

Abstract
Ethanol preconditioning (EtOH-PC) refers to a phenomenon in which tissues are protected from the deleterious effects of ischemia/reperfusion (I/R) by prior ingestion of ethanol at low to moderate levels. In this study, we tested whether prior (24 hrs) administration of ethanol as a single bolus that produced a peak plasma concentration of 42-46 mg/dl in gerbils would offer protective effects on neuronal damage due to cerebral I/R. In addition, we also tested whether reactive oxygen species (ROS)-derived from NADPH oxidase played a role as an initiator of these putative protective effects. Groups of gerbils were administered either ethanol or the same volume of water by gavage 24 hrs prior to transient global cerebral ischemia induced by occlusion of both common carotid arteries (CCA) for 5 min. In some experiments, apocynin, a specific inhibitor of NADPH oxidase, was administered (5 mg/kg body wt, i.p.) 10 min before ethanol administration. EtOH-PC ameliorated behavioral deficit induced by cerebral I/R and protected the brain against I/R-induced delayed neuronal death (DND), neuronal and dendritic degeneration, oxidative DNA damage, and glial cell activation. These beneficial effects were attenuated by apocynin treatment coincident with ethanol administration. Ethanol ingestion was associated with translocation of the NADPH oxidase subunit, p67phox, from hippocampal cytosol fraction to membrane, increased NADPH oxidase activity in hippocampus within the first hour after gavage, and increased lipid peroxidation (4-hydroxy-2-nonenal, HNE) in plasma and hippocampus within the first 2 hrs after gavage. These effects were also inhibited by concomitant apocynin treatment. Our data are consistent with the hypothesis that antecedent ethanol ingestion at socially-relevant levels induces neuroprotective effects in I/R by a mechanism that is triggered by ROS produced through NADPH oxidase. Our results further suggest the possibility that preconditioning with other pharmacological agents that induce a mild oxidative stress may have similar therapeutic value for suppressing stroke-mediated damage in brain.
Keywords: Ethanol preconditioning (EtOH-PC), neuroprotection, NADPH oxidase, reactive oxygen species, apocynin, cerebral ischemia/reperfusion, delayed neuronal death
INTRODUCTION
The results of a large number of epidemiologic studies indicate that consumption of red wine at low to moderate levels is cardioprotective, reducing the incidence and severity of myocardial infarction and stroke (1-3). Studies conducted in cultured cell and intact animal models attributed these beneficial effects to the polyphenolic antioxidants present in red wine (4-7). However, there is evidence that moderate consumption of ethanol alone also exerts protective effects against injury due to cerebral and myocardial ischemia (8-12). Furthermore, ethanol ingestion 24 hrs prior to ischemia/reperfusion (I/R) (ethanol preconditioning or EtOH-PC) completely prevented postischemic leukocyte-endothelial cell adhesive interactions by a mechanism that involves the ability for ethanol to activate an oxidant-dependent signaling pathway (13).
NADPH oxidase (nicotinamide adenine dinucleotide phosphate oxidase) is an important enzymatic source for the generation of reactive oxygen species (ROS) that damage cells in a variety of pathologic conditions, including I/R-induced injury in the brain and other tissues (14, 15). However, it is becoming increasingly apparent that these reactive molecules can also participate in a number of normal physiologic phenomena (16, 17). Indeed, we have demonstrated a role for ethanol-induced NADPH oxidase-derived oxidants in the appearance of anti-adhesive and anti-inflammatory phenotype in postcapillary venules that becomes apparent on subsequent exposure to I/R (13, 18, 19). However, whether ethanol also exerts neuroprotective effects in stroke is unknown.
In this study, we determined whether antecedent ethanol ingestion would confer protection against I/R in the gerbil brain and whether ROS derived from NADPH oxidase played a role as an initiator of these putative protective effects. Although NADPH oxidase has been identified in the cerebrovascular system (20-22) and in brain of the mouse and rat (23, 24), the presence of this oxidant-producing enzyme in the gerbil brain has not been evaluated. Therefore, this study included characterization of the NADPH oxidase subunits in hippocampal region of the gerbil brain.
MATERIALS AND METHODS
Production of ethanol preconditioning
Adult male Mongolian gerbils (60-80 g body wt) (Charles River, Wilmington, MA) were housed in the Small Animal Facilities of the University of Missouri-Columbia that was maintained at 22 ± 2°C with constant humidity under a 12:12 hrs light:dark cycle, and were allowed free access to water and lab chow. Experiments were carried out in accordance to the guidelines set forth by the NIH Guide for the Care and Use of Laboratory Animals.
Ethanol preconditioning (EtOH-PC) was induced by gavaging animals with a moderate dose of ethanol (volume ethanol in µl calculated from the relation: body weight (in g) × 0.6] + 0.3) 24 hrs before ischemia, as we have previously described (13, 18). This volume of ethanol (95% in µl) was mixed in 0.3 ml of sterile distilled water before gavage. Animals in the sham control group (no I/R) and the I/R alone group (no EtOH-PC) received similar volume of sterile distilled water by gavage. After administration, all animals were returned to their cages, and were provided free access to food and water.
Induction of global forebrain ischemia
Twenty-four hrs after ethanol or distilled water administration by gavage, animals were anesthetized with a mixture of 70% nitrous oxide, 30% oxygen and 2.5% isoflurane during preparation for surgical operation, after which isoflurane was reduced to 1% during surgery and ischemic insult. Transient global cerebral ischemia was induced by occlusion of both common carotid arteries (CCA) for 5 min and followed by reperfusion as described previously (5). Sham-operated animals underwent similar procedures except CCA were not occluded. To eliminate the complicating effects on cerebral blood flow changes that occur in the subset of gerbils with communicating arteries, regional cerebral blood flow (rCBF) was monitored before and after bilateral clamping the CCAs using a laser Doppler blood flow monitor (MBF3D, Moor Instruments, Devon, UK). After CCA occlusion, gerbils showing a decrease in rCBF of less than 80% were excluded from subsequent analyses (5). After surgical procedures, animals were allowed to regain consciousness and maintained in the Small Animal Facility with free access to food and water for four days. The animal protocol was approved by the University of Missouri-Columbia Animal Care and Use Committee (Protocol #1741).
Preparation of brain samples
Four days after ischemia, gerbils were anesthetized by inhaling 2.5% isoflurane and were transcardially perfused with heparinized saline followed by fixation with 4% (w/v) paraformaldehyde in 0.05 M phosphate buffered saline (PBS, pH 7.4). Brains were post-fixed for 3 days and embedded in paraffin. Six µm coronal sections were prepared from the dorsal hippocampus as described previously by Wang et al (5).
Identifying NADPH oxidase subunits in gerbil brain
p47phox and p67phox, two of the main cytosolic subunits of NADPH oxidase, were used as markers to identify NADPH oxidase in gerbil brain. p47phox and p67phox immunoreactivities were measured in coronal brain sections from the dorsal hippocampus in normal gerbils as previously described by Wang et al (5). Mouse anti-p47phox and p67phox antibodies (1;100 dilution, Upstate, Charlottesville, VA) were used as primary antibodies and Alexa Fluor 568 goat anti-mouse IgG was used as the secondary antibody (1:200 dilution, Molecular Probe, Eugene, OR). To assess specific staining, alternate sections were incubated with PBS instead of the primary antibody as the negative control.
Experimental protocols
In order to characterize the time course for changes in plasma ethanol concentration and lipid peroxidation in gerbils (n = 6 for each time point), fresh plasma and hippocampi were collected from gerbils prior to and at 30 min, 1 hr and 2 hrs after ethanol administration by gavages.
In the second study, apocynin, a selective inhibitor of NADPH oxidase, was tested for its ability to inhibit NADPH oxidase activation (cytosolic subunit translocation)(25), activity (NADPH consumption)(25, 26), and lipid peroxidation (HNE production)(27) in plasma and brain during the first two hours after ethanol ingestion. Gerbils were randomly divided into 3 groups; namely, control, EtOH alone, and EtOH plus apocynin pretreatment. Apocynin (Sigma, St Louis, MO) was dissolved in normal saline and administered by i. p. injection (5 mg/kg body wt) 10 min before administering ethanol or distilled water by gavage. Plasma and hippocampal samples were collected at 1hr and 2hrs after administration of ethanol or water.
To assess the neuroprotective effects of antecedent ethanol ingestion and the role of NADPH oxidase in these responses, gerbils were randomly divided into 4 groups: sham control (control, no I/R), I/R alone, antecedent ethanol ingestion 24 hrs prior to ischemia/reperfusion (EtOH-PC+I/R), and EtOH-PC coincident with apocynin treatment 24 hrs prior to induction of I/R (Apo+EtOH-PC+I/R). Apocynin was administered by intraperitoneal injection (i.p., 5 mg/kg body wt) 10 min before ethanol ingestion. Gerbils in the control, I/R alone (no EtOH-PC) and EtOH-PC+I/R groups received i.p.injections of saline at the volume and time as in the Apo+EtOH-PC+I/R group.
Measurement of ethanol concentration in plasma
Ready-to-use Q.E.D. A150 Alcohol Test kit (OraSure Technologies, Bethlehem, PA) was used to quantify alcohol levels in plasma samples obtained from gerbils under control conditions and 30 min, 1hr and 2hrs after ethanol administration by gavage. Prior to the study, we verified the accuracy and linearity of the readout over a range of ethanol concentrations (10 to 145 mg/dl or 0.01-0.145 mg %).
Measurement of lipid peroxidation in plasma and hippocampus
Generation of 4-hydroxy-2-nonenal (HNE) in plasma and in hippocampus was used as an indication of lipid peroxidation (27). Briefly, at different time points after ethanol ingestion, blood was collected in EDTA vacutainer tubes and centrifuged briefly to remove blood cells. Hippocampi (∼100mg) from both sides of the cerebrum were removed and homogenized in buffer (20 mM HEPES, pH 7.5, 0.25 M sucrose, 10 mM KCI, 1.5 mM MgCl2 1 mM EDTA, 1 mM EGTA, and 1 mM dithiothreitol). HNE in plasma and the hippocampus homogenates were detected using the Bioxytech HAE-586 spectrophotometric assay kit (OxisResearch, Portland, OR) as described by Wang et al. (15). Calculation of HNE in plasma or hippocampus was performed based on the standard curve generated by HNE standard and was expressed as µmol/dl in plasma or µmol/mg protein in hippocampus, respectively.
Western blotting to assess translocation of NADPH oxidase subunits
For preparation of membrane and cytosolic fractions, both sides of hippocampus were dissected and homogenized in 1 ml of homogenization buffer (10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 200 mM sucrose). The nuclei and cell debris were removed from the homogenate by centrifugation at 900 × g for 10 min at 4 °C. The resulting supernatant was centrifuged at 110,000 × g for 75 min at 4 °C. The resulting supernatant was collected as cytosolic fraction and the membrane pellet was solubilized in buffer (10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.5% Triton X-100) for 1 hr at 4 °C. Both of the membrane and cytosolic fractions were stored at −70 °C until use.
40 µg membrane or cytosolic proteins were subjected to SDS-PAGE. The separated proteins were transferred by electrophoresis to polyvinylidine difluoride membranes. The membranes were incubated with anti-p67phox antibody or anti-gp91phox antibodies (1:1,000 dilution; Upstate Biotechnology) at 4°C overnight and subsequently with a horseradish peroxidase-linked secondary antibody (1:5,000 dilution; Sigma) at room temperature for 90 min. The positive bands were revealed using Western blotting detection reagents (Pierce, Rochford, IL) and autoradiography film. The intensities of the immunoblot bands were quantified using the Quantity One software (Bio-Rad).
Measurement of NADPH oxidase activity in hippocampus
For measurement of NADPH oxidase activity, hippocampi from both side of the brain were homogenized together in Krebs-Ringer phosphate buffer at pH 7.4 (120 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 2.2 mM CaCl2, 0.1 M phospate buffer) with protease inhibitor cocktail (Sigma). Homogenates were centrifuged at 500 × g for 5 min at 4°C and the pellet discarded. Supernatants were spun at 100 000 × g for 1 hr at 4°C. Cytosolic (supernatant) and membrane (pellet) fractions were separated. The pellet was resuspended in 100 µL of buffer and kept at −70°C until analysis.
NADPH oxidase enzymatic activity was determined in membrane fraction using the protocol as described Wei and Whaley (25, 26). Briefly, aliquots of the membrane fractions (30 µg proteins) were incubated with NADPH (100 µM) at 37°C. NADPH activity was determined by measuring the conversion of NADPH to NADH using a Radical Detector kit (Cayman Chemical) and a plate reader spectrophotometer (450 nm) at every 10 min. NADPH oxidase activity was normalized by the amount of protein and the increase in optical density between 10-20 min. Activity was calculated as mOD/µg protein/min.
Assessment of locomotor activity
Locomotor activity in gerbils was assessed using a procedure described previously (28, 29). Locomotor activity was monitored automatically by the Med Associates Open Field Test Environments (ENV-515) (Georgia, VT). Each environment consisted of a 16 × 16 horizontal grid of infrared sensors and a bank of 16 vertical sensors. The sensor grids surrounded an acrylic cage (43.2 × 43.2 × 30.5 cm) and each sensor grid and cage was housed in a large sound-resistant cubicle. 24 hrs after ischemia, gerbils (n = 7 – 9 gerbils/group) were weighed and placed in the apparatus for 30 min. Data were collected in 5-min intervals by the Med Associates Open Field Activity Software (SOF-811), which recorded the number of sensor breaks and subsequently computed these data as distance traveled.
Histochemical and immunohistochemical staining for neurons and glial cells
Brain sections were stained with cresyl violet for neurons and immunohistochemical labeled with glial fibrillary acidic protein (GFAP) antibody for astrocytes according to the protocol described by Wang et al (5). Rabbit anti-human GFAP antibody (1:100 dilution, Sigma, St. Louis, MO) was used as primary antibody and Texas Red-conjugated goat-anti-rabbit IgG was used as secondary antibody (1:200 dilution; Jackson ImmunoResearch, West Grove, PA). Microglial cells were identified using FITC labeled isolectin B4 (20 µg/ml, Sigma, St. Louis, MO) according to the protocol outlined by (30) with modifications (5). The fluorescent dye, 4′, 6-diamidine-2′-phenylindole (DAPI) (0.1 µg/ml, Roche, Molecular Chemicals, Mannheim, Germany) was used to counter-stain the sections for identification of nuclear DNA in cells.
Fluoro-Jade B and MAP-2 staining for assessment of neuronal degeneration
Fluoro-Jade B is a polyanionic fluorescein derivative which sensitively and specifically binds to degenerating neurons (both apoptotic and necrotic). Its high affinity to degenerating neurons with green fluorescence has made it an excellent marker for detecting degenerating neurons. Fluoro-Jade B staining was performed according to the protocol described by Schmued et al (31).
MAP-2 is the major microtubule associated protein in brain tissue and MAP-2 is useful for identifying cytoskeletal structures in neurons. A mouse monoclonal antibody to MAP-2 (1:100, Sigma, St. Louis, MO) was used to mark the dendritic structures according to the protocol described by Monnerie et al (32). Alexa Fluor 568 goat anti-mouse IgG was used as the secondary antibody (1:200 dilution; Molecular Probe, Eugene, OR) and DAPI counter-staining was used to identify nuclear DNA damage in cells.
Detection of oxidized DNA by 8-OHdG immunohistochemistry
Formation of 8-OHdG (8-hydroxyl-deoxyguanosine) is regarded as a hallmark of oxidative DNA damage (33). A time course study by Hwang et al (34) showed an increase in immunoreactivity of 8-OHdG in the gerbil hippocampal CA1 region, with peaks at 12 hrs and 4 days after ischemic insult. In this study, determination of 8-OHdG was carried out with brain sections obtained at 4 days after I/R according to the protocol described previously by Wang et al (28). Briefly, brain sections were immunostained with mouse anti-8-OHdG antibody (diluted at 5 µg/ml, OxisResearch, Portland, OR) followed by goat anti-mouse Ig G labeled with horseradish peroxidase (HRP) (dilution 1: 200, Sigma, St. Louis, MO).
Quantitative assessment and statistical analysis
Neuronal damage, degeneration, glial activation and oxidative DNA were quantified by counting the number of live neurons and fluorescent or immuno-reactive positive cells of Fluoro-Jade B, GFAP, Isolectin-B4, 8-OHdG in the middle of a defined CA1 area (0.17 µm × 0.54 µm) in both sides of the hippocampi per high magnification field (magnification 400x) using the Bioquant Image Analysis System (Bioquant True Color Windows 95 Software Version 2.50, Nashville, TN) and the Mata Imaging Serials (Version 6.1, Molecular Devices Corporation, Downingtown, PA). The average values of live neurons and the above positive stained cells were obtained from left and right hippocampus of each gerbil.
The dendritic degeneration of neurons was assessed by quantifying the average intensity of the MAP-2 positive staining in the same defined area (0.17 µm × 0.54 µm) below the middle of CA1 in both sides of the hippocampi under the same magnification field (400x) using the Mata Imaging Serials (Version 6.1, Molecular Devices Corporation, Downingtown, PA.
Data (mean ± SEM) were subjected to a one-way ANOVA, followed by the Newman-Keuls post-hoc test using the GraphPad Prism program version 4.0. A p-value of less than 0.05 was considered to indicate significant difference.
In the assessment of locomotor activity, distance traveled data were analyzed via two-way repeated-measures ANOVA with treatment group as a between-groups factor and session time as a within-subjects factor. Tukey post-hoc tests were performed when appropriate (p < 0.05).
RESULTS
Plasma ethanol concentration
Using the same ethanol treatment regimen as described by Yamaguchi et al (18), we examined blood ethanol concentrations in gerbils. As shown in Fig. 1, ethanol administration resulted in an increase in blood ethanol concentration that reached a peak level of 42-46 mg/dl at 30-60 min after ingestion and returned to control levels 2 hrs after consumption. Thus, ethanol administration to gerbils by our dosing protocol produced an increase in plasma ethanol concentration similar to that noted in our earlier studies in mice (18).
Figure 1.

Changes of plasma ethanol concentrations in gerbils after ethanol administration by gavage (n=6 for each time point). The volume (in µl) of 95% ethanol instilled in each animal was calculated as follows: [body weight (in g) × 0.6] + 0.3), and was mixed in 0.3 ml sterile distilled water just before administration as a single bolus by gavage. *Values that are statistically different from corresponding values obtained during the control period (before ethanol gavage) and 2 hrs after ethanol ingestion at p < 0.05.
Detection of p47phox and p67phox immunoreactivity in the gerbil brain
Neuronal immunopositive staining for p47phox and p67phox were observed in both the cerebral cortex and hippocampus (Fig. 2). Replacement of primary antibodies with PBS did not exhibit any immunoreactivity, confirming the specificity of the p47phox and p67phox antibodies used in this experiment. In the cortex, p47phox and p67phox immunoreactivities were distributed in all cortical layers but were especially prominent in layer V (Fig. 2A and 2E). In the hippocampus, p47phox and p67phox immunoreactivities were prominently observed in the pyramidal neurons in CA1 and CA3 regions (Fig. 2B, 2C, 2F and 2G) and also in the molecular and polymorphic layers of dentate gyrus (DG) (Fig. 2D and 2F). These observations provide the first description of immunoreactivity of NADPH oxidase subunits in the gerbil brain.
Figure 2.
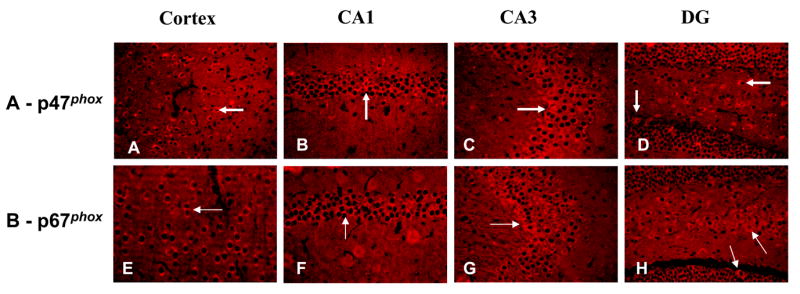
Immunoreactivities (IR) of p47phox and p67phox in normal gerbil brain. In cerebral cortex, IR of p47phox and p67phox were observed in all layers, but were especially prominent in layer V (Panel A and E). In the hippocampus, p47phox and p67phox IR were prominently observed in the pyramidal cell layer of the CA1 (Panel B and F) and CA3 (Panel C and G) regions and in both the molecular and polymorphic layers of dentate gyrus (DG) (Panel D and F). Arrows highlight individual cells that are positive for p47phox and p67phox IR. (Magnification, 200x).
Apocynin inhibited lipid peroxidation after EtOH-PC
Administration of a moderate dose of ethanol resulted in a transient increase in plasma HNE levels and reached a peak at 1 hr (Fig. 3A). This time course is in agreement with previous studies showing lipid peroxidation in plasma at 1-1.5 hrs following ethanol exposure (35). Ethanol administration also caused an increase in HNE levels in the hippocampus but unlike that in plasma, the levels of HNE in hippocampus remained elevated 2 hrs after ethanol administration (Fig. 3B). Apocynin pretreatment (i.p. 5 mg/kg body wt) 10 min before ethanol administration significantly attenuated the increases in both plasma (Fig. 3C) and hippocampal HNE (Fig. 3D) determined at 1 hr after ingestion, suggesting that increased ROS production due to ethanol administration was generated by NADPH oxidase. Pilot experiments using HPLC analysis demonstrated the ability of apocynin to cross the blood brain barrier (BBB) and reached the brain within 30 min after i.p. injection (data not shown).
Figure 3.

Moderate ethanol administration by gavage increased lipid peroxidation in plasma (Panel A) and hippocampus (Panel B), as assessed by changes in 4-hydroxy-2-nonenal (HNE) levels (n=6 for each time point and each sample type), and effects of apocynin on plasma (Panel C) and hippocampal (Panel D) lipid peroxidation after ethanol administration by gavage. Data depicted in Panels C and D illustrate the HNE levels in plasma and hippocampus in water control, ethanol ingestion alone (EtOH-PC), and i.p. injection of apocynin 10 min prior to ethanol gavage (Apo+EtOH-PC). * and # indicate values that were statistically different from corresponding values in control and EtOH-PC groups at p < 0.05, respectively.
Effects of apocynin on hippocampal NADPH oxidase subunit translocation and enzyme activity after EtOH-PC
NADPH oxidase activation requires translocation of cytosolic subunits to the plasma membrane (16, 17). To further determine whether NADPH oxidase contributed to EtOH-PC-induced ROS production, Western blotting of p67phox and gp91phox was used and a significant increase in p67phox expression in the membrane fraction and a decrease in the cytosolic fraction at 1hr after ethanol administration (Fig. 4A and 4B). The ethanol effects were were inhibited by apocynin administration 10 min before ethanol administration. These results suggest that p67phox subunit translocation from cytosol to membrane occurred in response to ethanol administration, and that this translocation was inhibited by the NADPH oxidase inhibitor, apocynin. The plasma membrane gp91phox subunit was strongly expressed in hippocampus but was not significantly affected by ethanol and apocynin treatment (Fig. 4A and 4C).
Figure 4.

Effects of apocynin on translocation of p67phox (Panel A, B and C) and on NADPH oxidase activity in hippocampus (Panel D) after ethanol administration by gavage. Data in Panels A to C illustrate the effects of control, ethanol ingestion alone (EtOH-PC), and i.p. injection of apocynin 10 min prior to ethanol gavage (Apo+EtOH-PC) on p67phox and gp91phox expression in membrane and cytosolic fractions. Data in Panel D show NADPH oxidase activity in hippocampus. See details in Method for the assay protocols. Samples were collected 1h after administration of ethanol or distilled water vehicle. Western blotting results are expressed as fold of control for three independent experiments (Panel B and C). * and # indicate values that are statistically different from corresponding values in control and EtOH alone groups at p < 0.05, respectively.
Measurement of NADPH oxidase activity in hippocampal membrane fraction showed a significant increase at 1 hr after EtOH administration and this effect was also significantly reduced by apocynin pretreatment (Fig. 4D). The increases in p67phox translocation and NADPH oxidase activity after EtOH administration and their reduction by apocynin pretreatment are positive data supporting the notion that NADPH oxidase contributed to EtOH-PC-induced ROS in brain.
EtOH-PC ameliorated I/R-induced hyperactivity that was attenuated by coincident apocynin treatment
A widely used behavioral test in animal models of stroke involves assessment of locomotor hyperactivity (Fig. 5). Analysis of distance traveled data revealed a significant main effect of session time (F(5,162) = 64.53, p < 0.001) and treatment group (F(3,162) = 9.07, p < 0.001). The interaction of session time by treatment group was not significant (F(15, 162) = 0.825, p = 0.65). Post-hoc tests determined that there was greater activity for gerbils that received ischemia (mean = 10,624 cm, S.E.M. = ±2185 cm) than for gerbils in the sham condition (mean = 7922 cm, S.E.M. = ±644 cm). There was no significant difference in activity between the gerbils that received apocynin pretreatment (mean = 12,037 cm, S.E.M. = ±1648 cm) and gerbils that received ischemia. However, there was significantly less activity for gerbils that received ethanol (mean = 7771 cm, S.E.M. = ±929 cm) than for gerbils that received only ischemia. There were no differences between the gerbils that received ethanol and gerbils under sham conditions.
Figure 5.

Effects of ethanol preconditioning and apocynin pretreatment on the spontaneous locomotor activity after cerebral I/R. Data represent mean (± S.E.M.) distance traveled (in cm) (n = 7 – 9 gerbils/group). Gerbils were placed in an automated locomotor activity monitor 24 hrs after cerebral I/R and locomotor activity was measured for 30 min. Statistical analysis revealed a significant main effect of session time (p <0.001) and treatment group (p < 0.001), but the session time by treatment group interaction was not significant (p = 0.65).
EtOH-PC induced neuroprotection in postischemic hippocampus was attenuated by coincident apocynin treatment
Cresyl violet staining revealed healthy neurons in the hippocampal CA1 area in the sham control group (Fig. 6A and 7B) and dead neurons 4 days after I/R (Fig. 6B and 1A). Fluoro-Jade B staining demonstrated very few degenerated neurons in the hippocampal CA1 region in sham control (Fig. 6E and 7B), but neuronal degeneration was markedly increased 4 days after I/R (Fig. 6F and 7B). Similar to description by Caceres et al. (36), we showed that the cytoskeletal protein MAP-2 is extensively localized in dendrites of hippocampal neurons. In the sham control group, immunoreactivity of MAP-2 was clearly visualized in the dendrites of pyramidal neurons in the hippocampal CA1 area (Fig. 6I and 7C) but was decreased significantly after I/R (Fig. 6J and 7C). When ethanol was administered 24 hrs before CCA occlusion, I/R-induced delayed neuronal death (DND) and neuronal and dendritic degeneration in the hippocampal CA1 area were significantly reduced as compared to the I/R group without prior ethanol treatment (Fig. 6C vs. 6B, 6G vs. 6F, 6K vs. 6J and Fig. 7A,B,C). Apocynin administration 10 min prior to ethanol ingestion significantly attenuated the neuroprotective effects of EtOH-PC in I/R (Fig. 6D vs. 6C, 6H vs. 6G, 6L vs. 6K and Fig. 7A,B,C).
Figure 6.
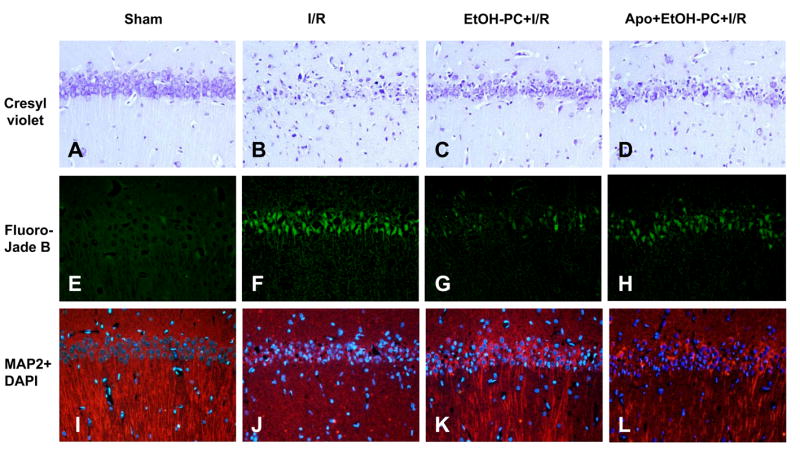
Representative micrographs depicting I/R-induced delayed neuronal death (DND) (cresyl violet) and neuronal (Fluoro-Jade B) and dendritic degeneration (MAP-2) in the hippocampal CA1 subfield of gerbils subjected to sham operation (Panels A, E, and I, respectively), 5 min occlusion of the common carotid arteries (ischemia) followed by 4 days reperfusion (I/R) (Panels B, F, and J, respectively), EtOH-PC 24 hrs prior to I/R (EtOH-PC+I/R) (Panels C, G, and K, respectively), and apocynin treatment coincident with ethanol administration 24 hrs prior to I/R (Apo+EtOH-PC+I/R) (Panels D, H, and L, respectively). (Magnification, 200x).
Figure 7.

Quantification of viable neurons (cresyl violet, Panel A) and neuronal (Fluoro-Jade B, Panel B) and dendritic (MAP-2, Panel 3) degeneration in the hippocampal CA1 region of gerbils subjected to sham, I/R alone, ethanol preconditioning 24 hrs prior to I/R (EtOH-PC+I/R), and apocynin treatment coincident with ethanol administration 24 hrs prior to I/R (Apo+EtOH-PC+I/R). *, #, and Δ = values statistically different from corresponding values in sham, I/R, and EtOH-PC+I/R groups, respectively, at p < 0.05.
Apocynin inhibited EtOH-PC-induced neuroprotection against glial activation and DNA oxidation after I/R
Immunohistochemical staining of GFAP showed few GFAP-positive astrocytes in the sham control group (Fig. 8A and 9A) but a large increase was observed in the I/R group (Fig. 8B and 9A). When brain sections were stained with isolectin B4 to identify microglial cells, very few microglial cells were observed in the sham control group (Fig. 8E and Fig. 9B). However, there was a substantial increase in the number of microglial cells in both the hippocampal CA1 area and in the surrounding area after I/R (Fig. 8F and Fig. 9B). EtOH-PC 24 hrs prior to I/R significantly reduced the number of reactive astrocytes and microglial cells as compared to I/R group (Fig. 8C vs. 8B, 8G vs. 8F and Fig. 9A and 9B). Apocynin administration 10 min before EtOH-PC significantly attenuated the inhibitory effects of EtOH-PC on activation of astrocytes and microglial cells after I/R (Fig. 8D vs. 8C, 8H vs. 8G and Fig. 9A and 9B).
Figure 8.

Representative micrographs depicting I/R-induced activation of astrocytes (GFAP, Panel A), microglial cells (isolectin-B4, Panel B) and DNA oxidation (8-OHdG, Panel C) in the hippocampal CA1 subfield of gerbils subjected to sham (Panels A, E, and I, respectively), I/R (Panels B, F, and J, respectively), ethanol preconditioning 24 hrs prior to I/R (EtOH-PC+I/R) (Panels C, G, and K, respectively), and apocynin treatment coincident with ethanol administration prior to I/R (Apo+EtOH-PC+I/R) (Panels D, H, and L, respectively). (Magnification, 200x).
Figure 9.

Quantification of astrocytic (GFAP, Panel A) and microglial activation (Isolectin-B4, Panel B) and DNA oxidation (8-OhdG, Panel C) in gerbils subjected to sham, 5 min occlusion of the common carotid arteries followed by 4 days of reperfusion (I/R), ethanol preconditioning 24 hrs prior to I/R (EtOH-PC+I/R), and apocynin treatment coincident with ETOH-PC 24 hrs prior to I/R (Apo+EtOH-PC+I/R). *, #, and Δ = values statistically different from corresponding values in sham, I/R, and EtOH-PC + I/R groups, respectively, at p < 0.05.
I/R-induced increases in DNA oxidation was assessed by immunohistochemical determination of 8-OHdG. In this study, weak 8-OHdG immunoreactivity was observed in the CA1 region of the sham control group (Fig. 8I and Fig. 9C) but was significantly increased by I/R (Fig. 8J). EtOH-PC significantly reduced the I/R-induced increase in 8-OHdG (Fig. 8K vs. 8J and Fig. 9C). Administration of apocynin prior to EtOH-PC significantly attenuated the ability of antecedent ethanol to reduce postischemic DNA oxidation (Fig. 8L vs. 8K and Fig. 9C).
DISCUSSION
Results from the present study demonstrate for the first time that moderate ethanol ingestion 24 hrs prior to global cerebral I/R exerts preconditioning-like protective effects, ameliorating postischemic behavioral deficit and reducing postischemic DND, neuronal and dendrite degeneration, oxidative DNA damage, and glial cell activation in the hippocampus. We further demonstrate that this moderate dose of ethanol is sufficient to cause a mild oxidative stress during the period of ethanol exposure as indicated by the increases in lipid peroxidation (HNE levels) and NADPH oxidase activity in the hippocampus concomitant with plasma ethanol elevation after ingestion. Moreover, treatment with the NADPH oxidase inhibitor apocynin 10 min prior to ethanol ingestion not only suppressed the ethanol-induced ROS production with increased NADPH activity, but also attenuated the beneficial actions of EtOH-PC to reduce postischemic DND, neuronal and dendrite degeneration, oxidative DNA damage, glial cell activation and behavioral deficit. Thus, our results also provide the first evidence that antecedent ethanol ingestion initiates a protective response in the brain that attenuates postischemic neurodegenerative processes by an oxidant-dependent triggering mechanism. Although NADPH oxidase has been recently identified as a major source of ROS production in the cerebrovascular system (20-22) and in brain of mouse and rat (23, 24), this study provides the first evidence for the presence of this oxidant-producing enzyme and action in the gerbil brain. Staining for NADPH oxidase subunits was observed in neurons in all regions of the gerbil brain, and particularly prominent in the hippocampus and cortex. This pattern of expression is similar to the distribution of NADPH oxidase reported in mouse and rat brain (23, 24). The expression of NADPH oxidase subunits in neurons suggests the possibility that superoxide produced by NADPH oxidase may play a role in normal as well as pathological function in the brain.
Several pharmacological inhibitors of NADPH oxidase are available including PR-39, diphenyliodonium, and apocynin. In prior studies, we have used PR-39 as a mechanistic probe for examining the role of NADPH oxidase-derived oxidants in the development of the anti-inflammatory phenotype induced by ischemic and ethanol preconditioning (13, 37). However, this agent is now known to produce a variety of other effects, in addition to inhibition of NADPH oxidase, which relate to its ability to enter cells and bind to SH3 domain-containing proteins such as p130Cas (38), an adapter protein that plays a role in several signaling pathways involved in cell survival (39). More relevant to preconditioning is the fact that it is now known that PR-39 inactivates NF-κB (40), a transcription factor that plays an essential role in the infarct-sparing effects of delayed ischemic preconditioning by promoting the expression of inducible NOS (iNOS) (41-43). Diphenyliodonium has been employed as an NADPH oxidase inhibitor in a large number of studies, but this agent is not specific and known to interfere with activities of flavor-proteins including endothelial nitric oxide synthase (eNOS) and mitochondrial respiratory chain activity (44). The latter effect is important because we have obtained strong evidence supporting a role for eNOS-derived NO as a trigger for the development of the anti-inflammatory effects of ethanol preconditioning (18, 45, 46).
In light of these non-specific effects of PR-39 and diphenyliodonium, we considered the use of other agents that inhibit NADPH oxidase activity for investigating the role of this enzyme in late EtOH-PC. Apocynin is a naturally occurring compound that effectively inhibits NADPH oxidase by reacting with thiol groups required for enzyme subunit assembly (15). The suggested action is in agreement with our observation that apocynin inhibited the translocation of cytosolic p67phox subunit to membrane. Apocynin is derived from the rhizome of the Himilayan herb Picrorhiza kurroa and has been used as a treatment for many inflammatory conditions for centuries (47). In contrast to PR-39 and diphenyliodonium, apocynin does not scavenge xanthine oxidase-derived superoxide, inhibit eNOS, or prevent NF-κB activation (40, 48-50). Coincident treatment with apocynin during the period of EtOH-PC attenuated the mild oxidative stress induced by ethanol ingestion, an effect that was associated with a concomitant reduction in NADPH activity that occurred during the time plasma ethanol rises and falls. Furthermore, apocynin treatment coincident with ethanol administration 24 hrs prior to induction of cerebral ischemia also markedly abrogated the postischemic neuroprotective action of EtOH-PC. These results suggest that moderate ethanol ingestion triggers entrance into this protected state through the formation of NADPH oxidase-derived oxidants during the period of ethanol exposure 24 hrs prior to I/R.
Some studies have correlated changes in cerebral I/R-induced biochemical indices of injury with neurological outcome in gerbils (51). Global cerebral ischemia in the gerbils produced a significant increase in locomotor activity which was correlated with a severe loss of hippocampal CA1 neurons after I/R (52). Our previous study demonstrated an increase in locomotor activity in gerbils after I/R, a behavorial effect of stroke that was reduced by curcumin treatment, a polyphenolic compound that also ameliorated hippocampal delayed neuronal death (29). We now extend these observations to the protective actions of EtOH-PC, which markedly inhibited the hyperlocomotion induced by I/R. Importantly, the reduction in the I/R-induced hyperactivity response by antecedent ethanol ingestion was abolished by coincident apocynin pretreatment.
It is important to note that while apocynin markedly decreased the oxidative stress induced by ethanol exposure and attenuated the neuroprotective effects that become apparent upon exposure to cerebral I/R, NADPH oxidase inhibition did not completely prevent these effects. It is possible that oxidative stress occurs secondary to ethanol metabolism by alcohol dehydrogenase and cytochrome P4502E1 could also play a role in triggering the development of EtOH-PC. However, the fact that nitric oxide (NO) inhibits the catalytic activity of both alcohol dehydrogenase and cytochrome P4502E1 (53, 54), when coupled with our observation that NO is formed during the period of ethanol preconditioning (18, 45, 46), argues against a role for either enzyme as additional sources of ROS in the EtOH-PC effects. Xanthine oxidase activity is increased by ethanol, although much higher ethanol doses were used to demonstrate this effect than were employed in the present study (13, 55-57). Nevertheless, xanthine oxidase has been implicated as an important instigating event for the development of the postischemic anti-inflammatory phenotype noted in postcapillary venules 24 hrs after ethanol ingestion (13). This may relate to the fact that xanthine oxidase produces superoxide when metabolizing acetaldehyde, the major product of ethanol catabolism (13, 57). It is also possible that production of mitochondrial oxidants at sites I and III contribute to the oxidative stress that precipitates the development of the neuroprotective phenotype in response to antecedent ethanol. This possibility is supported by the observation that ethanol stimulates the production of ROS in hepatocytes through mitochondria at complexes I and III (13, 58, 59), but again at ethanol concentrations higher than achieved in the present study. Although it is unclear whether this also occurs in the brain, especially at the concentrations achieved in our study, it is important to note that apocynin does not inhibit ischemia-induced mitochondrial oxidant production (13, 60).
Excessive production of ROS is an important event underlying cerebral I/R injury (61, 62) and the damaging effects can be ameliorated by administration of botanical antioxidants, including resveratrol from grape skin and curcumin from turmeric (5, 29). We recently demonstrated that apocynin similarly attenuated postischemic neuronal damage in the same gerbil stroke model (15). These findings provide additional support for the notion that ROS contribute to the genesis of I/R injury and indicate that that NADPH oxidase may be an important source of cytotoxic oxidants in stroke (15). Chronic excessive ethanol ingestion is also known to cause a number of pathophysiological manifestations that arise secondary to oxidative damage (6, 63). In light of this work, our demonstrated role for ethanol-induced oxidant formation by NADPH oxidase as an inaugural event that precipitates the development of a neuroprotective phenotype in I/R might be viewed as surprising. However, since the dose of ethanol used in this study raises plasma ethanol to a peak concentration that is only ∼50% of that required for legal intoxication, it seems reasonable to hypothesize that this moderate dose of ethanol produces only slight fluctuations in redox status that allow NADPH oxidase-derived ROS to subserve a signaling function as opposed to the biomolecular damage induced by the much higher oxidant flux that is produced during I/R or after excessive ethanol exposure. Indeed, previous studies have demonstrated that short bouts of preconditioning ischemia and reperfusion (ie, ischemic preconditioning) protect against the deleterious effects induced by subsequent exposure to prolonged I/R in stroke models (64, 65). More recent work has demonstrated that these protective effects are triggered by oxidants generated during ischemic preconditioning (66). In addition, we have previously demonstrated that ethanol preconditioning induces the development of an anti-inflammatory phenotype in postcapillary venules by a mechanism involved oxidants generated during the first hour after ethanol ingestion [13, 19]. Taken together, our studies indicate that NADPH-oxidase derived oxidants can play dual and opposing roles in I/R, serving to produce neuronal injury in brains exposed to I/R alone and by acting as important initiators in the signaling cascade that is activated by antecedent ethanol ingestion to produce a neuroprotective phenotype.
Identification of the downstream signaling elements that are activated by NADPH oxidase-derived ROS formed during the period of ethanol exposure and entrance into the preconditioned, neuroprotective state that becomes apparent when I/R is induced 24 hrs later are unknown. However, studies conducted by our group that have focused on the mechanisms whereby antecedent ethanol induces the development of an anti-inflammatory phenotype such that postcapillary venules fail to support leukocyte adhesion on subsequent exposure to I/R suggest the role for NO/superoxide interactions, release of calcitonin gene-related peptide, and activation of specific protein kinase C isoforms (13, 18, 45, 46, 67, 68). These observations, when coupled with the demonstration that leukocyte infiltration plays a critical role in the pathogenesis of cerebral I/R injury (69-72), suggest that these signaling elements may contribute to the protective actions of antecedent ethanol ingestion. In addition, mitochondrial dysfunction during cerebral I/R is well established as a contributor to stroke-induced injury (61, 73, 74), and amelioration of mitochondrial dysfunction reduces neuronal apoptosis and behavioral deficits after cerebral I/R (29). Clearly, much additional work will be required to identify the role of mitochondria and downstream mediators and ultimate effectors of the neuroprotective actions of antecedent ethanol in cerebral I/R.
The experimental design for the present study was based on earlier work by our group which demonstrated that ethanol ingestion 24 hrs prior to I/R completely prevented postischemic P-selectin expression and leukocyte/endothelial cell adhesion in postcapillary venules by an oxidant-dependent mechanism (13, 18, 75). The idea that moderate ethanol consumption induces a mild oxidative stress that provokes the development of a neuroprotective state in I/R is consistent with previous reports demonstrating that moderate ethanol consumption can limit the neurotoxicity and apoptosis induced by HIV-1 protein gp-120 and amyloid-β, respectively (76, 77). These studies suggest that the neuroprotective effects of moderate ethanol consumption may be of therapeutic value in other neurological disorders. However, we are not advocating the use of ethanol as a neuroprotective intervention. Rather, we suggest that elucidation of the underlying mechanisms that account for its remarkable protective actions may provide a rationale for the development of new treatment modalities that mimic its protective actions but avoid the untoward side effects associated with even moderate ethanol consumption.
In summary, the present study provides the first anatomical evidence for the presence of NADPH oxidase in gerbil brain and that antecedent ethanol ingestion 24 hrs prior to induction of cerebral I/R attenuates postischemic neuronal injury and behavioral deficit. Moreover, our work suggests that ethanol-induces the formation of ROS derived from NADPH oxidase occurs during the period of ethanol exposure, which serve a critical signaling function to inaugurate entrance into a preconditioned state that affords significant neuroprotection on subsequent exposure to I/R 24 hrs after ethanol ingestion.
Acknowledgments
This study is supported by DK P01 43785, AA R01 14945 and AG P01 18357 from NIH. The authors are grateful to Meifang Wang for technical assistance.
Abbreviations
- EtOH-PC
Ethanol preconditioning
- ROS
reactive oxygen species
- HNE
4-hydroxy-2-nonenal
- NADPH oxidase
nicotinamide adenine dinucleotide phosphate-oxidase
- CCA
common carotid arteries
- rCBF
regional cerebral blood flow
- BBB
blood brain barrier
- I/R
ischemia/reperfusion
- DND
delayed neuronal death
- GFAP
glial fibrillary acidic protein
- MAP-2
microtubule associated protein-2
- 8-OHdG
8-hydroxyl-deoxyguanosine
- DAPI
4′,6-diamidine-2′-phenylindole
- eNOS
endothelial nitric oxide synthase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Truelsen T, Gronbaek M. Wine consumption and cerebrovascular disease mortality in Spain. Stroke. 1999;30:186–188. doi: 10.1161/01.str.30.1.186. [DOI] [PubMed] [Google Scholar]
- 2.German JB, Walzem RL. The health benefits of wine. Annu Rev Nutr. 2000;20:561–593. doi: 10.1146/annurev.nutr.20.1.561. [DOI] [PubMed] [Google Scholar]
- 3.Rakotovao A, Berthonneche C, Guiraud A, de Lorgeril M, Salen P, de Leiris J, Boucher F. Ethanol, wine, and experimental cardioprotection in ischemia/reperfusion: role of the prooxidant/antioxidant balance. Antioxid Redox Signal. 2004;6:431–438. doi: 10.1089/152308604322899503. [DOI] [PubMed] [Google Scholar]
- 4.Ray PS, Maulik G, Cordis GA, Bertelli AA, Bertelli A, Das DK. The red wine antioxidant resveratrol protects isolated rat hearts from ischemia reperfusion injury. Free Radic Biol Med. 1999;27:160–169. doi: 10.1016/s0891-5849(99)00063-5. [DOI] [PubMed] [Google Scholar]
- 5.Wang Q, Xu J, Rottinghaus GE, Simonyi A, Lubahn D, Sun GY, Sun AY. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002;958:439–447. doi: 10.1016/s0006-8993(02)03543-6. [DOI] [PubMed] [Google Scholar]
- 6.Sun AY, Simonyi A, Sun GY. The “French Paradox” and beyond: neuroprotective effects of polyphenols. Free Radic Biol Med. 2002;32:314–318. doi: 10.1016/s0891-5849(01)00803-6. [DOI] [PubMed] [Google Scholar]
- 7.Shigematsu S, Ishida S, Hara M, Takahashi N, Yoshimatsu H, Sakata T, Korthuis RJ. Resveratrol, a red wine constituent polyphenol, prevents superoxide-dependent inflammatory responses induced by ischemia/reperfusion, platelet-activating factor, or oxidants. Free Radic Biol Med. 2003;34:810–817. doi: 10.1016/s0891-5849(02)01430-2. [DOI] [PubMed] [Google Scholar]
- 8.Sato M, Maulik N, Das DK. Cardioprotection with alcohol: role of both alcohol and polyphenolic antioxidants. Ann N Y Acad Sci. 2002;957:122–135. doi: 10.1111/j.1749-6632.2002.tb02911.x. [DOI] [PubMed] [Google Scholar]
- 9.Berger K, Ajani UA, Kase CS, Gaziano JM, Buring JE, Glynn RJ, Hennekens CH. Light-to-moderate alcohol consumption and risk of stroke among U.S. male physicians. N Engl J Med. 1999;341:1557–1564. doi: 10.1056/NEJM199911183412101. [DOI] [PubMed] [Google Scholar]
- 10.Reynolds K, Lewis B, Nolen JD, Kinney GL, Sathya B, He J. Alcohol consumption and risk of stroke: a meta-analysis. Jama. 2003;289:579–588. doi: 10.1001/jama.289.5.579. [DOI] [PubMed] [Google Scholar]
- 11.Miyamae M, Diamond I, Weiner MW, Camacho SA, Figueredo VM. Regular alcohol consumption mimics cardiac preconditioning by protecting against ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 1997;94:3235–3239. doi: 10.1073/pnas.94.7.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guiraud A, de Lorgeril M, Boucher F, Berthonneche C, Rakotovao A, de Leiris J. Cardioprotective effect of chronic low dose ethanol drinking: insights into the concept of ethanol preconditioning. J Mol Cell Cardiol. 2004;36:561–566. doi: 10.1016/j.yjmcc.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi T, Dayton CB, Ross CR, Yoshikawa T, Gute DC, Korthuis RJ. Late preconditioning by ethanol is initiated via an oxidant-dependent signaling pathway. Free Radic Biol Med. 2003;34:365–376. doi: 10.1016/s0891-5849(02)01292-3. [DOI] [PubMed] [Google Scholar]
- 14.Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006;1090:182–189. doi: 10.1016/j.brainres.2006.03.060. [DOI] [PubMed] [Google Scholar]
- 16.Paravicini TM, Sobey CG. Cerebral vascular effects of reactive oxygen species: recent evidence for a role of NADPH-oxidase. Clin Exp Pharmacol Physiol. 2003;30:855–859. doi: 10.1046/j.1440-1681.2003.03920.x. [DOI] [PubMed] [Google Scholar]
- 17.El-Benna J, Dang PM, Gougerot-Pocidalo MA, Elbim C. Phagocyte NADPH oxidase: a multicomponent enzyme essential for host defenses. Arch Immunol Ther Exp (Warsz) 2005;53:199–206. [PubMed] [Google Scholar]
- 18.Yamaguchi T, Dayton C, Shigematsu T, Carter P, Yoshikawa T, Gute DC, Korthuis RJ. Preconditioning with ethanol prevents postischemic leukocyte-endothelial cell adhesive interactions. Am J Physiol Heart Circ Physiol. 2002;283:H1019–1030. doi: 10.1152/ajpheart.00173.2002. [DOI] [PubMed] [Google Scholar]
- 19.Dayton C, Yamaguchi T, Kamada K, Carter P, Korthuis RJ. Antecedent ethanol ingestion prevents postischemic P-selectin expression in murine small intestine. Microcirculation. 2004;11:709–718. doi: 10.1080/10739680490521014. [DOI] [PubMed] [Google Scholar]
- 20.Paravicini TM, Drummond GR, Sobey CG. Reactive oxygen species in the cerebral circulation: physiological roles and therapeutic implications for hypertension and stroke. Drugs. 2004;64:2143–2157. doi: 10.2165/00003495-200464190-00001. [DOI] [PubMed] [Google Scholar]
- 21.Ishikawa M, Stokes KY, Zhang JH, Nanda A, Granger DN. Cerebral microvascular responses to hypercholesterolemia: roles of NADPH oxidase and P-selectin. Circ Res. 2004;94:239–244. doi: 10.1161/01.RES.0000111524.05779.60. [DOI] [PubMed] [Google Scholar]
- 22.Ago T, Kitazono T, Kuroda J, Kumai Y, Kamouchi M, Ooboshi H, Wakisaka M, Kawahara T, Rokutan K, Ibayashi S, Iida M. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke. 2005;36:1040–1046. doi: 10.1161/01.STR.0000163111.05825.0b. [DOI] [PubMed] [Google Scholar]
- 23.Serrano F, Kolluri NS, Wientjes FB, Card JP, Klann E. NADPH oxidase immunoreactivity in the mouse brain. Brain Res. 2003;988:193–198. doi: 10.1016/s0006-8993(03)03364-x. [DOI] [PubMed] [Google Scholar]
- 24.Kim MJ, Shin KS, Chung YB, Jung KW, Cha CI, Shin DH. Immunohistochemical study of p47Phox and gp91Phox distributions in rat brain. Brain Res. 2005;1040:178–186. doi: 10.1016/j.brainres.2005.01.066. [DOI] [PubMed] [Google Scholar]
- 25.Wei Y, Sowers JR, Nistala R, Gong H, Uptergrove GM, Clark SE, Morris EM, Szary N, Manrique C, Stump CS. Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells. J Biol Chem. 2006;281:35137–35146. doi: 10.1074/jbc.M601320200. [DOI] [PubMed] [Google Scholar]
- 26.Whaley-Connell AT, Morris EM, Rehmer N, Yaghoubian JC, Wei Y, Hayden MR, Habibi J, Stump CS, Sowers JR. Albumin activation of NAD(P)H oxidase activity is mediated via Rac1 in proximal tubule cells. Am J Nephrol. 2007;27:15–23. doi: 10.1159/000098432. [DOI] [PubMed] [Google Scholar]
- 27.Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Q, Simonyi A, Li W, Sisk BA, Miller RL, Macdonald RS, Lubahn DE, Sun GY, Sun AY. Dietary grape supplement ameliorates cerebral ischemia-induced neuronal death in gerbils. Mol Nutr Food Res. 2005;49:443–451. doi: 10.1002/mnfr.200500019. [DOI] [PubMed] [Google Scholar]
- 29.Wang Q, Sun AY, Simonyi A, Jensen MD, Shelat PB, Rottinghaus GE, MacDonald RS, Miller DK, Lubahn DE, Weisman GA, Sun GY. Neuroprotective mechanisms of curcumin against cerebral ischemia-induced neuronal apoptosis and behavioral deficits. J Neurosci Res. 2005;82:138–148. doi: 10.1002/jnr.20610. [DOI] [PubMed] [Google Scholar]
- 30.Streit WJ. An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I-B4) J Histochem Cytochem. 1990;38:1683–1686. doi: 10.1177/38.11.2212623. [DOI] [PubMed] [Google Scholar]
- 31.Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 32.Monnerie H, Shashidhara S, Le Roux PD. Effect of excess extracellular glutamate on dendrite growth from cerebral cortical neurons at 3 days in vitro: Involvement of NMDA receptors. J Neurosci Res. 2003;74:688–700. doi: 10.1002/jnr.10797. [DOI] [PubMed] [Google Scholar]
- 33.Won MH, Kang TC, Jeon GS, Lee JC, Kim DY, Choi EM, Lee KH, Choi CD, Chung MH, Cho SS. Immunohistochemical detection of oxidative DNA damage induced by ischemia-reperfusion insults in gerbil hippocampus in vivo. Brain Res. 1999;836:70–78. doi: 10.1016/s0006-8993(99)01611-x. [DOI] [PubMed] [Google Scholar]
- 34.Hwang IK, Yoo KY, Kim DS, Jeong YK, Kim JD, Shin HK, Lim SS, Yoo ID, Kang TC, Kim DW, Moon WK, Won MH. Neuroprotective effects of grape seed extract on neuronal injury by inhibiting DNA damage in the gerbil hippocampus after transient forebrain ischemia. Life Sci. 2004;75:1989–2001. doi: 10.1016/j.lfs.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 35.Schlorff EC, Husain K, Somani SM. Dose- and time-dependent effects of ethanol on plasma antioxidant system in rat. Alcohol. 1999;17:97–105. doi: 10.1016/s0741-8329(98)00039-1. [DOI] [PubMed] [Google Scholar]
- 36.Caceres A, Banker G, Steward O, Binder L, Payne M. MAP2 is localized to the dendrites of hippocampal neurons which develop in culture. Brain Res. 1984;315:314–318. doi: 10.1016/0165-3806(84)90167-6. [DOI] [PubMed] [Google Scholar]
- 37.Korthuis RJ, Gute DC, Blecha F, Ross CR. PR-39, a proline/arginine-rich antimicrobial peptide, prevents postischemic microvascular dysfunction. Am J Physiol. 1999;277:H1007–1013. doi: 10.1152/ajpheart.1999.277.3.H1007. [DOI] [PubMed] [Google Scholar]
- 38.Chan YR, Gallo RL. PR-39, a syndecan-inducing antimicrobial peptide, binds and affects p130(Cas) J Biol Chem. 1998;273:28978–28985. doi: 10.1074/jbc.273.44.28978. [DOI] [PubMed] [Google Scholar]
- 39.Zalewska T, Makarewicz D, Janik B, Ziemka-Nalecz M. Neonatal cerebral hypoxia-ischemia: involvement of FAK-dependent pathway. Int J Dev Neurosci. 2005;23:657–662. doi: 10.1016/j.ijdevneu.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 40.Bao J, Sato K, Li M, Gao Y, Abid R, Aird W, Simons M, Post MJ. PR-39 and PR-11 peptides inhibit ischemia-reperfusion injury by blocking proteasome-mediated I kappa B alpha degradation. Am J Physiol Heart Circ Physiol. 2001;281:H2612–2618. doi: 10.1152/ajpheart.2001.281.6.H2612. [DOI] [PubMed] [Google Scholar]
- 41.Korthuis RJ, Gute DC, Cepinskas G, Kvietys PR. Cellular mechanisms of acute versus delayed preconditioning. Pathophysiology. 1998;5:35–48. [Google Scholar]
- 42.Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol. 2001;33:1897–1918. doi: 10.1006/jmcc.2001.1462. [DOI] [PubMed] [Google Scholar]
- 43.Korthuis RJ, Gute DC. Adhesion molecule expression in postischemic microvascular dysfunction: activity of a micronized purified flavonoid fraction. J Vasc Res. 1999;36 1:15–23. doi: 10.1159/000054070. [DOI] [PubMed] [Google Scholar]
- 44.Stuehr DJ, Fasehun OA, Kwon NS, Gross SS, Gonzalez JA, Levi R, Nathan CF. Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs. Faseb J. 1991;5:98–103. doi: 10.1096/fasebj.5.1.1703974. [DOI] [PubMed] [Google Scholar]
- 45.Yamaguchi T, Kamada K, Dayton C, Gaskin FS, Yusof M, Yoshikawa T, Korthuis RJ. Role of eNOS-derived NO in the postischemic anti-inflammatory effects of antecedent ethanol ingestion in muring small intestine. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.00282.2006. Vol. Submitted. [DOI] [PubMed] [Google Scholar]
- 46.Yamaguchi T, Kamada K, Dayton C, Gaskin FS, Yusof M, Yoshikawa T, Carter P, Korthuis RJ. Role of eNOS-derived NO in the postischemic anti-inflammatory effects of antecedent ethanol ingestion in murine small intestine. Am J Physiol Heart Circ Physiol. 2007;292:H1435–1442. doi: 10.1152/ajpheart.00282.2006. [DOI] [PubMed] [Google Scholar]
- 47.Engels F, Renirie BF, Hart BA, Labadie RP, Nijkamp FP. Effects of apocynin, a drug isolated from the roots of Picrorhiza kurroa, on arachidonic acid metabolism. FEBS Lett. 1992;305:254–256. doi: 10.1016/0014-5793(92)80680-f. [DOI] [PubMed] [Google Scholar]
- 48.Abid MR, Kachra Z, Spokes KC, Aird WC. NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett. 2000;486:252–256. doi: 10.1016/s0014-5793(00)02305-x. [DOI] [PubMed] [Google Scholar]
- 49.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 50.Zhou J, Struthers AD, Lyles GA. Differential effects of some cell signalling inhibitors upon nitric oxide synthase expression and nuclear factor-kappaB activation induced by lipopolysaccharide in rat aortic smooth muscle cells. Pharmacol Res. 1999;39:363–373. doi: 10.1006/phrs.1998.0450. [DOI] [PubMed] [Google Scholar]
- 51.Delbarre B, Delbarre G, Calinon F. Effect of Daflon 500 mg, a flavonoid drug, on neurological signs, levels of free radicals and electroretinogram in the gerbil after ischemia-reperfusion injury. Int J Microcirc Clin Exp. 1995;15 1:27–33. doi: 10.1159/000179092. [DOI] [PubMed] [Google Scholar]
- 52.Katsuta K, Umemura K, Ueyama N, Matsuoka N. Pharmacological evidence for a correlation between hippocampal CA1 cell damage and hyperlocomotion following global cerebral ischemia in gerbils. Eur J Pharmacol. 2003;467:103–109. doi: 10.1016/s0014-2999(03)01573-5. [DOI] [PubMed] [Google Scholar]
- 53.Gergel D, Cederbaum AI. Inhibition of the catalytic activity of alcohol dehydrogenase by nitric oxide is associated with S nitrosylation and the release of zinc. Biochemistry. 1996;35:16186–16194. doi: 10.1021/bi962043r. [DOI] [PubMed] [Google Scholar]
- 54.Gergel D, Misik V, Riesz P, Cederbaum AI. Inhibition of rat and human cytochrome P4502E1 catalytic activity and reactive oxygen radical formation by nitric oxide. Arch Biochem Biophys. 1997;337:239–250. doi: 10.1006/abbi.1996.9765. [DOI] [PubMed] [Google Scholar]
- 55.Oei HH, Stroo WE, Burton KP, Schaffer SW. A possible role of xanthine oxidase in producing oxidative stress in the heart of chronically ethanol treated rats. Res Commun Chem Pathol Pharmacol. 1982;38:453–461. [PubMed] [Google Scholar]
- 56.Oei HH, Zoganas HC, McCord JM, Schaffer SW. Role of acetaldehyde and xanthine oxidase in ethanol-induced oxidative stress. Res Commun Chem Pathol Pharmacol. 1986;51:195–203. [PubMed] [Google Scholar]
- 57.Sultatos LG. Effects of acute ethanol administration on the hepatic xanthine dehydrogenase/oxidase system in the rat. J Pharmacol Exp Ther. 1988;246:946–949. [PubMed] [Google Scholar]
- 58.Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med. 1999;27:891–900. doi: 10.1016/s0891-5849(99)00138-0. [DOI] [PubMed] [Google Scholar]
- 59.Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- 60.Becker LB, vanden Hoek TL, Shao ZH, Li CQ, Schumacker PT. Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am J Physiol. 1999;277:H2240–2246. doi: 10.1152/ajpheart.1999.277.6.H2240. [DOI] [PubMed] [Google Scholar]
- 61.Chan PH. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem Res. 2004;29:1943–1949. doi: 10.1007/s11064-004-6869-x. [DOI] [PubMed] [Google Scholar]
- 62.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 63.Wu D, Cederbaum AI. Alcohol, oxidative stress, and free radical damage. Alcohol Res Health. 2003;27:277–284. [PMC free article] [PubMed] [Google Scholar]
- 64.Farwell W, Simonyi A, Scott H, Zhang JP, Carruthers V, Madsen R, Johnson J, Sun GY. Effects of ischemic tolerance on mRNA levels of IP3R1, beta-actin, and neuron-specific enolase in hippocampal CA1 area of the gerbil brain. Neurochem Res. 1998;23:539–542. doi: 10.1023/a:1022486619201. [DOI] [PubMed] [Google Scholar]
- 65.Schaller B, Graf R. Cerebral ischemic preconditioning. An experimental phenomenon or a clinical important entity of stroke prevention? J Neurol. 2002;249:1503–1511. doi: 10.1007/s00415-002-0933-8. [DOI] [PubMed] [Google Scholar]
- 66.Bell RM, Cave AC, Johar S, Hearse DJ, Shah AM, Shattock MJ. Pivotal role of NOX-2-containing NADPH oxidase in early ischemic preconditioning. Faseb J. 2005;19:2037–2039. doi: 10.1096/fj.04-2774fje. [DOI] [PubMed] [Google Scholar]
- 67.Dayton C, Yamaguchi T, Kamada K, Carter P, Korthuis RJ. Antecedent ethanol ingestion prevents postischemic leukocyte adhesion and P-selectin expression by a protein kinase C-dependent mechanism. Dig Dis Sci. 2005;50:684–690. doi: 10.1007/s10620-005-2557-1. [DOI] [PubMed] [Google Scholar]
- 68.Kamada K, Gaskin FS, Yamaguchi T, Carter P, Yoshikawa T, Yusof M, Korthuis RJ. Role of calcitonin gene-related peptide in the postischemic anti-inflammatory effects of antecedent ethanol ingestion. Am J Physiol Heart Circ Physiol. 2006;290:H531–537. doi: 10.1152/ajpheart.00839.2005. [DOI] [PubMed] [Google Scholar]
- 69.Mori E, del Zoppo GJ, Chambers JD, Copeland BR, Arfors KE. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke. 1992;23:712–718. doi: 10.1161/01.str.23.5.712. [DOI] [PubMed] [Google Scholar]
- 70.Gidday JM, Gasche YG, Copin JC, Shah AR, Perez RS, Shapiro SD, Chan PH, Park TS. Leukocyte-derived matrix metalloproteinase-9 mediates blood-brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- 71.Frijns CJ, Kappelle LJ. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke. 2002;33:2115–2122. doi: 10.1161/01.str.0000021902.33129.69. [DOI] [PubMed] [Google Scholar]
- 72.Zhang W, Stanimirovic D. Current and future therapeutic strategies to target inflammation in stroke. Curr Drug Targets Inflamm Allergy. 2002;1:151–166. doi: 10.2174/1568010023344689. [DOI] [PubMed] [Google Scholar]
- 73.Chan PH. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann N Y Acad Sci. 2005;1042:203–209. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- 74.Sugawara T, Chan PH. Reactive oxygen radicals and pathogenesis of neuronal death after cerebral ischemia. Antioxid Redox Signal. 2003;5:597–607. doi: 10.1089/152308603770310266. [DOI] [PubMed] [Google Scholar]
- 75.Kamada K, Dayton CB, Yamaguchi T, Korthuis RJ. Antecedent ethanol ingestion prevents postischemic microvascular dysfunction. Pathophysiology. 2004;10:131–137. doi: 10.1016/j.pathophys.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 76.Belmadani A, Neafsey EJ, Collins MA. Human immunodeficiency virus type 1 gp120 and ethanol coexposure in rat organotypic brain slice cultures: Curtailment of gp120-induced neurotoxicity and neurotoxic mediators by moderate but not high ethanol concentrations. J Neurovirol. 2003;9:45–54. doi: 10.1080/13550280390173409. [DOI] [PubMed] [Google Scholar]
- 77.Belmadani A, Kumar S, Schipma M, Collins MA, Neafsey EJ. Inhibition of amyloid-beta-induced neurotoxicity and apoptosis by moderate ethanol preconditioning. Neuroreport. 2004;15:2093–2096. doi: 10.1097/00001756-200409150-00019. [DOI] [PubMed] [Google Scholar]