

Abstract
In sympathetic neurons, unlike most nonneuronal cells, growth factor withdrawal–induced apoptosis requires the development of competence in addition to cytochrome c release to activate caspases. Thus, although most nonneuronal cells die rapidly with cytosolic cytochrome c alone, sympathetic neurons are remarkably resistant unless they develop competence. We have identified endogenous X-linked inhibitor of apoptosis protein (XIAP) as the essential postcytochrome c regulator of caspase activation in these neurons. In contrast to wild-type neurons that are resistant to injection of cytochrome c, XIAP-deficient neurons died rapidly with cytosolic cytochrome c alone. Surprisingly, the release of endogenous Smac was not sufficient to overcome the XIAP resistance in sympathetic neurons. In contrast, the neuronal competence pathway permitted cytochrome c to activate caspases by inducing a marked reduction in XIAP levels in these neurons. Thus, the removal of XIAP inhibition appears both necessary and sufficient for cytochrome c to activate caspases in sympathetic neurons. These data identify a critical function of endogenous XIAP in regulating apoptosis in mammalian cells.
Keywords: Smac; cytochrome c; nerve growth factor; IAP; neurons
Introduction
The ability of cells to induce apoptosis and die is important during development and is crucial for the maintenance of homeostasis (Vaux and Korsmeyer, 1999). Dysregulation of the apoptotic pathway is associated with many pathological conditions. For example, decreased apoptosis contributes to cancer progression (Green and Evan, 2002), whereas increased apoptosis leads to loss of neural cells during stroke, spinal cord injury and in many neurodegenerative diseases (Yuan and Yankner, 2000). Therefore, understanding how cells regulate apoptosis has important therapeutic implications in many pathological conditions.
The mechanism of neuronal apoptosis has been widely studied in sympathetic neurons that are dependent on NGF for survival. Deprivation of NGF induces an apoptotic neuronal death within 24–48 h after NGF removal in culture (Deckwerth and Johnson, 1993; Deshmukh and Johnson, 1997). This death is prevented by macromolecular synthesis inhibitors such as cycloheximide (Martin et al., 1988), is dependent on Bax (Deckwerth et al., 1996), and is executed by the caspase proteases (Deshmukh et al., 1996; Troy et al., 1996; McCarthy et al., 1997).
Activation of caspases is a crucial point at which cells become committed to die during apoptosis (Deshmukh et al., 2000; Denault and Salvesen, 2002). In most mammalian cells, including sympathetic neurons, caspase activation is triggered by the release of cytochrome c from mitochondria to cytosol (Deshmukh and Johnson, 1998; Neame et al., 1998; Wang, 2001). Data from several cell-free studies indicate that cytochrome c released from the mitochondria binds to Apaf-1 and promotes its oligomerization. Procaspase-9 is then recruited to bind oligomerized Apaf-1 to form the apoptosome complex where caspase-9 becomes activated. Activated caspase-9 then cleaves and activates other caspases, such as caspase-3, to induce rapid apoptosis and cell death (Wang, 2001).
Members of the inhibitor of apoptosis proteins (IAPs) can regulate caspase activity by binding directly to activated caspases and inhibiting their function (Salvesen and Duckett, 2002). The IAP family includes X-linked IAP (XIAP), cellular IAP-1 (cIAP-1), cIAP-2, neuronal apoptosis inhibitory protein, Survivin, melanoma IAP, and Bruce, all of which contain one or more repeats of the characteristic baculovirus IAP repeat domain. Overexpression of IAPs blocks apoptosis in many cells (Deveraux et al., 1998; Duckett et al., 1998; Simons et al., 1999), including in sympathetic neurons (Wiese et al., 1999; Yu et al., 2003). The IAPs themselves can potentially be regulated in cells by at least two mechanisms. First, mitochondrial proteins such as Smac/DIABLO and HtrA2/Omi, when translocated to the cytosol during apoptosis, can bind to and inhibit multiple IAPs (Du et al., 2000; Verhagen et al., 2000; Suzuki et al., 2001a). Second, several IAPs including XIAP, cIAP-1, and cIAP-2 contain a RING finger domain that can function as an E3 ubiquitin ligase and target themselves and other proteins for proteasome-mediated degradation (Yang et al., 2000; Suzuki et al., 2001b; MacFarlane et al., 2002). However, the mechanism by which degradation of IAPs is regulated in mammalian cells in not known.
In primary fibroblasts and many cell lines, cytosolic microinjection of cytochrome c induces a rapid, caspase-dependent apoptotic death, thus indicating that cytosolic accumulation of cytochrome c alone is sufficient to activate caspases in these nonneuronal cells (Li et al., 1997; Brustugun et al., 1998; Juin et al., 1999; Chang et al., 2000). In contrast, cytochrome c, although necessary, is not sufficient to induce cell death in sympathetic neurons. NGF-maintained sympathetic neurons are remarkably resistant to cytosolic microinjection of cytochrome c, thus pointing to a stringent postcytochrome c regulation of caspase activation in these neurons (Deshmukh and Johnson, 1998; Neame et al., 1998). Importantly, NGF deprivation activates a novel pathway, called development of competence, which is necessary to allow cytochrome c to activate caspases and induce cell death in sympathetic neurons (Deshmukh and Johnson, 1998). Therefore, NGF deprivation–induced apoptosis in sympathetic neurons requires the activation of two pathways: a cytochrome c release pathway that is dependent on protein synthesis and Bax function; and a development of the competence pathway that requires neither protein synthesis nor Bax function (Deshmukh and Johnson, 1998).
Recent data suggests that the inability of cytosolic cytochrome c alone to induce death in NGF-maintained sympathetic neurons is due to a block in caspase activation by the IAPs. Microinjection of excess exogenous Smac that inhibits IAPs overcomes this block and permits cytochrome c to induce a rapid, caspase-dependent death in these neurons (Deshmukh et al., 2002). Thus, the target of the competence pathway is likely to be IAPs, although which IAP may be important and the exact mechanism by which the competence pathway permits cytochrome c to induce neuronal apoptosis is not known. Here, we identify XIAP as the critical inhibitor of caspase activation in sympathetic neurons. We find that XIAP mRNA and protein are both selectively decreased when neurons develop competence and become permissive for cytochrome c–mediated caspase activation. Importantly, although cytochrome c alone is incapable of inducing cell death in wild-type neurons, it is remarkably sufficient to induce a rapid apoptotic death in XIAP-deficient neurons. These data identify an essential function for endogenous XIAP in regulating apoptosis in mammalian cells and indicate that removal of XIAP inhibition is both necessary and sufficient for cytochrome c to activate caspases during neuronal apoptosis.
Results
Removal of IAP inhibition is necessary for cytochrome c to induce apoptosis in sympathetic neurons
NGF deprivation–induced cytochrome c release and development of competence are both necessary for apoptosis in sympathetic neurons. These two pathways are distinct from each other because whereas cycloheximide addition and Bax deficiency block the cytochrome c release pathway, neither one blocks the development of the competence pathway (Deshmukh and Johnson, 1998). Thus, whereas microinjection of exogenous cytosolic cytochrome c is not sufficient to induce cell death in NGF-maintained neurons, it can do so in NGF-deprived, cycloheximide-treated neurons or NGF-deprived, Bax-deficient neurons, both of which develop competence (Fig. 1; Deshmukh and Johnson, 1998). Bax deficiency or cycloheximide addition itself does not induce competence; they are simply used as tools for blocking the cytochrome c release pathway, without which the NGF-deprived neurons would die, thereby precluding us from studying the competence pathway.
Figure 1.

The ability of exogenously microinjected Smac to inhibit IAPs is necessary for permitting cytochrome c to induce death in sympathetic neurons. NGF-maintained mouse sympathetic neurons were microinjected with either cytochrome c or wild-type AVPI-Smac alone, or cytochrome c along with wild-type AVPI or mutant MVPI-Smac. Parallel cultures of sympathetic neurons that were deprived of NGF in the presence of cycloheximide (−NGF+CHX) for 36 h were injected with cytochrome c as a positive control for competence. Viability of microinjected cells 3, 6, and 20 h after these injections is shown. Data are mean ± SEM for three experiments with ∼100 cells counted for each time point per experiment.
The resistance of NGF-maintained neurons to cytosolic cytochrome c is overcome with microinjection of excess mature Smac (Fig. 1; Deshmukh et al., 2002). To confirm that exogenous Smac permitted cytochrome c to induce apoptosis by inhibiting IAPs in these neurons, we examined whether a Smac mutant protein that cannot inhibit IAPs as a consequence of a single, alanine-to-methionine amino acid change in its mature NH2 terminus was capable of permitting cytochrome c to induce death in sympathetic neurons. Unlike wild-type AVPI-Smac, the mutant MVPI-Smac cannot bind to IAPs and relieve their inhibition of caspases (Chai et al., 2000). We found that unlike wild-type AVPI-Smac, the mutant MVPI-Smac was incapable of cooperating with cytochrome c to induce death in NGF-maintained sympathetic neurons (Fig. 1). Less than 5% of cells that were injected with wild-type AVPI-Smac and cytochrome c were viable 20 h after the injections, whereas almost 80% of cells that were injected with the mutant MVPI-Smac and cytochrome c were viable at that time (Fig. 1). Thus, cytochrome c was able to induce apoptosis in NGF-maintained sympathetic neurons only if the functions of one or more Smac-inhibitable IAPs were blocked in these neurons.
Levels of XIAP are reduced when sympathetic neurons develop competence
To identify the specific IAPs that regulated caspase activation during sympathetic neuronal apoptosis, we first examined which IAPs were expressed in sympathetic neurons. Western data show that NGF-maintained sympathetic neurons express multiple IAPs including XIAP, cIAP-1, and cIAP-2 (Fig. 2 a). Importantly, we examined whether the levels of any IAPs were altered under conditions where neurons develop competence. Sympathetic neurons that are deprived of NGF in the presence of cycloheximide for 24 h develop competence and become permissive for cytochrome c–mediated apoptosis (Fig. 1; Deshmukh and Johnson, 1998). Cycloheximide addition was used to block the cytochrome c release pathway specifically, thus maintaining the survival of competent neurons. Among the IAPs examined, we found the levels of XIAP protein to be most dramatically reduced in neurons that develop competence. Levels of XIAP protein in the competent, NGF-deprived, cycloheximide-treated neurons were reduced to 30% of NGF-maintained control neurons (Fig. 2, a and b). In contrast, the levels of cIAP-1 and cIAP-2 were reduced, but only marginally to 65–80% of NGF-maintained levels (Fig. 2, a and b).
Figure 2.

XIAP protein levels are reduced when sympathetic neurons develop competence. (a) Protein levels of XIAP, cIAP-1, and cIAP-2 were examined in NGF-maintained neurons (+NGF) and in competent neurons that were deprived of NGF in the presence of cycloheximide for 24 h (−NGF+CHX). Levels of α-tubulin were also examined as a loading control. Quantitation of this data (±SEM) from two representative experiments is shown in b. (c) Protein levels of XIAP and Apaf-1 were examined in NGF-maintained (+NGF) and NGF- deprived (24 h; −NGF) Bax-deficient neurons. (d) Proteins levels of XIAP and Apaf-1 were examined in NGF-maintained (+NGF) and NGF-deprived, zVAD-FMK (50 uM)-treated (−NGF+zVAD) sympathetic neurons. (e) Protein levels of XIAP, cIAP-1, c-IAP-2, and Apaf-1 were examined in NGF-maintained neurons (+NGF), in NGF-maintained neurons treated with cycloheximide for 24 h (+NGF+CHX), and in NGF-deprived neurons treated with cycloheximide for 24 h (−NGF+CHX). Quantitation of this data (±SEM) from two representative experiments is shown in f.
Another method to induce competence in sympathetic neurons, which does not include cycloheximide addition, is to deprive Bax-deficient neurons of NGF for 24 h. Bax deficiency, like cycloheximide addition, blocks the cytochrome c release pathway specifically and therefore allows us to maintain the survival of NGF-deprived neurons that develop competence (Deshmukh and Johnson, 1998). We compared the levels of XIAP in NGF-maintained and NGF-deprived conditions in Bax-deficient neurons. Levels of XIAP protein were markedly reduced in the NGF-deprived condition when neurons developed competence (Fig. 2 c). We also examined the levels of XIAP in NGF-deprived, caspase inhibitor (50 μM zVAD-FMK)-treated neurons. These neurons are arrested after cytochrome c release, at the point of caspase activation. A marked reduction in XIAP levels was also seen in the NGF-deprived-zVAD–treated neurons (Fig. 2 d). Thus, NGF deprivation induced the loss of XIAP in multiple conditions where the competence pathway was activated.
NGF deprivation is known to cause a global inhibition of protein synthesis in sympathetic neurons (Deckwerth and Johnson, 1993). Thus, the dramatic reduction in XIAP protein levels after NGF deprivation may simply reflect a rapid turnover of XIAP protein. To examine this possibility, we inhibited protein synthesis in NGF-maintained neurons with addition of cycloheximide and examined the levels of XIAP, cIAP-1, and cIAP-2 after 24 h of protein synthesis inhibition. Cycloheximide addition alone to NGF-maintained neurons reduced XIAP levels to ∼55% of the untreated, NGF-maintained control neurons (Fig. 2, e and f). Levels of cIAP-1 and cIAP-2 were reduced to ∼65% of control neurons, indicating that differences in the half-lives of these IAPs alone cannot account for the selective decrease in XIAP levels seen in the NGF-deprived competent conditions. Thus, although the marked reduction in XIAP levels after NGF deprivation could be partially accounted for by the global inhibition of protein synthesis seen after NGF withdrawal, NGF deprivation must also activate other mechanisms that cause the selective decrease in XIAP levels seen in competent neurons.
If the down-regulation of XIAP protein is important for the neuronal development of competence, then reduction in XIAP levels should always correlate with competent neurons. Conversely, XIAP levels should always be maintained in neurons that are not competent. First, we examined whether the time course of the reduction in XIAP protein levels after NGF deprivation correlated with the time course of development of competence. Sympathetic neurons develop competence and become permissible for cytochrome c–mediated caspase activation only after ∼24 h of NGF deprivation (Deshmukh and Johnson, 1998). Consistent with the time course of development of competence, significant reduction in XIAP protein levels were detected only after 20–30 h of NGF deprivation (Fig. 3 a). Second, depolarizing concentrations of potassium (with 35 mM KCl) or elevation of intracellular cyclic AMP levels (with 400 μM 8-(4-chlorophenylthio)adenosine-3′-5′-cyclic monophosphate; CPTcAMP) blocks the development of the competence pathway in NGF-deprived neurons (Deshmukh et al., 2002). Consistent with the ability of KCl and CPTcAMP to inhibit the development of competence, XIAP levels were maintained in both KCl- and CPTcAMP-treated neurons, despite 24 h of NGF deprivation (Fig. 3 b).
Figure 3.

Reduction in XIAP protein levels correlates with neuronal development of competence in multiple conditions. (a) Examination of the time course of reduction in XIAP levels during development of competence. Levels of XIAP or Apaf-1 (control) protein were examined in NGF-maintained neurons (+NGF) and in parallel cultures of neurons that were deprived of NGF in the presence of cycloheximide for the indicated times after NGF deprivation (−NGF+CHX). Arrow points to the band corresponding to XIAP. (b) Levels of XIAP or tubulin (control) protein were examined in NGF-maintained neurons (+NGF) and in NGF-deprived neurons (24 h) in which the competence pathway was blocked with the addition of 35 mM KCl (−NGF+KCl) or 400 μM CPTcAMP (−NGF+cAMP). For comparison, XIAP levels were also examined in the competent, NGF-deprived, cycloheximide-treated (−NGF+CHX; 24 h) neurons. These data are representative of multiple experiments.
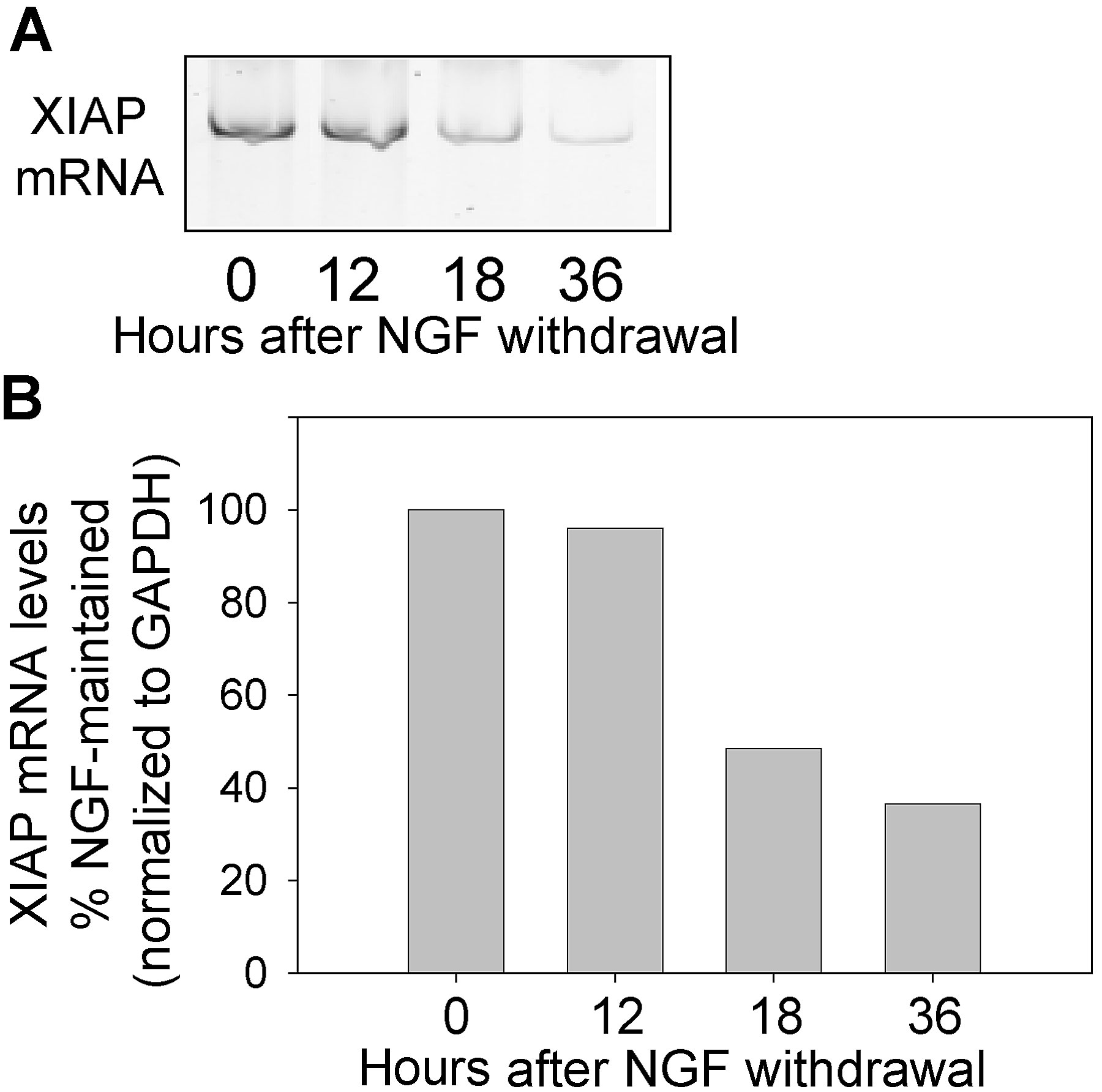
The observed changes in XIAP protein levels in competent neurons raised the possibility that perhaps XIAP mRNA levels were also regulated with the development of competence in sympathetic neurons. Steady-state levels of mRNAs were examined by a fluorescence-based, quantitative RT-PCR method in these neurons. In the competent, NGF-deprived, cycloheximide-treated neurons, the levels of XIAP mRNA were substantially reduced to <20% of NGF-maintained neurons (Fig. 4 a). Levels of the control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA in competent neurons were 75% of the NGF-maintained levels. The time course of the fall in XIAP levels indicate that the significant reduction in XIAP mRNA occurs only after 12 h of NGF deprivation (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200307130/DC1). We also examined whether levels of cIAP-1 and cIAP-2 were altered in competent neurons. In contrast to the marked reduction in XIAP mRNA levels in competent neurons, the levels of cIAP-1 mRNA were slightly increased to 110%, whereas those of cIAP-2 mRNA were slightly decreased to 70% in competent neurons as compared with the NGF-maintained neurons (Fig. 4 b). Finally, we examined whether XIAP mRNA levels were restored in conditions where competence is reversed with NGF readdition. Consistent with the observation that readdition of NGF for 24 h reverses competence (Deshmukh et al., 2002), we found XIAP mRNA levels to be restored to 85% of the NGF-maintained levels by 24 h after NGF readdition (Fig. 4 a).
Figure 4.
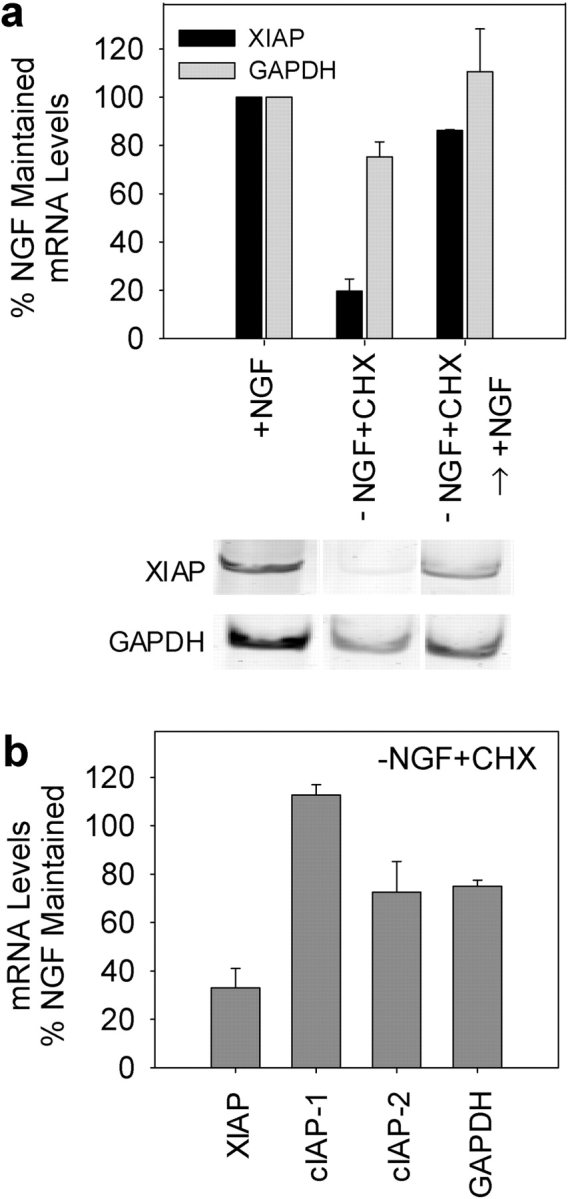
XIAP mRNA levels are significantly reduced in neurons that develop competence. (a) Levels of XIAP and GAPDH (control) mRNAs were examined with quantitative RT-PCR analysis in sympathetic neurons that were either maintained in NGF (+NGF) or deprived of NGF in the presence of cycloheximide for 36 h to develop competence (−NGF+CHX). Levels of these mRNAs were also examined in competent neurons that were treated with NGF readdition for 24 h to reverse the competence state (−NGF+CHX → +NGF). (b) Levels of mRNAs of cIAP-1 and cIAP-2 along with those of XIAP and GAPDH were determined in the competent NGF-deprived, cycloheximide-treated neurons (−NGF+CHX; 36 h) and expressed as a percentage of the mRNA levels in NGF-maintained neurons. Data are mean ± SEM of two to three experiments.
Thus, changes in XIAP levels, both mRNA and protein, correlated with the competence status of sympathetic neurons under all conditions that we examined. Among the IAPs that we examined, XIAP mRNA and protein levels were selectively reduced when neurons developed competence and became susceptible to cytochrome c–mediated apoptosis. In contrast, XIAP levels were maintained or restored under conditions when neurons were not competent and exhibit resistance to cytosolic cytochrome c.
XIAP is required for regulating caspase activation during sympathetic neuronal apoptosis
If the reduction in XIAP levels were sufficient to permit cytochrome c to induce apoptosis in sympathetic neurons, then one prediction of this model is that although cytosolic cytochrome c alone is not sufficient to induce apoptosis in wild-type neurons, it should be able to promote caspase activation and apoptosis in XIAP-deficient sympathetic neurons. To test this hypothesis, we isolated sympathetic neurons from XIAP-deficient mice (Harlin et al., 2001) and examined whether cytosolic microinjection of cytochrome c was sufficient to induce death in these neurons. In contrast to NGF-maintained wild-type neurons that are resistant to cytochrome c, the XIAP-deficient neurons were susceptible and died rapidly with cytosolic microinjection of cytochrome c (Fig. 5, a and b). Less than 10% of the XIAP-deficient neurons were alive within 4 h after cytochrome c microinjections, and only 1% remained alive by 24 h after the injections. In contrast, >80% of the wild-type neurons remained viable even 24 h after cytochrome c injections (Fig. 5 a). Injection of yeast cytochrome c, which does not promote caspase activation in mammalian cells (Ellerby et al., 1997), did not induce cell death in either the wild-type or XIAP-deficient neurons (Fig. 5 a).
Figure 5.
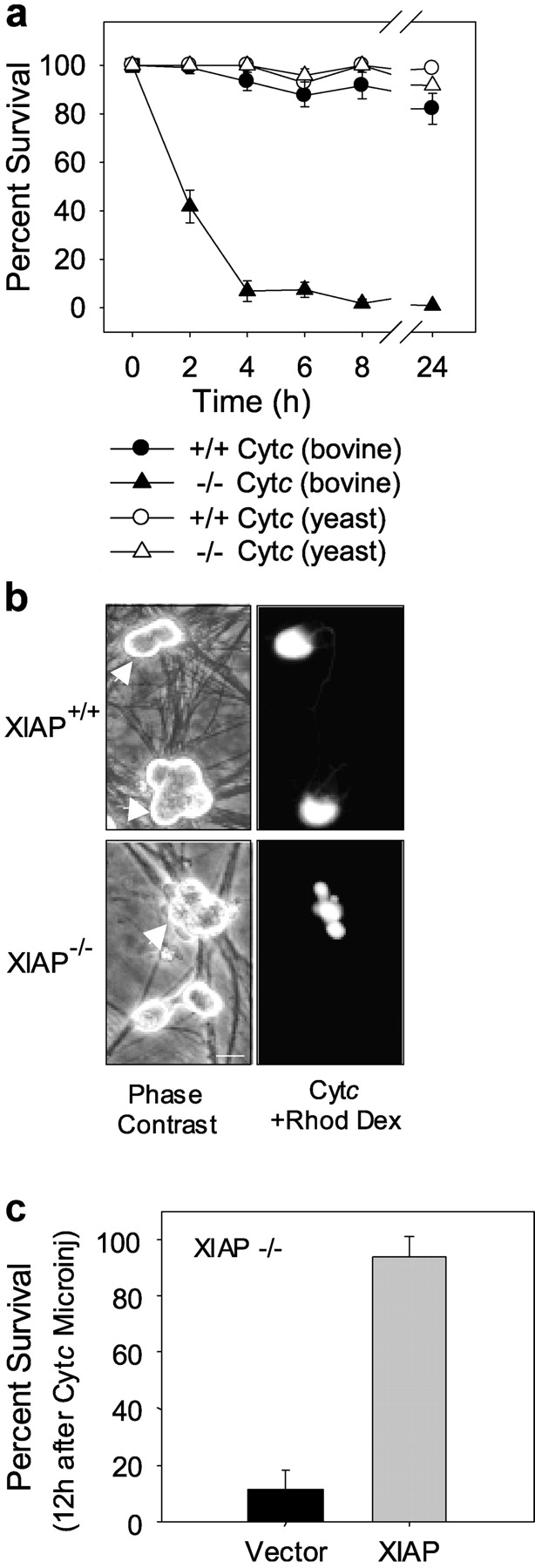
XIAP is necessary for the postcytochrome c regulation of caspase activation in sympathetic neurons. (a) NGF-maintained sympathetic neurons from XIAP-deficient (−/−) or wild-type (+/+) littermate mice were microinjected with either bovine or yeast cytochrome c. Survival of the injected neurons was assessed at the indicated times. Data shown are mean ± SEM of three independent experiments. (b) Phase contrast micrographs of wild-type (XIAP+/+) and XIAP-deficient (XIAP−/−) neurons are shown. Neurons that were injected with bovine cytochrome c (and rhodamine dextran) were identified by fluorescence micrographs for the same field and are marked with arrowheads. Bar, 10 μm. (c) Reintroduction of XIAP into XIAP-deficient neurons restores resistance to cytosolic microinjection of cytochrome c. NGF-maintained, XIAP-deficient sympathetic neurons were coinjected with plasmids expressing either XIAP or vector alone and EGFP. After 24 h to allow for expression, the injected neurons were reinjected with cytochrome c and survival of the cytochrome c–injected cells was determined 12 h after the injections. Data are mean ± SEM of three independent experiments.
We examined whether reintroduction of XIAP into the XIAP-deficient neurons was sufficient to restore resistance to cytosolic cytochrome c. Microinjection of XIAP into the XIAP-deficient neurons was sufficient to promote resistance to cytosolic cytochrome c as >90% of these cells were viable even 12 h after cytochrome c injections. In contrast, only 11% of the vector alone–injected XIAP-deficient neurons remained viable 12 h after cytochrome c injections (Fig. 5 c).
We also examined whether XIAP deficiency affected the overall time course of neuronal death after NGF deprivation. Cultures of sympathetic neurons from XIAP-deficient mice and their wild-type littermates were deprived of NGF, and their survival was assessed at 12, 24, and 48 h after NGF deprivation. No differences were observed in the time courses of death after NGF deprivation between the wild-type and XIAP-deficient neurons (Fig. 6). Thus, although removal of XIAP made neurons vulnerable to cytosolic cytochrome c, XIAP deficiency alone did not change the overall kinetics of apoptosis after NGF withdrawal. These results are consistent with the idea that XIAP functions as a critical safety brake to protect against aberrant caspase activation if cytochrome c is accidentally released from mitochondria. Whereas removal of XIAP was necessary for activating caspases in these neurons, the deletion of this safety mechanism by itself did not alter the kinetics of apoptosis after NGF deprivation.
Figure 6.

Wild-type and XIAP-deficient sympathetic neurons show no differences in the time course of death after NGF withdrawal. Equal numbers of wild-type and XIAP-deficient sympathetic neurons were either maintained in NGF or deprived of NGF for 12, 24, and 48 h. The number of neurons that became committed to die at those times was determined by replacing the media with NGF-containing media and counting the number of neurons that could be rescued after 7 d of NGF readdition. Rescued neurons show healthy, phase bright cell bodies. Data shown are mean ± SD of three experiments.
Hydrogen peroxide (H2O2)–induced release of endogenous cytochrome c and Smac are not sufficient to overcome XIAP in neurons
The results thus far indicate that removal of XIAP is both necessary and sufficient to allow cytochrome c to activate caspases and apoptosis in sympathetic neurons. Although our data show that the NGF deprivation–induced competence pathway promoted a marked reduction in XIAP levels, another potential mechanism by which XIAP function could be inhibited in these neurons is by the mitochondrial release of endogenous Smac. To examine whether endogenous Smac and cytochrome c are sufficient to overcome the XIAP inhibition and permit caspase activation in these neurons, we induced the release of these mitochondrial proteins by treating neurons with H2O2. H2O2 induces the release of mitochondrial cytochrome c in sympathetic neurons (Kirkland et al., 2002). First, we optimized conditions and found that an exposure of 3 mM H2O2 for 1 h induced the release of mitochondrial cytochrome c in >90% of the neurons by 3 h after the treatment (Fig. 7, a and b). Released cytochrome c is not detected in the cytosol because once translocated from the mitochondria, cytochrome c presumably gets degraded in these neurons (Deshmukh and Johnson, 1998; Neame et al., 1998). Second, we examined whether endogenous Smac was also released along with cytochrome c under these conditions. All detectable Smac was also released from the mitochondria in >90% of neurons with H2O2 treatment (Fig. 7, a and b). Like cytochrome c, the released Smac is also presumably degraded in the cytosol of these neurons. Thus, transient H2O2 treatment induced the loss of both cytochrome c and Smac from the mitochondria in sympathetic neurons.
Figure 7.
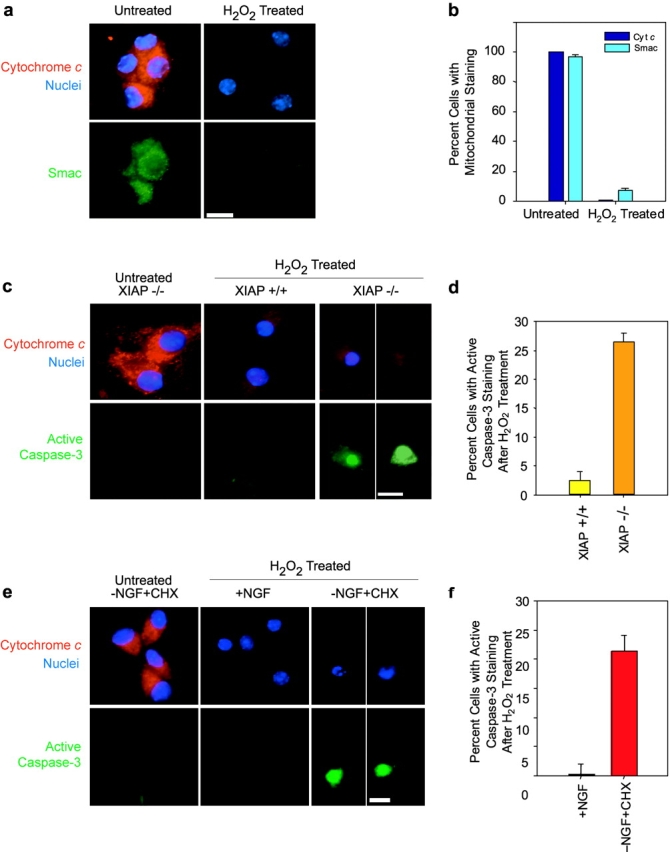
Release of endogenous cytochrome c and Smac are not sufficient to overcome the XIAP-mediated postcytochrome c inhibition in NGF-maintained sympathetic neurons. (a) NGF-maintained neurons were either left untreated or treated with 3 mM H2O2 for 1 h. The status of cytochrome c and Smac in these neurons was examined 3 h after the H2O2 exposure by immunohistochemical techniques. Micrographs show that whereas untreated, NGF-maintained neurons show a punctate, mitochondrial pattern of staining for both cytochrome c (red) and Smac (green), H2O2 exposure induces the loss of both cytochrome c and Smac staining from the mitochondria in these neurons. Bizbenzimide staining shows the nuclei (blue). (b) Quantitation of the loss of mitochondrial cytochrome c and Smac from these neurons in response to the transient H2O2 treatment. Approximately 100 neurons were counted to determine the status of cytochrome c and Smac in untreated or H2O2-treated neurons. (c) Wild-type (XIAP+/+) or XIAP-deficient (XIAP−/−) sympathetic neurons were maintained in NGF and either left untreated or transiently exposed to H2O2 as described in panel a. The status of cytochrome c (red) and activated caspase-3 (green) in these neurons was examined by immunohistochemical techniques. Bizbenzimide staining shows the nuclei (blue). For untreated neurons, the staining patterns for both XIAP+/+ and XIAP−/− conditions are identical but only the image for XIAP−/− is shown. The percentage of neurons showing activated caspase-3 immunostaining after transient H2O2 exposure in the wild-type (XIAP+/+) or XIAP-deficient (XIAP−/−) is quantitated in panel d. (e) Sympathetic neurons were either maintained in NGF (+NGF) or made competent by depriving them of NGF in the presence of cycloheximide for 24 h (−NGF+CHX). These neurons were either left untreated or treated with H2O2, and the status of cytochrome c (red) and activated caspase-3 (green) in these neurons was examined. For untreated neurons, the staining patterns for both +NGF and –NGF+CHX conditions are identical but only the image for −NGF+CHX is shown. The percentage of neurons showing activated caspase-3 immunostaining after transient H2O2 exposure in the NGF-maintained (+NGF) or competent, (−NGF+CHX) conditions is quantitated in panel f. Data shown in b, d, and f are mean ± SD of three experiments. Bars: (a, c, and e) 10 μm.
Next, we examined whether the H2O2-induced release of cytochrome c and Smac was sufficient to activate caspases in these neurons. Untreated neurons showed the expected punctate, mitochondrial distribution of cytochrome c and no staining for activated caspase-3. Surprisingly, even with H2O2 treatment, despite the release of all detectable endogenous cytochrome c and Smac, virtually none of the neurons (<2%) exhibited any caspase-3 activation in wild-type neurons (Fig. 7, c and d).
The inability of endogenous cytochrome c and Smac, which were released from mitochondria by H2O2 treatment, to activate caspases could be because of their inability to overcome XIAP inhibition in these neurons. To test this hypothesis, we examined whether the H2O2-induced release of endogenous cytochrome c and Smac were sufficient to activate caspases in XIAP-deficient neurons. In contrast to the results in wild-type neurons, H2O2 treatment induced a robust activation of caspase-3 in XIAP-deficient neurons in >25% of the cells (Fig. 7, c and d). The number of cells that show caspase activation at any given time is not expected to be very high because cells in which caspases become activated undergo rapid apoptosis and die. Thus, H2O2-induced release of endogenous cytochrome c and Smac, although not sufficient to overcome XIAP inhibition and permit caspase activation in wild-type sympathetic neurons, induced rapid caspase activation in XIAP-deficient neurons.
To determine whether the down-regulation of XIAP induced by the competence pathway was sufficient to permit caspase activation by endogenous cytochrome c and Smac, we examined whether H2O2 exposure induced caspase activation in competent, NGF-deprived cycloheximide-treated neurons. Like in the XIAP-deficient neurons, H2O2 treatment induced robust caspase activation in competent sympathetic neurons (Fig. 7, e and f). The inability of endogenous Smac release alone to overcome XIAP inhibition in NGF-maintained neurons underscores the importance of the competence pathway in removing XIAP and permitting endogenous cytochrome c to activate caspases in sympathetic neurons.
Discussion
Primary sympathetic neurons, unlike most mitotic cells, are remarkably resistant to cytosolic microinjection of cytochrome c. In this paper, we investigated the mechanism behind the stringent postcytochrome c regulation of caspase activation in sympathetic neurons. We report that the inability of cytochrome c to induce apoptosis in these neurons is because of an essential function of endogenous XIAP in regulating caspase activation postcytochrome c. Our results are consistent with the model in which removal of XIAP is both necessary and sufficient to allow cytochrome c–mediated caspase activation during sympathetic neuronal apoptosis. These results are the first to identify a mammalian cell type in which apoptosis is regulated postcytochrome c by endogenous XIAP.
Down-regulation of XIAP during sympathetic neuronal apoptosis
NGF deprivation–induced sympathetic neuronal apoptosis requires activation of both the cytochrome c release and the development of competence pathways. We found that upon NGF deprivation, the competence pathway induced a dramatic reduction in the levels of XIAP mRNA and protein in sympathetic neurons (Figs. 2 and 3). These data provide insight into how the competence pathway is able to overcome the neuronal resistance to cytosolic cytochrome c and permit caspase activation and apoptosis in these neurons. Other situations in which sympathetic neurons become competent and die with cytochrome c, such as after axotomy (Fletcher et al., 2000), may also induce similar mechanisms that remove XIAP. Decreases in XIAP levels have also been observed in other neurons undergoing cell death in vivo (Guegan et al., 2001; Korhonen et al., 2001; Ishigaki et al., 2002; Perrelet et al., 2002).
We found no evidence of XIAP cleavage during the development of competence as no XIAP cleavage product was found to accumulate in NGF-deprived, competent neurons (Fig. S3 a, available at http://www.jcb.org/cgi/content/full/jcb.200307130/DC1). Also, in contrast to the caspase-mediated cleavage of DIAP1 seen in Drosophila (Ditzel et al., 2003), the loss of XIAP could not be blocked with caspase inhibition in sympathetic neurons (Fig. 2 d). We also examined the possibility that XIAP might be targeted for proteasome-mediated degradation in these neurons, as is seen during thymocyte apoptosis (Yang et al., 2000). However, no ubiquitination of endogenous XIAP was detected in primary sympathetic neurons after NGF withdrawal (not depicted), and addition of the proteasome inhibitor lactacystin did not block the reduction in XIAP levels seen in NGF-deprived neurons (Fig. S3 b). A recent paper has identified the sites on XIAP that can be ubiquitinated in cells (Shin et al., 2003). The specific importance of the ubiquitin pathway in degrading XIAP in neurons may require examining whether mutant XIAP that cannot be ubiquitinated are also targeted for removal in NGF-deprived, competent sympathetic neurons.
Because NGF deprivation induces an overall reduction in neuronal metabolism (Deckwerth and Johnson, 1993), some decrease in the steady-state levels of all proteins is expected in NGF-deprived sympathetic neurons. However, although levels of cIAP-1, cIAP-2, and Apaf-1 protein were reduced (by 20–35%) after NGF deprivation, these decreases were small compared with the >70% decrease in XIAP levels observed under these conditions (Fig. 2). Importantly, total inhibition of protein synthesis with cycloheximide addition alone in NGF-maintained neurons induced a 45% reduction in XIAP levels (Fig. 2). Therefore, the turnover of XIAP protein after the fall in protein synthesis after NGF deprivation (Deckwerth and Johnson, 1993) could be one factor that contributes to the marked reduction in XIAP levels in NGF-deprived competent neurons. However, cycloheximide addition alone to NGF-maintained neurons is not sufficient to induce competence (Deshmukh and Johnson, 1998). The additional decrease in XIAP levels (25% above those seen with total inhibition of protein synthesis) that is seen in the NGF-deprived conditions, and is presumably important for the development of competence, could be mediated by other posttranslational mechanisms induced under the NGF deprivation conditions.
Essential function of endogenous XIAP in regulating caspase activation in sympathetic neurons
Our data show that whereas wild-type sympathetic neurons are remarkably resistant to cytosolic cytochrome c, the XIAP-deficient neurons exhibited no resistance and died rapidly with cytosolic cytochrome c (Figs. 5 and 7). Furthermore, restoring XIAP expression in the XIAP knockout neurons restored the resistance to cytosolic cytochrome c in these neurons (Fig. 5 c). These results identify XIAP as an essential postcytochrome c regulator of caspase activation in sympathetic neurons.
Although XIAP deficiency made sympathetic neurons vulnerable to cytosolic cytochrome c, it did not change the overall time course of apoptosis after NGF withdrawal (Fig. 6). Antisense-mediated depletion of XIAP also does not change the time course of sympathetic neuronal death after NGF deprivation (Troy et al., 2001). We have previously shown that the time courses of cytochrome c release and development of competence (removal of XIAP) after NGF deprivation are very similar, with 50% of neurons having released cytochrome c and developed competence by 18–20 h after NGF withdrawal (Deshmukh and Johnson, 1998). Thus, by the time the neuron reaches the point of cytochrome c release, it also removes XIAP, thereby permitting caspase activation and apoptosis to occur in these neurons. Apoptosis in XIAP-deficient neurons, although no longer needing the competence pathway to remove XIAP, is still dependent on the release of cytochrome c and therefore occurs with a time course that is indistinguishable from wild-type neurons.
Removal of XIAP during sympathetic neuronal apoptosis: Relative importance of the competence pathway versus mitochondrial release of Smac
We found that H2O2-induced mitochondrial release of endogenous Smac and cytochrome c was not sufficient to overcome XIAP inhibition and activate caspases in NGF-maintained sympathetic neurons (Fig. 7). Thus, although exogenously microinjected, excess Smac is capable of removing XIAP inhibition in these neurons (Fig. 1; Deshmukh et al., 2002), levels of endogenous Smac may simply be insufficient to overcome XIAP in these cells. However, in competent neurons, which have markedly decreased XIAP levels, and in XIAP-deficient neurons, the H2O2-induced release of endogenous cytochrome c and Smac induced robust caspase activation (Fig. 7). Consistent with this observation, recent data show that NGF-deprived Bax −/− competent neurons are more susceptible to H2O2 exposure than NGF-maintained sympathetic neurons (Kirkland et al., 2002). These results suggest that the competence pathway, and not the release of endogenous mitochondrial Smac, is likely to be the predominant mechanism that removes XIAP and permits caspase activation in sympathetic neurons.
This critical function of XIAP in sympathetic neurons is presumably to be a “safety brake” that protects against any accidental caspase activation if cytochrome c is unexpectedly released into the cytosol of these neurons. Arguably, for such a safety mechanism to be effective, XIAP must also withstand any inhibition from other proteins that may be released along with cytochrome c, such as Smac, in such accidental situations. Such a safety brake would be particularly important in postmitotic cells such as sympathetic neurons that are not replaceable and need to last for the lifetime of the organism. Similar postcytochrome c protection from apoptosis is also seen in Xenopus eggs, presumably because ensuring the survival of gametes is advantageous to the organism (Tashker et al., 2002). In neurons, physiologically appropriate apoptotic stimuli such as NGF deprivation would activate not only the cytochrome c release pathway, but also the competence pathway to ensure the removal of the XIAP brake and permit caspase activation.
The regulation of apoptosis in mammalian sympathetic neurons is conceptually similar to the “gas and brake” model in Drosophila, where apoptosis requires both the removal of IAPs and activation of caspases (Rodriguez et al., 2002). However, in Drosophila, activation of caspases appears to be constitutive, as removal of DIAP1 alone results in increased apoptosis and lethality (Wang et al., 1999). In contrast, in mammalian sympathetic neurons, caspase activation is dependent on both the removal of endogenous XIAP (the brake) and the initiation of the cytochrome c release pathway (the gas). These results are consistent with the observation that XIAP deficiency alone does not cause increased apoptosis in mammalian cells under normal physiological conditions (Harlin et al., 2001). However, the XIAP-deficient neurons, like neuronal apoptosis inhibitory protein-deficient neurons (Holcik et al., 2000), may be predicted to be more vulnerable if exposed to toxic stimuli that cause mitochondrial damage and cytochrome c release because they lack the postcytochrome c safety brake.
Materials and methods
Reagents
All reagents were purchased from Sigma-Aldrich or Fisher Scientific, unless otherwise stated. Collagenase and trypsin were purchased from Worthington Biochemical Corporation and cycloheximide was purchased from Tocris Cookson. The pan-caspase inhibitor zVAD-FMK was purchased from Enzyme Systems Products.
Sympathetic neuronal cultures
Sympathetic neurons were dissected from superior cervical ganglia of postnatal day zero to one mice (P0–P1) and maintained in culture as described previously (Deshmukh et al., 2002). Cells were plated on collagen-coated dishes at a density of 30,000 cells per well for protein lysates, or 2,500 cells per 35-mm dish for microinjection, survival counts, or immunofluorescence experiments. Sympathetic neurons were grown for 4–5 d in the NGF-containing media before treating them with experimental conditions. For NGF deprivation, cultures were rinsed twice with medium lacking NGF followed by addition of goat anti-NGF neutralizing antibody to this media. Other conditions required the addition of 1 μg/ml cycloheximide, 400 μM CPTcAMP, or 35 mM KCl to anti-NGF–containing media. Sympathetic neurons from Bax-deficient mice were isolated as described previously (Deckwerth et al., 1996).
Bacterial expression of recombinant proteins
A COOH-terminal His-tagged bacterial expression plasmid (pET15b) containing mature Smac cDNA (AVPI-Smac; corresponding to amino acids 56–236 of the full-length Smac protein) was provided by C. Du (Stowers Institute, Kansas City, MO). The initiating Met in the “AVPI-Smac” protein is removed during bacterial expression by the aminopeptidase activity because of the presence of Ala as the second residue (Chai et al., 2000). To generate the “MVPI-Smac” protein, the first Ala residue was simply deleted by site directed mutagenesis. Because the second residue now is Val instead of Ala, the initiating Met remains intact, thus generating the MVPI-Smac protein. Primer sequences used for this site directed mutagenesis were: forward 5′-AGAAGGAGATATACCATGGTTCCTATTGCAGAG-3′ and reverse 5′-CTGTGCAATAGGAACCATGGTATATCTCCTTCT-3′. Escherichia coli BL21-(DE3) bacteria strain (Stratagene) were transformed with either the AVPI-Smac– or MVPI-Smac–containing plasmids. Bacterial cultures were grown in Luria Bertani media until reaching a density of OD600 = 0.6. Expression of protein was induced with 1 mM IPTG for 3 h at 37°C and cells were harvested by centrifugation. The bacterial pellet was lysed by sonication and Ni-NTA agarose beads (QIAGEN) were used to purify the His-tagged protein according to the manufacturer's instructions. The eluted fractions were analyzed by SDS-PAGE and fractions containing the desired protein were pooled and dialyzed overnight at 4°C into dialysis buffer (20 mM Hepes, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM EDTA, 0.5 mM EGTA, and 1 mM DTT, pH 7.4) and stored in aliquots at −80°C until needed.
Microinjection and quantitation of cell survival
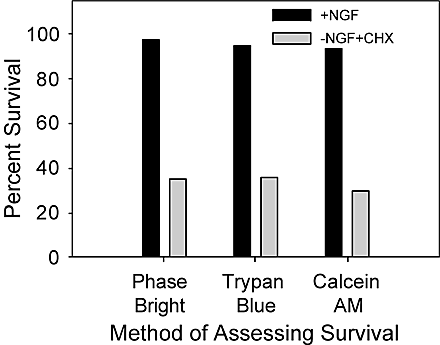
Our method for microinjecting sympathetic neurons with cytochrome c has been described previously (Deshmukh and Johnson, 1998). In brief, sympathetic neurons were microinjected using Femtotip II needles (Eppendorf Inc.) and a Narashigi micromanipulator mounted on an inverted fluorescence microscope (Leica). The microinjection solution (100 mM KCl and 10 mM KPi, pH 7.4) contained 4 mg/ml of rhodamine dextran dye to mark the injected cells. The concentration of bovine cytochrome c injected was 10–15 mg/ml, and that of mature Smac was 1 mg/ml. Immediately after the injections, the number of viable cells injected was determined by counting the number of rhodamine-positive cells that had intact, phase-bright cell bodies. This method of assessing neuronal survival correlates well with other cell survival assays such as trypan blue exclusion and staining with calcien AM (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200307130/DC1). At various times after injections, the number of viable, injected neurons remaining was determined by using the same counting criteria and expressed as a percentage of the original number of microinjected cells.
In experiments involving microinjection of the XIAP-expressing plasmid (pEBB-XIAP; a gift provided by C. Duckett, University of Michigan, Ann Arbor, MI), neurons were injected in the nucleus with 200 ng/μl of the plasmid DNA along with 50 ng/μl EGFP-expressing DNA (CLONTECH Laboratories, Inc.) in microinjection buffer. After 24 h to allow for expression, GFP-expressing cells were identified by fluorescence microscopy and reinjected with cytochrome c (as described in the previous paragraph). The survival of these double injected cells was assessed 12 h after cytochrome c injections.
Western blot analysis
Sympathetic neuronal cultures were washed in PBS and harvested in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 8.0, and protease inhibitor cocktail), and subjected to SDS-PAGE analysis as described previously (Deshmukh et al., 2002). For quantitation, the Western blots were developed with ECL-plus reagents (Amersham Biosciences) and analyzed on a Typhoon fluorescent imager (Amersham Biosciences) using the ImageQuant software (Amersham Biosciences). The specific antibodies used in experiments are listed in the Online supplemental materials and methods section, available at http://www.jcb.org/cgi/content/full/jcb.200307130/DC1.
Quantitation of mRNA expression
Our method of quantitative RT-PCR analysis is a modification of a previously published protocol (Estus et al., 1994), where we substituted the radioactivity-based detection method with a fluorescence-based detection technique. In brief, parallel cultures of sympathetic neurons with equal cell numbers plated were grown under the appropriate conditions and total RNA was harvested using QIAGEN RNAeasy kit. RNA was converted to cDNA by Superscript II reverse transcriptase (Invitrogen) using random hexamers (Invitrogen) as primer templates. Primer sets for GAPDH, XIAP, cIAP-1, and cIAP-2 (see Online supplemental material) were first analyzed for their linear range of amplification by varying the number of cycles and amount of input cDNA. The optimal cycle number that yielded a detectable, linearly amplified product was found to be 18 cycles for GAPDH and 22 cycles for XIAP, cIAP-1, and cIAP-2. PCR was performed for the indicated number of cycles for each primer pair and the PCR products were separated on an 8% polyacrylamide nondenaturing gel. The gel was stained with Sybr Green I (Molecular Probes) for 45 min on a rocker and the stained PCR products were visualized on a Typhoon fluorescence imager and quantitated using ImageQuant software.
Genotyping of XIAP −/− mice
Genotyping was performed by digesting tail DNA overnight in genotyping buffer (100 mM Tris-HCl, pH 8.5, 200 mM NaCl, 5 mM EDTA, 0.2% SDS, 10 mg/ml proteinase K) at 55°C. DNA was precipitated by gently adding three volumes of ice-cold ethanol to the supernatant and spooling at the interface. The PCR primers used for genotyping are listed in the Online supplemental materials and methods section.
Quantitation of neuronal survival after NGF deprivation
Sympathetic neuronal survival after 0, 12, 24, and 48 h of NGF deprivation was assessed by determining the percentage of cells that could be rescued by adding back NGF to the cultures at those times. Equal numbers of sympathetic neurons plated in multiple dishes were deprived of NGF for the indicated times, rinsed three times, and incubated in fresh NGF-containing media. After 7 d of NGF readdition, the rescued neurons were clearly identifiable with large and phase-bright cell bodies, whereas the nonrescued neurons atrophied and degenerated. All rescued cells in the dish were counted and expressed as a percent of the number of cells in the 0 h, untreated condition.
Immunofluorescence analysis
Immunofluorescence analysis on sympathetic neuronal cultures was performed essentially as described previously (Deshmukh and Johnson, 1998). The specific antibodies used in experiments are listed in the Online supplemental materials and methods section.
Transient exposure of sympathetic neurons to H2O2 was done by treatment of neurons with 3 mM H2O2 for 1 h, with a change into fresh H2O2 containing media after 30 min. Cells were washed three times with NGF-containing (AM50) media and allowed to incubate at 37°C for three additional hours in AM50 media. Cells were fixed and subjected to immunofluorescence analysis.
Online supplemental material
Fig. S1 shows the assessment of neuronal survival after cytochrome c microinjections using multiple techniques. Fig. S2 shows the time course of decrease in XIAP mRNA levels after NGF deprivation. Fig. S3 shows a full-length Western blot of extracts from NGF-maintained and NGF-deprived, cycloheximide-treated sympathetic neurons probed for XIAP. This figure also shows the inability of lactacystin to block the decrease in XIAP levels in the NGF-deprived, competent neurons. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200307130/DC1.
Supplemental Material
Acknowledgments
We thank Colin Duckett for the XIAP plasmid and Chunying Du for the Smac plasmid. We also thank Eugene Johnson and members of the Deshmukh laboratory for critical review of this manuscript.
This work was supported by National Institutes of Health grant NS42197 and the Burroughs Wellcome Fund New Investigator Award to M. Deshmukh.
P.R. Potts and S. Singh contributed equally to this work.
The online version of this article includes supplemental material.
Abbreviations used in this paper: cIAP, cellular inhibitor of apoptosis protein; CPTcAMP, 8-(4-chlorophenylthio)adenosine-3′-5′-cyclic monophosphate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; H2O2, hydrogen peroxide; IAP, inhibitor of apoptosis protein; XIAP, X-linked IAP.
References
- Brustugun, O.T., K.E. Fladmark, S.O. Doskeland, S. Orrenius, and B. Zhivotovsky. 1998. Apoptosis induced by microinjection of cytochrome c is caspase-dependent and is inhibited by Bcl-2. Cell Death Differ. 5:660–668. [DOI] [PubMed] [Google Scholar]
- Chai, J., C. Du, J.W. Wu, S. Kyin, X. Wang, and Y. Shi. 2000. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature. 406:855–862. [DOI] [PubMed] [Google Scholar]
- Chang, S.H., P.C. Phelps, I.K. Berezesky, M.J. Ebersberger, and B.F. Trump. 2000. Studies on the mechanisms and kinetics of apoptosis induced by microinjection of cytochrome c in rat kidney tubule epithelial cells (NRK-52E). Am. J. Pathol. 156:637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth, T.L., and E.M. Johnson, Jr. 1993. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J. Cell Biol. 123:1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth, T.L., J.L. Elliott, C.M. Knudson, E.M. Johnson, Jr., W.D. Snider, and S.J. Korsmeyer. 1996. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 17:401–411. [DOI] [PubMed] [Google Scholar]
- Denault, J.B., and G.S. Salvesen. 2002. Caspases: keys in the ignition of cell death. Chem. Rev. 102:4489–4500. [DOI] [PubMed] [Google Scholar]
- Deshmukh, M., and E.M. Johnson, Jr. 1997. Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol. Pharmacol. 51:897–906. [DOI] [PubMed] [Google Scholar]
- Deshmukh, M., and E.M. Johnson, Jr. 1998. Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 21:695–705. [DOI] [PubMed] [Google Scholar]
- Deshmukh, M., J. Vasilakos, T.L. Deckwerth, P.A. Lampe, B.D. Shivers, and E.M. Johnson, Jr. 1996. Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE-family proteases. J. Cell Biol. 135:1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh, M., K. Kuida, and E.M. Johnson, Jr. 2000. Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J. Cell Biol. 150:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh, M., C. Du, X. Wang, and E.M. Johnson, Jr. 2002. Exogenous smac induces competence and permits caspase activation in sympathetic neurons. J. Neurosci. 22:8018–8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux, Q.L., N. Roy, H.R. Stennicke, T. Vanarsdale, Q. Zhou, S.M. Srinivasula, E.S. Alnemri, G.S. Salvesen, and J.C. Reed. 1998. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 17:2215–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditzel, M., R. Wilson, T. Tenev, A. Zachariou, A. Paul, E. Deas, and P. Meier. 2003. Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat. Cell Biol. 5:467–473. [DOI] [PubMed] [Google Scholar]
- Du, C., M. Fang, Y. Li, L. Li, and X. Wang. 2000. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 102:33–42. [DOI] [PubMed] [Google Scholar]
- Duckett, C.S., F. Li, Y. Wang, K.J. Tomaselli, C.B. Thompson, and R.C. Armstrong. 1998. Human IAP-like protein regulates programmed cell death downstream of Bcl-xL and cytochrome c. Mol. Cell. Biol. 18:608–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellerby, H.M., S.J. Martin, L.M. Ellerby, S.S. Naiem, S. Rabizadeh, G.S. Salvesen, C.A. Casiano, N.R. Cashman, D.R. Green, and D.E. Bredesen. 1997. Establishment of a cell-free system of neuronal apoptosis: comparison of premitochondrial, mitochondrial, and postmitochondrial phases. J. Neurosci. 17:6165–6178. [PMC free article] [PubMed] [Google Scholar]
- Estus, S., W.J. Zaks, R.S. Freeman, M. Gruda, R. Bravo, and E.M. Johnson, Jr. 1994. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 127:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher, G.C., L. Xue, S.K. Passingham, and A.M. Tolkovsky. 2000. Death commitment point is advanced by axotomy in sympathetic neurons. J. Cell Biol. 150:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, D.R., and G.I. Evan. 2002. A matter of life and death. Cancer Cell. 1:19–30. [DOI] [PubMed] [Google Scholar]
- Guegan, C., M. Vila, G. Rosoklija, A.P. Hays, and S. Przedborski. 2001. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J. Neurosci. 21:6569–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlin, H., S.B. Reffey, C.S. Duckett, T. Lindsten, and C.B. Thompson. 2001. Characterization of XIAP-deficient mice. Mol. Cell. Biol. 21:3604–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcik, M., C.S. Thompson, Z. Yaraghi, C.A. Lefebvre, A.E. MacKenzie, and R.G. Korneluk. 2000. The hippocampal neurons of neuronal apoptosis inhibitory protein 1 (NAIP1)-deleted mice display increased vulnerability to kainic acid-induced injury. Proc. Natl. Acad. Sci. USA. 97:2286–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki, S., Y. Liang, M. Yamamoto, J. Niwa, Y. Ando, T. Yoshihara, H. Takeuchi, M. Doyu, and G. Sobue. 2002. X-linked inhibitor of apoptosis protein is involved in mutant SOD1-mediated neuronal degeneration. J. Neurochem. 82:576–584. [DOI] [PubMed] [Google Scholar]
- Juin, P., A.O. Hueber, T. Littlewood, and G. Evan. 1999. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 13:1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland, R.A., J.A. Windelborn, J.M. Kasprzak, and J.L. Franklin. 2002. A Bax-induced pro-oxidant state is critical for cytochrome c release during programmed neuronal death. J. Neurosci. 22:6480–6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen, L., N. Belluardo, and D. Lindholm. 2001. Regulation of X-chromosome-linked inhibitor of apoptosis protein in kainic acid-induced neuronal death in the rat hippocampus. Mol. Cell. Neurosci. 17:364–372. [DOI] [PubMed] [Google Scholar]
- Li, F., A. Srinivasan, Y. Wang, R.C. Armstrong, K.J. Tomaselli, and L.C. Fritz. 1997. Cell-specific induction of apoptosis by microinjection of cytochrome c. Bcl-XL has activity independent of cytochrome c release. J. Biol. Chem. 272:30299–30305. [DOI] [PubMed] [Google Scholar]
- MacFarlane, M., W. Merrison, S.B. Bratton, and G.M. Cohen. 2002. Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J. Biol. Chem. 277:36611–36616. [DOI] [PubMed] [Google Scholar]
- Martin, D.P., R.E. Schmidt, P.S. DiStefano, O.H. Lowry, J.G. Carter, and E.M. Johnson, Jr. 1988. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 106:829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, M.J., L.L. Rubin, and K.L. Philpott. 1997. Involvement of caspases in sympathetic neuron apoptosis. J. Cell Sci. 110:2165–2173. [DOI] [PubMed] [Google Scholar]
- Neame, S.J., L.L. Rubin, and K.L. Philpott. 1998. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J. Cell Biol. 142:1583–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrelet, D., A. Ferri, P. Liston, P. Muzzin, R.G. Korneluk, and A.C. Kato. 2002. IAPs are essential for GDNF-mediated neuroprotective effects in injured motor neurons in vivo. Nat. Cell Biol. 4:175–179. [DOI] [PubMed] [Google Scholar]
- Rodriguez, A., P. Chen, H. Oliver, and J.M. Abrams. 2002. Unrestrained caspase-dependent cell death caused by loss of Diap1 function requires the Drosophila Apaf-1 homolog, Dark. EMBO J. 21:2189–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen, G.S., and C.S. Duckett. 2002. IAP proteins: blocking the road to death's door. Nat. Rev. Mol. Cell Biol. 3:401–410. [DOI] [PubMed] [Google Scholar]
- Shin, H., K. Okada, J.C. Wilkinson, K.M. Solomon, C.S. Duckett, J.C. Reed, and G.S. Salvesen. 2003. Identification of ubiquitination sites on the X-linked inhibitor of apoptosis protein. Biochem. J. 373:965–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, M., S. Beinroth, M. Gleichmann, P. Liston, R.G. Korneluk, A.E. MacKenzie, M. Bahr, T. Klockgether, G.S. Robertson, M. Weller, and J.B. Schulz. 1999. Adenovirus-mediated gene transfer of inhibitors of apoptosis protein delays apoptosis in cerebellar granule neurons. J. Neurochem. 72:292–301. [DOI] [PubMed] [Google Scholar]
- Suzuki, Y., Y. Imai, H. Nakayama, K. Takahashi, K. Takio, and R. Takahashi. 2001. a. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell. 8:613–621. [DOI] [PubMed] [Google Scholar]
- Suzuki, Y., Y. Nakabayashi, and R. Takahashi. 2001. b. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. USA. 98:8662–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashker, J.S., M. Olson, and S. Kornbluth. 2002. Post-cytochrome c protection from apoptosis conferred by a MAPK pathway in Xenopus egg extracts. Mol. Biol. Cell. 13:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy, C.M., L. Stefanis, A. Prochiantz, L.A. Greene, and M.L. Shelanski. 1996. The contrasting roles of ICE family proteases and interleukin-1β in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc. Natl. Acad. Sci. USA. 93:5635–5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy, C.M., S.A. Rabacchi, J.B. Hohl, J.M. Angelastro, L.A. Greene, and M.L. Shelanski. 2001. Death in the balance: alternative participation of the caspase-2 and -9 pathways in neuronal death induced by nerve growth factor deprivation. J. Neurosci. 21:5007–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux, D.L., and S.J. Korsmeyer. 1999. Cell death in development. Cell. 96:245–254. [DOI] [PubMed] [Google Scholar]
- Verhagen, A.M., P.G. Ekert, M. Pakusch, J. Silke, L.M. Connolly, G.E. Reid, R.L. Moritz, R.J. Simpson, and D.L. Vaux. 2000. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 102:43–53. [DOI] [PubMed] [Google Scholar]
- Wang, S.L., C.J. Hawkins, S.J. Yoo, H.A. Muller, and B.A. Hay. 1999. The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell. 98:453–463. [DOI] [PubMed] [Google Scholar]
- Wang, X. 2001. The expanding role of mitochondria in apoptosis. Genes Dev. 15:2922–2933. [PubMed] [Google Scholar]
- Wiese, S., M.R. Digby, J.M. Gunnersen, R. Gotz, G. Pei, B. Holtmann, J. Lowenthal, and M. Sendtner. 1999. The anti-apoptotic protein ITA is essential for NGF-mediated survival of embryonic chick neurons. Nat. Neurosci. 2:978–983. [DOI] [PubMed] [Google Scholar]
- Yang, Y., S.Y. Fang, J.P. Jensen, A.M. Weissman, and J.D. Ashwell. 2000. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 288:874–877. [DOI] [PubMed] [Google Scholar]
- Yu, L.Y., L. Korhonen, R. Martinez, E. Jokitalo, Y. Chen, U. Arumae, and D. Lindholm. 2003. Regulation of sympathetic neuron and neuroblastoma cell death by XIAP and its association with proteasomes in neural cells. Mol. Cell. Neurosci. 22:308–318. [DOI] [PubMed] [Google Scholar]
- Yuan, J., and B.A. Yankner. 2000. Apoptosis in the nervous system. Nature. 407:802–809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}