

Abstract
Apoptosis is defined on the basis of morphological changes like nuclear fragmentation and chromatin condensation, which are dependent on caspases. Many forms of caspase-independent cell death have been reported, but the mechanisms are still poorly understood. We found that hypoxic cell death was independent of caspases and was associated with significant nuclear shrinkage. Neither Bcl-2 nor Apaf-1 deficiency prevented hypoxic nuclear shrinkage. To understand the molecular mechanism of the nuclear shrinkage, we developed an in vitro system using permeabilized cells, which allowed us to purify a novel member of the phospholipase A2 (PLA2) family that induced nuclear shrinkage. Purified PLA2 induced nuclear shrinkage in our permeabilized cell system. PLA2 inhibitors prevented hypoxic nuclear shrinkage in cells and cell death. Hypoxia caused elevation of PLA2 activity and translocation of intracellular PLA2s to the nucleus. Knockdown of the Ca2+-independent PLA2 delayed nuclear shrinkage and cell death. These results indicate that Ca2+-independent PLA2 is crucial for a caspase-independent cell death signaling pathway leading to nuclear shrinkage.
Keywords: hypoxia; phospholipase A; caspases; cell nucleus; cell death
Introduction
Apoptosis, or programmed cell death, is a fundamental biological process that eliminates unwanted cells; it plays a crucial role in various events, including morphogenesis during normal development and maintenance of homeostasis. Apoptosis is defined on the basis of morphological changes such as chromatin condensation, nuclear fragmentation, and formation of apoptotic bodies, all of which are driven by a specific family of proteases called caspases (Salvesen and Dixit, 1997). Several molecules involved in apoptotic nuclear changes have been identified, including CAD (Enari et al., 1998; Samejima et al., 2001), acinus (Sahara et al., 1999), apoptosis-inducing factor (AIF; Susin et al., 1999), and caspase-6 (Ruchaud et al., 2002), although the molecular mechanisms underlying these morphological changes have not been elucidated.
Caspase-independent cell death has recently been attracting more interest. This type of cell death can be induced by agents that also induce apoptosis under different circumstances, such as nitric oxide (Okuno et al., 1998), Bax overexpression (Xiang et al., 1996), NGF deprivation (Chang and Johnson, 2002), and hypoxia (Shimizu et al., 1996a). Cells that are deficient in Apaf-1 or caspases, which are both essential for apoptosis, undergo death in a caspase-independent manner (Cheng et al., 2001). However, the morphological features of caspase-independent cell death vary depending on the death stimulus or cell type, and several descriptive terms have been suggested such as necrosis, oncosis, paraptosis, aponecrosis, and autophagic cell death (Leist and Jaattela, 2001; Lockshin and Zakeri, 2002). We found that nuclear shrinkage is one of the features of caspase-independent cell death caused by hypoxia. Nuclear shrinkage (pyknosis) has already been noticed as a feature of hypoxic or ischemic caspase-independent cell death associated with swelling of organelles, vacuolization, and increased membrane permeability (Grune et al., 1997; Hossmann et al., 2001). EM has revealed that nuclear pyknosis is associated with condensation of the chromatin into many uneven and irregular clumps that are readily distinguishable from the small number of uniformly dense and regular aggregates produced by apoptotic chromatin condensation (Martin et al., 1998).
Phospholipase A2 (PLA2) comprises a family of esterases that hydrolyze the sn-2 ester bond in phospholipids to release free fatty acids and lysophospholipids. A number of mammalian PLA2 isotypes have been identified, and are divided into three major subfamilies: secretory PLA2 (sPLA2), cytosolic Ca2+-dependent PLA2 (cPLA2), and Ca2+-independent PLA2 (iPLA2; Six and Dennis, 2000; Ma and Turk, 2001). sPLA2s are extracellular low molecular mass enzymes (14 kD) that require millimolar concentrations of Ca2+ for activation. sPLA2s are thought to be potent mediators of inflammation and also show antibacterial activity. cPLA2s are intracellular enzymes that specifically target arachidonic acid (AA) at the sn-2 position of phospholipids. Their activity is regulated by submicromolar levels of Ca2+, and these enzymes are believed to play a pivotal role in the production of AA metabolites, such as eicosanoids. The activity of iPLA2 is Ca2+-independent, and it is thought to be a remodeling enzyme that maintains the composition of membrane phospholipids. Although the molecular basis of caspase-independent cell death is largely unknown, PLA2s have been implicated in ischemic cell death; however, their actual role remains unclear (Bazan and Rodriguez de Turco, 1980; Edgar et al., 1982). In addition, it has been reported that some PLA2 inhibitors can prevent ischemic cell death (Wang et al., 1996; Arai et al., 2001; Michiels et al., 2002; Williams and Gottlieb, 2002), and that cPLA2α-deficient mice show partial resistance to ischemic cell death (Bonventre et al., 1997). However, the molecular mechanism of the morphological changes that occur during hypoxia is unknown. Here, we report that PLA2 is responsible for nuclear shrinkage in the process of caspase-independent cell death.
Results
Hypoxia induces nuclear shrinkage in a caspase- and Apaf-1–independent and Bcl-2–insensitive manner
To understand the molecular basis of caspase-independent cell death, we focused on nuclear shrinkage as a starting point for analysis because it is known to be often associated with caspase-independent cell death. By screening various culture conditions, we found that PC12 cells subjected to hypoxia in the presence of a low glucose concentration (such as 2.2 g/l) reproducibly showed nuclear shrinkage without chromatin fragmentation (Fig. 1 A). Therefore, we cultured the cells with 2.2 g/l glucose for this experiment. When stained with propidium iodide, shrunken nuclei also incorporated the dye (Fig. 1 A, b), demonstrating the loss of membrane integrity that is a characteristic of necrotic death. A pan-caspase inhibitor (zVAD-fmk) had no effect on nuclear shrinkage (Fig. 1 A, c and d), whereas apoptotic nuclear changes such as chromatin condensation and fragmentation induced by staurosporine (STS) were completely prevented by zVAD-fmk (Fig. 1 A, e and f), indicating that caspases were not involved in the process of hypoxia-induced nuclear shrinkage. Also, we could not detect caspase-3 activity and caspase-dependent cleavage of lamin B1 to a 30-kD fragment under hypoxia (Fig. 1 B). To further confirm the caspase independence of nuclear shrinkage, we used mouse embryonic fibroblasts (MEFs) from Apaf-1–deficient mice. Apaf-1 has been shown to be essential for mitochondria-dependent caspase activation in the intrinsic death pathway (Cecconi et al., 1998; Yoshida et al., 1998). Upon exposure to hypoxia, Apaf-1−/− MEFs showed nuclear shrinkage (Fig. 1 C), confirming the independence of hypoxic nuclear shrinkage from the caspase cascade. This result also demonstrated the occurrence of hypoxic nuclear shrinkage in a different type of cell. Because it has been reported that Bcl-2 not only inhibits apoptosis, but also caspase-independent cell death (Monney et al., 1998; Okuno et al., 1998; Haraguchi et al., 2000), we examined whether Bcl-2 could protect against hypoxic nuclear shrinkage using Bcl-2–overexpressing PC12 cells. As shown in Fig. 1 D (e), Bcl-2 had no inhibitory effect on nuclear shrinkage, although apoptotic nuclear changes were completely prevented in the Bcl-2–overexpressing cells (Fig. 1 D, f). These results showed that hypoxic nuclear shrinkage was induced by a caspase- and Apaf-1–independent mechanism that was Bcl-2–insensitive.
Figure 1.
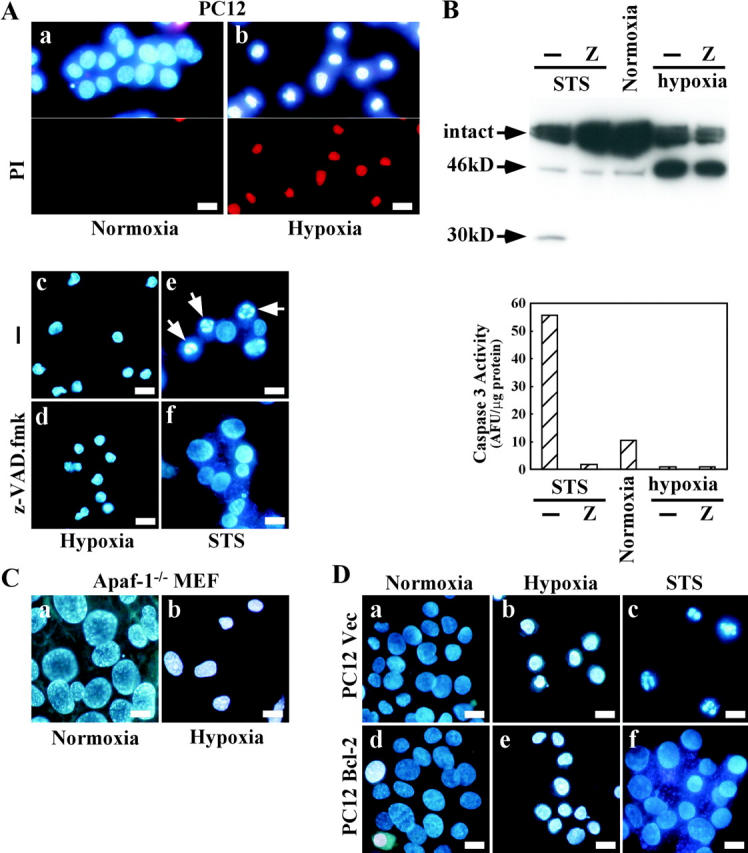
Hypoxic nuclear shrinkage is caspase- and Apaf-1–independent, and Bcl-2–insensitive. (A) PC12 cells were incubated under normoxic conditions (a) or hypoxic conditions for 36 h in the absence (b and c) or presence (d) of 50 μM zVAD-fmk. PC12 cells were also treated with 1 μM staurosporine (STS) for 14 h in the absence (e) or presence (f) of 50 μM zVAD-fmk. (B) PC12 cells were treated as in A. Cell lysates were subjected to Western blot analysis with an antibody for lamin B1 (top), and caspase activity was measured (bottom). (C) Apaf-1−/− MEFs were cultured under normoxic (a) or hypoxic (b) conditions for 40 h. (D) PC12 cells stably transfected with Bcl-2 DNA (d–f) or control vector DNA (a–c) were incubated under normoxic (a and d) or hypoxic (b and e) conditions for 36 h, or were treated with 1 μM STS for 14 h (c and f). Nuclei were stained with Hoechst 33342 together with (or without) propidium iodide and then visualized under a fluorescence microscope. Arrows in A indicate apoptotic nuclei. Bars, 10 μm.
Nuclear shrinkage assay and purification of a nuclear shrinkage factor from rat liver
To identify a molecule that induced nuclear shrinkage, we developed an in vitro assay system using digitonin-permeabilized cells. Permeabilized HeLa cells were incubated with a lysate prepared from hypoxic PC12 cells. As shown in Fig. 2 A, the lysate induced nuclear shrinkage, indicating that this permeabilized cell system could reproduce the shrinkage that occurred in cultured cells. However, a lysate from normoxic PC12 cells also induced nuclear shrinkage to a similar extent (Fig. 2 A, c). This was due to activation of PLA2 by disruption of the cells (see Fig. 6). To obtain a large volume of starting material, we examined whether rat liver S100 lysate could induce nuclear shrinkage using the permeabilized cell system. We found that this lysate induced nuclear shrinkage in a caspase-independent manner (Fig. 2 B, b and c). The lysate-induced nuclear shrinkage was morphologically distinct from the nuclear changes induced by rat mitochondrial supernatant, which possibly contained AIF activity (Fig. 2 B, d). AIF induced peripheral chromatin condensation, as described elsewhere (Susin et al., 2000), whereas liver S100 or PC12 lysate induced homogeneous nuclear condensation, indicating the presence of an unidentified factor. A nuclear shrinkage factor was partially purified by seven steps of column chromatography, as shown in Fig. 2 C. Two proteins, shown by arrows in Fig. 2 E, were found to be closely associated with nuclear shrinkage (Fig. 2 D). After lysine endopeptidase digestion and separation on a reverse-phase HPLC column, the amino acid sequences of proteins 1 and 2 were determined. Protein 1 was rat UDP-N-acetylglucosamine 2-epimerase, which is a liver-specific protein with a predicted molecular mass of 79 kD that is associated with the biosynthesis of N-acetylneuraminic acid (Stasche et al., 1997). However, the fact that hypoxic nuclear shrinkage was not tissue specific suggested that UDP-N-acetylglucosamine 2-epimerase was not the nuclear shrinkage factor. Although the sequences of four peptides of protein 2 with a predicted molecular mass of 66 kD were determined, we could not find any gene in the database that encoded these four sequences. Therefore, we attempted to isolate part of the gene by RT-PCR using various combinations of degenerative primers based on the amino acid sequences of the four peptides. The amino acid sequence deduced from the PCR product, which included three of the four peptides (peptides 1, 3, and 4), is shown in Fig. 3. The sequences showed significant homology with human cPLA2γ (cytosolic PLA2 type γ) and included the lipase consensus sequence (GXSXS; Fig. 3), but the carboxy-terminal portion (aa 255–329) had no homology with human cPLA2γ. Human cPLA2γ is reported to possess calcium-independent PLA2 activity (Underwood et al., 1998; Pickard et al., 1999). We have not determined the sequence of the full-length DNA of protein 2. Subsequently, we found that the mouse counterpart of protein 2, which was inferred from the mouse genome database of Celera Genomics, contained the peptide 2 sequence outside the region amplified by PCR.
Figure 2.
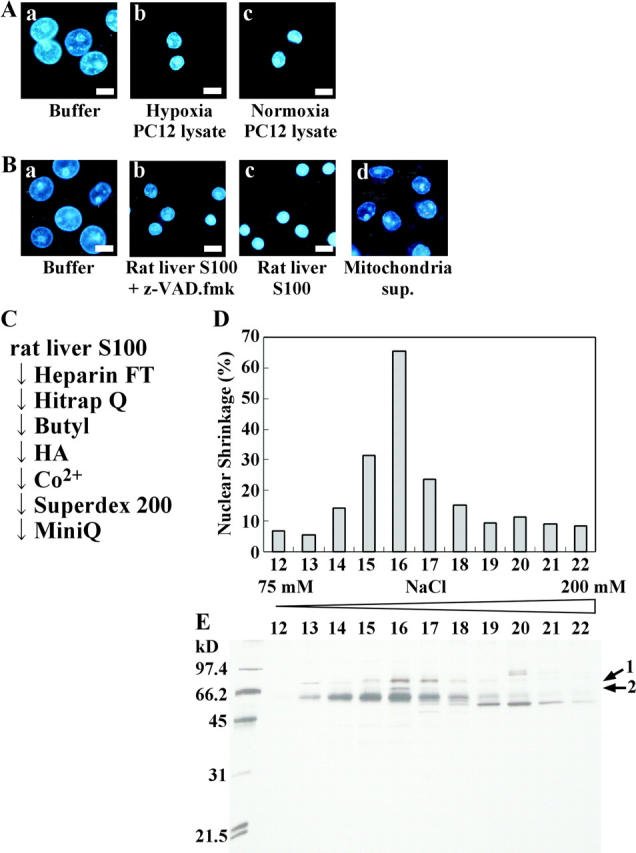
Detection of nuclear shrinkage factor in cell lysates using the permeabilized cell system. (A) HeLa cells were permeabilized with digitonin as described in Materials and methods, and then were incubated for 2 h with buffer (a), hypoxic PC12 cell lysate (b), or normoxic PC12 cell lysate (c). (B) Permeabilized HeLa cells were incubated for 2 h with buffer (a), rat liver S100 with (b) or without (c) zVAD-fmk, or rat mitochondrial supernatant (d). Nuclei were visualized under a fluorescence microscope after staining with Hoechst 33342. Bars, 10 μm. (C) Illustration of the purification steps for nuclear shrinkage factor. Proteins bound to each column were applied to the next column, except that the flow-through (FT) fractions were used in the case of the heparin column. (D) Aliquots of Mini Q™ column fractions were added to digitonin-permeabilized HeLa cells and incubated for 2 h at 37°C, followed by Hoechst 33342 staining. Under a fluorescence microscope, the morphology of >500 nuclei was observed, and the percentage of cells showing nuclear shrinkage was assessed. (E) Aliquots of Mini Q™ column fractions were subjected to 15% SDS-PAGE and silver staining. The amino acid sequences of the proteins indicated by arrows were determined.
Figure 3.

Alignment of the amino acid sequences of protein 2 and human cPLA2γ. The amino acid sequences of the four peptides are shown at the top. The amino acid sequence of protein 2, which was predicted from the cDNA sequence generated by PCR using degenerative primers, and that of human cPLA2γ (hcPLA2γ) were aligned. Identical and similar amino acid residues are shown by asterisks and dots, respectively. Three sequenced peptides that are part of the known sequence of protein 2 are underlined. The lipase consensus sequence is boxed. The degenerative PCR primers used to amplify protein 2 cDNA are shown by arrows. These sequence data are available from GenBank/EMBL/DDBJ under accession no. AB113213.
PLA2 activity is necessary for nuclear shrinkage in the permeabilized cell system
To test whether or not PLA2 activity was a prerequisite for nuclear shrinkage in the permeabilized cell system, we examined the effect of intracellular PLA2 inhibitors on the occurrence of shrinkage. We used three kinds of intracellular PLA2 inhibitors: methyl arachidonyl fluorophosphonate (MAFP), bromoenol lactone (BEL), and arachidonyltrifluoromethyl ketone (AACOCF3). These inhibitors were originally thought to be selective for a specific PLA2, but are now known to also inhibit other isoforms (Balsinde et al., 1999). When permeabilized HeLa cells were treated with the HiTrap™ Q Sepharose column-bound fraction of rat liver S100 lysate, all the inhibitors blocked nuclear shrinkage in a dose-dependent manner (Fig. 4 A). These results indicated that PLA2 activity in the rat liver S100 lysate was responsible for nuclear shrinkage in our permeabilized cell system.
Figure 4.
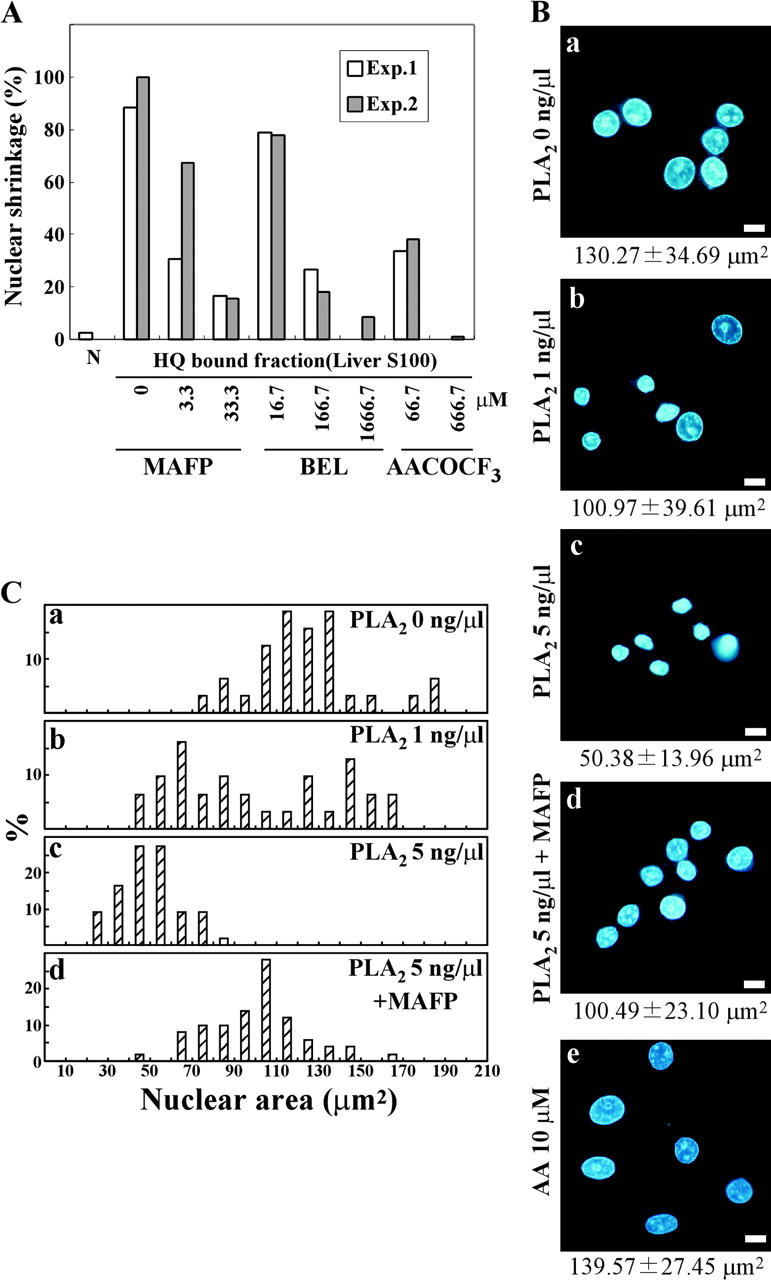
PLA2 activity is required for nuclear shrinkage of permeabilized cells. (A) PLA2 inhibitors prevent rat liver S100-induced nuclear shrinkage of permeabilized cells. An aliquot of the fraction bound to the HiTrap™ Q column was incubated with permeabilized HeLa cells for 2 h at 37°C in the presence or absence of the indicated PLA2 inhibitors. The percentage of cells with nuclear shrinkage in two independent experiments is shown. (B) Permeabilized HeLa cells were exposed to bee venom PLA2 at (a) 0, (b) 1.0, (c) 5.0, or (d) 5.0 ng/μl together with 66.7 μM MAFP, or at 10 μM AA (e) for 2 h. Nuclei were visualized under a fluorescence microscope after Hoechst 33342 staining. The average nuclear area is shown under the photograph. Bars, 10 μm. (C) The areas of shrunken nuclei were individually determined from the fluorescence microscopy images, and the percentage of cells with the indicated nuclear size was plotted.
PLA2 induces nuclear shrinkage in the permeabilized cell system
To investigate whether PLA2 activity alone was sufficient to induce nuclear shrinkage, bee venom PLA2 (a commercially available form of purified PLA2) was used. When added to permeabilized HeLa cells, bee venom PLA2 induced nuclear shrinkage in a concentration-dependent manner (Fig. 4, B and C). MAFP inhibited bee venom PLA2-induced nuclear shrinkage (Fig. 4 B, d; Fig. 4 C, d), although only partially due to its incomplete inhibition of PLA2 activity (MAFP inhibited PLA2 activity by 69.9%). Quantitative measurement of the nuclear area showed that it was 130.27 ± 34.69 μm2 for control nuclei and 50.38 ± 13.96 μm2 for the nuclei of cells treated with 5 ng/μl PLA2. Another type of PLA2 (porcine pancreatic sPLA2) also caused nuclear shrinkage like bee venom PLA2 (unpublished data). Because PLA2 hydrolyzes phospholipids at the sn-2 position to liberate fatty acids, we tested whether fatty acids (saturated and unsaturated) could cause nuclear shrinkage in the permeabilized cell system. AA (Fig. 4 B, e) and other fatty acids (stearic, linoleic, palmitic, and oleic acids; unpublished data) had no effect, suggesting that PLA2 induced nuclear shrinkage itself and that its metabolites were ineffective.
PLA2 activity is essential for nuclear shrinkage in hypoxic cells
Because our results showed that PLA2 activity was necessary and sufficient to induce nuclear shrinkage in permeabilized cells, we next examined the effect of PLA2 inhibitors on nuclear shrinkage in PC12 cells. Some cells developed typical apoptotic nuclear features in response to hypoxia and PLA2 inhibitor treatment (Fig. 5 A, d), so zVAD-fmk was also added to the culture medium to prevent apoptosis. The four PLA2 inhibitors tested (BEL, AACOCF3, MAFP, and palmitoyl trifluoromethyl ketone [PACOCF3]) prevented the nuclear shrinkage of PC12 cells induced by hypoxia to a varying extent (Fig. 5 A), indicating that PLA2 activity plays an important role in hypoxic nuclear shrinkage. BEL almost completely inhibited nuclear shrinkage, whereas PACOCF3 only caused partial inhibition (Fig. 5 B). BEL and PACOCF3 (iPLA2 inhibitors) were more potent than MAFP and AACOCF3 (cPLA2 inhibitors; Fig. 5 A), suggesting that iPLA2 plays a major role in hypoxic nuclear shrinkage.
Figure 5.
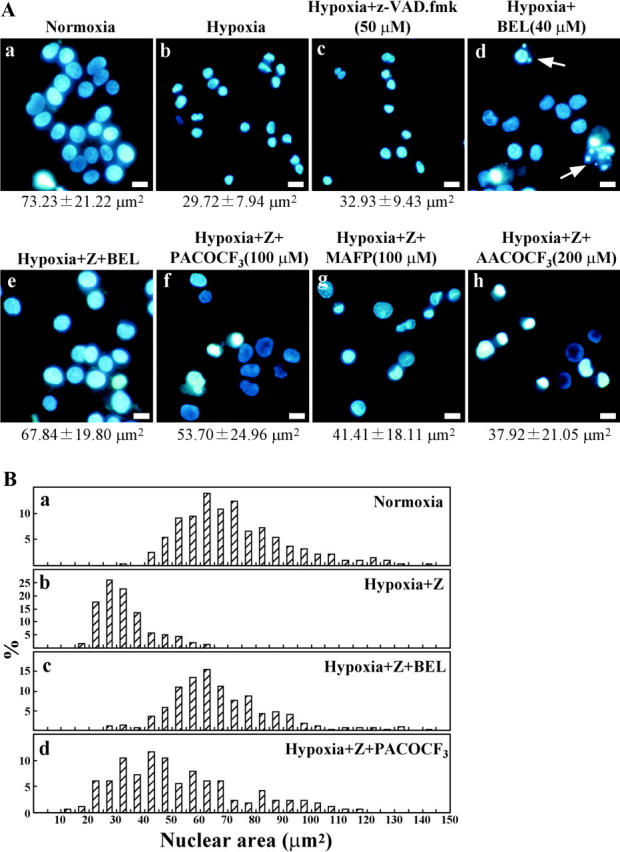
PLA2 is essential for nuclear shrinkage in hypoxic cells. (A) PC12 cells were incubated under normoxic (a) or hypoxic (b–h) conditions for 36 h. Before hypoxia, 50 μM zVAD-fmk (Z) and the indicated PLA2 inhibitors were added to the culture medium. Nuclei were visualized under a fluorescence microscope after Hoechst 33342 staining. The average nuclear area is shown under the photographs. Arrows indicate apoptotic nuclei. Bars, 10 μm. (B) As described in Fig. 4 C, the percentage of cells with the indicated nuclear size was plotted.
Activation and nuclear translocation of PLA2 during hypoxia
It has been reported that intracellular PLA2s, including cPLA2α and iPLA2, undergo translocation from the cytoplasm to the nuclear membrane, and PLA2 activity is elevated in ATP-depleted cells (Arai et al., 2001; Sheridan et al., 2001; Michiels et al., 2002; Williams and Gottlieb, 2002). We examined whether PLA2 activity was increased during hypoxia using the fluorescent PLA2 reporter N-((6-(2,4-dinitrophenyl)amino)hexanoyl)-2-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-pentanoyl)-sn-glycero-3-phosphoethanolamine (PED6). In this lipid, the fluorescent BODIPY® moiety at the sn-2 position is quenched by the DNP group in the head group, but emits fluorescence when released by PLA2 catalysis (Farber et al., 2001). As shown in Fig. 6 A, strong BODIPY® fluorescence was detected in hypoxic cells with shrunken nuclei, but only weak fluorescence was found in control cells and BEL/zVAD-fmk–treated cells. However, when cells were lysed, PLA2 activity was spontaneously increased (Fig. 6 B), explaining that a lysate of normal PC12 also induced nuclear shrinkage (Fig. 2 A, c). To further confirm the increase of PLA2 activity during hypoxia, AA release into the culture medium was measured. AA release was induced by hypoxia, and was inhibited by BEL/zVAD-fmk, but not by zVAD-fmk (Fig. 6 C). These results showed that PLA2 activity was increased during hypoxia and then induced nuclear shrinkage.
Figure 6.
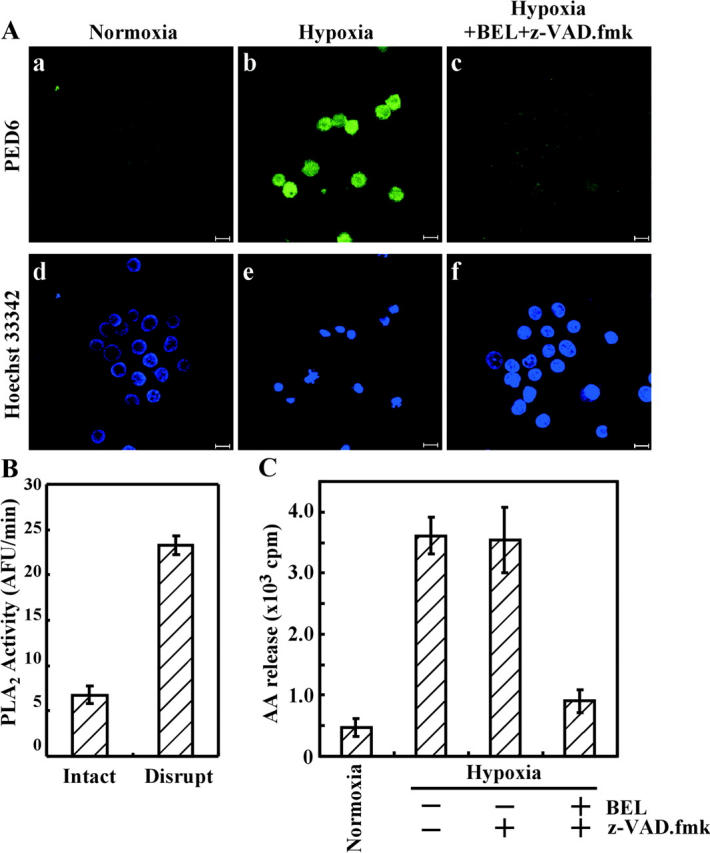
Intracellular PLA2 is activated by hypoxia. PC12 cells were exposed to hypoxia for 30 h in the absence or presence of a PLA2 inhibitor and a caspase inhibitor. Cells were also incubated under normoxic conditions. (A) Liposomes containing PED6 (a fluorescent PLA2 reporter) were prepared as described in Materials and methods, added to the culture medium, and incubated for 10 min. After nuclear staining, fluorescence arising from Hoechst 33342 (d–f) and from PED6 (a–c) was visualized using a confocal microscope. Bars, 10 μm. (B) Before and after disruption of normoxic cells, PLA2 activity was measured as described in Materials and methods. (C) AA released into the culture medium by hypoxia was measured as described in Materials and methods.
Because the distribution of PED6 was not necessarily correlated with the subcellular localization of PLA2 activity, we investigated the localization of intracellular PLA2 proteins during hypoxia. CHO-k1 cells were used because of the reactivity of the available anti-iPLA2 antibody. After exposure to hypoxia, CHO-k1 cells were treated with Triton X-100 for separation into supernatant and pellet fractions that contained cytosolic and nuclear proteins, respectively. Analysis of the localization of iPLA2 and cPLA2α during hypoxia showed that these PLA2s translocated to the nucleus from the cytoplasm within 24 h of hypoxic exposure (Fig. 7 A). Although a large amount of cPLA2α remained in the cytoplasm after exposure to hypoxia, most iPLA2 underwent translocation to the nucleus. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein was observed in the nuclear fraction at 35 h, but this may have been due to accumulation of unfolded GAPDH caused by the depletion of ATP. Accumulation of iPLA2 in the nucleus during hypoxia was also confirmed by immunostaining with anti-iPLA2 antibody (Fig. 7 B). These results demonstrated that iPLA2 underwent translocation to the nucleus during hypoxia and that PLA2 activity showed a concomitant increase in the shrunken nuclei.
Figure 7.
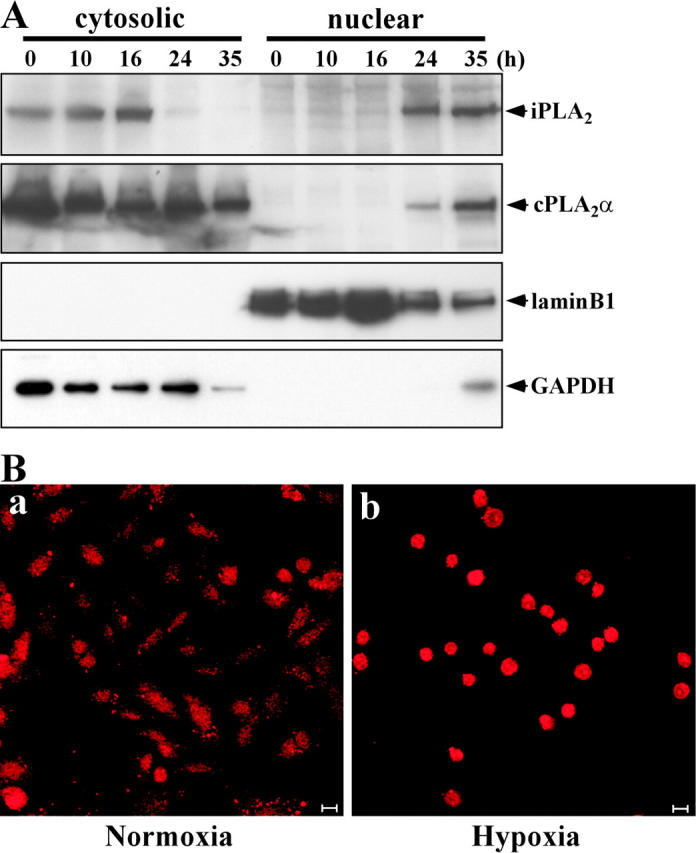
Translocation of iPLA2 from cytosol to nucleus during hypoxia. (A) CHO-k1 cells were incubated under hypoxic conditions for the indicated time. Cells were separated into cytosolic and nuclear fractions, and then were subjected to 15% SDS-PAGE followed by Western blot analysis. The filter was probed with antibodies for iPLA2, cPLA2α, lamin B1, and GAPDH. Lamin B1 (a nuclear protein) and GAPDH (a cytoplasmic protein) were used as the controls for cell fractionation. (B) CHO-k1 cells were exposed to normoxia (a) or hypoxia (b) for 36 h. Then, the cells were immunostained with anti-iPLA2 antibody, as described in Materials and methods, and visualized under a confocal microscope. Bars, 10 μm.
Reduction of iPLA2 inhibits hypoxic nuclear shrinkage
To further investigate the importance of iPLA2 for hypoxic nuclear shrinkage, we reduced the iPLA2 level using small interfering RNA (siRNA). CHO-k1 cells were treated with siRNA for iPLA2 or GFP (negative control), and the expression of iPLA2 protein was assessed by Western blotting. After transfection with siRNA for iPLA2, the level of iPLA2 protein decreased significantly in a dose-dependent manner (Fig. 8 A). As shown in Fig. 8 B, hypoxic nuclear shrinkage was suppressed in cells treated with siRNA for iPLA2, but not in cells treated with siRNA for GFP. These results suggested that iPLA2 plays a major role in hypoxic nuclear shrinkage.
Figure 8.
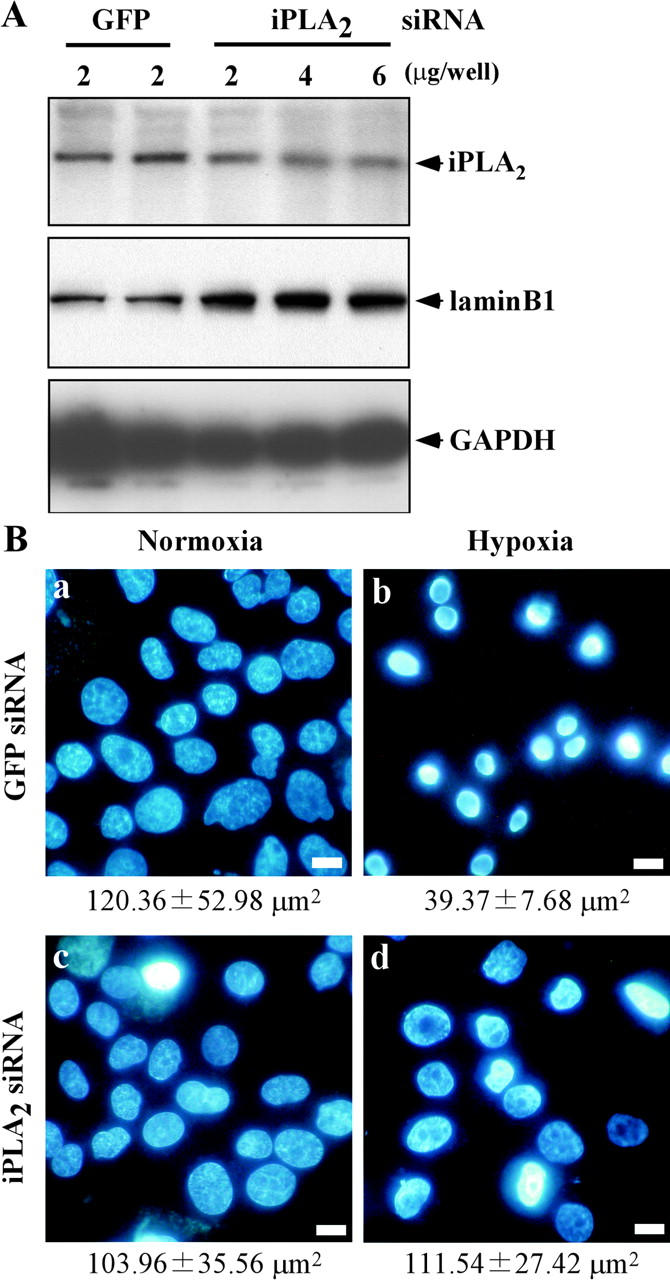
A reduced iPLA2 level inhibits hypoxic nuclear shrinkage. The indicated dose of siRNA for GFP or iPLA2 was transfected into CHO-k1 cells three times as described in Materials and methods. GFP was used as the negative control. (A) Expression of iPLA2 protein was assessed by Western blotting. The lysates from GFP siRNA- or iPLA2 siRNA-treated CHO-k1 cells were subjected to 15% SDS-PAGE followed by Western blotting with an antibody for iPLA2, lamin B1, or GAPDH (Lamin B1 and GAPDH were loading controls). (B) CHO-k1 cells were transfected with siRNA for GFP (2 μg/well) or siRNA for iPLA2 (6 μg/well), and then were exposed to normoxia or hypoxia for 36 h. Subsequently, the cells were stained with Hoechst 33342 and visualized under a fluorescence microscope. Bars, 10 μm. Note that siRNA for iPLA2 (2 μg/well) showed similar results (not depicted). The average nuclear area is shown under the photograph.
PLA2 inhibitors and reduction of iPLA2 prevent hypoxic cell death
Next, we examined whether PLA2 inhibitors or a reduction of the iPLA2 level could influence hypoxic cell death. Cell death was assessed by lactate dehydrogenase (LDH) release into the culture medium, as described in Materials and methods. Treatment with BEL partially inhibited hypoxic cell death in the absence of zVAD-fmk (Fig. 9 A); the partial inhibition was due to the occurrence of apoptosis (Fig. 5 A, d). In the presence of zVAD-fmk, BEL and PACOCF3 effectively prevented hypoxic cell death, whereas MAFP and AACOCF3 only partially prevented it (Fig. 9 A). Moreover, reduction of the iPLA2 level by siRNA delayed the onset of hypoxic cell death (Fig. 9 B). These results demonstrated that PLA2 (mainly iPLA2) not only regulated nuclear shrinkage, but also mediated the death of hypoxic cells.
Figure 9.
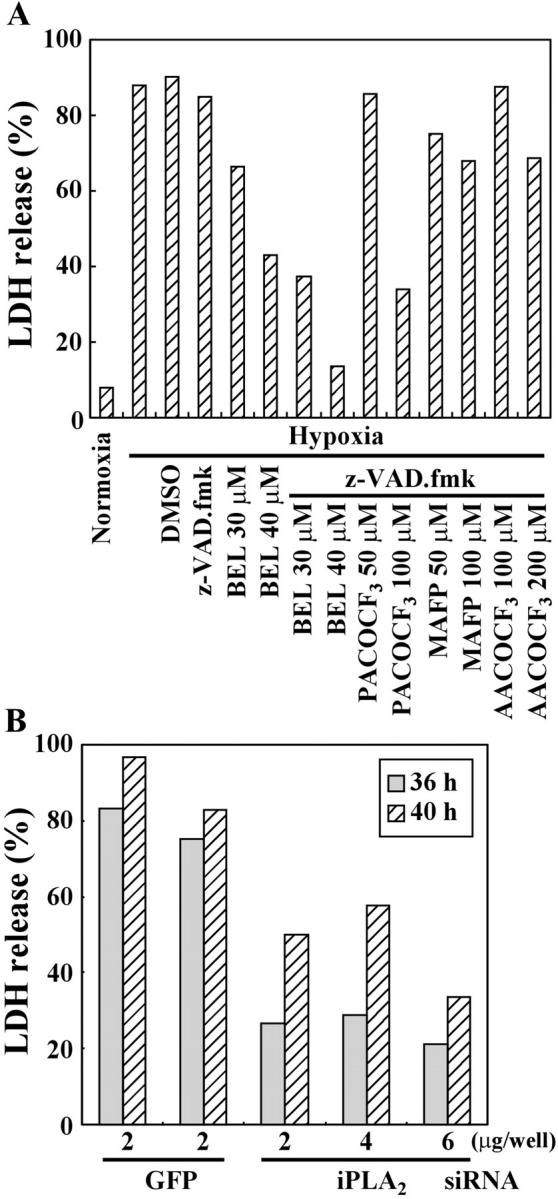
PLA2 inhibitors or reduction of iPLA2 protein prevents hypoxic cell death in the presence of a caspase inhibitor. (A) PC12 cells were exposed to normoxia or hypoxia for 36 h. The indicated inhibitors were added to the culture medium before hypoxia. LDH release from the cells was measured as described in Materials and methods. (B) As described in Fig. 8, CHO-k1 cells were transfected with siRNA for GFP or iPLA2 and were exposed to hypoxia for the indicated times. Cell death was then monitored by LDH release.
BEL also prevents caspase-independent nuclear shrinkage induced by other stimuli
We examined whether the inhibitory effect of the PLA2 inhibitor BEL on nuclear shrinkage was also applicable to other types of caspase-independent cell death. PC12 cells were exposed to STS in the presence of zVAD-fmk. To accelerate caspase-independent death, the glucose concentration of the culture medium was decreased to 0.9 g/l. Under these conditions, almost all of the cells showed shrunken nuclei after STS and zVAD-fmk treatment for 24 h (Fig. 10 A, b). BEL inhibited STS-induced nuclear shrinkage, but marginal chromatin condensation still occurred (Fig. 10 A, c).
Figure 10.

Effect of PLA2 inhibitor on nuclear shrinkage induced in a caspase-independent manner by other stimuli. (A) Caspase-independent death of PC12 cells was induced by incubation for 24 h with 500 nM STS and 50 μM zVAD-fmk in the presence (c) or absence (b) of 35 μM BEL at a low glucose concentration (0.9 g/l). (B) Cerebellar granule neurons were harvested as described in Materials and methods. Neurons were cultured for 7 d in medium containing 10% serum and 25 mM KCl, and caspase-independent cell death was induced by replacing this medium with serum-free medium containing 5 mM KCl (low K+) and incubation for 24 h in the presence (c) or absence (b) of 40 μM BEL. Nuclei were visualized under a fluorescence microscope after Hoechst 33342 staining. Bars, 10 μm. The average nuclear area is shown under the photograph.
Next, primary cultured mouse cerebellar granule neurons were exposed to a low K+ medium, which induced caspase-independent death. BEL also partially inhibited nuclear shrinkage in these cells (Fig. 10 B). The architecture of euchromatin and heterochromatin appeared to remain intact in BEL/low K+-treated neurons, but not in low K+-treated neurons. These results showed that the PLA2 inhibitor BEL could prevent nuclear shrinkage in various forms of caspase-independent death.
Discussion
Nuclear shrinkage associated with caspase-independent cell death
A great deal of attention has been focused on caspase-independent cell death, which is associated with cell death paradigms related to nitric oxide, Bax, hypoxia, and NGF deprivation (Shimizu et al., 1996a; Xiang et al., 1996; Okuno et al., 1998; Chang and Johnson, 2002). We focused on hypoxic nuclear shrinkage to investigate the molecular pathway of caspase-independent cell death. Nuclear shrinkage termed pyknosis occurs in organ ischemia (Grune et al., 1997; Hossmann et al., 2001), and we confirmed that almost all cells exposed to hypoxia developed significant nuclear shrinkage that was mediated by a caspase-independent, Apaf-1–independent, and Bcl-2–insensitive pathway (Fig. 1). This nuclear shrinkage was also induced by STS in the presence of a caspase inhibitor for PC12 cells or low K+ for cerebellar granule neurons (Fig. 10), indicating that the occurrence of nuclear shrinkage is not specific to hypoxia. Because caspase-independent cell death that is not accompanied by nuclear shrinkage has also been reported (Kawahara et al., 1998; Miyazaki et al., 2001), nuclear shrinkage is not universal to all types of caspase-independent cell death. Instead, it is a feature of some kinds of caspase-independent cell death, including that due to hypoxia, suggesting that there is more than one mechanism of caspase-independent death.
Essential role of PLA2 in nuclear shrinkage
Using HeLa cells permeabilized by digitonin, we established an in vitro assay system that allowed the purification of a factor causing nuclear shrinkage, and we identified a member of the PLA2 family as a nuclear shrinkage factor in rat liver lysate. The evidence for an essential role of PLA2 in hypoxic nuclear shrinkage was as follows: (1) rat cPLA2γ and bee venom PLA2 induced nuclear shrinkage in our permeabilized cell system (Fig. 2 D and Fig. 4 B); (2) there was inhibition of hypoxic nuclear shrinkage by several PLA2 inhibitors (Fig. 5); and (3) there was inhibition of hypoxic nuclear shrinkage by reduction of the iPLA2 level (Fig. 8). The mechanism through which PLA2 induces nuclear shrinkage remains to be determined. Addition of various fatty acids to permeabilized cells had no effect on nuclear shrinkage, excluding the direct involvement of its metabolites. Significant loss of nuclear membrane phospholipids has been reported during myocardial ischemia, which probably contributes to nuclear shrinkage (Williams et al., 2000). It has also been reported that bee venom PLA2 induces the shrinkage of erythrocytes independently of an osmotic effect (Rudenko et al., 1997), and such erythrocyte shrinkage may occur by the same mechanism as nuclear shrinkage. The nuclear lamina is important for maintaining the stability of the nucleus and is cleaved to a 30-kD fragment during apoptosis, leading to nuclear changes (Ruchaud et al., 2002). During hypoxic death, the lamina was also cleaved (although at a different site; Fig. 1 B), and the ring of the nuclear lamina was smaller in shrunken nuclei (unpublished data). Cleavage of the lamina during hypoxic cell death might have some role in nuclear shrinkage. Alternatively, nuclear shrinkage may result from the loss of mobile components of the nucleus after breakdown of nuclear membrane by PLA2. EM indicated that the density of hypoxic shrunken nuclei was decreased (unpublished data).
AIF was initially purified as a factor released from the mitochondria when the mitochondrial permeability transition occurred that induced apoptotic changes of the nucleus in a caspase-independent manner (Susin et al., 1999). Addition of recombinant AIF protein to purified nuclei induces DNA loss, peripheral chromatin condensation, DNA fragmentation into pieces ∼50 kbp in size, and nuclear shrinkage. However, the effects of PLA2 and AIF on the nucleus are significantly different: AIF induced peripheral chromatin condensation (Susin et al., 2000), whereas PLA2 induces homogeneous nuclear condensation. Furthermore, the release of AIF from the mitochondria is inhibited by Bcl-2, but nuclear shrinkage still occurred in the presence of Bcl-2. These results suggest that PLA2 plays a different role in nuclear changes from that of AIF.
iPLA2 is responsible for nuclear shrinkage
A number of mammalian PLA2 isotypes have been identified; they are divided into three major subfamilies: sPLA2, cPLA2, and iPLA2 (Six and Dennis, 2000; Ma and Turk, 2001).
We purified rat cPLA2γ as a factor causing nuclear shrinkage (Fig. 2). We were successful in purifying cPLA2γ protein from among many PLA2 isotypes for the following reasons: (1) we assayed nuclear shrinkage in the presence of 0.5 mM EGTA to avoid nonspecific activation of Ca2+-dependent proteases, and there are only three known intracellular Ca2+-independent PLA2 genes (iPLA2β, iPLA2γ, and cPLA2γ); (2) MAFP was most the effective inhibitor of liver S100 lysate-induced nuclear shrinkage (Fig. 4 A); and (3) 10 μM MAFP has been shown to completely suppress recombinant human cPLA2γ (Stewart et al., 2002). Thus, cPLA2γ might be most stable or abundant in the rat liver S100 fraction.
Which isotype of the PLA2 family is responsible for nuclear shrinkage in cells? Among several PLA2 inhibitors tested, BEL was the most potent inhibitor of hypoxic nuclear shrinkage in PC12 cells (Fig. 5). Previously, it has been reported that BEL is a potent and selective inhibitor of all iPLA2 isotypes, including iPLA2β, iPLA2γ, and cPLA2γ (Balsinde et al., 1999; Mancuso et al., 2000; Stewart et al., 2002). Here, we showed that reduction of iPLA2β using siRNA could inhibit nuclear shrinkage, and that most iPLA2β underwent translocation to the nucleus during hypoxia. Based on these results, we concluded that iPLA2, but not cPLA2, plays a major role in hypoxic nuclear shrinkage, although we could not exclude the possibility that other PLA2 isoforms may have a role in different circumstances.
PLA2 activation
We found that PLA2 activity was increased and that iPLA2 protein accumulated in the nucleus during hypoxia (Fig. 6 and Fig. 7), changes that probably contributed to nuclear shrinkage. An increase of PLA2 activity and loss of nuclear membrane phospholipids during hypoxia or ischemia has been reported previously (McHowat et al., 1998; Williams et al., 2000; Arai et al., 2001), but the mechanism of PLA2 activation remains to be elucidated. A decrease of ATP may trigger PLA2 activation during hypoxia because the activity of iPLA2 purified from cardiac myocytes was inhibited in the presence of ATP (McHowat et al., 1998), although there is a contradictory report that the activity of recombinant iPLA2β (which has an ATP-binding motif) was augmented in the presence of ATP (Ma et al., 1999). Another possibility is that PLA2 activity remains unchanged, but membrane phospholipids might be modified more susceptible to PLA2.
Cell death
A number of authors have reported that PLA2 inhibitors have a cytoprotective role against various death paradigms (Wissing et al., 1997; Cummings et al., 2000; Arai et al., 2001; Williams and Gottlieb, 2002). However, none of these analyses rigorously assessed whether PLA2 was specifically involved in caspase-dependent cell death (apoptosis) or in caspase-independent cell death. Here, we showed that PLA2 inhibitors could prevent hypoxic cell death in the presence of a pan-caspase inhibitor (Fig. 9), although the pan-caspase inhibitor itself was ineffective. How does PLA2 induce cell death? Activated PLA2 may disrupt the nuclear architecture, thus eventually causing death. Alternatively, PLA2 releases free fatty acids from phospholipid membranes, and these may induce the mitochondrial permeability transition, leading to mitochondrial dysfunction. It is also possible that PLA2 may directly target and disrupt cellular membranes, thus killing cells.
Materials and methods
Reagents
zVAD-fmk and Ac-DEVD-MCA were purchased from Peptide Institute, Inc. BEL and MAFP were obtained from Cayman Chemical. AACOCF3 and PACOCF3 were purchased from Calbiochem. Bee venom PLA2 and AA were obtained from Sigma-Aldrich. Primary antibodies for iPLA2, cPLA2α (N-216), GAPDH, and lamin B1 were from Cayman Chemical, Santa Cruz Biotechnology, Inc., Biogenesis, or Zymed Laboratories, respectively.
Cell culture and hypoxia
PC12 cells transfected with Bcl-2 and the control vector were described previously (Shimizu et al., 1996a). PC12 and HeLa cells were maintained in RPMI 1640 medium with 10% FBS, whereas MEF and CHO-k1 cells were cultured in DME with 10% FBS. 8.0 × 105 cells were seeded in 3.5-cm dishes. After the cells became attached to the dishes, the medium was changed to DME with 2.2 g/l glucose and 10% dialyzed FBS, and the cells were exposed to normoxia or hypoxia at 37°C. Hypoxia was achieved by using a BBL GasPak Plus™ in an anaerobic jar (Becton Dickinson), and the atmosphere was monitored by the white color of indicator strips (BBL). After trypsinization, cells were collected and resuspended in PBS. After staining with 10 μM Hoechst 33342, nuclear changes were assessed under a fluorescence microscope (model BX50; Olympus). Cerebellar granule neurons were prepared as described previously (Tanabe et al., 1997). In brief, the cerebellum was removed from 7-d-old mice, digested with trypsin and DNase I, and cells were plated on poly-d-lysine–coated dishes. After elimination of nonneuronal cells by incubation with cytosine arabinofranoside, cells were grown for 7 d in DME containing 10% horse serum and 25 mM KCl. Cell death was induced by changing the medium to serum-free DME containing 5 mM KCl.
Caspase activity
PC12 cells were harvested and suspended in buffer A as described below. Caspase activity was measured as described previously (Shimizu et al., 1996b).
Cell-free system
HeLa cells were attached to 12-well slide glasses. After washing twice with transportation buffer (TB; 20 mM Hepes-KOH, pH 7.3, 110 mM CH3COOK, 2 mM (CH3COO)2 Mg, and 0.5 mM sodium EGTA), cells were incubated with TB containing 20 μg/μl digitonin and 2 mM DTT for exactly 3 min at RT. After washing twice with TB containing 2 mM DTT, permeabilized cells were incubated with 30 μl cell lysates for 2 h at 37°C. Nuclear changes were assessed under a fluorescence microscope after staining with 10 μM Hoechst 33342. To prepare PC12 cell lysates, cells were cultured in a hypoxic or normoxic atmosphere for 24 h. Then, the cells were harvested and suspended in buffer A (5 mM Hepes-KOH, pH 7.3, 10 mM CH3COOK, 2 mM (CH3COO)2 Mg, 0.5 mM sodium EGTA, and 1 mM DTT) supplemented with 0.1 mM PMSF, 10 μg/ml pepstatin, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Cells were homogenized and sonicated. After centrifugation at 39,000 rpm for 1 h, the supernatant was used for the nuclear shrinkage assay. Mitochondrial supernatant was prepared as described previously (Shimizu et al., 1998). In brief, isolated rat liver mitochondria were incubated with 50 μM Ca2+ for 30 min. After centrifugation at 15,000 rpm for 5 min, the supernatant was used for the nuclear shrinkage assay. The rat liver S100 fraction was prepared as described below.
Purification of nuclear shrinkage factor
All purification steps were performed at 4°C. Livers from 25 male rats were perfused with PBS via the portal vein and were homogenized in 5 vol of buffer A (supplemented with 0.1 mM PMSF, 10 μg/ml pepstatin, 10 μg/ml leupeptin, and 10 μg/ml aprotinin) using a motorized homogenizer. The homogenates were centrifuged at 39,000 rpm for 1 h in a rotor (60Ti; Beckman Coulter) to obtain the supernatant (the S100 lysate). After filtering through a 0.45-μm filter (MILLEX-HA; Millipore), the S100 lysate was loaded onto a 50-ml heparin Sepharose column and the flow-through fractions were pooled. The pooled fractions were loaded onto a 50-ml HiTrap™ Q Sepharose column equilibrated with buffer A, then was eluted with a 600-ml linear salt gradient (0–0.25 M NaCl), and 15-ml fractions were collected. The active fractions (0.16–0.22 M NaCl) were pooled, 1 M ammonium sulfate was added directly, and the precipitate was removed by centrifugation. The resulting supernatant was loaded on a 25-ml butyl Sepharose column, followed by washing with buffer A containing 0.5 M ammonium sulfate and subsequent elution with buffer A. Active fractions (75 ml) were pooled, and directly loaded onto a 10-ml hydroxyapatite column equilibrated with 0.01 M potassium phosphate (pH 6.8), followed by washing with 0.25 M potassium phosphate (pH 6.8) and elution with a 30-ml linear gradient (0.25–1.0 M) of potassium phosphate. The active fractions (7 ml) were directly loaded onto a 5-ml chelating Sepharose column charged with 0.1 M Co2+, and eluted with 0.02 M Na2PO4, pH 7.2, 0.5 M NaCl, and 0.05 M EDTA. The active fractions were concentrated and loaded onto a 24-ml Superdex™ 200 gel filtration column equilibrated with buffer A containing 0.05 M NaCl. Active fractions (1 ml, 15.3 μg) were loaded onto a 0.24-ml Mini Q™ Sepharose column equilibrated with buffer A containing 0.05 M NaCl. Proteins bound to the column was eluted with a 0.9-ml linear NaCl gradient (0.05–0.25 M NaCl), and 50 μl fractions were collected. These fractions (0.075–0.2 M NaCl) were used for nuclear shrinkage analysis and for SDS-PAGE with silver staining. All of the equipment and column resins for purification were purchased from Amersham Biosciences. After lysyl endopeptidase digestion and reverse HPLC, peptide sequences were determined by AproScience Co.
Degenerative PCR
The degenerative PCR primers including inosine (I) were designed from four peptides. The single-letter designation for each combination of nucleotides was as follows: R (A or G), S (G or C), H (A, C, or T), N (A, G, C, or T), Y (C or T), M (A or C), W (A or T), and D (A, G, or T).
The peptides and their primers are listed as follows: peptide 1 D/AVYFATGLQEE: sense primer 1 5′-GMIGTITAYTTYGCIACIGG-3′, sense primer 2 5′-TAYTTYGCIACIGGIYTICARG-3′, antisense primer 1 5′-YTCYTCYTGIARICCIGTIGC-3′, antisense primer 2 5′-ARICCIGTIGCRAARTAIAC-3′; peptide 2 PFPEVSEDQLK: sense primer 1 5′-CCITTYCCIGARGTIWSIGARG-3′, sense primer 2 5′-GAIGTIISIGARGAYCARYT-3′, antisense primer 1 5′-TTIAIYTGRTCYTCISWIAC-3′, antisense primer 2 5′-YTGRTCITCISWIACYTCIGG-3′; peptide 3 QFALGDGTGEF: sense primer 1 5′-CARTTYGCIYTIGGICCIGG-3′, sense primer 2 5′-GCIYTIGGICCIGGNACNGG-3′, antisense primer 1 5′-RAAYTCICCIGTICCIGGNCC-3′, antisense primer 2 5′-CCIGGICCIARIGCRAAYTG-3′; peptide 4 MAEIIQNWNEI: sense primer 1 5′-ATGGCIGARATHATHCARAA-3′, sense primer 2 5′-RATIATICARAAYTGGAAYG-3′, antisense primer 1 5′-DATITCITTCCARTTYTGDA-3′, and antisense primer 2 5′-CARTTITGIATDATYTCNGC-3′
Template cDNA was synthesized from total rat liver RNA using reverse transcriptase and oligo dT. cDNA was amplified with every combination of sense and antisense primer 1 by PCR (94°C for 3 min and 30 cycles of 94°C for 30 s, 45°C for 30 s, and 72°C for 1 min). PCR products were diluted and amplified with each combination of sense and antisense primer 2 under the same conditions. The amplified products were cloned into the pGEM-T vector (Promega), and the nucleotide sequence was determined.
Measurement of the nuclear area
After staining of the nucleus with 10 μM Hoechst 33342, the fluorescence image was recorded using a digital CCD camera (model C303; Olympus). Then the area of each nucleus was measured with Quantity One software (Bio-Rad Laboratories).
PLA2 activity
A mixture of DPPS (1,2-dipalmitoylphosphatidylserine), cholesterol, phosphatidylglycerol, and PED6 (107:31:20:1; a total of 287 nmol) was dried and resuspended in 1 ml PBS, and liposomes were prepared by sonication on ice for 5 min. Then, the liposomes were added to cultures at a concentration of 50 μl/ml of medium and incubation was done for 10 min at 37°C. The cells were washed with PBS, stained with 10 μM Hoechst 33342, and placed on a glass slide. Then, fluorescence was visualized using a confocal microscope (model 510; Carl Zeiss MicroImaging, Inc.). In the experiment shown in Fig. 6 B, cells were treated with the liposome (50 μl/ml) for 1 min and collected by centrifugation. The cells were resuspended in PBS and disrupted by freezing at −80°C and thawing, followed by sonication. Fluorescence intensity was measured using a spectrofluorometer with excitation at 485 nm and emission at 525 nm.
AA release assay
Cells were labeled for 24 h with 0.1 μCi/ml [3H]AA in serum-free DME containing 0.2% (wt/vol) fatty acid-free BSA. After washing three times with DME, the cells were exposed to hypoxia for 36 h. After centrifugation, the radioactivity of the supernatant was measured using a scintillation counter.
LDH release
LDH released into the culture medium was determined using a commercial LDH assay kit (Wako Pure Chemical Industries, Ltd.).
Cell fractionation
CHO-k1 cells exposed to hypoxia or normoxia were homogenized in hypotonic buffer (10 mM Tris-Cl, pH 7.8, 150 mM NaCl, and 1 mM EDTA) containing 0.1% Triton X-100. The lysates were centrifuged at 3,000 rpm for 10 min at 4°C and separated into pellet and supernatant fractions. The pellet fraction was resuspended in hypotonic buffer containing 0.1% Triton X-100, recentrifuged, and used as the nuclear fraction. The supernatant fraction was recentrifuged at 15,000 rpm for 20 min at 4°C and was used as the cytosolic fraction.
Western blot analysis and immunohistochemistry of iPLA2
Immunoblot analysis was performed with an HRP-conjugated secondary antibody using ECL Western blotting detection reagents (Amersham Biosciences). For immunohistochemistry, CHO-k1 cells were cultured in a glass-bottomed chamber. After exposure to hypoxia for 36 h, the cells were fixed with 2% PFA and 0.1% Triton X-100. After washing with PBS, the cells were incubated with polyclonal anti-iPLA2 antiserum, and then with biotin-conjugated anti–rabbit IgG and streptavidin rhodamine. Fluorescence was visualized using a confocal microscope (model 510, Carl Zeiss MicroImaging, Inc.).
siRNA
siRNA for GFP and iPLA2β mRNA was obtained from Dharmacon Research, Inc. The targeted region of GFP and hamster iPLA2β cDNA was 5′-GGCTACGTCCAGGAGCGCACC-3′ and 5′-AACGTGTTCGAGAGGAAGGGC-3′, respectively. Transfection of siRNA into CHO-k1 cells in 6-well dishes was performed with siFECTOR (B-Bridge International, Inc.) according to the supplier's protocol. In brief, 6 μg siFECTOR was diluted in 100 μl Opti-MEM®, mixed with 2, 4, or 6 μg siRNA diluted in 100 μl of Opti-MEM®, and incubated for 15 min. Then, the whole mixture was added to cells in the culture medium (serum-free DME) in one well of a 6-well dish. After 10 h of incubation, 2 ml DME supplemented with 10% FBS was added to each well, and the cells were incubated for another 16 h. This transfection procedure was repeated three times before performing immunoblotting and hypoxia treatment.
Acknowledgments
We are grateful to Dr. Xiaodong Wang (University of Texas Southwestern Medical Center, Dallas, TX) for providing Apaf-1+/+ and −/− MEF, and Drs. Yutaka Eguchi and Shigeomi Shimizu for their helpful advice.
This work was supported in part by a grant to Y. Tsujimoto for Scientific Research on Priority Areas; a grant for Center of Excellence Research; a grant for Scientific Research from the Ministry of Education, Science, Sports, and Culture of Japan; and by Special Coordination Funds for Promoting Science and Technology from the Science and Technology Agency of Japan.
Abbreviations used in this paper: AA, arachidonic acid; AACOCF3, arachidonyltrifluoromethyl ketone; AIF, apoptosis-inducing factor; BEL, bromoenol lactone; cPLA2, cytosolic Ca2+-dependent PLA2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; iPLA2, Ca2+-independent PLA2; LDH; lactate dehydrogenase; MAFP, methyl arachidonyl fluorophosphonate; MEF, mouse embryonic fibroblast; PACOCF3, palmitoyl trifluoromethyl ketone; PED6, (N-((6-(2,4-dinitrophenyl)amino)hexanoyl)-2-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-pentanoyl)-sn-glycero-3-phosphoethanolamine); PLA2, phospholipase A2; siRNA, small interfering RNA; sPLA2, secretory PLA2; STS, staurosporine.
References
- Arai, K., Y. Ikegaya, Y. Nakatani, I. Kudo, N. Nishiyama, and N. Matsuki. 2001. Phospholipase A2 mediates ischemic injury in the hippocampus: a regional difference of neuronal vulnerability. Eur. J. Neurosci. 13:2319–2323. [DOI] [PubMed] [Google Scholar]
- Balsinde, J., M.A. Balboa, P.A. Insel, and E.A. Dennis. 1999. Regulation and inhibition of phospholipase A2. Annu. Rev. Pharmacol. Toxicol. 39:175–189. [DOI] [PubMed] [Google Scholar]
- Bazan, N.G., and E.B. Rodriguez de Turco. 1980. Membrane lipids in the pathogenesis of brain edema: phospholipids and arachidonic acid, the earliest membrane components changed at the onset of ischemia. Adv. Neurol. 28:197–205. [PubMed] [Google Scholar]
- Bonventre, J.V., Z. Huang, M.R. Taheri, E. O'Leary, E. Li, M.A. Moskowitz, and A. Sapirstein. 1997. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 390:622–625. [DOI] [PubMed] [Google Scholar]
- Cecconi, F., G. Alvarez-Bolado, B.I. Meyer, K.A. Roth, and P. Gruss. 1998. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 94:727–737. [DOI] [PubMed] [Google Scholar]
- Chang, L.K., and E.M. Johnson, Jr. 2002. Cyclosporin A inhibits caspase-independent death of NGF-deprived sympathetic neurons: a potential role for mitochondrial permeability transition. J. Cell Biol. 157:771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, E.H., M.C. Wei, S. Weiler, R.A. Flavell, T.W. Mak, T. Lindsten, and S.J. Korsmeyer. 2001. BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell. 8:705–711. [DOI] [PubMed] [Google Scholar]
- Cummings, B.S., J. McHowat, and R.G. Schnellmann. 2000. Phospholipase A2s in cell injury and death. J. Pharmacol. Exp. Ther. 294:793–799. [PubMed] [Google Scholar]
- Edgar, A.D., J. Strosznajder, and L.A. Horrocks. 1982. Activation of ethanolamine phospholipase A2 in brain during ischemia. J. Neurochem. 39:1111–1116. [DOI] [PubMed] [Google Scholar]
- Enari, M., H. Sakahira, H. Yokoyama, K. Okawa, A. Iwamatsu, and S. Nagata. 1998. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 391:43–50. [DOI] [PubMed] [Google Scholar]
- Farber, S.A., M. Pack, S.Y. Ho, I.D. Johnson, D.S. Wagner, R. Dosch, M.C. Mullins, H.S. Hendrickson, E.K. Hendrickson, and M.E. Halpern. 2001. Genetic analysis of digestive physiology using fluorescent phospholipid reporters. Science. 292:1385–1388. [DOI] [PubMed] [Google Scholar]
- Grune, T., K. Muller, S. Zollner, R. Haseloff, I.E. Blasig, H. David, and W. Siems. 1997. Evaluation of purine nucleotide loss, lipid peroxidation and ultrastructural alterations in post-hypoxic hepatocytes. J. Physiol. 498:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi, M., S. Torii, S. Matsuzawa, Z. Xie, S. Kitada, S. Krajewski, H. Yoshida, T.W. Mak, and J.C. Reed. 2000. Apoptotic protease activating factor 1 (Apaf-1)-independent cell death suppression by Bcl-2. J. Exp. Med. 191:1709–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossmann, K.A., U. Oschlies, W. Schwindt, and H. Krep. 2001. Electron microscopic investigation of rat brain after brief cardiac arrest. Acta Neuropathol. (Berl.). 101:101–113. [DOI] [PubMed]
- Kawahara, A., Y. Ohsawa, H. Matsumura, Y. Uchiyama, and S. Nagata. 1998. Caspase-independent cell killing by Fas-associated protein with death domain. J. Cell Biol. 143:1353–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leist, M., and M. Jaattela. 2001. Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2:589–598. [DOI] [PubMed] [Google Scholar]
- Lockshin, R.A., and Z. Zakeri. 2002. Caspase-independent cell deaths. Curr. Opin. Cell Biol. 14:727–733. [DOI] [PubMed] [Google Scholar]
- Ma, Z., and J. Turk. 2001. The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog. Nucleic Acid Res. Mol. Biol. 67:1–33. [DOI] [PubMed] [Google Scholar]
- Ma, Z., X. Wang, W. Nowatzke, S. Ramanadham, and J. Turk. 1999. Human pancreatic islets express mRNA species encoding two distinct catalytically active isoforms of group VI phospholipase A2 (iPLA2) that arise from an exon-skipping mechanism of alternative splicing of the transcript from the iPLA2 gene on chromosome 22q13.1. J. Biol. Chem. 274:9607–9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso, D.J., C.M. Jenkins, and R.W. Gross. 2000. The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A2. J. Biol. Chem. 275:9937–9945. [DOI] [PubMed] [Google Scholar]
- Martin, L.J., N.A. Al-Abdulla, A.M. Brambrink, J.R. Kirsch, F.E. Sieber, and C. Portera-Cailliau. 1998. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain Res. Bull. 46:281–309. [DOI] [PubMed] [Google Scholar]
- McHowat, J., S. Liu, and M.H. Creer. 1998. Selective hydrolysis of plasmalogen phospholipids by Ca2+-independent PLA2 in hypoxic ventricular myocytes. Am. J. Physiol. 274:C1727–C1737. [DOI] [PubMed] [Google Scholar]
- Michiels, C., P. Renard, N. Bouaziz, N. Heck, F. Eliaers, N. Ninane, R. Quarck, P. Holvoet, and M. Raes. 2002. Identification of the phospholipase A2 isoforms that contribute to arachidonic acid release in hypoxic endothelial cells: limits of phospholipase A2 inhibitors. Biochem. Pharmacol. 63:321–332. [DOI] [PubMed] [Google Scholar]
- Miyazaki, K., H. Yoshida, M. Sasaki, H. Hara, G. Kimura, T.W. Mak, and K. Nomoto. 2001. Caspase-independent cell death and mitochondrial disruptions observed in the Apaf1-deficient cells. J. Biochem. (Tokyo). 129:963–969. [DOI] [PubMed]
- Monney, L., I. Otter, R. Olivier, H.L. Ozer, A.L. Haas, S. Omura, and C. Borner. 1998. Defects in the ubiquitin pathway induce caspase-independent apoptosis blocked by Bcl-2. J. Biol. Chem. 273:6121–6131. [DOI] [PubMed] [Google Scholar]
- Okuno, S., S. Shimizu, T. Ito, M. Nomura, E. Hamada, Y. Tsujimoto, and H. Matsuda. 1998. Bcl-2 prevents caspase-independent cell death. J. Biol. Chem. 273:34272–34277. [DOI] [PubMed] [Google Scholar]
- Pickard, R.T., B.A. Strifler, R.M. Kramer, and J.D. Sharp. 1999. Molecular cloning of two new human paralogs of 85-kDa cytosolic phospholipase A2. J. Biol. Chem. 274:8823–8831. [DOI] [PubMed] [Google Scholar]
- Ruchaud, S., N. Korfali, P. Villa, T.J. Kottke, C. Dingwall, S.H. Kaufmann, and W.C. Earnshaw. 2002. Caspase-6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. EMBO J. 21:1967–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudenko, S.V., G.A. Bojok, and E.E. Nipot. 1997. Bee venom-induced shrinkage of erythrocyte ghosts. Biochemistry (Mosc.). 62:104–109. [PubMed]
- Sahara, S., M. Aoto, Y. Eguchi, N. Imamoto, Y. Yoneda, and Y. Tsujimoto. 1999. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature. 401:168–173. [DOI] [PubMed] [Google Scholar]
- Salvesen, G.S., and V.M. Dixit. 1997. Caspases: intracellular signaling by proteolysis. Cell. 91:443–446. [DOI] [PubMed] [Google Scholar]
- Samejima, K., S. Tone, and W.C. Earnshaw. 2001. CAD/DFF40 nuclease is dispensable for high molecular weight DNA cleavage and stage I chromatin condensation in apoptosis. J. Biol. Chem. 276:45427–45432. [DOI] [PubMed] [Google Scholar]
- Sheridan, A.M., A. Sapirstein, N. Lemieux, B.D. Martin, D.K. Kim, and J.V. Bonventre. 2001. Nuclear translocation of cytosolic phospholipase A2 is induced by ATP depletion. J. Biol. Chem. 276:29899–29905. [DOI] [PubMed] [Google Scholar]
- Shimizu, S., Y. Eguchi, W. Kamiike, Y. Itoh, J. Hasegawa, K. Yamabe, Y. Otsuki, H. Matsuda, and Y. Tsujimoto. 1996. a. Induction of apoptosis as well as necrosis by hypoxia and predominant prevention of apoptosis by Bcl-2 and Bcl-XL. Cancer Res. 56:2161–2166. [PubMed] [Google Scholar]
- Shimizu, S., Y. Eguchi, W. Kamiike, H. Matsuda, and Y. Tsujimoto. 1996. b. Bcl-2 expression prevents activation of the ICE protease cascade. Oncogene. 12:2251–2257. [PubMed] [Google Scholar]
- Shimizu, S., Y. Eguchi, W. Kamiike, Y. Funahashi, A. Mignon, V. Lacronique, H. Matsuda, and Y. Tsujimoto. 1998. Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc. Natl. Acad. Sci. USA. 95:1455–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six, D.A., and E.A. Dennis. 2000. The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochim. Biophys. Acta. 1488:1–19. [DOI] [PubMed] [Google Scholar]
- Stasche, R., S. Hinderlich, C. Weise, K. Effertz, L. Lucka, P. Moormann, and W. Reutter. 1997. A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Molecular cloning and functional expression of UDP-N-acetyl-glucosamine 2-epimerase/N-acetylmannosamine kinase. J. Biol. Chem. 272:24319–24324. [DOI] [PubMed] [Google Scholar]
- Stewart, A., M. Ghosh, D.M. Spencer, and C.C. Leslie. 2002. Enzymatic properties of human cytosolic phospholipase A2γ. J. Biol. Chem. 277:29526–29536. [DOI] [PubMed] [Google Scholar]
- Susin, S.A., H.K. Lorenzo, N. Zamzami, I. Marzo, B.E. Snow, G.M. Brothers, J. Mangion, E. Jacotot, P. Costantini, M. Loeffler, et al. 1999. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 397:441–446. [DOI] [PubMed] [Google Scholar]
- Susin, S.A., E. Daugas, L. Ravagnan, K. Samejima, N. Zamzami, M. Loeffler, P. Costantini, K.F. Ferri, T. Irinopoulou, M.C. Prevost, et al. 2000. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 192:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe, H., Y. Eguchi, S. Kamada, J.C. Martinou, and Y. Tsujimoto. 1997. Susceptibility of cerebellar granule neurons derived from Bcl-2-deficient and transgenic mice to cell death. Eur. J. Neurosci. 9:848–856. [DOI] [PubMed] [Google Scholar]
- Underwood, K.W., C. Song, R.W. Kriz, X.J. Chang, J.L. Knopf, and L.L. Lin. 1998. A novel calcium-independent phospholipase A2, cPLA2-γ, that is prenylated and contains homology to cPLA2. J. Biol. Chem. 273:21926–21932. [DOI] [PubMed] [Google Scholar]
- Wang, H., D.C. Harrison-Shostak, J.J. Lemasters, and B. Herman. 1996. Contribution of pH-dependent group II phospholipase A2 to chemical hypoxic injury in rat hepatocytes. FASEB J. 10:1319–1325. [DOI] [PubMed] [Google Scholar]
- Williams, S.D., and R.A. Gottlieb. 2002. Inhibition of mitochondrial calcium-independent phospholipase A2 (iPLA2) attenuates mitochondrial phospholipid loss and is cardioprotective. Biochem. J. 362:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, S.D., F.F. Hsu, and D.A. Ford. 2000. Electrospray ionization mass spectrometry analyses of nuclear membrane phospholipid loss after reperfusion of ischemic myocardium. J. Lipid Res. 41:1585–1595. [PubMed] [Google Scholar]
- Wissing, D., H. Mouritzen, M. Egeblad, G.G. Poirier, and M. Jaattela. 1997. Involvement of caspase-dependent activation of cytosolic phospholipase A2 in tumor necrosis factor-induced apoptosis. Proc. Natl. Acad. Sci. USA. 94:5073–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, J., D.T. Chao, and S.J. Korsmeyer. 1996. BAX-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA. 93:14559–14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, H., Y.Y. Kong, R. Yoshida, A.J. Elia, A. Hakem, R. Hakem, J.M. Penninger, and T.W. Mak. 1998. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 94:739–750. [DOI] [PubMed] [Google Scholar]