

Abstract
Flowing leukocytes roll on P-selectin that is mobilized from secretory granules to the surfaces of endothelial cells after stimulation with histamine or thrombin. Before it is internalized, P-selectin clusters in clathrin-coated pits, which enhances its ability to support leukocyte rolling. We found that thrombin and histamine induced comparable exocytosis of P-selectin on endothelial cells. However, compared with histamine, thrombin decreased the recruitment of P-selectin into clathrin-coated pits, slowed the internalization of P-selectin, and reduced the number and stability of neutrophils rolling on P-selectin. Significantly more RhoA was activated in thrombin- than in histamine-stimulated endothelial cells. Inhibitors of RhoA or its effector, Rho kinase, reversed thrombin's ability to inhibit the internalization and adhesive function of P-selectin in endothelial cells. Experiments with transfected cells confirmed that the inhibitory actions of thrombin and Rho kinase on P-selectin required its cytoplasmic domain. Thus, a signaling event affects both the function and clearance of a protein that enters the constitutive clathrin-mediated endocytic pathway.
Keywords: selectin; endocytosis; endothelial cell; thrombin; RhoA
Introduction
Lymphocyte homing and leukocyte trafficking into inflamed tissues require that cells first tether to and roll on vascular surfaces. Interactions of selectins with cell surface glycoconjugates mediate rolling, which is a prerequisite for subsequent integrin-dependent firm adhesion and transendothelial migration (Vestweber and Blanks, 1999; McEver, 2002). L-selectin, expressed on leukocytes, binds to ligands on lymph node endothelial cells and on other leukocytes. P- and E-selectin, expressed on activated platelets or endothelial cells, bind to ligands on leukocytes, platelets, and some endothelial cells. The membrane-distal C-type lectin domain of each selectin recognizes glycans with terminal sialylated and fucosylated components. The dominant leukocyte ligand for P- and L-selectin is P-selectin glycoprotein ligand-1 (PSGL-1), a homodimeric mucin (McEver, 2001).
Rolling properties depend on the densities of selectins and their ligands and on the intrinsic kinetic and mechanical properties of the bonds that they form (McEver, 2001, 2002). Rolling is further modulated by cellular features, which may include extrusion of thin membrane tethers that reduce force on bonds (Schmidtke and Diamond, 2000; Yago et al., 2002). The cell surface distributions of selectins and their ligands also affect rolling under flow. For example, both P-selectin and PSGL-1 form dimers, which favor formation of dimeric bonds that enhance tether duration and strength (Ramachandran et al., 2001). Clustering of L-selectin or PSGL-1 through cytoskeletal interactions may promote multivalent interactions that further stabilize rolling (Dwir et al., 2001; Snapp et al., 2002).
Physiological inflammation requires that leukocyte adhesion be initiated and then terminated on demand. A key control is the regulated expression of P- and E-selectin (McEver, 1997; Vestweber and Blanks, 1999). Inflammatory cytokines such as TNF-α and interleukin-1 β cause endothelial cells to transiently synthesize E-selectin, which is transported directly to the cell surface. Megakaryocytes and endothelial cells constitutively synthesize P-selectin, but it is stored in membranes of α granules in platelets and Weibel-Palade bodies in endothelial cells. During injury or infection, mediators such as thrombin or histamine induce rapid mobilization of P-selectin to the cell surface. P- and E-selectin are cleared from the endothelial cell surface by endocytosis in clathrin-coated pits (von Asmuth et al., 1992; Setiadi et al., 1995, 1998). Both proteins recycle to the cell surface, although they move relatively early from endosomes to lysosomes for degradation (Green et al., 1994). P-selectin also recycles from endosomes to the TGN, where it is incorporated into new Weibel-Palade bodies (Subramaniam et al., 1993). The internalization of P-selectin is particularly rapid (Setiadi et al., 1995). Wild-type P-selectin clusters in coated pits of transfected CHO cells, whereas P-selectin with deletions or substitutions in the cytoplasmic domain that impair internalization does not cluster (Setiadi et al., 1998). Significantly, leukocytes roll in greater numbers and with more regular motions on transfected cells expressing wild-type P-selectin than on cells expressing equal densities of internalization-defective P-selectin (Setiadi et al., 1998). Therefore, recruitment to clathrin-coated membrane microdomains enhances the adhesive function of P-selectin before it enters the cell.
Like that of the transferrin receptor or the low density lipoprotein receptor, the clathrin-mediated endocytosis of P-selectin is constitutive (Setiadi et al., 1995). Ligand engagement is not required to initiate receptor uptake, as is necessary for tyrosine kinase receptors and G protein–coupled receptors (McPherson et al., 2001). Nevertheless, some evidence suggests that extracellular signals may regulate the constitutive clathrin-mediated pathway. Binding of NGF to its tyrosine kinase receptor, TrkA, enhances clathrin-mediated endocytosis of transferrin (Beattie et al., 2000). The stress-induced activation of p38 MAPK accelerates endocytosis by stimulating formation of the guanyl nucleotide dissociation inhibitor–Rab5 complex (Cavalli et al., 2001). In contrast, microinjection of activated forms of the small G protein Rho or Rac inhibits endocytosis of the transferrin and low density lipoprotein receptors (Lamaze et al., 1996; Hrboticky et al., 2002). Whether physiological or pathological signals use activation of Rho or Rac to diminish endocytosis has not been studied.
Thrombin and histamine stimulate endothelial cells through their respective G protein–coupled receptors (Coughlin, 2001; Schneider et al., 2002). Ligand engagement propagates multiple signaling cascades, including activation of p38 and RhoA (Essler et al., 1998; Vouret-Craviari et al., 1998; Marin et al., 2001; Wojciak-Stothard et al., 2001; Kaur et al., 2003). We asked whether histamine or thrombin, after mobilizing P-selectin to the endothelial cell surface, might also modulate its recruitment into clathrin-coated pits. We observed that thrombin, but not histamine, retarded clathrin-mediated endocytosis of P-selectin through activation of RhoA and its effector, Rho kinase. Reduced clustering of P-selectin in coated pits of thrombin-activated cells was associated with less effective support of leukocyte rolling under flow.
Results
Thrombin stimulation of human endothelial cells reduces the internalization rate of P-selectin and the number of neutrophils rolling on P-selectin
Human umbilical vein endothelial cells (HUVEC) treated with optimal concentrations of histamine or thrombin mobilized equivalent levels of P-selectin from Weibel-Palade bodies to the cell surface (Fig. 1 A). However, cell surface P-selectin was internalized much slower in thrombin- than in histamine-stimulated HUVEC (Fig. 1 B). This suggests that histamine-stimulated cells rapidly recycle P-selectin from endosomes to the cell surface to compensate for the higher internalization rate. HUVEC treated with both histamine and thrombin exhibited the same slow internalization rate as cells treated only with thrombin. This indicates that thrombin signaling impairs internalization even in cells stimulated with histamine. The internalization rate of P-selectin was equivalent in HUVEC treated with thrombin or with a thrombin receptor-activating peptide (TRAP; Fig. 1 B). Thus, thrombin acted directly through the dominant G protein–coupled receptor in HUVEC, protease-activated receptor 1 (Coughlin, 2001).
Figure 1.

Thrombin stimulation of HUVEC reduces the internalization rate of P-selectin and the number of neutrophils rolling on P-selectin. (A) HUVEC were either unstimulated (time 0) or were stimulated with thrombin or histamine for 4, 9, or 14 min. The cells were rapidly chilled, and the steady-state levels of P-selectin on the cell surface were measured by specific binding of a saturating concentration of 125I-labeled mAb S12. The data represent the mean ± SEM of three experiments. (B) HUVEC were stimulated with histamine, thrombin, TRAP, or both thrombin and histamine. After 4 min, the internalization rate of P-selectin was determined by the ability of acidic buffer to remove surface-bound 125I-mAb G1 as described in Materials and methods. The cell-bound radioactivity remaining at each time point represents the amount of internalized P-selectin, and is plotted as a percentage of the initial cell-bound radioactivity. The data represent the mean ± SEM of at least four experiments. The internalization rate determined from the initial slope was significantly higher for HUVEC stimulated with histamine than for HUVEC stimulated with thrombin or with thrombin plus histamine (P < 0.02). (C) Neutrophils were perfused over confluent HUVEC. At the indicated time, histamine, thrombin, TRAP, or both thrombin and histamine was perfused into the flow chamber. The number of rolling neutrophils was measured at 1-min intervals. The data represent the mean ± SEM of 10 experiments. At each time point after addition of agonist, the number of rolling neutrophils was significantly greater on HUVEC stimulated with histamine than on HUVEC stimulated with thrombin or with thrombin plus histamine (P < 0.05). (D) HUVEC were stimulated with histamine or thrombin, and neutrophils were immediately perfused into the flow chamber at the indicated wall shear stress. After 5 min, the number of rolling neutrophils was measured. The data represent the mean ± SEM of at least four experiments. (E) HUVEC were stimulated with histamine or thrombin, and neutrophils were immediately perfused into the flow chamber at 1 dyn/cm2 in the presence or absence of the indicated mAb. After 5 min, the number of rolling neutrophils was measured. The data represent the mean ± SEM of at least seven experiments. (F) Neutrophils incubated with histamine or thrombin were perfused over adsorbed sP-selectin (60 sites/μm2) in the presence or absence of the indicated mAb. After 5 min, the number of rolling neutrophils was measured. The data represent the mean ± SEM of three experiments. Significant p-values as measured by the unpaired t test are indicated.
The similar densities of P-selectin on histamine- and thrombin-stimulated HUVEC allowed us to ask whether differential signaling affected the adhesive function of P-selectin at matched surface densities. Flowing neutrophils rapidly began to roll on HUVEC after stimulation with histamine, thrombin, or TRAP (Fig. 1 C). However, significantly more neutrophils rolled on histamine- than on thrombin- or TRAP-stimulated HUVEC during the first 10 min after addition of agonist (Fig. 1 C) and at later intervals (unpublished data). The reduced number of neutrophils rolling on thrombin-activated endothelium did not result from thrombin-mediated proteolytic damage of the endothelial cell surface because fewer neutrophils also rolled on TRAP-activated HUVEC. More neutrophils rolled on histamine- than on thrombin-stimulated HUVEC over a range of wall shear stresses (Fig. 1 D). The number of neutrophils rolling on HUVEC treated with both histamine and thrombin was similar to that on HUVEC treated only with thrombin (Fig. 1 C). This suggests a dominant effect of thrombin on inhibiting rolling even in HUVEC treated with histamine. Rolling on both histamine- and thrombin-stimulated HUVEC was mediated by interactions of PSGL-1 on neutrophils with P-selectin on HUVEC, because anti–PSGL-1 mAb PL1 or anti–P-selectin mAb G1 eliminated rolling (Fig. 1 E). Furthermore, anti–β2 integrin mAb IB4 did not affect the number of cells rolling on histamine- or thrombin-activated HUVEC (Fig. 1 E). Importantly, equal numbers of neutrophils rolled on purified recombinant soluble P-selectin (sP-selectin) in the presence of histamine or thrombin (Fig. 1 F). This result demonstrates that the differential effects of histamine and thrombin on rolling resulted from signaling in endothelial cells rather than in neutrophils.
To compare the stability of neutrophil rolling on histamine- and thrombin-activated HUVEC, we tracked the displacements of neutrophils between successive video frames. Each displacement was divided by the time interval of 0.033 s to derive the velocity. Fig. 2 A shows the velocity at each frame for a representative neutrophil rolling on histamine- or thrombin-activated HUVEC in the presence or absence of anti–β2 integrin mAb IB4. The neutrophil rolling on histamine-activated HUVEC exhibited smaller fluctuations in velocity than the neutrophil rolling on thrombin-activated HUVEC. Although treatment with IB4 did not change the total number of neutrophils rolling on HUVEC (Fig. 1 E), it caused greater velocity fluctuations (Fig. 2 A), which is consistent with a previously demonstrated contribution of β2 integrins to rolling stability on activated endothelial cells (Jones et al., 1993; Jung et al., 1998; Dunne et al., 2002). Even in the presence of IB4, greater velocity fluctuations were observed for the neutrophil rolling on thrombin-activated HUVEC than for the neutrophil rolling on histamine-activated HUVEC. To quantify the rolling behavior, the frame by frame velocity data were used to calculate the mean velocity and the variance in velocity for each cell as it rolled for a period up to 5 s. The pooled data from at least 20 cells were used to calculate the mean velocity and variance of velocity for a cell population. The variance is a quantitative measure of the irregularities in rolling velocities. Higher variances correlate with greater sensitivity to detachment by increasing shear stress and with shorter rolling periods before detachment (Setiadi et al., 1998; Ramachandran et al., 2001; Yago et al., 2002). Neutrophils rolled with significantly greater mean velocities and with significantly greater variances of velocity on thrombin- than on histamine-activated HUVEC in the presence or absence of IB4 (Fig. 2, B and C). These data demonstrate that neutrophils roll less stably and more irregularly on P-selectin expressed on thrombin- than on histamine-activated HUVEC. This difference is maintained even after eliminating the contribution of β2 integrins to slowing rolling.
Figure 2.

Neutrophils roll less stably on thrombin- than on histamine-stimulated HUVEC. (A) Frame by frame velocities of representative neutrophils rolling on thrombin- or histamine-stimulated HUVEC at 1 dyn/cm2 in the presence or absence of anti–β2 integrin mAb IB4. (B and C) Mean velocities and variances of velocities for neutrophil populations rolling on thrombin- or histamine-activated HUVEC at 1 dyn/cm2 in the presence or absence of IB4. Significant p-values as measured by the unpaired t test are indicated. The data represent the mean ± SEM for 20–50 cells, each measured for up to 5 s.
Less P-selectin colocalizes with α-adaptin, a component of clathrin-coated pits, on thrombin-activated HUVEC than on histamine-activated HUVEC
Membrane proteins must be recruited into clathrin-coated pits before they undergo clathrin-mediated endocytosis (Conner and Schmid, 2003). In transfected CHO cells, a subset of P-selectin molecules colocalizes with α-adaptin, a subunit of the AP2 complex of clathrin-coated pits (Setiadi et al., 1998). The lower internalization rate of P-selectin in thrombin- than in histamine-activated HUVEC suggested that fewer P-selectin molecules might cluster in clathrin-coated pits of thrombin-activated endothelial cells at steady state. To address this possibility, we used dual-label immunofluorescence confocal microscopy to examine the colocalization of cell surface P-selectin with α-adaptin (Fig. 3 A). After the clathrin-coated pit buds to form a vesicle, α-adaptin dissociates as the vesicle loses its coat. Therefore, the total pool of α-adaptin is distributed between clathrin-coated pits and the cytoplasm. Green or red staining illustrated the respective distribution of P-selectin or α-adaptin. In HUVEC stimulated with histamine, there was partial colocalization of α-adaptin with P-selectin, as manifested by the punctate yellow staining in the merged images. In HUVEC stimulated with thrombin, there was much less colocalization. Quantification in multiple cells of the percentage of P-selectin pixels that colocalized with α-adaptin pixels confirmed the conclusions from the representative images (Fig. 3 B). Significantly more P-selectin colocalized with α-adaptin in histamine-activated HUVEC than in thrombin-activated HUVEC. Incubation of HUVEC in hypertonic medium, which reversibly disrupts clathrin-coated lattices (Heuser and Anderson, 1989), abrogated the observed colocalization, confirming that it represented colocalization in clathrin-coated pits (Fig. 3 B). Furthermore, P-selectin did not significantly colocalize with caveolin-1, a component of caveolae, in histamine- or thrombin-activated HUVEC. These results demonstrate that less P-selectin clusters in clathrin-coated pits of thrombin- than of histamine-activated HUVEC, consistent with the lower internalization rate in thrombin-activated cells.
Figure 3.


Less cell surface P-selectin colocalizes with α-adaptin in thrombin- than in histamine-stimulated HUVEC. HUVEC preincubated with isotonic or hypertonic medium were stimulated with thrombin or histamine for 4 min. They were fixed and incubated with biotinylated polyclonal antibodies to P-selectin, followed by streptavidin conjugated to Alexa-488. After permeabilization, the cells were incubated with an mAb to α-adaptin or to caveolin-1, followed by donkey anti–mouse Ig conjugated to Cy-3.Using a confocal microscope, an optical section at the middle and apical portion of each cell was examined for staining of P-selectin (green) or α-adaptin or caveolin-1 (red). (A) Representative images revealed partial colocalization of α-adaptin with P-selectin (yellow) in histamine-stimulated HUVEC but less colocalization in thrombin-stimulated HUVEC. (B) The degree of colocalization was quantified by measuring the percentage of green pixels (P-selectin) that colocalized with red pixels (α-adaptin or caveolin-1) as described in Materials and methods. The data represent the mean ± SEM of at least three experiments, with at least five cells counted in each experiment. Significant p-values as measured by the unpaired t test are indicated.
Disruption of clathrin-coated pits with hypertonic medium decreases rolling of selectin ligand-coupled microspheres on both histamine- and thrombin-activated HUVEC
In transfected CHO cells, disruption of clathrin-coated pits with hypertonic medium reversibly blocks endocytosis of P-selectin and significantly impairs neutrophil rolling on P-selectin (Setiadi et al., 1998). The lesser clustering of P-selectin in clathrin-coated pits of thrombin- than of histamine-activated HUVEC might explain the reduced ability of thrombin-activated HUVEC to support neutrophil rolling. If so, the differences in rolling on thrombin- and histamine-activated HUVEC should disappear after disrupting clathrin-coated pits. To test this hypothesis, thrombin- or histamine-activated HUVEC were treated with hypertonic or isotonic medium. Hypertonic medium blocked internalization of P-selectin in activated HUVEC for prolonged periods (Fig. 4 A). Neutrophil rolling could not be measured in hypertonic medium because it altered the shape and rolling characteristics of neutrophils. Therefore, we used rigid microspheres bearing 2-GSP-6, a glycosulfopeptide modeled after the P-selectin–binding region of PSGL-1 (Yago et al., 2002). These microspheres roll on P-selectin, although they roll less stably on matched P-selectin densities than neutrophils do. Microspheres roll less stably because they lack cellular features that favor bond formation and prolongation of tether lifetimes; these features may include deformation at the contact zone and extrusion of membrane tethers (Yago et al., 2002). Ligand-coupled microspheres can be used as sensitive indicators of how alterations in selectin density or distribution affect rolling adhesion. Like neutrophils, more 2-GSP-6–coupled microspheres rolled on histamine- than on thrombin-activated HUVEC maintained in isotonic medium (Fig. 4 B). Anti–P-selectin mAb G1 blocked microsphere rolling, confirming its specificity (unpublished data). Strikingly, virtually no microspheres rolled on histamine- or thrombin-activated HUVEC maintained in hypertonic medium (Fig. 4 B). These results demonstrate that disruption of clathrin-coated pits on activated HUVEC impairs the ability of P-selectin to support rolling of ligand-coupled microspheres under flow, and suggest that the diminished neutrophil rolling on thrombin-activated HUVEC results from thrombin-impaired clustering of P-selectin in clathrin-coated pits.
Figure 4.
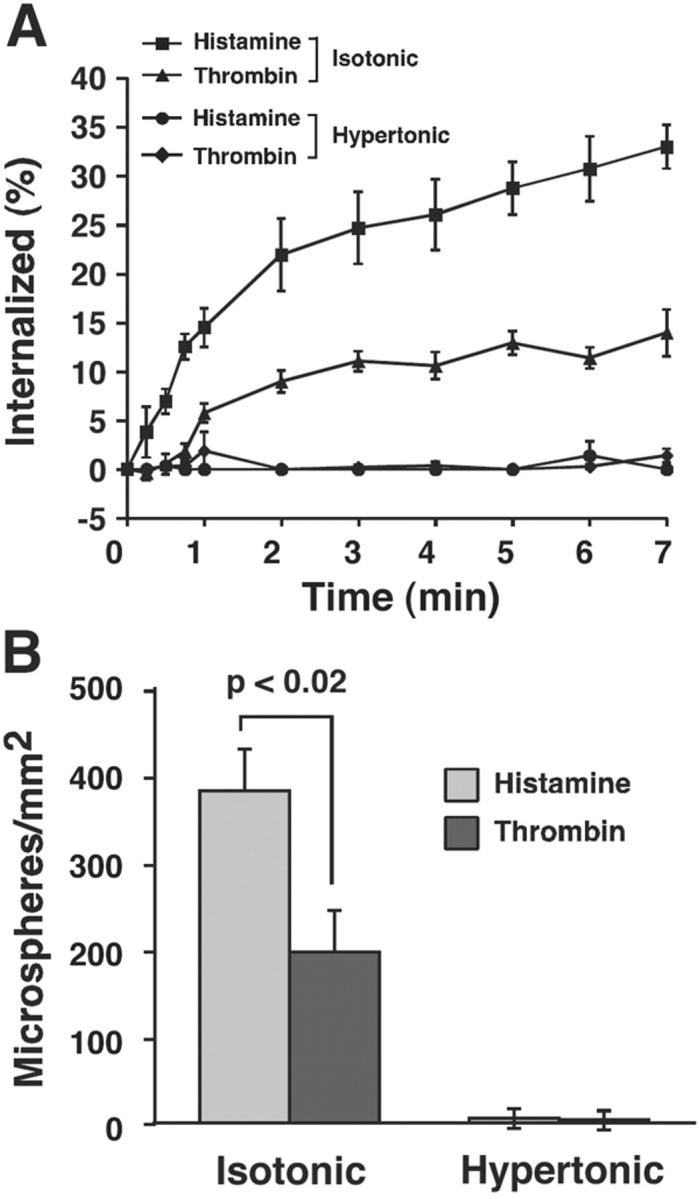
Disruption of clathrin-coated pits with hypertonic medium decreases rolling of selectin ligand-coupled microspheres on both histamine- and thrombin-activated HUVEC. HUVEC preincubated with isotonic or hypertonic medium for 15 min were stimulated with thrombin or histamine for 4 min. The cells were maintained in the same buffer. The internalization rate of P-selectin (A) or the number of rolling 2-GSP-6–coupled microspheres (B) was measured. The data represent the mean ± SEM of at least three experiments. Significant p-values as measured by the unpaired t test are indicated.
Thrombin-stimulated HUVEC activate more RhoA but not more p38 than histamine-stimulated HUVEC
The ability of thrombin to reduce internalization of P-selectin in histamine-treated HUVEC suggests that thrombin engagement of its receptor propagates a signal that impairs clathrin-mediated endocytosis. Both histamine and thrombin activate p38 and RhoA in HUVEC and other cells (Essler et al., 1998; Vouret-Craviari et al., 1998; Marin et al., 2001; Wojciak-Stothard et al., 2001; Kaur et al., 2003). In some cells, activated p38 MAPK augments clathrin-mediated endocytosis (Cavalli et al., 2001), whereas activated RhoA suppresses endocytosis (Lamaze et al., 1996; Hrboticky et al., 2002). To determine whether either signaling molecule was a candidate to explain the differential internalization rate of P-selectin, we measured the relative activation of p38 or RhoA in HUVEC stimulated with histamine or thrombin, normalized to that observed in untreated HUVEC. Representative immunoblots revealed that histamine- and thrombin-treated HUVEC generated equivalent amounts of activated phospho-p38 (Fig. 5 A), whereas thrombin-treated HUVEC activated more RhoA than histamine-treated HUVEC (Fig. 5 C). Quantification of multiple experiments revealed that stimulation with thrombin or histamine only modestly increased phospho-p38 in HUVEC (Fig. 5 B). In marked contrast, thrombin-treated HUVEC produced significantly more activated RhoA than histamine-activated HUVEC did (Fig. 5 D). The toxin exoenzyme C3 transferase (C3T) from Clostridium botulinum inhibits RhoA activation by ADP-ribosylating its GTP-binding site (Wilde et al., 2000). Treatment of HUVEC with C3T partially inhibited the thrombin-mediated increase in activated RhoA, although the level of inhibition did not reach statistical significance (Fig. 5, C and D).
Figure 5.


Thrombin-stimulated HUVEC activate more RhoA but not more p38 than histamine-stimulated HUVEC. (A) Confluent HUVEC were untreated or stimulated with thrombin or histamine for 5 min. Cell lysates were subjected to Western blotting with antibodies to p38 MAPK or to phospho-p38 MAPK. A representative immunoblot from one experiment is depicted. For clarity, only the relevant bands on the blot are shown. (B) Densitometry of stained protein bands was used to quantify the levels of total p38 and phospho-p38. The percentage of phospho-p38 relative to total p38 in stimulated HUVEC was normalized to that of untreated cells and expressed as relative p38 activity. The data represent the mean ± SEM of three experiments. No statistically significant differences (P < 0.05 with unpaired t test) among treatment groups were observed. (C) Confluent HUVEC preincubated with or without 2.5 μg/ml C3T for 16 h were stimulated with thrombin or histamine for 10 min. The amount of activated and total RhoA in cell lysates was measured as described in Materials and methods. A representative immunoblot from one experiment is depicted. For clarity, only the relevant bands on the blot are shown. (D) The percentage of activated RhoA relative to total RhoA in stimulated HUVEC was normalized to that of untreated cells and expressed as relative RhoA activity. The data represent the mean ± SEM of five experiments. Significant p-values as measured by the unpaired t test are indicated.
Inhibiting Rho kinase in thrombin-stimulated HUVEC increases the internalization rate of P-selectin and the number of neutrophils rolling on P-selectin
The higher levels of RhoA generated in thrombin-activated HUVEC might inhibit clustering of P-selectin into clathrin-coated pits and thereby decrease its adhesive function under flow. To test this possibility, we treated HUVEC with the RhoA inhibitor C3T or with the small molecule Y27632, which inhibits Rho kinase, a downstream effector of RhoA (Ishizaki et al., 2000). C3T did not affect endocytosis of P-selectin in histamine-stimulated HUVEC. It accelerated endocytosis of P-selectin in thrombin-stimulated HUVEC but not to the level observed in histamine-stimulated HUVEC (Fig. 6 A). The effect was most pronounced during the first minute of internalization. Y27632 did not affect the internalization rate of P-selectin in histamine-stimulated HUVEC, but it accelerated internalization of P-selectin in thrombin-stimulated HUVEC to the level observed in histamine-stimulated HUVEC (Fig. 6 B). Y27632 increased the number of neutrophils rolling on thrombin-activated HUVEC to the levels seen on histamine-activated HUVEC (Fig. 6 C). Y27632 also stabilized rolling as measured by a reduction in mean rolling velocity and in the variance of rolling velocity (unpublished data). C3T did not increase the number of neutrophils rolling on thrombin-activated HUVEC (Fig. 6 C), but it did reduce the mean rolling velocity (Fig. 6 D) and the variance of rolling velocity (Fig. 6 E). The more modest effect of C3T probably reflects its failure to completely inhibit RhoA activation (Fig. 5 D). These data demonstrate that the thrombin-generated activation of RhoA in HUVEC impairs both the internalization rate of P-selectin and the adhesive function of P-selectin, suggesting a causal relationship.
Figure 6.

Inhibiting Rho kinase in thrombin-stimulated HUVEC increases the internalization rate of P-selectin and the number of neutrophils rolling on P-selectin. (A) HUVEC preincubated with or without 2.5 μg/ml C3T for 16 h were stimulated with histamine or thrombin. After 4 min, the internalization rate of P-selectin was measured. The internalization rate determined from the initial slope was significantly higher for HUVEC stimulated with thrombin in the presence of C3T than for HUVEC stimulated with thrombin in the absence of C3T (P < 0.02). (B) HUVEC preincubated with or without 10 μM Y27632 for 1 h were stimulated with histamine or thrombin. After 4 min, the internalization rate of P-selectin was measured. The internalization rate determined from the initial slope was significantly higher for HUVEC stimulated with thrombin in the presence of Y27632 than for HUVEC stimulated with thrombin in the absence of Y27632 (P < 0.01). (C) HUVEC preincubated with or without C3T or Y27632 were stimulated with histamine or thrombin. After 5 min, the number of rolling neutrophils was measured. (D and E) HUVEC preincubated with or without C3T or Y27632 were stimulated with thrombin. After 5 min, the mean rolling velocity and the variance of velocity of neutrophils was measured. For all panels, the data represent the mean ± SEM of at least three experiments.
To test more directly the link between the RhoA-mediated decrease in the adhesive function of P-selectin and the RhoA-mediated decreased recruitment of P-selectin into clathrin-coated pits, we studied the effects of thrombin on transfected CHO cells expressing matched densities of wild-type P-selectin or of tail-less P-selectin. The latter protein contains only first seven amino acids of the 35-residue cytoplasmic domain and is not recruited into clathrin-coated pits (Setiadi et al., 1995). CHO cells express thrombin receptors, as manifested by their ability to generate Ca2+ fluxes when exposed to thrombin (unpublished data). As in HUVEC, thrombin stimulation of transfected CHO cells diminished the internalization rate of wild-type P-selectin. The Rho kinase inhibitor Y27632 reversed this effect of thrombin (Fig. 7 A). More neutrophils rolled on transfected CHO cells expressing wild-type P-selectin than tail-less P-selectin (Fig. 7 B), confirming previous observations (Setiadi et al., 1998). Anti–P-selectin mAb G1 or anti–PSGL-1 mAb eliminated rolling, demonstrating its specificity (unpublished data). As in HUVEC, thrombin stimulation of transfected CHO cells diminished neutrophil rolling on wild-type P-selectin, whereas Y27632 reversed this effect of thrombin. In contrast, thrombin stimulation did not further diminish neutrophil rolling on CHO cells expressing tail-less P-selectin. These results demonstrate that RhoA inhibits the adhesive function of P-selectin only if it contains its cytoplasmic domain, which is required for clathrin-mediated endocytosis. Therefore, the RhoA-mediated decreased recruitment of P-selectin into clathrin-coated pits is likely to impair directly the ability of P-selectin to mediate leukocyte rolling under flow.
Figure 7.

Thrombin- and Rho kinase–mediated inhibition of the internalization rate and adhesive function of P-selectin requires the cytoplasmic domain of P-selectin. Transfected CHO cells expressing matched densities of wild-type or tail-truncated P-selectin (50–60 sites/μm2) were preincubated with or without 10 μM Y27632 for 1 h and treated with 4 U/ml thrombin for 4 min. (A) The internalization rate of P-selectin was measured. The internalization rate determined from the initial slope was significantly higher for CHO cells stimulated with thrombin in the presence of Y27632 than for CHO cells stimulated with thrombin in the absence of Y27632 (P < 0.05). (B) The number of rolling neutrophils was measured. (A and B) The data represent the mean ± SEM of at least five experiments. Significant p-values as measured by the unpaired t test are indicated.
Discussion
The regulated expression of P-selectin on endothelial cells serves to initiate and to limit the inflammatory response. Mediators such as thrombin or histamine mobilize P-selectin from Weibel-Palade bodies to the cell surface, where it supports the initial rolling of leukocytes on the vessel wall. P-selectin is cleared from the cell surface by clathrin-mediated endocytosis and by trafficking of P-selectin from endosomes to lysosomes for degradation or to the TGN for reincorporation into Weibel-Palade bodies. Here, we show that two distinct inflammatory signals differentially affect the recruitment of P-selectin into clathrin-coated pits. This changes not only the internalization rate of P-selectin but also the ability of cell surface P-selectin to mediate leukocyte rolling under flow.
The internalization rate of P-selectin was significantly lower in thrombin- than in histamine-stimulated HUVEC. Thrombin directly inhibited endocytosis because it diminished the internalization rate of P-selectin in HUVEC costimulated with histamine and in transfected CHO cells. The reduced endocytic rate correlated with reduced colocalization of P-selectin with α-adaptin, a component of clathrin-coated pits. This suggests that thrombin impairs the initial recruitment of P-selectin into these pits. In transfected cells, clustering of P-selectin in coated pits augments its ability to support leukocyte rolling under flow (Setiadi et al., 1998). Fewer neutrophils rolled on thrombin-stimulated HUVEC or transfected CHO cells, and the neutrophils that rolled did so with less uniform motions. The thrombin-induced decrease in P-selectin clustering appeared to be directly responsible for the impaired neutrophil rolling on P-selectin. Disruption of clathrin-coated pits with hypertonic medium markedly inhibited rolling of 2-GSP-6–coupled microspheres on both histamine- and thrombin-stimulated HUVEC. Furthermore, thrombin did not further decrease neutrophil rolling on CHO cells expressing P-selectin that lacked the cytoplasmic sequences required for recruitment into coated pits. Thrombin signaling reduced but did not eliminate recruitment of P-selectin into clathrin-coated pits, whereas hypertonic medium completely blocked recruitment. Thrombin also impaired the adhesive function of P-selectin, but not as severely as truncation of the cytoplasmic domain of P-selectin or disruption of clathrin-coated pits by hypertonic medium. Clathrin-mediated clustering of P-selectin likely cooperates with cytoskeletal-mediated clustering of PSGL-1 on leukocytes (Moore et al., 1995; Snapp et al., 2002) and with molecular self-associations that drive dimerization of P-selectin and of PSGL-1 (Ramachandran et al., 2001). The local cell surface concentrations of selectins and ligands, especially on cells where global densities are not high, might increase bond numbers in tethers and thus increase the lifetimes of the tethers. Signal-mediated alterations in recruitment of P-selectin into coated pits provide an additional mechanism to modulate the tether lifetimes and rolling stabilities of leukocytes.
Growth factor binding to some receptor tyrosine kinases increases recruitment of clathrin and AP2 adaptors to the plasma membrane and accelerates the constitutive internalization of the transferrin receptor (Beattie et al., 2000). The signaling pathways for this process include Src-dependent phosphorylation of the clathrin heavy chain and rapid activation of Rab5, a small GTPase that increases clathrin-mediated internalization (Wilde et al., 1999; Barbieri et al., 2000). In contrast, thrombin engagement of its G protein–coupled receptor slowed the constitutive, clathrin-mediated internalization of P-selectin. This signaling pathway involved activation of RhoA, which extends previous observations that microinjection of active RhoA inhibits clathrin-mediated endocytosis of the transferrin and low density lipoprotein receptors (Lamaze et al., 1996; Hrboticky et al., 2002). The net balance of signals received by a cell might be critical for determining the efficiency of clathrin-mediated endocytosis in the constitutive pathway. Different members of the Rho GTPase family regulate each other's activities. In fibroblasts, one striking mechanism is the Rac-mediated generation of reactive oxygen species that inhibit Rho activity (Nimnual et al., 2003). Thrombin signals endothelial cells to activate Rac1 (Vouret-Craviari et al., 1998) and to generate reactive oxygen species (Takano et al., 2002), but these are apparently not made in sufficient quantities to inhibit RhoA. However, ischemia-reperfusion injury and other inflammatory responses generate reactive oxygen species that mobilize P-selectin to the cell surface (Johnston et al., 1996; Akgur et al., 2000). Perhaps higher levels of these reactive oxygen species inhibit RhoA; this might allow more P-selectin to cluster into clathrin-coated pits, augmenting its adhesive activity. Stress-activated inflammatory signals also activate p38 MAPK, which accelerates clathrin-mediated endocytosis through a Rab5-dependent mechanism (Cavalli et al., 2001). Thrombin and histamine both activate p38 in endothelial cells (Marin et al., 2001; Kaur et al., 2003), but the greater activation of RhoA in thrombin-stimulated cells appears to counter any p38-mediated acceleration of clathrin-mediated endocytosis. This is consistent with the failure of p38 MAPK inhibitors to reduce P-selectin–dependent neutrophil rolling on thrombin-activated HUVEC (Kaur et al., 2003). Although RhoA impairs P-selectin-mediated rolling of leukocytes on endothelial cells, it augments subsequent leukocyte adhesion-dependent interactions of integrin ligands with the cytoskeleton (Wojciak-Stothard et al., 1999). These combined data suggest that an interplay of signals regulates the cell surface distributions and functions of adhesion molecules on the vascular surface.
The mechanism by which RhoA inhibits clathrin-mediated endocytosis of P-selectin and other proteins is not known. The effector Rho kinase is involved, because the Rho kinase inhibitor Y27632 blocked the thrombin inhibition of endocytosis of P-selectin. The best characterized actions of Rho, Rac, and cdc42 GTPases are to modulate the actin-based cytoskeleton (Etienne-Manneville and Hall, 2002). Rho, through Rho kinase, promotes actin–myosin interactions that develop stress fibers. Rac forms lamellipodia and cdc42 forms filopodia. HUVEC develop significantly more stress fibers after stimulation with thrombin than with histamine. The Rho inhibitor C3T, a dominant negative RhoA, or the Rho kinase inhibitor Y27632 prevent stress fiber formation, confirming the contribution of Rho and its effector Rho kinase to this process (Wojciak-Stothard et al., 2001; unpublished data). A rapidly enlarging literature suggests that the actin-based cytoskeleton affects clathrin-mediated endocytosis (Schafer, 2002). Inhibitors of actin polymerization have only variable effects on endocytosis (Fujimoto et al., 2000). However, evidence from videomicroscopy suggests that coated pits dynamically attach to specific regions of the cortical cytoskeleton (Gaidarov et al., 1999; Merrifield et al., 2002). AP2 adaptor proteins align in a linear array with cortical actin; inhibiting myosin association with actin or overexpression of the Hub fragment of clathrin heavy chain disrupts this alignment (Bennett et al., 2001). Many cytosolic proteins bind directly or indirectly to components of both clathrin-coated pits and the actin cytoskeleton (Schafer, 2002; Conner and Schmid, 2003). For example, Huntingtin interacting protein 1 related binds to both clathrin and actin (Engqvist-Goldstein et al., 2001). Intersectins 1 and 2 bind both to several clathrin-associated proteins and to neural Wiskott-Aldrich syndrome protein and cdc42, thereby catalyzing Arp2/3-dependent actin polymerization (Hussain et al., 2001; McGavin et al., 2001). Perhaps Rho kinase–dependent stress fiber formation diverts actin or myosin proteins from the cortical cytoskeleton that are needed for alignment or function of clathrin-coated pits. Nevertheless, no direct evidence indicates that Rho and Rho kinase act through the cytoskeleton to inhibit clathrin-mediated endocytosis. Phosphorylation and dephosphorylation of critical coat proteins might control other essential aspects of clathrin-mediated endocytosis (Conner and Schmid, 2003). For instance, a serine/threonine kinase phosphorylates the μ2 chain of AP2, which markedly enhances its affinity for tyrosine-based internalization motifs of membrane proteins (Conner and Schmid, 2003). Rho kinase might phosphorylate another coat protein that negatively regulates endocytosis. In at least some cells, RhoA is concentrated in caveolae, which are specialized membrane domains that are associated with actin-rich regions (Michaely et al., 1999). RhoA signaling might provide a mechanism for one membrane domain, caveolae, to regulate another, clathrin-coated pits.
Our results, in conjunction with other observations, demonstrate that extracellular signals can either increase or decrease the rate of constitutive clathrin-mediated endocytosis. This dynamic regulation allows alterations in the uptake of soluble ligands that bind to particular receptors and in the degradation or storage of the receptors themselves. For membrane proteins such as P-selectin, regulating local densities in clathrin-coated pits also provides a mechanism to control cell adhesion.
Materials and methods
Cells, proteins, and other materials
Cultured HUVEC were passaged two times or less (Setiadi et al., 1998). Transfected CHO cells expressed wild-type human P-selectin or a truncated P-selectin containing only the first seven residues of the 35-residue cytoplasmic domain (Setiadi et al., 1995). Human neutrophils were isolated as described previously (Setiadi et al., 1998). The anti–human P-selectin mAbs G1 and S12 and the anti–human PSGL-1 mAb PL1 were prepared as described previously (Yago et al., 2002). Goat polyclonal antibodies to human P-selectin were biotinylated (Setiadi et al., 1995). mAbs to α-adaptin and caveolin-1 were from Transduction Laboratory. Human thrombin was a gift from C. Esmon (Oklahoma Medical Research Foundation, Oklahoma City, OK). TRAP with the sequence SFLLRN was provided by R. Cummings (University of Oklahoma Health Sciences Center). The PSGL-1–derived glycosulfopeptide 2-GSP-6 was provided by R. Cummings and A. Leppänen (University of Oklahoma Health Sciences Center; Yago et al., 2002). sP-selectin was purified (Ushiyama et al., 1993). The Rho inhibitor C3T was purchased from List Biological Laboratory. The Rho kinase inhibitor Y27632 was provided by Welfide Corporation.
Internalization assay
The internalization rate of P-selectin in HUVEC and transfected CHO cells was measured by the ability of an acidic buffer to remove 125I-labeled mAb G1, prebound to the cell surface at 4°C, after warming to 37°C for various intervals (Setiadi et al., 1995). The internalization rate of P-selectin in HUVEC was measured 4 min after stimulation with 10−4 M histamine, 1 U/ml thrombin, or 100 μM TRAP.
Adhesion assay under flow
Rolling adhesion of neutrophils on cells or on adsorbed sP-selectin was assayed in a 35-mm dish incorporated into a parallel-plate flow chamber (Setiadi et al., 1998). Site densities of P-selectin were measured by binding of mAb G1 (Moore et al., 1995). Neutrophils (106/ml in HBSS containing 0.5% human serum albumin) were perfused over cells or sP-selectin at defined wall shear stresses. The number of rolling cells was quantified by videomicroscopy (30 frames/s) using imaging software (Inovision). Rolling on HUVEC was measured continuously for 10 min after administration of 10−4 M histamine, 1 U/ml thrombin, or 100 μM TRAP, or was measured at a single point 5 min after addition of agonist. Rolling on transfected CHO cells or on sP-selectin was measured 4 min after neutrophils were perfused. In some experiments, rolling was conducted in the presence of 10−4 M histamine or 4 U/ml thrombin. Rolling velocities were measured by tracking an individual cell frame by frame, an interval of 0.033 s, in the direction of flow. For each experimental condition, 20–50 cells were tracked, each for up to 5 s, to yield a total observation time of 100–250 s. The frame by frame velocity data were used to calculate the mean velocity and the variance of velocity for each cell. The pooled data were used to calculate the mean velocity and variance of velocity for the cell population (Ramachandran et al., 2001; Yago et al., 2002).
Effect of hypertonic medium on the internalization rate and adhesive function of P-selectin
Hypertonic medium containing sucrose was used to block clathrin-mediated endocytosis (Setiadi et al., 1995, 1998). HUVEC preincubated with isotonic or hypertonic medium for 15 min were stimulated with thrombin or histamine for 4 min. The cells were maintained in the same medium. The internalization rate of P-selectin or the rolling of 10-μm-diam polystyrene microspheres coupled with 2-GSP-6 (provided by T. Yago, Oklahoma Medical Research Foundation) was measured (Yago et al., 2002).
Immunofluorescence confocal microscopy
HUVEC stimulated for 4 min with thrombin or histamine or transfected CHO cells were cultured on glass coverslips in a 12-well plate. Immunofluorescence confocal microscopy was used to quantify the extent of colocalization of cell surface P-selectin with α-adaptin or caveolin-1 (Setiadi et al., 1998).
Quantification of p38 and RhoA activity
The amount of phosphorylated p38 MAPK in HUVEC was measured using a kit from Cell Signaling Technologies. In brief, HUVEC were untreated or were stimulated with histamine or thrombin for 5 min. Cell lysates were resolved by SDS-PAGE, transferred to an Immobilon-P membrane (Millipore), and probed with rabbit polyclonal antibodies to phospho-p38 or to total p38. Bound antibodies were detected with HRP-conjugated anti–rabbit IgG using ECL. Densitometry of stained protein bands was used to quantify the levels of total p38 and phospho-p38. The percentage of phospho-p38 relative to total p38 in stimulated HUVEC was normalized to that of untreated cells and expressed as relative p38 activity.
The amount of activated RhoA in HUVEC was measured using a kit from Cytoskeleton, Inc. In brief, HUVEC were untreated or were stimulated with histamine or thrombin for 10 min. Cell lysates were incubated with beads coated with the Rho-binding domain of the Rho effector protein, rhotekin, for 1 h at 4°C, and centrifuged. Bound activated RhoA was eluted from the beads, resolved by SDS-PAGE, and transferred to an Immobilon-P membrane. Total RhoA in an aliquot of cell lysate was also resolved by SDS-PAGE and transferred to the membrane. The membrane was probed with a murine anti-RhoA mAb. Bound mAb was detected with HRP-conjugated goat anti–mouse IgG using ECL. Densitometry of stained bands was used to quantify the levels of total and activated RhoA. The percentage of activated RhoA relative to total RhoA in stimulated HUVEC was normalized to that of untreated cells and expressed as relative RhoA activity.
Acknowledgments
We thank Anne Leppänen and Richard Cummings for 2-GSP-6, Tadayuki Yago for 2-GSP-6–coupled microspheres, Sougata Karmakar for assistance with Ca2+ flux measurements, Cindy Carter, Lisa Mayer, and Todd Walker for technical assistance, and Mark Coggeshall, Brian Ceresa, and Cheng Zhu for critical reading of the manuscript. We are grateful to Mercy Lab Oklahoma, Pathology Department for providing umbilical cords. Confocal microscopy was performed in the Flow Cytometry and Confocal Microscopy Laboratory, University of Oklahoma Health Sciences Center. Ca2+ flux measurements were performed in the Oklahoma Medical Research Foundation signal transduction core facility, supported by the National Institutes of Health grant RR 015577.
This work was supported by the National Institutes of Health grant HL 34363 (to R.P. McEver) and by a Beginning Grant-in-Aid from the Heartland Affiliate of the American Heart Association (to H. Setiadi).
Abbreviations used in this paper: C3T, exoenzyme C3 transferase; HUVEC, human umbilical vein endothelial cells; PSGL-1, P-selectin glycoprotein ligand-1; sP-selectin, recombinant soluble P-selectin; TRAP, thrombin receptor-activating peptide.
References
- Akgur, F.M., M.F. Brown, G.B. Zibari, J.C. McDonald, C.J. Epstein, C.R. Ross, and D.N. Granger. 2000. Role of superoxide in hemorrhagic shock-induced P-selectin expression. Am. J. Physiol. Heart Circ. Physiol. 279:H791–H797. [DOI] [PubMed] [Google Scholar]
- Barbieri, M.A., R.L. Roberts, A. Gumusboga, H. Highfield, C. Alvarez-Dominguez, A. Wells, and P.D. Stahl. 2000. Epidermal growth factor and membrane trafficking. EGF receptor activation of endocytosis requires Rab5a. J. Cell Biol. 151:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie, E.C., C.L. Howe, A. Wilde, F.M. Brodsky, and W.C. Mobley. 2000. NGF signals through TrkA to increase clathrin at the plasma membrane and enhance clathrin-mediated membrane trafficking. J. Neurosci. 20:7325–7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, E.M., C.Y. Chen, A.E. Engqvist-Goldstein, D.G. Drubin, and F.M. Brodsky. 2001. Clathrin hub expression dissociates the actin-binding protein Hip1R from coated pits and disrupts their alignment with the actin cytoskeleton. Traffic. 2:851–858. [DOI] [PubMed] [Google Scholar]
- Cavalli, V., F. Vilbois, M. Corti, M.J. Marcote, K. Tamura, M. Karin, S. Arkinstall, and J. Gruenberg. 2001. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol. Cell. 7:421–432. [DOI] [PubMed] [Google Scholar]
- Conner, S.D., and S.L. Schmid. 2003. Regulated portals of entry into the cell. Nature. 422:37–44. [DOI] [PubMed] [Google Scholar]
- Coughlin, S.R. 2001. Protease-activated receptors in vascular biology. Thromb. Haemost. 86:298–307. [PubMed] [Google Scholar]
- Dunne, J.L., C.M. Ballantyne, A.L. Beaudet, and K. Ley. 2002. Control of leukocyte rolling velocity in TNF-α-induced inflammation by LFA-1 and Mac-1. Blood. 99:336–341. [DOI] [PubMed] [Google Scholar]
- Dwir, O., G.S. Kansas, and R. Alon. 2001. Cytoplasmic anchorage of L-selectin controls leukocyte capture and rolling by increasing the mechanical stability of the selectin tether. J. Cell Biol. 155:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engqvist-Goldstein, A.E., R.A. Warren, M.M. Kessels, J.H. Keen, J. Heuser, and D.G. Drubin. 2001. The actin-binding protein Hip1R associates with clathrin during early stages of endocytosis and promotes clathrin assembly in vitro. J. Cell Biol. 154:1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essler, M., M. Amano, H.J. Kruse, K. Kaibuchi, P.C. Weber, and M. Aepfelbacher. 1998. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J. Biol. Chem. 273:21867–21874. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2002. Rho GTPases in cell biology. Nature. 420:629–635. [DOI] [PubMed] [Google Scholar]
- Fujimoto, L.M., R. Roth, J.E. Heuser, and S.L. Schmid. 2000. Actin assembly plays a variable, but not obligatory role in receptor-mediated endocytosis in mammalian cells. Traffic. 1:161–171. [DOI] [PubMed] [Google Scholar]
- Gaidarov, I., F. Santini, R.A. Warren, and J.H. Keen. 1999. Spatial control of coated-pit dynamics in living cells. Nat. Cell Biol. 1:1–7. [DOI] [PubMed] [Google Scholar]
- Green, S.A., H. Setiadi, R.P. McEver, and R.B. Kelly. 1994. The cytoplasmic domain of P-selectin contains a sorting determinant that mediates rapid degradation in lysosomes. J. Cell Biol. 124:435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser, J.E., and R.G.W. Anderson. 1989. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J. Cell Biol. 108:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrboticky, N., T. Feldmeer, M. Essler, A. Wiedemann, and M. Aepfelbacher. 2002. Involvement of the GTPase Rho in the cellular uptake of low density lipoprotein by human skin fibroblasts. Biochim. Biophys. Acta. 1580:123–132. [DOI] [PubMed] [Google Scholar]
- Hussain, N.K., S. Jenna, M. Glogauer, C.C. Quinn, S. Wasiak, M. Guipponi, S.E. Antonarakis, B.K. Kay, T.P. Stossel, N. Lamarche-Vane, and P.S. McPherson. 2001. Endocytic protein intersectin-l regulates actin assembly via Cdc42 and N-WASP. Nat. Cell Biol. 3:927–932. [DOI] [PubMed] [Google Scholar]
- Ishizaki, T., M. Uehata, I. Tamechika, J. Keel, K. Nonomura, M. Maekawa, and S. Narumiya. 2000. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol. Pharmacol. 57:976–983. [PubMed] [Google Scholar]
- Johnston, B., S. Kanwar, and P. Kubes. 1996. Hydrogen peroxide induces leukocyte rolling: modulation by endogenous antioxidant mechanisms including NO. Am. J. Physiol. 271:H614–H621. [DOI] [PubMed] [Google Scholar]
- Jones, D.A., O. Abbassi, L.V. McIntire, R.P. McEver, and C.W. Smith. 1993. P-selectin mediates neutrophil rolling on histamine-stimulated endothelial cells. Biophys. J. 65:1560–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, U., K.E. Norman, K. Scharffetter-Kochanek, A.L. Beaudet, and K. Ley. 1998. Transit time of leukocytes rolling through venules controls cytokine-induced inflammatory cell recruitment in vivo. J. Clin. Invest. 102:1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur, J., R.C. Woodman, and P. Kubes. 2003. P38 MAPK: critical molecule in thrombin-induced NF-κB-dependent leukocyte recruitment. Am. J. Physiol. Heart Circ. Physiol. 284:H1095–H1103. [DOI] [PubMed] [Google Scholar]
- Lamaze, C., T.H. Chuang, L.J. Terlecky, G.M. Bokoch, and S.L. Schmid. 1996. Regulation of receptor-mediated endocytosis by Rho and Rac. Nature. 382:177–179. [DOI] [PubMed] [Google Scholar]
- Marin, V., C. Farnarier, S. Gres, S. Kaplanski, M.S. Su, C.A. Dinarello, and G. Kaplanski. 2001. The p38 mitogen-activated protein kinase pathway plays a critical role in thrombin-induced endothelial chemokine production and leukocyte recruitment. Blood. 98:667–673. [DOI] [PubMed] [Google Scholar]
- McEver, R.P. 1997. Regulation of expression of E-selectin and P-selectin. The Selectins: Initiators of Leukocyte Endothelial Adhesion. D. Vestweber, editor. Harwood Academic Publishers, Amsterdam. 31–47.
- McEver, R.P. 2001. Adhesive interactions of leukocytes, platelets, and the vessel wall during hemostasis and inflammation. Thromb. Haemost. 86:746–756. [PubMed] [Google Scholar]
- McEver, R.P. 2002. Selectins: lectins that initiate cell adhesion under flow. Curr. Opin. Cell Biol. 14:581–586. [DOI] [PubMed] [Google Scholar]
- McGavin, M.K., K. Badour, L.A. Hardy, T.J. Kubiseski, J. Zhang, and K.A. Siminovitch. 2001. The intersectin 2 adaptor links Wiskott Aldrich Syndrome protein (WASp)-mediated actin polymerization to T cell antigen receptor endocytosis. J. Exp. Med. 194:1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson, P.S., B.K. Kay, and N.K. Hussain. 2001. Signaling on the endocytic pathway. Traffic. 2:375–384. [DOI] [PubMed] [Google Scholar]
- Merrifield, C.J., M.E. Feldman, L. Wan, and W. Almers. 2002. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 4:691–698. [DOI] [PubMed] [Google Scholar]
- Michaely, P.A., C. Mineo, Y.S. Ying, and R.G. Anderson. 1999. Polarized distribution of endogenous Rac1 and RhoA at the cell surface. J. Biol. Chem. 274:21430–21436. [DOI] [PubMed] [Google Scholar]
- Moore, K.L., K.D. Patel, R.E. Bruehl, L. Fugang, D.A. Johnson, H.S. Lichenstein, R.D. Cummings, D.F. Bainton, and R.P. McEver. 1995. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J. Cell Biol. 128:661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimnual, A.S., L.J. Taylor, and D. Bar-Sagi. 2003. Redox-dependent downregulation of Rho by Rac. Nat. Cell Biol. 5:236–241. [DOI] [PubMed] [Google Scholar]
- Ramachandran, V., T. Yago, T.K. Epperson, M.M.A. Kobzdej, M.U. Nollert, R.D. Cummings, C. Zhu, and R.P. McEver. 2001. Dimerization of a selectin and its ligand stabilizes cell rolling and enhances tether strength in shear flow. Proc. Natl. Acad. Sci. USA. 98:10166–10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, D.A. 2002. Coupling actin dynamics and membrane dynamics during endocytosis. Curr. Opin. Cell Biol. 14:76–81. [DOI] [PubMed] [Google Scholar]
- Schmidtke, D.W., and S.L. Diamond. 2000. Direct observation of membrane tethers formed during neutrophil attachment to platelets or P-selectin under physiological flow. J. Cell Biol. 149:719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, E., M. Rolli-Derkinderen, M. Arock, and M. Dy. 2002. Trends in histamine research: new functions during immune responses and hematopoiesis. Trends Immunol. 23:255–263. [DOI] [PubMed] [Google Scholar]
- Setiadi, H., M. Disdier, S.A. Green, W.M. Canfield, and R.P. McEver. 1995. Residues throughout the cytoplasmic domain affect the internalization efficiency of P-selectin. J. Biol. Chem. 270:26818–26826. [DOI] [PubMed] [Google Scholar]
- Setiadi, H., G. Sedgewick, S.L. Erlandsen, and R.P. McEver. 1998. Interactions of the cytoplasmic domain of P-selectin with clathrin-coated pits enhance leukocyte adhesion under flow. J. Cell Biol. 142:859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp, K.R., C.E. Heitzig, and G.S. Kansas. 2002. Attachment of the PSGL-1 cytoplasmic domain to the actin cytoskeleton is essential for leukocyte rolling on P-selectin. Blood. 99:4494–4502. [DOI] [PubMed] [Google Scholar]
- Subramaniam, M., J.A. Koedam, and D.D. Wagner. 1993. Divergent fates of P- and E-selectins after their expression on the plasma membrane. Mol. Biol. Cell. 4:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano, M., A. Meneshian, E. Sheikh, Y. Yamakawa, K.B. Wilkins, E.A. Hopkins, and G.B. Bulkley. 2002. Rapid upregulation of endothelial P-selectin expression via reactive oxygen species generation. Am. J. Physiol. Heart Circ. Physiol. 283:H2054–H2061. [DOI] [PubMed] [Google Scholar]
- Ushiyama, S., T.M. Laue, K.L. Moore, H.P. Erickson, and R.P. McEver. 1993. Structural and functional characterization of monomeric soluble P-selectin and comparison with membrane P-selectin. J. Biol. Chem. 268:15229–15237. [PubMed] [Google Scholar]
- Vestweber, D., and J.E. Blanks. 1999. Mechanisms that regulate the function of the selectins and their ligands. Physiol. Rev. 79:181–213. [DOI] [PubMed] [Google Scholar]
- von Asmuth, E.J.U., E.F. Smeets, L.A. Ginsel, J.J.M. Onderwater, J.F.M. Leeuwenberg, and W.A. Buurman. 1992. Evidence for endocytosis of E-selectin in human endothelial cells. Eur. J. Immunol. 22:2519–2526. [DOI] [PubMed] [Google Scholar]
- Vouret-Craviari, V., P. Boquet, J. Pouyssegur, and E. Van Obberghen-Schilling. 1998. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. Mol. Biol. Cell. 9:2639–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde, A., E.C. Beattie, L. Lem, D.A. Riethof, S.H. Liu, W.C. Mobley, P. Soriano, and F.M. Brodsky. 1999. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 96:677–687. [DOI] [PubMed] [Google Scholar]
- Wilde, C., H. Genth, K. Aktories, and I. Just. 2000. Recognition of RhoA by Clostridium botulinum C3 exoenzyme. J. Biol. Chem. 275:16478–16483. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard, B., L. Williams, and A.J. Ridley. 1999. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J. Cell Biol. 145:1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciak-Stothard, B., S. Potempa, T. Eichholtz, and A.J. Ridley. 2001. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J. Cell Sci. 114:1343–1355. [DOI] [PubMed] [Google Scholar]
- Yago, T., A. Leppänen, H. Qiu, W.D. Marcus, M.U. Nollert, C. Zhu, R.D. Cummings, and R.P. McEver. 2002. Distinct molecular and cellular contributions to stabilizing selectin-mediated rolling under flow. J. Cell Biol. 158:787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]