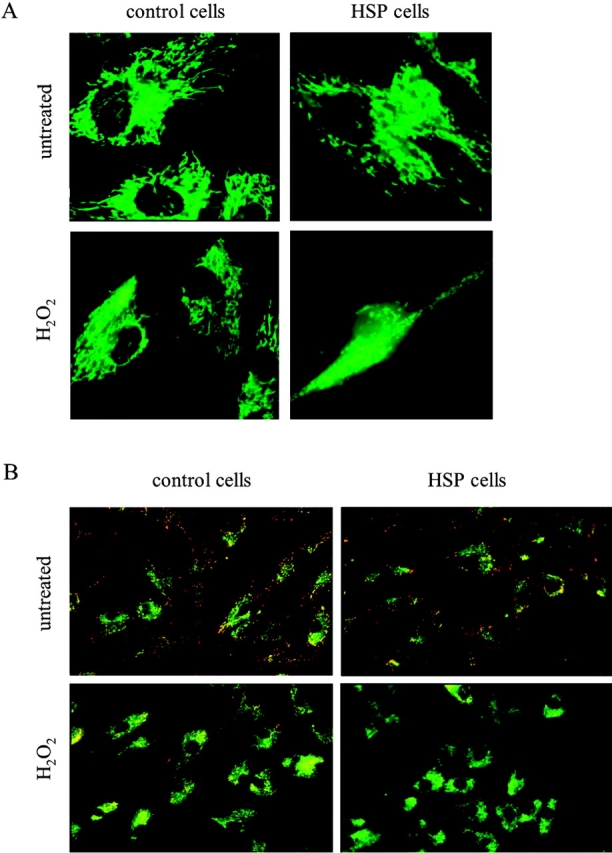
Mitochondrial depolarization of cells subjected to oxidative stress. Control and HSP fibroblasts are visualized by Rh123 (A) and JC-1 (B) staining.
The increased sensitivity towards ROS of HSP mitochondria lacking the paraplegin–AFG3L2 complex was confirmed by measurements of mitochondrial ATP synthesis (Fig. 3 A). ATP synthesis after H2O2 challenge (at 400 μM for 4 h) was remarkably reduced in HSP fibroblasts compared with controls (from 27.7 ± 4 nmol ATP/mg protein in untreated cells to 4.9 ± 2.8 nmol ATP/mg protein in H2O2 -treated cells; P < 0.0005).
Figure 3.
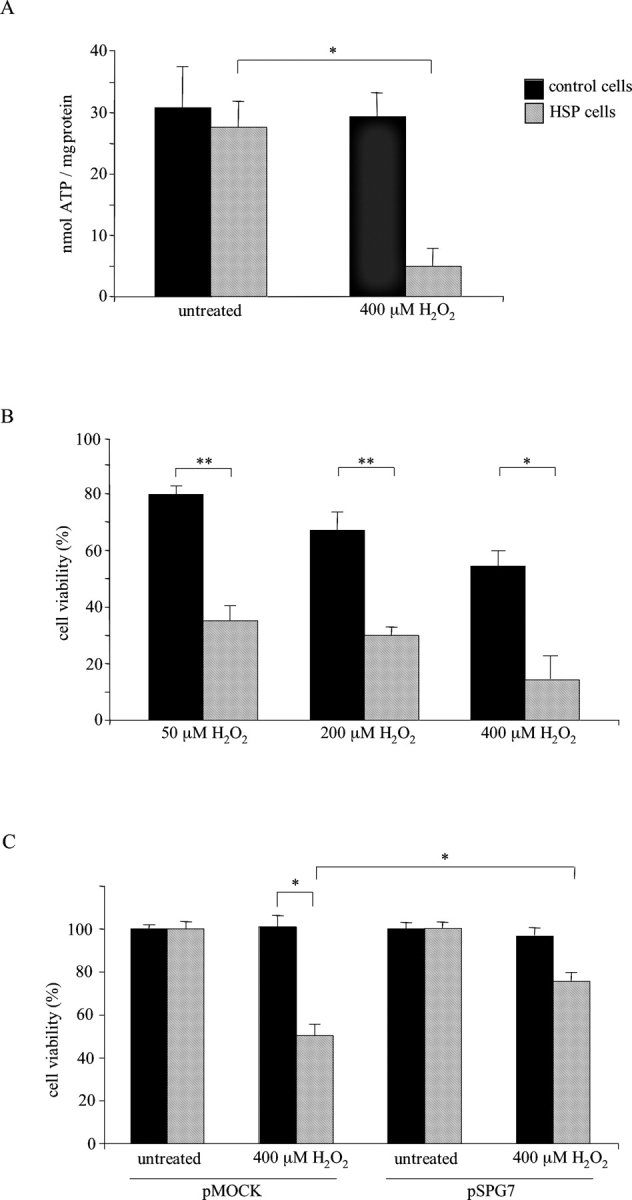
Hydrogen peroxide challenge and phenotype rescue. (A) ATP depletion after oxidative stress. Data represent means of six independent experiments. (B) MTT reduction assay, data are expressed as percentage between the absorbance (570 nm) of treated and untreated cells; values, in triplicates, represent means of four independent experiments. (C) HSP and control cells transfections by paraplegin expressing vector pSPG7. Data, in triplicates, are indicated as mean of three independent experiments. *, P < 0.0005; **, P < 0.000001. Bars represent ±SEM.
Cell vitality ascertained by MTT reduction assay confirmed the dose-dependent vulnerability of HSP cells to ROS (upon exposure to 50 μM H2O2 for 8 h, 35.1 ± 4.9% HSP cells vs. 79.7 ± 2.5% controls; at 200 μM, 30.0 ± 2.6% vs. 67.0 ± 6.2%, P < 0.000001; at 400 μM, 16.3 ± 4.7% vs. 52.3 ± 7.7%, P < 0.004; Fig 3 B). Catalase administration showed a protective action both in HSP and control cells exposed to H2O2 (unpublished data).
To examine whether the increased sensitivity of HSP fibroblasts towards oxidative stress is directly caused by a lack of paraplegin, we set up paraplegin cDNA transfection experiments. HSP and control cells were transfected with constructs harboring the full-length paraplegin cDNA or the empty vector, and then were exposed to different concentrations of H2O2. Cell damage was evaluated by assessing the MTT metabolism (Fig. 3 C). The increased sensitivity of HSP cells to oxidative stress was almost completely reversed to control values by exogenous expression of wild-type paraplegin. Trypan blue exclusion assays revealed identical results (unpublished data). Together, these results demonstrate an increased susceptibility to ROS of HSP patients' fibroblasts lacking paraplegin, which can be reversed by transfecting cells with paraplegin.
Impaired respiratory complex I activity in HSP fibroblasts
The decrease of ATP levels and mitochondrial transmembrane potential, together with the increased vulnerability of HSP cells under oxidative stress, prompted us to explore specific impairments of mitochondrial respiratory chain complexes in HSP fibroblasts under standard growth conditions. When compared with control cells, the growth rate of HSP cells was strongly reduced in galactose medium (Fig. 4 A). This is characteristic of cells with defective mitochondrial metabolism (Robinson et al., 1992) and, therefore, indicates an impaired respiratory activity in HSP cells even in the absence of an induced oxidative stress.
Figure 4.

Complex I deficiency in HSP fibroblasts. (A) Ratio of cell viability measured in galactose or glucose media; data, two replicates, represent means of three independent experiments. (B) ATP synthesis rates in permeabilized fibroblasts. 1, no substrate (basal activity); 2, pyruvate and l-malate (complexes I, II, III, IV, and V); 3, glutamate and l-malate (complexes I, II, III, IV, and V); 4, rotenone and succinate (complexes II, III, IV, and V); 5, antimycin, ascorbate, and TMPD (complexes IV and V). *, P < 0.05; **, P < 0.001; ***, P < 0.0001. Bars represent ±SEM.
To dissect the contribution of the different respiratory chain complexes to mitochondrial ATP synthesis, we quantified ATP production in permeabilized cells in the presence of different substrates and inhibitors of respiratory complexes. The basal activity of the respiratory chain in control and HSP cells is similar (Fig. 4 B, group 1). By providing pyruvate/malate or glutamate/malate (Fig. 4 B, groups 2 and 3), we stimulated ATP synthesis that is dependent on complexes I, II, III, IV, and V. Using a complex I inhibitor, rotenone, and succinate, we measured ATP production that is dependent on complexes II, III, IV, and V (Fig. 4 B, group 4). Finally, incubating cells with antimycin A, an inhibitor of complex III, and TMPD/ascorbate, we evaluated the ATP level due to the activity of complexes IV and V (Fig. 4 B, group 5). The ATP level measured in the presence of succinate and ascorbate was the same in HSP and control cells, suggesting that the activity of complexes II, III, IV, and V was comparable. In contrast, we detected a substantial reduction of ATP synthesis in HSP cells using pyruvate and glutamate as substrates, which stimulate the activity of the overall respiratory chain complexes (i.e., complexes I, II, III, IV, and V). These results correlate the observed reduction of ATP synthesis with an impairment of complex I activity in HSP cells. Similar experiments using isolated mitochondria also revealed a significant decrease of complex I activity in the absence of paraplegin (unpublished data).
To confirm this data, we used blue native PAGE (BN-PAGE) to isolate enzymatically active membrane proteins from mitochondria. In agreement with previous estimates (Schagger et al., 1994; Klement et al., 1995), complex I of both HSP and control cells exhibited a molecular mass of ∼750 kD (Fig. 5 A). By combining BN-PAGE with an in situ activity staining reaction that specifically detects enzyme activity of complex I, we confirmed a decrease in complex I activity in HSP cells (Fig. 5 B, HSP) when compared with control fibroblasts (Fig. 5 B, control). Transfection of HSP cells with paraplegin partially recovered the activity of complex I (Fig. 5 C, HSP/pSPG7), pointing to a direct role of paraplegin in the regulation of this respiratory complex.
Figure 5.
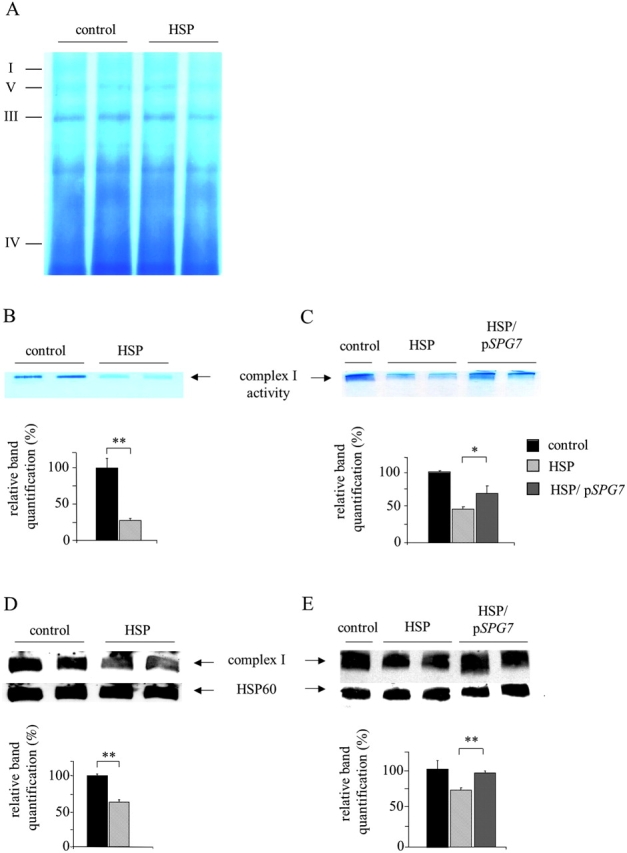
Blue native gel electrophoresis of mitochondrial complexes. (A) Coomassie brilliant blue staining of mitoplast preparations. (B) Complex I in situ activity staining by nitro blue tetrazolium reduction assay. (C) Rescue of complex I in situ activity in HSP cells after pSPG7 transfection. (D and E) Western blot analysis of complex I revealed by anti–39-kD antibody. Immunoblotting with HSP60 antibody was used to verify equal loading. Activity and protein amount of complex I were quantified by densitometric analysis. *, P < 0.05; **, P < 0.001. Bars represent ±SEM.
The reduced activity of complex I in HSP fibroblasts could reflect either a reduced specific activity of the enzymatic complex or a smaller number of complex I units per mitochondrion. To distinguish between these possibilities, we performed BN-PAGE followed by immunoblotting with an antibody directed against the 39-kD subunit of complex I. This experiment revealed a decreased amount of the assembled complex I in HSP cells (Fig. 5 D, HSP), which can be rescued to a normal level upon transfection with paraplegin cDNA (Fig. 5 E, HSP/pSPG7).
The reduced amount of complex I may have different causes: a defective expression, or import of nuclear-encoded mitochondrial preproteins, or an inefficient synthesis of mitochondrially encoded proteins. We addressed these points by examining the mitochondrial import of the 39-kD protein in mitochondria from HSP and control fibroblasts. After protein synthesis in a cell-free system in the presence of [35S]methionine and incubation of the radiolabeled protein with isolated mitochondria, the 39-kD protein accumulated at similar levels in HSP and control fibroblasts, suggesting that the protein import machinery is not impaired in HSP cells (Fig. 6 A). Consistently, the whole amount of the 39-kD protein, including both the unassembled and the complex I–assembled fractions, was comparable in HSP and control mitoplasts as visualized by Western blot analysis (Fig. 6 B). The mitochondrially encoded protein synthesis was measured by metabolic labeling of control and HSP cells in the presence of an inhibitor of cytoplasmic protein synthesis. Similar amounts of proteins are synthesized under these conditions (Fig. 6 C), indicating that the decreased activity of complex I is not due to an impaired synthesis of mitochondrially encoded subunits. Therefore, we conclude from these experiments that the paraplegin–AFG3L2 complex is required for the efficient assembly of complex I.
Figure 6.
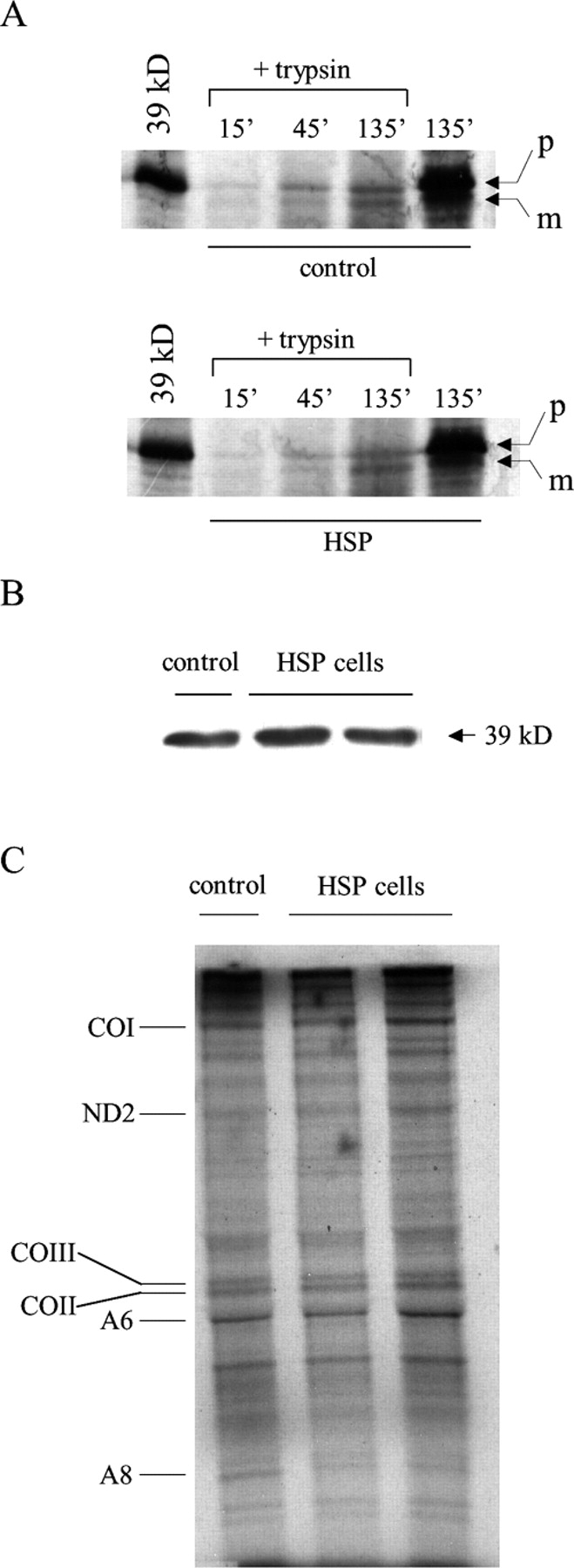
Mitochondrial import and protein synthesis in HSP and control fibroblasts. (A) Mitochondrial protein import efficiency of the 39-kD protein in control and HSP mitochondria (p, precursor; m, mature). (B) Western blot analysis of the 39-kD protein in control and HSP mitoplasts. (C) Mitochondrial protein synthesis in control and HSP cells (CO1, cytochrome c oxidase subunit I; ND2, NADH dehydrogenase subunit 2; COIII, cytochrome c oxidase subunit III; COII, cytochrome c oxidase subunit II; A6, ATPase subunit 6; A8, ATPase subunit 8).
Because the chaperone-like and the proteasic activities of the yeast mitochondrial metalloprotease complex are both essential for the respiratory competence (Arlt et al., 1998), we tested whether the proteolytic domain of paraplegin is responsible for the complex I rescue obtained on paraplegin mutant fibroblast. Therefore, we mutagenized the glutamic acid residue in position 575, which is conserved in the yeast and human metal binding site, and when changed to glutamine (E575Q; Fig. 7 A), is known to neutralize the proteasic activity (Arlt et al., 1996). As shown in Fig. 7 B, by expressing the mutagenized construct pSPG7 Q575, we were able to revert the reduced activity of complex I of HSP fibroblasts in a comparable manner to the construct expressing wild-type paraplegin. Again, in our system, the complex I activity is directly correlated to the assembled complex I amount (Fig. 7 C). Hence, the integrity of the proteolytic domain of paraplegin is not indispensable for the correct assembly of complex I. The same pattern of results was obtained when we analyzed the sensitivity to ROS. The mutagenized construct pSPG7 Q575 rescued the increased sensitivity to oxidant stress of HSP cells comparably to the wild-type construct (unpublished data).
Figure 7.
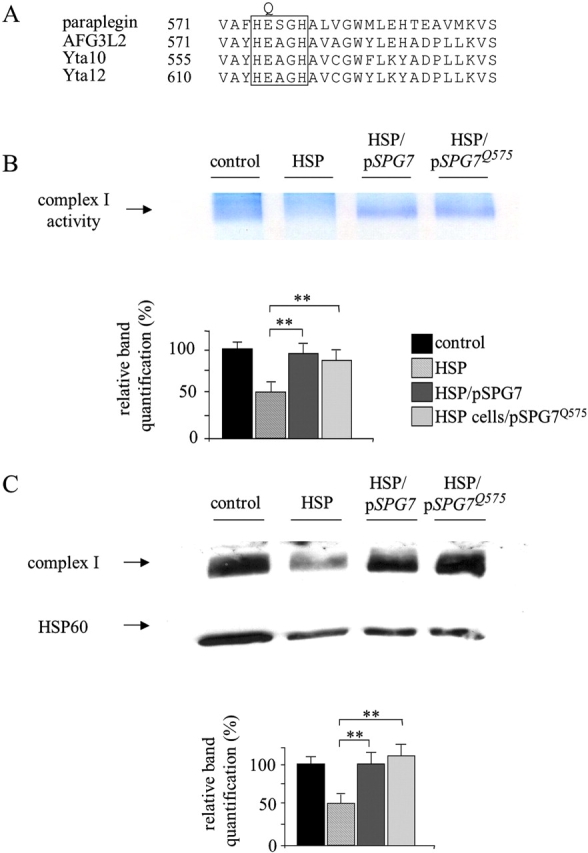
Integrity of the proteasic site of paraplegin is not essential for complex I rescue. (A) Partial sequence alignment of human and yeast metalloproteases surrounding the divalent metal binding site (boxed area); the glutamic acid residue, E, replaced by glutamine, Q, is indicated. (B) Rescue of complex I in situ activity in HSP cells after pSPG7 and pSPG7Q575 transfection. (C) Western blot analysis of complex I revealed by anti–39-kD antibody. Immunoblotting with HSP60 antibody was used to verify equal loading. Activity and protein amount of complex I were quantified by densitometric analysis. **, P < 0.001. Bars represent ±SEM.
Coexpression of paraplegin and AFG3L2 restores the respiratory competence of yta10yta12 mutant yeast cells
Paraplegin shares high sequence identity with subunits of the m-AAA protease in the yeast Saccharomyces cerevisiae, Yta10 (Afg3) and Yta12 (Rca1; Casari et al., 1998). To examine functional conservation, we performed a complementation analysis in yeast. Mitochondrial targeting of paraplegin and AFG3L2 in the heterologous organism was ensured by replacing their putative mitochondrial targeting sequences (amino acid residues 1–58 and amino acid residues 1–35, respectively) with the sorting signal of Yta10 (amino acid residues 1–63). The resulting hybrid proteins were expressed in yeast cells lacking Yta10, Yta12, or both. Expression and mitochondrial localization were confirmed by immunoblotting of cell extracts (unpublished data). Yeast cells lacking the m-AAA protease are respiratory deficient and cannot use nonfermentable carbon sources like glycerol (Tauer et al., 1994; Tzagoloff et al., 1994; Arlt et al., 1998). Expression of paraplegin or AFG3L2 did not restore the respiratory competence of Δyta10Δyta12 cells (Fig. 8 A) nor of Δyta10 or Δyta12 (unpublished data). However, the respiratory deficiency of Δyta10Δyta12 cells was suppressed upon coexpression of both paraplegin and AFG3L2 (Fig. 8 A). Thus, in agreement with the experiments in the human system, paraplegin and AFG3L2 functionally interact in yeast mitochondria. Complex formation of both proteins in yeast was subsequently observed by coimmunoprecipitation experiments (unpublished data) and BN-PAGE (Fig. 8 B), confirming that paraplegin and AFG3L2 act in a cooperative manner in a 900-kD complex, and, thereby, can substitute for the m-AAA protease in yeast.
Figure 8.

Complementation assays of Δyta10Δyta12 yeast cells with paraplegin–AFG3L2 complex. (A) Suppression of the respiratory deficiency of Δyta10Δyta12 yeast cells by paraplegin and AFG3L2 coexpression. Fivefold serial dilutions of yeast cells were spotted onto YP plates containing either 2% glucose (left, YPD) or 3% glycerol (right, YPG). (B) Western blot analysis of paraplegin–AFG3L2 complex on BN-PAGE: the human m-AAA protease has been revealed by c-Myc antibody, recognizing the AFG3L2 fusion protein; the same band was observed in an identical blot decorated by HA antibody, recognizing the paraplegin fusion protein (not depicted). (C) Proteolytic activity of paraplegin and AFG3L2 is required for respiratory competence of Δyta10Δyta12 cells; yeast cells harboring a proteolytic site mutation in paraplegin, AFG3L2, or both were cultured and spotted on YP plates containing 3% glycerol.
These results demonstrate functional conservation of the human paraplegin–AFG3L2 complex and the yeast m-AAA protease. They also provide first evidence for a proteolytic activity of paraplegin and AFG3L2 because maintenance of the respiratory competence of yeast cells requires proteolysis by the m-AAA protease (Arlt et al., 1998).
To substantiate this conclusion, point mutations were introduced into the predicted proteolytic center of both paraplegin and AFG3L2. Glutamic acid residues 575 in the consensus metal binding sites of both proteins were replaced by glutamine residues by site-directed mutagenesis (Fig. 7 A). The mutant proteins (fused to the mitochondrial targeting sequence of Yta10) were expressed in Δyta10Δyta12 cells, and the respiratory competence was examined. In contrast to the wild-type forms, coexpression of mutant paraplegin and AFG3L2 did not confer respiratory competence to Δyta10Δyta12 cells, demonstrating that the integrity of their proteolytic center is required for functional complementation (Fig. 8 C). We analyzed the expression of both subunits in these cells by Western blotting and verified their correct targeting to mitochondria by cell fractionation (unpublished data). Notably, coexpression of either mutant paraplegin or mutant AFG3L2 with the respective wild-type counterpart already impaired the respiratory competence (Fig. 8 C). Thus, in contrast to the yeast m-AAA protease (Arlt et al., 1998), the catalytic activity of both subunits of the human paraplegin–AFG3L2 complex is required for proteolytic activity.
Discussion
Mutations in paraplegin are known to cause neurodegeneration in an autosomal recessive form of HSP that approximately accounts for the 12% of recessive cases (unpublished data). However, the pathogenetic mechanism of this disorder, particularly the role of mitochondria, is presently not understood.
Here, we have identified paraplegin and its interactor, the homologous protein AFG3L2, as part of a high molecular mass complex in the inner membrane of mitochondria. They form a complex of about 900 kD, which is aberrant in HSP patients' cells lacking paraplegin, where a smaller complex of 250 kD is present. Our data identify the paraplegin–AFG3L2 complex as a functionally active structure whose absence plays a key role in the establishment of a cellular phenotype in HSP fibroblasts. We observed a remarkable reduction of complex I (NADH–ubiquinone oxidoreductase) activity in HSP cells compared with control cells. Furthermore, blue native gels confirmed this result and showed that the reduction of complex I activity is due to stoichiometric limitation of the enzymatic complex.
Our results reveal the first functional role of the paraplegin–AFG3L2 complex in the biogenesis of the respiratory chain. The deficiency is apparently caused by neither an impaired import of nuclear-encoded proteins nor an inefficient synthesis of mitochondrially encoded subunits, suggesting a role for the paraplegin–AFG3L2 complex in effectively assembling complex I. This defect was restored upon transfection of HSP patients' cells with a paraplegin-expressing vector.
A complementation study in yeast revealed, for the first time, the functional conservation of mitochondrial m-AAA proteases between distantly related species such as human and yeast. In yeast cells, inactivation of m-AAA protease by deleting one or both Yta10 and Yta12 causes pleiotropic phenotypes because maintenance of the respiratory competence requires proteolysis by the yeast m-AAA protease (Arlt et al., 1998). The respiratory deficiency of Δyta10Δyta12 cells (Langer, 2000) was suppressed by coexpression of paraplegin and AFG3L2, providing the first evidence for their proteolytic activity. This result was substantiated by the observation that the integrity of the proteolytic center of both paraplegin and AFG3L2 is required for functional complementation. Thus, the complementation experiments in yeast unambiguously demonstrate the proteolytic activity of the paraplegin–AFG3L2 complex.
We expressed the proteolytic site variant of paraplegin in paraplegin-deficient fibroblasts, and observed both a rescue of complex I activity and an increased resistance towards oxidative stress. This is reminiscent of findings in the yeast system where the expression of a proteolytic site variant of one homologous subunit of the m-AAA protease restores respiratory competence (Arlt et al., 1998). The control experiment demonstrating the inability of the mutant paraplegin–AFG3L2 complex (i.e., coexpression of proteolytic site variants of both paraplegin and AFG3L2) to rescue the described HSP phenotypes is not feasible in human cells because AFG3L2-deficient fibroblasts are currently not available. However, we envision the following scenario explaining these findings in the two systems. Maintenance of respiration in yeast requires only very low specific activity of the m-AAA protease; hence, a single proteolytic site mutant of a yeast subunit is sufficient for complementation. Although we see complementation of the yeast double mutant by the human paraplegin–AFG3L2 complex, the rescue of the respiratory competence is at the lower limit. In fact, a further decrease of the specific activity of the protease complex (for instance by introducing a point mutation into the proteolytic site of one subunit) abolishes complementation, explaining the effect of a single proteolytic site mutation of the human protease in yeast. However, paraplegin–AFG3L2 complex is in the presence of the natural mitochondrial substrates in the autologous human system. Lowering the proteolytic activity by introducing a point mutation in the proteolytic site of paraplegin does not completely inactivate the protease (as it happens in yeast, when autologous elements are involved. This is due to the presence of the wild-type copy of AFG3L2; therefore, we still observe complementation of an assembly defect (as in yeast; Arlt et al., 1998).
In view of the observed functional conservation, we propose to term the paraplegin–AFG3L2 complex the human m-AAA protease, which combines proteolytic and chaperone-like activities. The detected complex I deficiency in HSP cells is very likely due to an inefficient folding process in the absence of a physiological paraplegin–AFG3L2 complex. Impaired complex I activity is frequently accompanied by enhanced ROS production and lipid peroxidation (Barrientos and Moraes, 1999; Li et al., 2003). Indeed, an intimate link between ROS and defects in the electron transport function leading to a further increase in ROS production plays an important role in the development and progression of neurodegenerative diseases (Schapira et al., 1989; Wong et al., 2002; Taylor et al., 2003). Therefore, we analyzed the sensitivity of HSP patients' cells towards oxidative stress, and observed a significantly increased vulnerability of these cells by ROS. This effect was restored, again, upon exogenous expression of paraplegin.
In HSP, complex I deficiency could directly contribute to neurodegeneration via a free radical mechanism by direct ROS production and by a decreased ATP synthesis leading to energy failure. In particular, mitochondrial alterations have been documented in neuronal aging (Beal, 1996), and excessive production of ROS has been associated with different forms of neurodegeneration including: Parkinson's disease (Jenner and Olanow, 1996); Alzheimer's disease (Behl, 1999); amyotrophic lateral sclerosis (Rosen et al., 1993; Aguirre et al., 1998); Friedreich's ataxia (Wong et al., 1999); and, more recently, neurodegeneration related to prion infection (Milhavet et al., 2000).
Neurons are particularly susceptible to oxidative stress due to the high rate of oxidative metabolic activity, low level of antioxidant enzymes, catalase and glutathione peroxidase, extended axonal morphology prone to peripheral injury, and impairment of the respiratory chain enzymes. In particular, complex I is considered to be one of the most severely affected by age-related increases in oxidative stress (Wong et al., 2002). In synaptic mitochondria, this effect is even worse (Davey et al., 1998). In addition, the effects of ROS on mitochondrial respiratory activity limits the ability of neuronal cells to respond appropriately to increase of ATP demand (Sims et al., 2000; Mattson and Liu, 2002). These findings, together with the evidence of a different complex I distribution among types of neurons (Pettus et al., 2000), may account for the selective vulnerability of a restricted subset of neurons, and applies, in particular, to the retrograde axonopathy typical of HSP.
Our findings shed new light on the molecular pathogenesis of HSP. We showed how paraplegin mutations suppress the human m-AAA protease formation and generate complex I deficiency in HSP cells, which may lead to additional production of ROS causing oxidative stress in the particular subset of neurons affected by HSP. Therefore, the initial pathogenic mechanism is likely to stem from the combined effects of misfolded peptide accumulation and the lack of chaperone activity on complex I assembling. Specifically, our results link this mitochondriopathy to the increasing group of misfolding diseases such as Alzheimer's disease, Parkinson's disease, prion encephalopathies, and expanded polyglutamine disease (Dobson, 1999; Temussi et al., 2003). HSP commonly has a typical progressive course and onset in the third or forth decade of life. This implies that the phenotypic expression of this disease may involve two factors, a predisposing mutation and an age-related factor that causes a decline in mitochondrial function, which manifests the inherited defect.
Materials and methods
Reagents
Cell culture media and chemicals were purchased from Life Technologies (Invitrogen) and Sigma-Aldrich, respectively.
Cell cultures
Primary fibroblasts were obtained from skin biopsies of two controls and two HSP patients belonging to the original SPG7 family, homozygous for a 9.5-kb deletion in the SPG7 gene (patients no. IV-9 and IV-11; De Michele et al., 1998). In addition, we obtained fibroblasts from a healthy heterozygous individual belonging to the same family, which was used as an internal control (patient no. IV-3). Primary fibroblasts and HEK293 were cultured in α minimal essential medium supplemented with 2 mM l-glutamine, 200 U/ml penicillin, 200 mg/ml streptomycin, and 20% FBS at 37°C in 95% humidified air and 5% CO2.
Transfections
The paraplegin expression plasmid (pSPG7) was prepared by cloning the full-length SPG7 cDNA (GenBank/EMBL/DDBJ accession no. Y16610) in a modified pMT21 vector. The resulting recombinant plasmid was transiently transfected by the calcium phosphate (Gualberto et al., 1998) and metafectene (Biontex Laboratories) methods into cultured primary fibroblasts.
pSPG7 was mutagenized to obtain pSPG7Q575 as described later for the analogous yeast constructs (see Yeast expression constructs section).
Antibodies
To generate rabbit polyclonal antisera directed against paraplegin and AFG3L2, we generated GST fusion proteins (pGEX2T; Amersham Biosciences) with protein fragments spanning amino acids 243–844 of SPG7 gene product (GenBank/EMBL/DDBJ accession no. AAH07692) and amino acids 413–828 of AFG3L2 gene product (GenBank/EMBL/DDBJ accession no. Q9Y4W6). After expression in Escherichia coli, proteins were purified according to standard procedures and used to immunize rabbits. Anti-Hsp60 mAb was from StressGen Biotechnologies; anti–OxPhos complex I 39-kD subunit and mouse antiporin monoclonal antibodies from Molecular Probes were used. Anti–cytochrome c mAb was from Promega.
Isolation of mitochondria and mitoplasts
Mitochondria were isolated by differential centrifugation of HEK293 or fibroblast cell homogenates (Smith, 1967). In brief, cells were washed in PBS, resuspended in an appropriate isotonic buffer (0.25 M sucrose, 5 mM Tris-HCl, pH 7.5, and 0.1 mM PMSF) and homogenized using a glass teflon homogenizer. Unbroken cells and nuclei were pelleted by centrifugation at 600 g for 15 min. Supernatants were centrifuged at 10,000 g for 25 min, and the mitochondrial pellet was washed once with the isotonic buffer containing 1 mM EDTA, pH 8.
Mitoplasts were obtained by incubating mitochondria to a final protein concentration of 1.0 mg/ml in PBS with 2.7 mg/ml digitonin for 20 min on ice. The sample was centrifuged at 10,000 g for 10 min, and the final pellet (mitoplast fraction) was washed twice with PBS.
Isolation of integral proteins of the inner mitochondrial membrane
Isolation of integral proteins of the inner mitochondrial membrane was performed by incubating mitoplasts in 100 mM Na2CO3 and 1 mM PMSF (final solution pH 11.5) for 60 min on ice. Separation of the pellet (integral protein of the inner membrane) and supernatant (soluble and matrix proteins) was achieved by centrifugation at 70,000 g for 60 min at 4°C in a ultracentrifuge (model Tl 100; Beckman Coulter; Pajic et al., 1994).
Coimmunoprecipitation
100 μg of mitochondria isolated from HEK293 cells were resuspended at a concentration of 500 μg/ml in immunoprecipitation buffer (PBS containing 0.5% NP-40, 0.5 mM PMSF, 30 μg/ml aprotinin, and 1 mM ATP) and incubated with antiparaplegin, anti-AFG3L2 antisera, or preimmune sera for 2 h at 4°C in constant agitation. Subsequently, protein A–Sepharose was added. Precipitates were used for Western blot analysis.
Gel filtration analysis
1 mg of isolated mitochondria was solubilized at 5 mg/ml with 0.25% NP-40 in PBS supplemented with 1 mM ATP. Mitochondrial extracts were centrifuged for 30 min at 109,000 g in an ultracentrifuge. Supernatants were applied onto a Superose 6-gel filtration column (Amersham Biosciences) and chromatographed at a flow rate of 0.5 ml/min in solubilization buffer. Calibration standards included ferritin (monomer: 450 kD; dimer: 900 kD), catalase (232 kD), and myoglobin (17.5 kD). 0.5-ml fractions were collected; TCA was precipitated and analyzed by SDS-PAGE and Western blotting using AFG3L2- or paraplegin-specific antisera.
Oxidant challenge
Primary fibroblasts were plated at a density of 105 cells/cm2 and allowed to grow for 24 h before treatments. Cultures showing ≥98% viable cells were treated under different H2O2 concentrations (0–400 μM); in separated experiments, cells were incubated with 400 μM H2O2 and 400 U/ml catalase. After 8 h of incubation, cell vitality was evaluated by trypan blue exclusion assay (Reese and Byard, 1981) or MTT reduction assay (Liu et al., 1997).
To evaluate the effect of H2O2 on the ATP level, after 5 h of incubation, cells were lysed with the ATP lysis buffer (see Luminometric assay of ATP section), and ATP was measured using the Luciferin-Luciferase method (Ronner et al., 1999).
Mitochondrial membrane potential
Primary fibroblasts were stained with 0.5 μM JC-1 (Molecular Probes; Diaz et al., 1999) and with 5 μM Rh123 (Johnson et al., 1980) in culture medium at 37°C for 30 min. JC-1 produces two emissions depending on the mitochondrial membrane potential (orange signal, high polarized mitochondria; green signal, low polarized mitochondria; green cytoplasmic signal, depolarized mitochondria), whereas active mitochondria stained with Rh123 appear yellow-green. The picture acquisition system was Eclipse E600 with a digital camera (model DXM1200; Nikon).
Respiratory chain activity
Tests for respiratory chain defects were essentially performed as described by Robinson (1996). In brief, digitonin-permeabilized cells or isolated mitochondria were incubated at 37°C for 30 min in a respiratory buffer (0.25 M sucrose, 20 mM MOPS, 1 mM EDTA, 5 mM inorganic phosphate, 0.1% BSA fatty acid free, and 1 mM ADP, pH 7.4) containing specific substrates and inhibitors of the respiratory chain complexes. By providing pyruvate/malate (5 mM and 1 mM, respectively) and glutamate/malate (5 mM and 1 mM, respectively), we stimulated ATP synthesis dependent on complexes I, II, III, IV, and V. Using a complex I inhibitor, 1 μM rotenone, and 10 mM succinate, we measured ATP production that is dependent on complexes II, III, IV, and V. Finally, incubating cells with 20 μg/ml antimycin A, an inhibitor of complex III, and TMPD/ascorbate (0.1 mM and 2 mM, respectively), we evaluated the ATP level due to the activity of complexes IV and V. ATP production was measured by luminometric assay. A t test was applied for significance calculation.
Luminometric assay of ATP
ATP concentration was determined with the Luciferin-Luciferase method as described previously (Ronner et al., 1999); in brief, the assay solution was prepared as follows: 250 mM glycilglycine, 2 mM EGTA, 2 mM MgCl2, 0.4 g/liter BSA fatty acid free, 7.5 mM DTT, 15 μM luciferin, and 10 μg/ml luciferase. Cells were lysed with the ATP lysis buffer (0.2 M NaOH and 0.5 mM EDTA), and an aliquot of the obtained extract was diluted with the ATP dilution buffer (0.1 M NaOH and 0.5 mM EDTA). In the luminometer, 20 μl of this mixture was added to 100 μl of the assay solution, and the ATP content was measured. Data were expressed as nanomoles of ATP per milligram of protein (Bradford, 1976).
BN-PAGE and complex I in situ activity staining
Isolated mitoplasts were solubilized at a protein concentration of 5 mg/ml by dodecyl maltoside (2% final concentration) and centrifuged, and supernatants were loaded on a linear 5–13% gradient polyacrylamide gel. Gels were stained with coomassie brilliant blue or transblotted on a nitrocellulose membrane and immunodecorated with anti–39-kD antibody (Schagger et al., 1994). Complex I in situ activity staining was performed by incubation of the gel in 0.1 M Tris-HCl, 0.14 mM NADH, and 1 mg/ml nitro blue tetrazolium, pH 7.4; the reaction was carried out at RT and stopped after 30 min by 45% methanol and 10% acetic acid (Jung et al., 2000).
Band quantifications relative to in situ activity staining gels and Western blots have been performed by densitometric analysis (densitometer, Epson Twain Pro); a t test was used for significant calculation.
Import of 39-kD subunit of respiratory complex I in mitochondria isolated from control and HSP fibroblasts
Mitochondria from control and HSP fibroblasts were isolated as described previously, and import assay was performed (Yano et al., 2000). Precursor protein 39 kD was synthesized by in vitro transcription and translation in the presence of [35S]methionine (TNT-coupled retuculocyte lysate system; Promega). The mitochondrial import assay was performed as follows: 660 μg of mitochondria was resuspended in 35 μl of mannitol buffer (225 mM mannitol, 25 mM sucrose, 10 mM Tris-HCl, pH 7.8, and 0.1 mM EDTA) and incubated with 70 μl of translate, 0.6 μl of 180 mM malate, and 1.1 μl of 1 M pyruvate. After 15, 45, and 135 min, 20-μl aliquots were removed, transferred to a fresh tube, incubated with trypsin, and centrifuged at 13,000 g for 5 min at 4°C. Pellets were washed once with mannitol buffer and resuspended in sample buffer for electrophoretic analysis in 12% SDS-PAGE. The imported protein was analyzed by fluorography.
Mitochondrial protein synthesis analysis
Primary fibroblasts were labeled with [35S]methionine (1,000 Ci/mmol and 100 μCi/ml) for 2 h in presence of 200 μg/ml cycloeximide as described previously (Chomyn, 1996). After labeling, the cells were washed and directly lysed in a sample buffer containing 62.5 mM Tris/HCl, pH 6.8, 2% (vol/vol) 2-mercaptoethanol, 1% (wt/vol) SDS, and 0.01% bromophenol blue. The mitochondrial translation products were analyzed by 15% SDS-PAGE and autoradiography.
Yeast expression constructs
Mature forms of paraplegin and AFG3L2 were fused to a mitochondrial leader peptide to ensure efficient mitochondrial import in the yeast. Precisely, the mature paraplegin (amino acids 59–795; GenBank/EMBL/DDBJ accession no. AAH07692) and AFG3L2 (amino acids 36–798; GenBank/EMBL/DDJB accession no. Q9Y4W6) were tagged with the HA and c-Myc epitopes, respectively, and fused to the mitochondrial targeting sequence of YTA10 (amino acids 1–63; GenBank/EMBL/DDJB accession no. S46611). The constructs were cloned into the vectors YcpLac111 (paraplegin) and pRS316 (AFG3L2) under the ADH1 promoter. Paraplegin and AFG3L2 were mutagenized in the yeast expression constructs using a site-directed mutagenesis kit (model QuickChange XL; Stratagene) and the following oligonucleotides: 5'-GTG GTT GCG TTT CAT CAG TCG GGC CAC GCC-3' (forward primer) and 5'-GGC GTG GCC CGA CTG ATG AAA CGC AAC CAC-3' (reverse primer) for paraplegin; 5'-CTG TGG CAT ACC ACC AAG CAG GCC ATG CGG-3' (forward primer) and 5'-CCG CAT GGC CTG CTT GGT GGT ATG CCA CAG-3' (reverse primer) for AFG3L2. The codon GAA coding for glutamic acid residue 575 of paraplegin was replaced by CAG coding for glutamine. In case of AFG3L2, glutamic acid residue 575 was replaced by glutamine, mutating the respective codon GAA to CAA. Mutagenesis was verified by DNA sequencing.
Yeast strains were transformed with the described vectors and cultivated on synthetic medium supplemented with 2% glucose (YPD) or 3% glycerol (YPG). To allow adaptation to respiratory conditions, cells grown on YPD plates were shifted to glycerol-containing YP medium and incubated for 6 d before spotting on YPG plates, as described previously (Arlt et al., 1996). BN-PAGE and Western blot analysis on yeast mitochondria were performed as described in BN-PAGE and complex I in situ activity staining; human paraplegin–AFG3L2 complex was revealed by the anti–c-Myc (9E10; 10 μg/ml).
Acknowledgments
We want to thank Paolo Bernardi, Peter Bross, Eugenio Monti, Alessandro Bulfone, and Elena Rugarli for critical discussion.
We are grateful to the Italian Telethon Foundation (grant F1), the National Institutes of Health (grant RO1 NS38713-01), the Armenise-Harvard and the Agarini Foundations, and the Italian Minister of Health (grant RF00263). Work in the laboratory of T. Langer was supported by the Center for Molecular Medicine Cologne and by grants from the Deutsche Forschungsgemeinschaft.
L. Atorino and L. Silvestri contributed equally to this work.
Abbreviations used in this paper: BN-PAGE, blue native PAGE; HSP, hereditary spastic paraplegia; ROS, reactive oxygen species.
References
- Aguirre, T., L. Van Den Bosch, K. Goetschalckx, P. Tilkin, G. Mathijs, J.J. Cassiman, and W. Robberecht. 1998. Increased sensitivity of fibroblasts from amyotrophic lateral sclerosis patients to oxidative stress. Ann. Neurol. 43:452–457. [DOI] [PubMed] [Google Scholar]
- Arlt, H., R. Tauer, H. Feldmann, W. Neupert, and T. Langer. 1996. The YTA10-12 complex, an AAA protease with chaperone-like activity in the inner membrane of mitochondria. Cell. 85:875–885. [DOI] [PubMed] [Google Scholar]
- Arlt, H., G. Steglich, R. Perryman, B. Guiard, W. Neupert, and T. Langer. 1998. The formation of respiratory chain complexes in mitochondria is under the proteolytic control of the m-AAA protease. EMBO J. 17:4837–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfi, S., M.T. Bassi, G. Andolfi, A. Marchitiello, S. Zanotta, A. Ballabio, G. Casari, and B. Franco. 1999. Identification and characterization of AFG3L2, a novel paraplegin-related gene. Genomics. 59:51–58. [DOI] [PubMed] [Google Scholar]
- Barrientos, A., and C.T. Moraes. 1999. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J. Biol. Chem. 274:16188–16197. [DOI] [PubMed] [Google Scholar]
- Beal, M.F. 1996. Mitochondria, free radicals, and neurodegeneration. Curr. Opin. Neurobiol. 6:661–666. [DOI] [PubMed] [Google Scholar]
- Behl, C. 1999. Alzheimer's disease and oxidative stress: implications for novel therapeutic approaches. Prog. Neurobiol. 57:301–323. [DOI] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. [DOI] [PubMed] [Google Scholar]
- Casari, G., and E. Rugarli. 2001. Molecular basis of inherited spastic paraplegias. Curr. Opin. Genet. Dev. 11:336–342. [DOI] [PubMed] [Google Scholar]
- Casari, G., M. De Fusco, S. Ciarmatori, M. Zeviani, M. Mora, P. Fernandez, G. De Michele, A. Filla, S. Cocozza, R. Marconi, et al. 1998. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 93:973–983. [DOI] [PubMed] [Google Scholar]
- Charvin, D., C. Cifuentes-Diaz, N. Fonknechten, V. Joshi, J. Hazan, J. Melki, and S. Betuing. 2003. Mutations in SPG4 are responsible for a loss of function of spastin, an abundant neuronal protein localized in the nucleus. Hum. Mol. Genet. 12:71–78. [DOI] [PubMed] [Google Scholar]
- Chomyn, A. 1996. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol. 264:197–210. [DOI] [PubMed] [Google Scholar]
- Coppola, M., A. Pizzigoni, S. Banfi, M.T. Bassi, G. Casari, and B. Incerti. 2000. Identification and characterization of YME1L1, a novel paraplegin-related gene. Genomics. 66:48–54. [DOI] [PubMed] [Google Scholar]
- Crosby, A.H., and C. Proukakis. 2002. Is the transportation highway the right road for hereditary spastic paraplegia? Am. J. Hum. Genet. 71:1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey, G.P., S. Peuchen, and J.B. Clark. 1998. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J. Biol. Chem. 273:12753–12757. [DOI] [PubMed] [Google Scholar]
- De Michele, G., M. De Fusco, F. Cavalcanti, A. Filla, R. Marconi, G. Volpe, A. Monticelli, A. Ballabio, G. Casari, and S. Cocozza. 1998. A new locus for autosomal recessive hereditary spastic paraplegia maps to chromosome 16q24.3. Am. J. Hum. Genet. 63:135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz, G., M.D. Setzu, A. Zucca, R. Isola, A. Diana, R. Murru, V. Sogos, and F. Gremo. 1999. Subcellular heterogeneity of mitochondrial membrane potential: relationship with organelle distribution and intercellular contacts in normal, hypoxic and apoptotic cells. J. Cell Sci. 112:1077–1084. [DOI] [PubMed] [Google Scholar]
- Dobson, C.M. 1999. Protein misfolding, evolution and disease. Trends Biochem. Sci. 24:329–332. [DOI] [PubMed] [Google Scholar]
- Errico, A., A. Ballabio, and E.I. Rugarli. 2002. Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum. Mol. Genet. 11:153–163. [DOI] [PubMed] [Google Scholar]
- Gualberto, A., K. Aldape, K. Kozakiewicz, and T.D. Tlsty. 1998. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc. Natl. Acad. Sci. USA. 95:5166–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, J.J., A. Durr, I. Cournu-Rebeix, C. Georgopoulos, D. Ang, M.N. Nielsen, C.S. Davoine, A. Brice, B. Fontaine, N. Gregersen, and P. Bross. 2002. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am. J. Hum. Genet. 70:1328–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, A.E. 1983. Classification of the hereditary ataxias and paraplegias. Lancet. 1:1151–1155. [DOI] [PubMed] [Google Scholar]
- Hazan, J., N. Fonknechten, D. Mavel, C. Paternotte, D. Samson, F. Artiguenave, C.S. Davoine, C. Cruaud, A. Durr, P. Wincker, et al. 1999. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 23:296–303. [DOI] [PubMed] [Google Scholar]
- Jenner, P., and C.W. Olanow. 1996. Oxidative stress and the pathogenesis of Parkinson's disease. Neurology. 47:S161–S170. [DOI] [PubMed] [Google Scholar]
- Johnson, L.V., M.L. Walsh, and L.B. Chen. 1980. Localization of mitochondria in living cells with rhodamine 123. Proc. Natl. Acad. Sci. USA. 77:990–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, C., C.M. Higgins, and Z. Xu. 2000. Measuring the quantity and activity of mitochondrial electron transport chain complexes in tissues of central nervous system using blue native polyacrylamide gel electrophoresis. Anal. Biochem. 286:214–223. [DOI] [PubMed] [Google Scholar]
- Klement, P., L.G. Nijtmans, C. Van den Bogert, and J. Houstek. 1995. Analysis of oxidative phosphorylation complexes in cultured human fibroblasts and amniocytes by blue-native-electrophoresis using mitoplasts isolated with the help of digitonin. Anal. Biochem. 231:218–224. [DOI] [PubMed] [Google Scholar]
- Langer, T. 2000. AAA proteases: cellular machines for degrading membrane proteins. Trends Biochem. Sci. 25:247–251. [DOI] [PubMed] [Google Scholar]
- Leonhard, K., J.M. Herrmann, R.A. Stuart, G. Mannhaupt, W. Neupert, and T. Langer. 1996. AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP-dependent degradation of inner membrane proteins in mitochondria. EMBO J. 15:4218–4229. [PMC free article] [PubMed] [Google Scholar]
- Li, N., K. Ragheb, G. Lawler, J. Sturgis, B. Rajwa, J.A. Melendez, and J.P. Robinson. 2003. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 278:8516–8525. [DOI] [PubMed] [Google Scholar]
- Liu, Y., D.A. Peterson, H. Kimura, and D. Schubert. 1997. Mechanism of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J. Neurochem. 69:581–593. [DOI] [PubMed] [Google Scholar]
- Mattson, M.P., and D. Liu. 2002. Energetics and oxidative stress in synaptic plasticity and neurodegenerative disorders. Neuromolecular Med. 2:215–231. [DOI] [PubMed] [Google Scholar]
- Milhavet, O., H.E. McMahon, W. Rachidi, N. Nishida, S. Katamine, A. Mange, M. Arlotto, D. Casanova, J. Riondel, A. Favier, and S. Lehmann. 2000. Prion infection impairs the cellular response to oxidative stress. Proc. Natl. Acad. Sci. USA. 97:13937–13942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura, T., and A.J. Wilkinson. 2001. AAA+ superfamily ATPases: common structure—diverse function. Genes Cells. 6:575–597. [DOI] [PubMed] [Google Scholar]
- Pajic, A., R. Tauer, H. Feldmann, W. Neupert, and T. Langer. 1994. Yta10p is required for the ATP dependent degradation of polypeptides in the inner membrane of mitochondria. FEBS Lett. 353:201–206. [DOI] [PubMed] [Google Scholar]
- Patel, H., H. Cross, C. Proukakis, R. Hershberger, P. Bork, F.D. Ciccarelli, M.A. Patton, V.A. McKusick, and A.H. Crosby. 2002. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat. Genet. 31:347–348. [DOI] [PubMed] [Google Scholar]
- Pettus, E.H., R. Betarbet, B. Cottrell, D.C. Wallace, V. Madyastha, and J.T. Greenamyre. 2000. Immunocytochemical characterization of the mitochondrially encoded ND1 subunit of complex I (NADH : ubiquinone oxidoreductase) in rat brain. J. Neurochem. 75:383–392. [DOI] [PubMed] [Google Scholar]
- Polo, J.M., J. Calleja, O. Combarros, and J. Berciano. 1993. Hereditary “pure” spastic paraplegia: a study of nine families. J. Neurol. Neurosurg. Psychiatry. 56:175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese, J.A., and J.L. Byard. 1981. Isolation and culture of adult hepatocytes from liver biopsies. In Vitro. 17:935–940. [DOI] [PubMed] [Google Scholar]
- Reid, E. 2003. Science in motion: common molecular pathological themes emerge in the hereditary spastic paraplegias. J. Med. Genet. 40:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, B.H. 1996. Use of fibroblast and lymphoblast cultures for detection of respiratory chain defects. Methods Enzymol. 264:454–464. [DOI] [PubMed] [Google Scholar]
- Robinson, B.H., R. Petrova-Benedict, J.R. Buncic, and D.C. Wallace. 1992. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 48:122–126. [DOI] [PubMed] [Google Scholar]
- Ronner, P., E. Friel, K. Czerniawski, and S. Frankle. 1999. Luminometric assays of ATP, phosphocreatine, and creatine for estimation of free ADP and free AMP. Anal. Biochem. 275:208–216. [DOI] [PubMed] [Google Scholar]
- Rosen, D.R., T. Siddique, D. Patterson, D.A. Figlewicz, P. Sapp, A. Hentati, D. Donaldson, J. Goto, J.P. O'Regan, H.X. Deng, et al. 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 362:59–62. (published erratum appears in Nature. 1993. 364:362) [DOI] [PubMed] [Google Scholar]
- Schagger, H., W.A. Cramer, and G. von Jagow. 1994. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217:220–230. [DOI] [PubMed] [Google Scholar]
- Schapira, A.H., J.M. Cooper, D. Dexter, P. Jenner, J.B. Clark, and C.D. Marsden. 1989. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1:1269. [DOI] [PubMed] [Google Scholar]
- Schwarz, G.A., and C.N. Liu. 1956. Hereditary (familial) spastic paraplegia: further clinical and pathological observations. AMA Arch. Neurol. Psychiatry. 75:144–162. [DOI] [PubMed] [Google Scholar]
- Shah, Z.H., G.A. Hakkaart, B. Arku, L. de Jong, H. van der Spek, L.A. Grivell, and H.T. Jacobs. 2000. The human homologue of the yeast mitochondrial AAA metalloprotease Yme1p complements a yeast yme1 disruptant. FEBS Lett. 478:267–270. [DOI] [PubMed] [Google Scholar]
- Sims, N.R., M.F. Anderson, L.M. Hobbs, J.Y. Kong, S. Phillips, J.A. Powell, and E. Zaidan. 2000. Impairment of brain mitochondrial function by hydrogen peroxide. Brain Res. Mol. Brain Res. 77:176–184. [DOI] [PubMed] [Google Scholar]
- Smith, A.L. 1967. Preparation, properties, and conditions for assay of mitochondria: slaughterhouse matherial, small-scale. Methods Enzymol. 10:81–86. [Google Scholar]
- Tauer, R., G. Mannhaupt, R. Schnall, A. Pajic, T. Langer, and H. Feldmann. 1994. Yta10p, a member of a novel ATPase family in yeast, is essential for mitochondrial function. FEBS Lett. 353:197–200. [DOI] [PubMed] [Google Scholar]
- Taylor, E.R., F. Hurrell, R. Shannon, T.K. Lin, J. Hirst, and M.P. Murphy. 2003. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 278:19603–19610. [DOI] [PubMed] [Google Scholar]
- Temussi, P.A., L. Masino, and A. Pastore. 2003. From Alzheimer to Huntington: why is a structural understanding so difficult? EMBO J. 22:355–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzagoloff, A., J. Yue, J. Jang, and M.F. Paul. 1994. A new member of a family of ATPases is essential for assembly of mitochondrial respiratory chain and ATP synthetase complexes in Saccharomyces cerevisiae. J. Biol. Chem. 269:26144–26151. [PubMed] [Google Scholar]
- Wong, A., J. Yang, P. Cavadini, C. Gellera, B. Lonnerdal, F. Taroni, and G. Cortopassi. 1999. The Friedreich's ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 8:425–430. [DOI] [PubMed] [Google Scholar]
- Wong, A., L. Cavelier, H.E. Collins-Schramm, M.F. Seldin, M. McGrogan, M.L. Savontaus, and G.A. Cortopassi. 2002. Differentiation-specific effects of LHON mutations introduced into neuronal NT2 cells. Hum. Mol. Genet. 11:431–438. [DOI] [PubMed] [Google Scholar]
- Yano, M., N. Hoogenraad, K. Terada, and M. Mori. 2000. Identification and functional analysis of human Tomm22 for protein import into mitochondria. Mol. Cell. Biol. 20:7205–7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X., D. Alvarado, S. Rainier, R. Lemons, P. Hedera, C.H. Weber, T. Tukel, M. Apak, T. Heiman-Patterson, L. Ming, et al. 2001. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet. 29:326–331. [DOI] [PubMed] [Google Scholar]