

Abstract
Caspase-directed apoptosis usually fragments cells, releasing nonfunctional, prothrombogenic, membrane-bound apoptotic bodies marked for rapid engulfment by macrophages. Blood platelets are functional anucleate cells generated by specialized fragmentation of their progenitors, megakaryocytes (MKs), but committed to a constitutive caspase-independent death. Constitutive formation of the proplatelet-bearing MK was recently reported to be caspase-dependent, apparently involving mitochondrial release of cytochrome c, a known pro-apoptogenic factor. We extend those studies and report that activation of caspases in MKs, either constitutively or after Fas ligation, yields platelets that are functionally responsive and evade immediate phagocytic clearance, and retain mitochondrial transmembrane potential until constitutive platelet death ensues. Furthermore, the exclusion from the platelet progeny of caspase-9 present in the progenitor accounts for failure of mitochondrial release of cytochrome c to activate caspase-3 during platelet death. Thus, progenitor cell death by apoptosis can result in birth of multiple functional anucleate daughter cells.
Keywords: mitochondria; Fas; apoptosis; caspases; thrombopoiesis
Introduction
Blood platelets are anucleate cells vital for hemostasis, generated within bone marrow by a highly specialized form of progenitor cell fragmentation (Tavassoli and Aoki, 1989). Platelet progenitors, the megakaryocytes (MKs),* undergo a dramatic morphological change in which the MK extends long bifurcating filamentous projections. These “proplatelet”-bearing MKs then develop platelet-sized nodes through the specific delivery of platelet material along the processes, which appear similar to beads on a string. Mature platelets are then released in large numbers from the tip of such processes (Stenberg and Levin, 1989; Cramer et al., 1997; Italiano et al., 1999). This process is markedly different from the blebbing or zeiosis of cells dying by apoptosis, in which membrane-bound bodies bud individually from the cell surface (Kerr et al., 1972; Robertson et al., 1978). Cultured MK cells have also been observed to undergo apoptosis in vitro (Zauli et al., 1997), and remnant MK cell bodies containing nuclei with typical apoptotic chromatin condensation are evident in bone marrow in vivo (Radley and Haller, 1983). Furthermore, studies have recently revealed that pro-apoptotic stimuli such as nitric oxide can, in concert with potentially pro-apoptotic cytokines such as TNF-α and IFN-γ, trigger increased release of platelet-like bodies from cultured MK cells (Battinelli et al., 2001). More recently, evidence was provided that proplatelet formation in response to thrombopoietin (TPO) was a consequence of caspase activation dependent on mitochondrial release of the pro-apoptogenic factor cytochrome c, indicative of an intrinsic program of cell death (De Botton et al., 2002). However, no data has yet demonstrated that such specialized MK cell death gives rise to mature functional progeny.
For some time, we have been interested in the relationship between MK apoptosis and platelet production. In particular, we were intrigued by the possible role of caspases in both processes, as we had previously shown that mature blood platelets were committed to a caspase-independent program of cell death (Brown et al., 2000). Thus, when deprived of plasma as a source of survival factors, cultured platelets exhibited morphological condensation accompanied by exposure of phosphatidylserine (PS), maintenance of plasma membrane integrity, and specific recognition and engulfment by monocyte-derived macrophages (MDMs) using the class-A scavenger receptor (Brown et al., 2000). Although these events were strongly reminiscent of constitutive apoptosis in cultured leukocytes, a key difference was that platelet death was not inhibited by broad-spectrum caspase inhibitors, nor was there evidence of activation by cleavage of effector caspase-3 (Brown et al., 2000), which is found in abundance in platelets.
The current study supports the hypothesis that MKs produce platelets by a novel compartmentalized form of caspase-directed apoptosis (De Botton et al., 2002), but importantly extends those studies to show that such proplatelet MKs yield functional platelets that retain inner mitochondrial membrane potential (ΔψM) and membrane PS asymmetry. By contrast, the MK cell body exhibited typical nuclear morphological features of apoptosis. Further evidence of compartmentalized progenitor cell death was the absence from viable platelets of caspase-9 present in MKs, accounting for the caspase-independent nature of constitutive platelet death.
Results
Constitutive apoptosis in MKs is associated with production of functional platelets
Production of platelets from their progenitors, MKs, occurs through an intermediate proplatelet phenotype consisting of thin cytoplasmic processes along which platelet-sized nodes develop before platelets are released by multiple detachment events from the process tip (Stenberg and Levin, 1989; Cramer et al., 1997; Italiano et al., 1999). Using cultures of the well validated MEG-01 human MK cell line (Ogura et al., 1985), which spontaneously produces functional platelets under normal culture conditions (Takeuchi et al., 1998), we observed proplatelet MKs at a frequency of 0.6 ± 0.1% (Table I). Intriguingly, both proplatelet MK frequency and production of functional platelets were significantly reduced by the broad-spectrum caspase inhibitor zVAD-fmk (Table I), implying a role for caspase-directed apoptosis in the formation of proplatelet extensions by MKs and the subsequent production of functional platelets. Furthermore, in studies focusing on the important initial event in platelet production, formation of proplatelet extensions by MKs, we also observed inhibition by zVAD-fmk and by the more specific peptide inhibitor of caspase-3, zDEVD-fmk, at only 10 μM (Table II). To confirm this inhibitor evidence of caspase activation driving platelet production, we used a quenched fluorescent reagent of broad specificity for caspases, CaspaTag™, finding that the cell body of proplatelet-bearing MKs contained active caspases, whereas the processes did not (Fig. 1 A). Similar results were obtained with SET-2, another MK cell line (unpublished data).
Table I. Fas ligation augments platelet production by MKs in a zVAD-fmk–inhibitable manner.
| Condition | Proplatelet MKs per 2 × 105 MKs |
Functional platelet yield × 104 per 2 × 105 MKs |
|---|---|---|
| Control | 1,193 ± 183 | 26 ± 3 |
| ZVAD-fmk (100 μM) | 5 ± 8a | 13 ± 1a |
| CH11 (50 ng/ml) | 8,024 ± 1,777a | 234 ± 38a |
| CH11;ZB4 (50 ng/ml; 1 μg/ml) | ND | 31 ± 2 |
| CH11;zVAD-fmk (50 ng/ml; 100 μM) | 784 ± 115a | 28 ± 4 |
| sFas-L (5 ng/ml) | ND | 134 ± 28a |
| sFas-L;zVAD-fmk (5 ng/ml; 100 μM) | ND | 29 ± 9 |
| TNFα (25 ng/ml) | ND | 30 ± 5 |
Proplatelet MK count represents total number of proplatelet-bearing MKs following 8 h of culture, scored by phase microscopy. Functional platelet yield represents total number of platelets produced following 18 h of culture, enumerated as detailed in Fig. 3. CH11 is an anti-Fas agonistic mAb, and ZB4 an antagonistic mAb. Data represent mean ± SD of a minimum of three determinations.
P < 0.01 compared to control.
Table II. Caspases direct constitutive and Fas-induced proplatelet formation.
| Proplatelet MKs per 2 × 105 MKs
|
||
|---|---|---|
| Condition | Constitutive | CH11 induced |
| Control | 1,340 ± 147 | 7,930 ± 923 |
| zVAD-fmk (10 μM) | 82 ± 36a | 245 ± 47a |
| zDEVD-fmk (10 μM) | 227 ± 55a | 990 ± 247a |
| zIETD-fmk (10 μM) | 699 ± 97a | 536 ± 103a |
| BioPORTER® control | 1,516 ± 48 | 4,756 ± 372 |
| BioPORTER® crmA | 1,387 ± 156 | 2,396 ± 240b |
| BioPORTER® HI crmA | ND | 4,400 ± 500 |
| BioPORTER® active caspase-8 | 2,502 ± 368b | ND |
Proplatelet MK count represents total number of proplatelet-bearing MKs following 8 h of culture, scored by phase microscopy. zDEVD-fmk has enhanced specificity over zVAD-fmk as a caspase-3 inhibitor. HI, heat inactivated; Data represent mean ± SD of a minimum of three determinations.
P < 0.01 compared to control.
P < 0.01 compared to BioPORTER® control.
Figure 1.

Proplatelet-extending MKs exhibit nuclear pyknosis and contain active caspases. (a) A mature MEG-01 MK displaying proplatelet extensions, stained positive for active caspases with CaspaTag™ (green), which remained localized within the main cell body. Note that MKs without proplatelet extensions (left and right) lacked caspase activity. (b) Dual staining of nuclear material with Hoechst 33342 (blue) and active caspases reveals MKs bearing proplatelets to contain pyknotic nuclei, with nuclear material remaining within the cell body along with active caspases. Bars, 20 μm.
Because our own data confirmed the report by De Botton et al. (2002) that caspase activation was important in formation of proplatelet MKs, we went on to seek the morphological hallmarks of apoptosis in MKs extending proplatelet extensions. The main cell body of proplatelet MKs failed to exclude the vital dye Hoechst 33342, revealing chromosomal condensation and nuclear fragmentation typical of apoptosis (Fig. 1 B). Indeed, transmission electron microscopy (TEM) revealed that MKs undergoing the very earliest stages of cytoplasmic projection exhibited clear heterochromatin condensation typical of early apoptosis (Kerr et al., 1972), proceeding to extensive condensation in those bearing proplatelet extensions (Fig. 2). These data demonstrated that caspase-mediated MK apoptosis was responsible for the production of mature platelets, and were further supported by constitutive internucleosomal chromatin cleavage (DNA laddering) typical of apoptosis in MKs cultured under control conditions (Wyllie, 1980; unpublished data).
Figure 2.
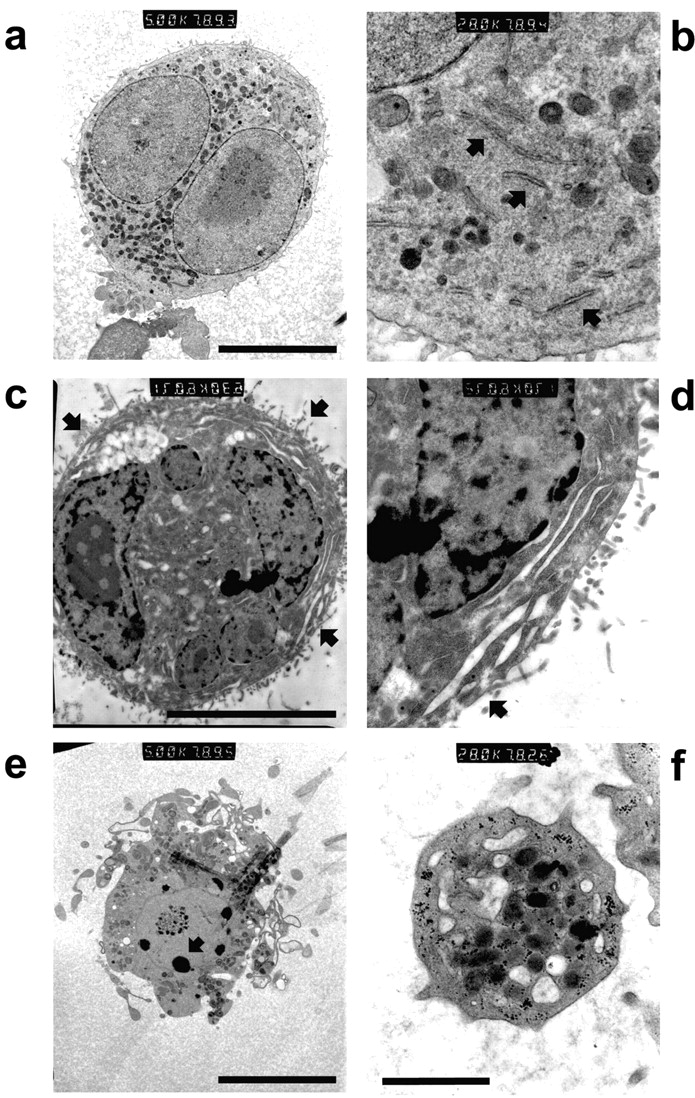
Mature MKs exhibit nuclear condensation typical of apoptosis, producing bodies indistinguishable from blood platelets. Purified mature MEG-01 MKs where examined by TEM. (a) MKs, shown at higher magnification in b to demonstrate a typical distribution of α-granules and demarcation membranes (arrows), contain nuclei with evenly dispersed heterochromatin. (c) MK, shown at higher magnification in d, exhibiting early cytoplasmic rearrangements consistent with platelet formation (arrows), with a nucleus displaying heterochromatin condensation typical of early apoptosis. (e) MK-bearing proplatelets show extensive condensation and fragmentation of the nuclear material (arrow). (f) TEM of MEG-01 MK culture supernatants reveals platelets with morphology consistent with that typically observed for blood platelets. Bars: 20 μm (a–e) and 1 μm (f).
The anucleate cells produced by MEG-01 MKs were morphologically indistinguishable from blood platelets; TEM revealing a discoid shape and a characteristic distribution of α and dense granules (Fig. 2 F). Flow cytometry further revealed that MK-derived platelets, identified by positive staining for GpIIb/IIIa (unpublished data), did not bind annexin-V (a marker of PS exposure), excluded propidium iodide, were able to undergo the key functional response of shape change (Otterdal et al., 2001) (Fig. 3), and displayed a characteristic transient calcium flux, detected with Fluo3 (unpublished data), when stimulated by either thrombin or ADP. By contrast, MK fragments bound annexin-V and propidium iodide and did not undergo agonist-induced shape change (Fig. 3). Furthermore, the functional nature of MK-derived platelets was emphasized by the thrombin-stimulated conformational change of GpIIb/IIIa to a fibrinogen-binding competent state, as assessed by flow cytometry using labeled fibrinogen (t + 20 s = 21.6%; t + 40 s = 40.1%).
Figure 3.

Culture-derived platelets are functional. (a) Freshly isolated blood platelets undergo an agonist-induced shape change in response to thrombin (as indicated), detected as an increase in side scatter with events moving from gate R1 to R2. Absolute counts of platelets were obtained by spiking samples with a fixed number of 10-μM beads (R3). (b) Freshly isolated blood platelets treated with or without agonist (as indicated) rarely bound annexin-V under the conditions used. (c) In contrast, culture-derived platelets contained an annexin- V–positive population, with only annexin-V–negative cells undergoing thrombin-induced shape change. (d) Culture-derived platelets incubated with MDMs result in clearance of debris with the remaining population able to shape change on stimulation. Functional platelets were defined as being annexin-V–negative and capable of undergoing shape change in response to agonist.
Fas ligation augments platelet production by MKs
In view of this and other evidence (De Botton et al., 2002) that constitutive caspase-mediated apoptosis of proplatelet MKs was associated with platelet production, we hypothesized that the classical pro-apoptotic stimulus of Fas ligation might be sufficient to increase platelet production. Ligation of MK Fas by either agonistic anti-Fas antibody (CH.11) or soluble Fas ligand over an 18-h period resulted in a significant increase in proplatelet extensions and production of viable (annexin-V–negative) platelets, shown to be functional as detailed above (Fig. 3). This increase in proplatelet formation and functional platelet production was shown to be caspase-dependent by the inhibitory effects of zVAD-fmk and zIETD-fmk (Tables I and II). However, ligation of the TNF-α death receptor with TNF-α did not increase platelet production (Table I). Furthermore, an essential role for Fas-directed activation of initiator caspases in driving MK proplatelet extension was provided by direct protein transduction of the protein CrmA (Table II), a potent inhibitor of such initiator caspases (Garcia-Calvo et al., 1998). Importantly, CrmA had no effect on constitutive formation of proplatelet MKs. Indeed, introduction of active caspase-8, which is directly activated by Fas ligation, specifically increased proplatelet extension (Table II). Epifluorescent microscopy of Fas-stimulated MK cultures further revealed that 95 ± 3% (mean ± SD of n = 3) of the proplatelet-bearing cells exhibited nuclear condensation typical of apoptosis, and that 89 ± 3% (mean ± SD of n = 3) exhibited staining for active caspases within the main cell body. These observations were reproduced with primary murine MKs differentiated from bone marrow. Using Jo-2, an anti-murine Fas agonistic mAb, we again observed that the number of functional platelets produced increased in a caspase-dependent manner after 18 h of treatment (Fig. 4 A).
Figure 4.
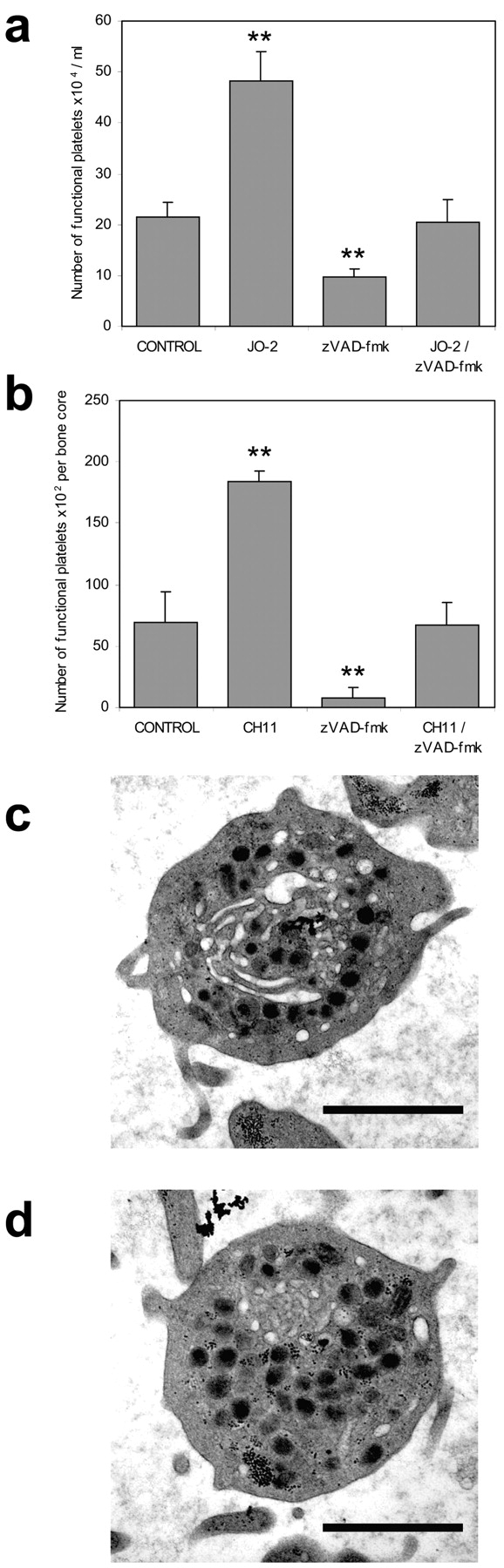
Caspase-dependent production of platelets by primary murine MKs and human bone cores is augmented by Fas ligation. (a) Mature primary murine MKs produce functional platelets, inhibitable with zVAD-fmk and augmented with the anti-murine Fas ligating antibody JO-2. Functional platelets were enumerated as for Fig. 3. Data represent mean ± SD, n = 4. ** denotes P < 0.02. (b) Ex vivo human bone core explants, maintained within a Zetos™ bone perfusion chamber, were exposed to fresh media containing CH.11 and/or zVAD-fmk as indicated. Perfused media was collected over 18 h and GpIIb/IIIa platelets were enumerated by flow cytometry. Data represent mean ± SD. ** denotes P < 0.05. (c) TEM of perfused media components showing presence of platelets with morphology consistent with blood-derived platelets (d). Bars in c and d , 1 μm.
Furthermore, identical results were obtained using a novel human bone core explant bio-culture system (Smith and Jones, 1998). Human trabecular bone from femoral heads, removed at surgery, contained viable bone marrow and constitutively produced platelets after 4 d that stained positive for the lineage specific fibrinogen receptor (GpIIb/IIIa). Such platelet production was robustly inhibited by zVAD-fmk and augmented in a zVAD-fmk inhibitable manner by Fas ligation (Fig. 4 B). Importantly, and although the relatively small number of platelet-like particles generated in this system precluded any functional assessment, ultrastructural analysis by TEM again showed morphology consistent with blood platelets (Brown et al., 2000; Fig. 4, C and D).
Platelets generated by MK apoptosis are not ingested by macrophages
To confirm the morphological and functional evidence that platelets produced by MEG-01 MKs undergoing apoptosis were viable, we investigated whether MDMs would selectively clear nonfunctional platelets and MK fragments. Incubating MK culture supernatants with MDMs resulted in the selective clearance of all PS-positive bodies, leaving a population of functional platelets demonstrating agonist-induced shape change (Fig. 3 D).
Mitochondrial permeability transition is not observed in proplatelet MK extensions and occurs only as mature platelets die
The foregoing data strongly implied that a compartmentalized form of apoptosis in proplatelet MKs gave rise to viable platelets and an apoptotic remnant body. Because mitochondrial permeability transition is a prominent feature of caspase-mediated apoptosis, we investigated proplatelet MK ΔψM using JC-1, a mitochondrial dye that fluoresces orange in respiring mitochondria that maintain ΔψM (Petit et al., 1995; Salvioli et al., 1997). Importantly, we found that mitochondria, localized to platelet-sized nodes along the cytoplasmic extensions of proplatelet MKs, had not undergone permeability transition despite double staining with Hoechst 33342 showing clear morphological evidence of nuclear condensation and fragmentation within the main cell body (Fig. 5, A and B). In addition, confocal microscopy revealed that mitochondria remaining within the cell body were polarized to the MK edge with the remaining proplatelet “bridge” still attached (Fig. 5 C). Furthermore, viable MK culture-derived platelets that were allowed to adhere and spread on glass also showed no evidence of mitochondrial permeability transition (Fig. 5 D). This was only observed when mature human blood platelets were cultured in the absence of plasma-derived survival factors for 16 h to allow constitutive death (Brown et al., 2000), or when fresh blood platelets were treated with the respiratory chain uncoupler mCCCP (Fig. 6 A).
Figure 5.
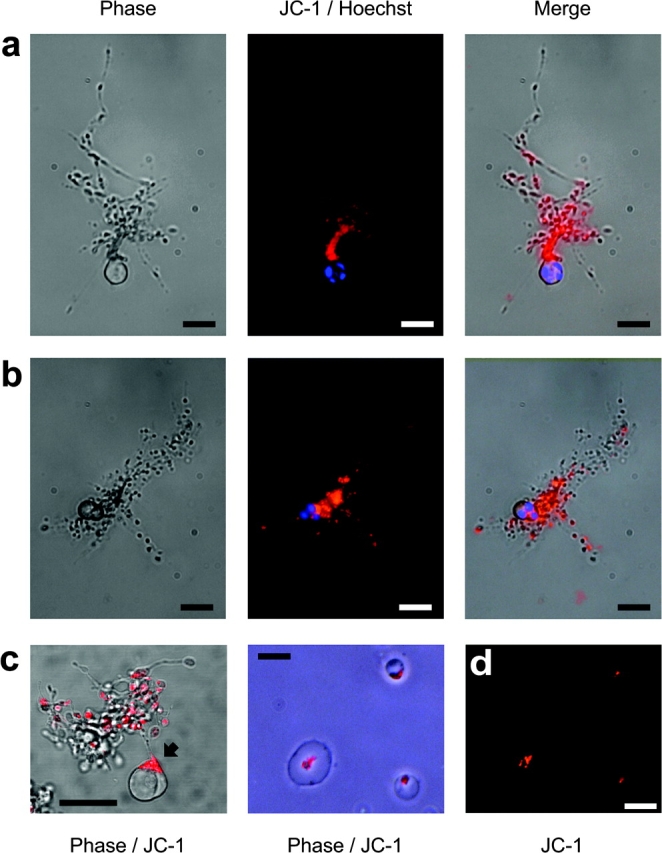
Functional platelet production is associated with the maintenance of ΔψM. (a and b) Mature MEG-01 MKs were double stained with the ΔψM-sensitive dye JC-1 (orange), and the nuclear staining vital dye Hoechst 33342 (blue). Mitochondria with an intact ΔψM are seen localized within platelet-sized nodes along extended proplatelets, whereas the cell bodies display nuclear condensation. (c) An MK stained with JC-1 only and analyzed by confocal microscopy reveals functional mitochondria with an intact ΔψM to be polarized within the cell body toward the remaining attached proplatelet (arrow). Bars in a–c, 20 μm. (d) Culture-derived platelets were also stained with JC-1 and allowed to adhere and spread on glass. Again, mitochondria with an intact ΔψM are seen coalesced within the body of the spreading platelet. Bar in d, 5 μm.
Figure 6.

Senescent platelets show loss of ΔψM and release of mitochondrial cytochrome c . (a) Fresh PRP, aged PRP, or aged washed platelets, as indicated, were stained with the ΔψM-sensitive dye JC-1. (i) Flow cytometric analysis of fresh platelets display a typical fluorescent profile of high orange fluorescence, indicative of an intact ΔψM. (ii) Platelets aged in the presence of plasma survival factors show a small (∼10%) distinct subpopulation characteristic of a collapsed ΔψM, i.e., loss of FL2-H fluorescence with concomitant increase in FL1-H. (iii) By contrast, aged washed platelets display ∼100% loss of ΔψM. (iv) Treatment of fresh platelets with the protonophore mCCCP resulted in a collapse of the ΔψM, equivalent to that seen with aged platelets. (b) Aged platelets release mitochondrial cytochrome c into the cytoplasm. Fresh (viable) and aged washed (nonviable) platelets, as indicated, were lysed and cells were fractionated by centrifugation to yield a cytosolic supernatant and a subcellular pellet. Both supernatant and solubilized pellet were precleared with a control IgG before immunoprecipitating cytochrome c. Cytochrome c was detected by Western blot analysis, in which Jurkats treated with cycloheximide are provided as a positive control.
ΔψM collapse and the opening of the permeability transition pore is associated with mitochondrial release of cytochrome c, a prerequisite for the formation of cytoplasmic apoptosome complexes (Liu et al., 1996; Marchetti et al., 1996; Kluck et al., 1997; Li et al., 1997; Yang et al., 1997), normally comprising coaggregated APAF-1, caspase-9, cytochrome c, and dATP. In keeping with this sequence of events, we found cytochrome c to be contained within the mitochondrial-enriched pellet of viable platelets, but to have been released into the cytosolic fraction of senescent platelets or apoptotic Jurkat cells, used as a control (Fig. 6 B).
Unlike MKs, freshly isolated platelets lack caspase-9, which is required for caspase-3 activation in platelet lysates
We were intrigued by mitochondrial permeability transition and cytochrome c release in senescent platelets because our previous work had clearly shown that constitutive death in such platelets was caspase-independent, with no evidence of the caspase-3 activation normally seen in caspase-mediated apoptosis (Brown et al., 2000). Together, these data implied that freshly isolated mature blood platelets lacked either APAF-1 or caspase-9, key components of the apoptosome that cleaves caspase-3 to its active p12/17 form (Liu et al., 1996; Li et al., 1997).
Two lines of evidence demonstrated that platelets (but not MKs) lacked caspase-9. First, Western blot analysis of blood platelets confirmed that APAF-1 was present and at comparable levels to Jurkat T cells (Fig. 7 A). However, although caspase-9 was readily identified in Jurkat T cells, migrating with an apparent molecular size of 48 kD, caspase-9 could not be detected in blood platelets. Importantly, the MK cell lines MEG-01 and SET-2 expressed caspase-9, and at levels equivalent to Jurkat T cells using both a polyclonal (Fig. 7 B) and a monoclonal antibody (unpublished data), indicating exclusion of the enzyme during platelet formation.
Figure 7.

Platelets contain APAF-1, but do not contain caspase-9. (a) Whole-cell lysates prepared from fresh platelets (i), Jurkat T cells (ii), and MEG-01 MKs (iii) were probed with an anti-APAF-1 pAb. All cells contained an immunodetectable protein with an apparent molecular mass of ∼130 kD, consistent with it being APAF-1. (b) Whole-cell lysates prepared from Jurkat T cells (i), fresh platelets (ii and iii), MEG-01 MKs (iv), and SET-2 MKs (v) were probed by Western blot for caspase-9 with a pAb that recognized both pro- and activated forms. A major band was detected with an apparent molecular mass approximating that of caspase-9 (48 kD) within Jurkat T cells and MKs. The Western blot was stripped and reprobed for β-actin, where β-actin accounts for 30% of platelet protein.
Second, we attempted to activate caspase-9 in fresh platelet lysates by adding exogenous cytochrome c and dATP, which should have reconstituted an active apoptosome complex capable of processing caspase-3 to its active p12/17 form (Liu et al., 1996; Li et al., 1997). However, pro-caspase-3 remained unprocessed (Fig. 8 A), whether control platelet lysates or those treated with exogenous cytochrome c/dATP were examined. This was in stark contrast to caspase-3 activation by Jurkat T cell lysates where exposure to cytochrome c/dATP revealed a progressive processing of procaspase-3 to the p12/17 active fragment (Fig. 8 B). Similar processing was obtained using lysates from MEG-01 MKs (unpublished data). As the only proteins required for cell-free caspase-3 processing are caspase-9 and APAF-1, we examined whether addition of recombinant caspase-9 could reconstitute caspase-3 cleavage in lysates from fresh platelets. Using the same preparations of fresh platelet lysates as above, we observed that the addition of hrCaspase-9 (0.1 U/ml) resulted in the processing of caspase-3, presumably by reconstitution and activation of the apoptosome complex (Fig. 8 C). Therefore, two lines of evidence indicate that a further manifestation of the apparent compartmentalization of apoptosis in proplatelet MKs is exclusion of caspase-9 from the platelet progeny, committing them to a caspase-independent program of constitutive cell death.
Figure 8.

Exogenous cytochrome c /dATP fails to promote caspase-3 processing within platelet lysates. Cytosolic extracts were treated with cytochrome c and dATP or buffer alone and incubated at 37°C. (a) Western blot analysis of platelet lysates revealed a failure of cytochrome c/dATP to promote the processing of caspase-3. (b) By contrast, Jurkat T cell lysates showed progressive processing of the enzyme to the proteolytically active p17/p12 fragments. (c) On addition of human recombinant caspase-9 and cytochrome c/dATP to fresh platelet lysates, derived from the same preparation used above, caspase-3 was now processed to its active p17/p12 subunits. Each Western blot had 50 μg of protein loaded per lane and was repeated twice.
Discussion
Our key conclusion, in agreement with that of De Botton et al. (2002), is that compartmentalized apoptosis of a progenitor cell is a hitherto unrecognized mechanism for generation of multiple, functional anucleate daughter cells. Thus, proplatelet MKs exhibited clear morphological evidence of nuclear changes of apoptosis and caspase activation in the main cell body, but bore processes that retained ΔψM and yielded functional anucleate progeny (platelets) that very importantly were not marked for immediate clearance by MDMs. Strong mechanistic evidence also supported caspase-directed MK apoptosis as critical to both formation of proplatelet MKs and production of functional platelets. First, both parameters were inhibited in constitutive MK cultures by caspase inhibition with the poly-caspase inhibitor zVAD-fmk, with caspase-3 implicated by use of a well-established peptide inhibitor zDEVD-fmk. Second, platelet production could be driven in the human MEG-01 MK cell line, primary murine MKs and a novel human bone marrow tissue culture system by the classical pro-apoptotic stimulus of Fas ligation, which induced proplatelet MKs exhibiting nuclear condensation of apoptosis. Third, Fas-directed proplatelet MK formation was specifically inhibited by the use of the potent inhibitor of initiator caspases, CrmA, and pointed to the involvement of effector caspases in the formation of proplatelet MKs, mimicked by introduction of active caspase-8 into cultured MKs. However, the compartmentalized nature of MK apoptosis leading to proplatelet and platelet production was further emphasized by our finding that caspase-9, a key component of the apoptosome after mitochondrial cytochrome c release, was present in progenitor MKs but was excluded from platelets, accounting for the caspase-independent nature of constitutive platelet death.
Our findings differ importantly from the probable role of caspases in generation of the erythrocyte, another anucleate cell (Zermati et al., 2001). Rather than production of multiple daughter cells from a single progenitor (Stenberg and Levin 1989; Cramer et al., 1997; Italiano et al., 1999), the data suggest that viable erythrocytes transiently activate caspases, resulting in cleavage of a subset of structural proteins that may help lead to the enucleation of the erythroblast (Zermati et al., 2001) to produce a single erythrocyte (Gregory and Eaves, 1978). Nevertheless, it seems clear that evolution has achieved adaptations of the basic program of cell death by apoptosis to produce anucleate cells of critical importance in blood. However, such adaptations do not yield immortal cells; viable platelets arising from compartmentalized MK death are committed to a short life span and later death (Brown et al., 2000), in keeping with nucleated blood cells such as neutrophils (Savill et al., 1989).
Our data also reinforce the concept that plasma membrane changes of apoptosis may be dissociated from the caspase-directed program of nuclear condensation and fragmentation (Knepper-Nicolai et al., 1998; Harper et al., 2001). Clearly, further work will be required to define the mechanisms by which caspase activation in one part of the cell can lead to nuclear changes, whereas mitochondria in a different area of the cell retain their ΔψM, even when cell death is induced by Fas ligation. Nevertheless, because it is believed that there is specific delivery of progenitor cell cytoplasmic components into the extensions of proplatelet MKs (Italiano et al., 1999), it is possible that there is specific exclusion of death pathway components upstream of mitochondria analogous to the exclusion of caspase-9 suggested by our data. Unfortunately, we could not directly test this possibility because platelet yields from MK cultures were insufficient for blotting studies, and the delicate nature of proplatelet-bearing MKs, which tended to lose their processes on manipulation, excluded direct immunofluorescent localization of caspase-9. However, despite our biochemical and functional evidence that platelets lack caspase-9, others have reported its presence (Wolf et al., 1999), even though our data would suggest this should have resulted in caspase-3 cleavage, which we were unable to detect. Indeed, the possibility that we might have artifactually “missed” caspase-9 activity, because calcium-dependent calpains can indirectly block caspase-3 activation by inactivating caspase-9 (Wolf et al., 1999; Chua et al., 2000; Lankiewicz et al., 2000), appeared most unlikely in our studies given the lack of Ca2+, and presence of EGTA, EDTA, and calpain inhibitors within the lysis buffer. Moreover, it should be noted that we were particularly careful to ensure that our platelet preparations were free of low grade contamination by leukocytes, which may have served as an artifactual source of caspase-9 (Webb et al., 2000; Bantel et al., 2001).
Future studies should also examine whether retention of ΔψM and plasma membrane asymmetry (denying recognition by phagocytes) in those parts of the dying MKs destined to become platelets reflect special expression of anti-apoptotic members of the Bcl-2 family. Such proteins have been implicated in platelet production because knockout of the pro-apoptotic Bcl-2–relative Bim results in reduced circulating platelet counts, whereas other circulating blood cells are increased (Bouillet et al., 1999), with identical effects being observed when the balance of Bcl-2 family members is similarly shifted toward survival of hemopoietic cells by targeted overexpression of Bcl-2 (Ogilvy et al., 1999). Although such observations provide indirect support for MK apoptosis being involved in platelet production, such data require careful interpretation in the light of intriguing observations suggesting special patterns of antiapoptotic Bcl-XL expression in MKs producing platelets (Sanz et al., 2001). Thus, although Bcl-XL was greatly up-regulated (Terui et al., 1998; Sanz et al., 2001) as MKs differentiated from CD34+ progenitors and was detectable in proplatelet fragments and mature platelets, potentially explaining the maintenance of ΔψM, Bcl-XL was apparently absent from senescent/apoptotic MK cell bodies (Sanz et al., 2001). A further degree of complexity is found with overexpression of Bcl-2 in CD34+ progenitor cells cultured in vitro where proplatelet extensions were inhibited (De Botton et al., 2002). This further suggests that Bcl-2 members may be differentially targeted to different populations of mitochondria, where Bcl-2 is known to be absent from mature blood platelets (Brown et al., 2000; Sanz et al., 2001). Such data may support sorting of cytoplasmic proteins during formation of proplatelet extensions by MKs and emphasize the future need to overcome technical and logistical problems to follow location of Bcl-XL, caspase-9, and other relevant proteins in proplatelet forming MKs in real time.
Nevertheless, our findings contrast with those of De Botton et al. (2002) in assigning relative importance to cytochrome c and the intrinsic cell death program in the initiation of proplatelet formation. Mitochondrial staining with the ΔψM-sensitive dye JC-1 suggested that during platelet formation the mitochondria retained an active electron-transport chain and were actively sorted to the proplatelets, consistent with the high energy demands that are placed on platelet formation (Watanabe et al., 1990) and the subsequent utilization of oxidative phosphorylation by platelets throughout their life span (Doery et al., 1970; Akahori et al., 1995; Parker and Gralnick, 1997). Given the intensity of the JC-1 stain, we also failed to observe mitochondria with altered fluorescence properties, indicative of a loss in ΔψM even when distinctive chromosomal margination of the attached MK body was apparent. The ability of mitochondria to resist participation in an apoptotic program that results in proplatelet formation would also be assisted by the high levels of reported Bcl-XL (Terui et al., 1998; Sanz et al., 2001), thus able to inhibit an intrinsic (but not extrinsic) cell death program. This is in keeping with further results where the enforced release of cytochrome c by antagonism of anti-apoptotic Bcl-XL with “BH3 mimetics” resulted in the loss of ΔψM and formation of platelet-like progeny that were dysfunctional (unpublished data). Therefore, it is unlikely that cytochrome c would be released into the cytosol of MKs destined to extend processes and form functional platelets. Furthermore, to our knowledge there is no precedent for the long-term maintenance of ΔψM after release of mitochondrial cytochrome c.
Our findings further support the idea that abnormalities of circulating platelet numbers, important in disorders of hemostasis and in thrombotic diseases such as stroke and myocardial infarction, could reflect abnormalities in control of MK apoptosis. Indeed, in keeping with our findings, mice deficient in components of the Fas death pathway exhibit thrombocytopenia (Rieux-Laucat et al., 1995; Le Deist et al., 1996), which has largely been attributed to autoimmune thrombocytopenia. Indeed, one might speculate that pharmacological manipulation of MK apoptosis, and the Fas pathway in particular, might provide a novel therapeutic strategy for the management and control of thrombostasis.
Materials and methods
All chemicals were of analytical reagent grade and purchased from Sigma-Aldrich, and all culture media and supplements were obtained from GIBCO BRL, unless stated otherwise.
Cell culture
The human megakaryoblastic cell line MEG-01 (Ogura et al., 1985; European Collection of Cell Cultures) and Jurkat T cells were routinely maintained in RPMI 1640 supplemented with 10% FCS, 2 mM l-glutamine, and 0.1 mg/ml penicillin/streptomycin. The human megakaryoblastic cell line SET-2 (Uozumi et al., 2000; provided by K. Uozumi, Kagoshima University, Japan) was maintained in DME supplemented with 10% FCS, 2 mM l-glutamine, 0.1 mg/ml penicillin/streptomycin, 10 μM 2-mercaptoethanol, and 10% nonessential amino acids. Generation of primary murine MKs is described elsewhere (Drachman et al., 1997; Rojnuckarin and Kaushansky, 2001). In brief, bone marrow was flushed from femurs of Balb-C mice and a single cell suspension was obtained by gentle pipetting. Cells were incubated overnight in StemSpan™ H2000 (StemCell Technologies, Inc.) with 40 ng/ml human recombinant TPO (PeproTech), and all nonadherent cells and media were transferred to fresh wells and incubated for a further 72 h. Enrichment of primary or cell line–derived mature MKs was by velocity sedimentation through a discontinuous 1.5%, 3.0% BSA gradient at 1 g, collecting those cells reaching the bottom within 30 min. Purified primary MKs were replated and cultured with 30 ng/ml TPO and 10% normal human plasma. MKs were treated with the following reagents: 100 or 10 μM zVAD-fmk (Bachem), 10 μM zDEVD-fmk, 10 μM zIETD-fmk (Calbiochem); 50 ng/ml anti-Fas agonistic antibody CH.11 (Upstate Biotechnology), 1 μg/ml anti-Fas antagonistic antibody ZB4 (Upstate Biotechnology), 5 ng/ml soluble Fas ligand and enhancer (Qbiogene), 25 ng/ml TNF-α (R&D Systems), and 250 ng/ml JO-2 (BD Biosciences). Phagocytic assays with MDMs were as described previously (Brown et al., 2000). Direct protein transfection was achieved using BioPORTER® II (Gene Therapy Systems) as instructed. In brief, CrmA (Kamiya Biomedical) or caspase-8 (CHEMICON International) in PBS was complexed with the BioPORTER® reagent for 5 min before the addition of cells in serum-free media. After 3 h of incubation, an equal volume of 20% serum media was applied, and cells were cultured for a further 8 h before enumeration of proplatelet-bearing MKs.
Ex vivo bone culture
Trabecular bone from femoral heads was isolated from a 58-yr old male patient undergoing hip surgery, machined with high precision to cylindrical cores under sterile conditions, and inserted into the loading chambers of a Zetos™ bone perfusion system (Smith and Jones, 1998; EOBM, Philipps-University, Marburg, Germany). Each core was maintained at 37°C /5% CO2 and perfused with 5 ml DME, recirculated at a rate of 5 ml/h. On day 5, the circulating media was replaced with 5 ml of fresh media containing 50 ng/ml Fas agonistic antibody CH.11 and/or 100 μM zVAD-fmk. The fresh media was then perfused and recirculated for 18 h at 5 ml/h before the eluent was collected and the bone core flushed with 5 ml of PBS. Large cellular material was removed by centrifugation (200 g for 5 min) and remaining supernatant components concentrated by centrifugation (1,500 g for 15 min). Resuspended pellets were stained for GpIIb (Serotec) and GpIIIa (CALTAG Laboratories), and analyzed on a FACScan™ (Becton Dickinson), with reference to a fixed count of 10-μm beads (Polysciences, Inc.) for platelet enumeration.
Flow cytometry
Culture supernatants containing platelets were separated by centrifugation at 300 g for 2 min, and platelets were enumerated by reference to a fixed count of 10-μm polystyrene beads (Polysciences, Inc.). Functional platelets were distinguished from cellular debris by a characteristic increase in side scatter on shape change after 0.5 U/ml thrombin or 3 μM ADP treatment, and an absence of 0.3 μl/ml annexin-V binding (Boehringer) to the shape changed population (Otterdal et al., 2001). All conditions contained 10 μg/ml propidium iodide as a dead cell gate. Transient calcium flux was detected using 10 μm Fluo3-AM (Molecular Probes, Inc.). Agonist-induced activation of the fibrinogen receptor (GpIIb/IIIa) by 3 U/ml thrombin was assessed by increased binding of 2.5 μg/ml Fibrinogen-Alexa Fluor (Molecular Probes, Inc.). Samples were analyzed on a FACScalibur™ system (Becton Dickinson) using CellQuest software. Immunofluorescent labeling of intact platelets for ΔψM was performed by adding 5 μl of platelets to 400 μl of PBS w/o Ca2+ containing 10 μg/ml JC-1 (Molecular Probes, Inc.), followed by incubation for 20 min at 37°C, before sampling by flow cytometry. Deliberate uncoupling of the respiratory chain with loss of platelet ΔψM was achieved by direct addition of 10 μg/ml of the protonophore mCCCP.
Microscopy
Images were captured on an inverted microscope (Axiovert S100; Carl Zeiss MicroImaging, Inc.) equipped with a CoolSNAP CCD camera and OpenLab 3.0 image analysis software (ImproVision). Confocal images were captured on a confocal microscope and software system (TCS NT; Leica). Active caspases were detected using CaspaTag™ (Serologicals) as instructed. ΔψM was qualitatively determined using JC-1 (Molecular Probes, Inc.) at a final concentration of 5 μg/ml. Nuclear morphology was detected using 2 μg/ml Hoechst 33342. For TEM cells or culture supernatants were fixed with excess 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (EM grade I), followed by embedding within a fibrin plug as described previously (Brown et al., 2000). Plugs were subsequently treated as normal resected tissue and processed with osmium tetroxide, lead citrate, and araldite embedding, followed by 80-nm ultra thin sectioning. Samples were analyzed on an electron microscope (CM12; Philips).
Platelet isolation and culture
Freshly drawn venous blood was obtained from aspirin-free healthy donors, citrated (0.33%; Pharma Hameln), and PRP was prepared by centrifugation (350 g, 20 min). Washed platelets were prepared by diluting PRP with 5 vol of HBSS, pH 6.4, containing EDTA (4 mM final) into a round-bottom capped polystyrene centrifuge tube before centrifugation (280 g for 20 min). After washing, platelets were resuspended in HBSS, pH 6.4, and maintained at 37°C in a closed (capped) tube at 3 × 108 ml. Low levels of contaminating leukocytes were removed from platelet preparations after two successive rounds of centrifugation (220 g for 5 min) in which the supernatant was retained each time.
Cell free apoptosis and cytochrome c immunoprecipitation
Jurkat T cell and platelet lysates were prepared by resuspending cells in lysis buffer (20 mM Hepes, pH 7.5, 10 mM KCl, 1.5 mM MgCl, 1mM EDTA, 1 mM EGTA, 1 mM DTT, 100 μm PMSF, 10 μg/ml leupeptin, and 2 μg/ml aprotinin) and were incubating for 10 min on ice. Platelets were subjected to three cycles of freeze-thaw. Cells were further disrupted by passing through a 25 G needle 10 times. Cell free apoptosis was initiated by addition of 10 μg/ml cytochrome c and 1 mM dATP, and in some experiments, 0.1 U/ml human recombinant caspase-9 (CHEMICON International), followed by incubation at 37°C. At time points indicated aliquots were removed and directly added to x4 SDS Laemmli sample buffer, and immediately boiled for 3 min. For cytochrome c, washed platelets were resuspended in ice-cold 10 mM Hepes buffer (containing 1 mM EDTA, 1 mM 1,10-phenanthroline, 1 mM PMSF, 1 mM benzamidine, 10 μM pepstatin, 10 μM leupeptin, and 10 μM antipain, pH 8.0) and lysed after two rounds of freeze-thaw. The lysed platelets were centrifuged (13,000 g for 10 min) to yield a cytosolic supernatant, whereas the pellet was resuspended in lysis buffer before centrifuging again and dissolving the pellet in 1% TX-100 to release mitochondrial cytochrome c. The soluble fractions were then precleared with a control IgG and Protein G agarose before immunoprecipitating cytochrome c with clone 6H2.B4 (BD Biosciences) and boiling in SDS sample buffer. Cycloheximide (20 μg/ml)-treated Jurkats were used as a positive control and prepared identically.
SDS-PAGE and immunoblotting
SDS-PAGE and immunodetection of transblotted protein to PVDF membranes was performed as described previously (Brown et al., 1997). Western blots were probed with anti-caspase-3 pAb, anti-caspase-9 pAb and mAb (clone B40), anti-APAF-1 pAb, and anti-cytochrome c mAb (clone 7H8.2C12), all purchased from BD Biosciences. Molecular weight markers used were Rainbow markers, SDS-7B, or BenchMark (GIBCO BRL).
Acknowledgments
We thank Steve Mitchell for his technical assistance with TEM preparation, Marita Kratz for technical assistance with the bone core work, Jeremy Hughes for helpful discussions, and Everett Smith for development of the bone core system with D. Jones.
This work was supported in part by the Wellcome Trust (047273, to J. Savill), Chief Scientists Office of the Scottish Executive (CZB/4/8, to S.B. Brown), European Space Agency Microgravity Applications Programme (AO 99-122, to D.B. Jones), the AO ASIF Davos (200J and O2 J7, to D.B. Jones), and a National Kidney Research Foundation Studentship (JS1, to M.C.H. Clarke).
Footnotes
Abbreviations used in this paper: ΔψM, inner mitochondrial membrane potential; MDM, monocyte-derived macrophage; MK, megakaryocyte; PRP, platelet-rich plasma; PS, phosphatidylserine; TEM, transmission electron microscopy; TPO, thrombopoietin.
References
- Akahori, M., Y. Uedono, K. Yamagami, N. Takeyama, Y. Kitazawa, and T. Tanaka. 1995. Hypoxia alters the energy metabolism and aggregation of washed human platelets. Haematologia (Budap). 26:191–198. [PubMed] [Google Scholar]
- Bantel, H., B. Sinha, W. Domschke, G. Peters, K. Schulze-Osthoff, and R.U. Janicke. 2001. alpha-Toxin is a mediator of Staphylococcus aureus-induced cell death and activates caspases via the intrinsic death pathway independently of death receptor signaling. J. Cell Biol. 155:637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battinelli, E., S.R. Willoughby, T. Foxall, C.R. Valeri, and J. Loscalzo. 2001. Induction of platelet formation from megakaryocytoid cells by nitric oxide. Proc. Natl. Acad. Sci. USA. 98:14458–14463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillet, P., D. Metcalf, D.C. Huang, D.M. Tarlinton, T.W. Kay, F. Kontgen, J.M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 286:1735–1738. [DOI] [PubMed] [Google Scholar]
- Brown, S.B., K. Bailey, and J. Savill. 1997. Actin is cleaved during constitutive apoptosis. Biochem. J. 323:233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, S.B., M.C. Clarke, L. Magowan, H. Sanderson, and J. Savill. 2000. Constitutive death of platelets leading to scavenger receptor-mediated phagocytosis. A caspase-independent cell clearance program. J. Biol. Chem. 275:5987–5996. [DOI] [PubMed] [Google Scholar]
- Chua, B.T., K. Guo, and P. Li. 2000. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J. Biol. Chem. 275:5131–5135. [DOI] [PubMed] [Google Scholar]
- Cramer, E.M., F. Norol, J. Guichard, J. Breton-Gorius, W. Vainchenker, J.M. Masse, and N. Debili. 1997. Ultrastructure of platelet formation by human megakaryocytes cultured with the Mpl ligand. Blood. 89:2336–2346. [PubMed] [Google Scholar]
- De Botton, S., S. Sabri, E. Daugas, Y. Zermati, J.E. Guidotti, O. Hermine, G. Kroemer, W. Vainchenker, and N. Debili. 2002. Platelet formation is the consequence of caspase activation within megakaryocytes. Blood. 100:1310–1317. [DOI] [PubMed] [Google Scholar]
- Doery, J.C., J. Hirsh, and I. Cooper. 1970. Energy metabolism in human platelets: interrelationship between glycolysis and oxidative metabolism. Blood. 36:159–168. [PubMed] [Google Scholar]
- Drachman, J.G., D.F. Sabath, N.E. Fox, and K. Kaushansky. 1997. Thrombopoietin signal transduction in purified murine megakaryocytes. Blood. 89:483–492. [PubMed] [Google Scholar]
- Garcia-Calvo, M., E.P. Peterson, B. Leiting, R. Ruel, D.W. Nicholson, and N.A. Thornberry. 1998. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 273:32608–32613. [DOI] [PubMed] [Google Scholar]
- Gregory, C.J., and A.C. Eaves. 1978. Three stages of erythropoietic progenitor cell differentiation distinguished by a number of physical and biologic properties. Blood. 51:527–537. [PubMed] [Google Scholar]
- Harper, L., P. Cockwell, D. Adu, and C.O. Savage. 2001. Neutrophil priming and apoptosis in anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Kidney Int. 59:1729–1738. [DOI] [PubMed] [Google Scholar]
- Italiano, J.E., Jr., P. Lecine, R.A. Shivdasani, and J.H. Hartwig. 1999. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J. Cell Biol. 147:1299–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr, J.F., A.H. Wyllie, and A.R. Currie. 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 26:239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck, R.M., E. Bossy-Wetzel, D.R. Green, and D.D. Newmeyer. 1997. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 275:1132–1136. [DOI] [PubMed] [Google Scholar]
- Knepper-Nicolai, B., J. Savill, and S.B. Brown. 1998. Constitutive apoptosis in human neutrophils requires synergy between calpains and the proteasome downstream of caspases. J. Biol. Chem. 273:30530–30536. [DOI] [PubMed] [Google Scholar]
- Lankiewicz, S., L.C. Marc, B.N. Truc, A.J. Krohn, M. Poppe, G.M. Cole, T.C. Saido, and J.H. Prehn. 2000. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J. Biol. Chem. 275:17064–17071. [DOI] [PubMed] [Google Scholar]
- Le Deist, F., J.F. Emile, F. Rieux-Laucat, M. Benkerrou, I. Roberts, N. Brousse, and A. Fischer. 1996. Clinical, immunological, and pathological consequences of Fas-deficient conditions. Lancet. 348:719–723. [DOI] [PubMed] [Google Scholar]
- Li, P., D. Nijhawan, I. Budihardjo, S.M. Srinivasula, M. Ahmad, E.S. Alnemri, and X. Wang. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 91:479–489. [DOI] [PubMed] [Google Scholar]
- Liu, X., C.N. Kim, J. Yang, R. Jemmerson, and X. Wang. 1996. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 86:147–157. [DOI] [PubMed] [Google Scholar]
- Marchetti, P., M. Castedo, S.A. Susin, N. Zamzami, T. Hirsch, A. Macho, A. Haeffner, F. Hirsch, M. Geuskens, and G. Kroemer. 1996. Mitochondrial permeability transition is a central coordinating event of apoptosis. J. Exp. Med. 184:1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvy, S., D. Metcalf, C.G. Print, M.L. Bath, A.W. Harris, and J.M. Adams. 1999. Constitutive Bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc. Natl. Acad. Sci. USA. 96:14943–14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura, M., Y. Morishima, R. Ohno, Y. Kato, N. Hirabayashi, H. Nagura, and H. Saito. 1985. Establishment of a novel human megakaryoblastic leukemia cell line, MEG- 01, with positive Philadelphia chromosome. Blood. 66:1384–1392. [PubMed] [Google Scholar]
- Otterdal, K., T.M. Pedersen, and N.O. Solum. 2001. Platelet shape change induced by the peptide YFLLRNP. Thromb. Res. 103:411–420. [DOI] [PubMed] [Google Scholar]
- Parker, R.I., and H.R. Gralnick. 1997. Energy-dependent expression of platelet-von Willebrand factor on the surface of unstimulated and stimulated platelets. J. Lab. Clin. Med. 130:520–529. [DOI] [PubMed] [Google Scholar]
- Petit, P.X., H. Lecoeur, E. Zorn, C. Dauguet, B. Mignotte, and M.L. Gougeon. 1995. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J. Cell Biol. 130:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley, J.M., and C.J. Haller. 1983. Fate of senescent megakaryocytes in the bone marrow. Br. J. Haematol. 53:277–287. [DOI] [PubMed] [Google Scholar]
- Rieux-Laucat, F., F. Le Deist, C. Hivroz, I.A. Roberts, K.M. Debatin, A. Fischer, and J.P. de Villartay. 1995. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 268:1347–1349. [DOI] [PubMed] [Google Scholar]
- Robertson, A.M., C.C. Bird, A.W. Waddell, and A.R. Currie. 1978. Morphological aspects of glucocorticoid-induced cell death in human lymphoblastoid cells. J. Pathol. 126:181–187. [DOI] [PubMed] [Google Scholar]
- Rojnuckarin, P., and K. Kaushansky. 2001. Actin reorganization and proplatelet formation in murine megakaryocytes: the role of protein kinase calpha. Blood. 97:154–161. [DOI] [PubMed] [Google Scholar]
- Salvioli, S., A. Ardizzoni, C. Franceschi, and A. Cossarizza. 1997. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 411:77–82. [DOI] [PubMed] [Google Scholar]
- Sanz, C., I. Benet, C. Richard, B. Badia, E.J. Andreu, F. Prosper, and J.L. Fernandez-Luna. 2001. Antiapoptotic protein Bcl-xL is up-regulated during megakaryocytic differentiation of CD34+ progenitors but is absent from senescent megakaryocytes. Exp. Hematol. 29:728–735. [DOI] [PubMed] [Google Scholar]
- Savill, J.S., A.H. Wyllie, J.E. Henson, M.J. Walport, P.M. Henson, and C. Haslett. 1989. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest. 83:865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, E., and D.B. Jones, inventors and assignees. 1998 July 6. A combined perfusion and mechanical loading system for explanted bone. US patent 6,171,812.
- Stenberg, P.E., and J. Levin. 1989. Mechanisms of platelet production. Blood Cells. 15:23–47. [PubMed] [Google Scholar]
- Takeuchi, K., M. Satoh, H. Kuno, T. Yoshida, H. Kondo, and M. Takeuchi. 1998. Platelet-like particle formation in the human megakaryoblastic leukaemia cell lines, MEG-01 and MEG-01s. Br. J. Haematol. 100:436–444. [DOI] [PubMed] [Google Scholar]
- Tavassoli, M., and M. Aoki. 1989. Localization of megakaryocytes in the bone marrow. Blood Cells. 15:3–14. [PubMed] [Google Scholar]
- Terui, Y., Y. Furukawa, J. Kikuchi, S. Iwase, K. Hatake, and Y. Miura. 1998. Bcl-x is a regulatory factor of apoptosis and differentiation in megakaryocytic lineage cells. Exp. Hematol. 26:236–244. [PubMed] [Google Scholar]
- Uozumi, K., M. Otsuka, N. Ohno, T. Moriyama, S. Suzuki, S. Shimotakahara, I. Matsumura, S. Hanada, and T. Arima. 2000. Establishment and characterization of a new human megakaryoblastic cell line (SET-2) that spontaneously matures to megakaryocytes and produces platelet-like particles. Leukemia. 14:142–152. [DOI] [PubMed] [Google Scholar]
- Watanabe, Y., M. Taniguchi, N. Fukamachi, M. Sakuma, and B. Kobayashi. 1990. Effects of gangliosides on cell maturation of murine megakaryocytes in a liquid culture system. Cell Struct. Funct. 15:79–84. [DOI] [PubMed] [Google Scholar]
- Webb, P.R., K.Q. Wang, D. Scheel-Toellner, J. Pongracz, M. Salmon, and J.M. Lord. 2000. Regulation of neutrophil apoptosis: a role for protein kinase C and phosphatidylinositol-3-kinase. Apoptosis. 5:451–458. [DOI] [PubMed] [Google Scholar]
- Wolf, B.B., J.C. Goldstein, H.R. Stennicke, H. Beere, G.P. Amarante-Mendes, G.S. Salvesen, and D.R. Green. 1999. Calpain functions in a caspase-independent manner to promote apoptosis-like events during platelet activation. Blood. 94:1683–1692. [PubMed] [Google Scholar]
- Wyllie, A.H. 1980. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 284:555–556. [DOI] [PubMed] [Google Scholar]
- Yang, J., X. Liu, K. Bhalla, C.N. Kim, A.M. Ibrado, J. Cai, T.I. Peng, D.P. Jones, and X. Wang. 1997. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 275:1129–1132. [DOI] [PubMed] [Google Scholar]
- Zauli, G., M. Vitale, E. Falcieri, D. Gibellini, A. Bassini, C. Celeghini, M. Columbaro, and S. Capitani. 1997. In vitro senescence and apoptotic cell death of human megakaryocytes. Blood. 90:2234–2243. [PubMed] [Google Scholar]
- Zermati, Y., C. Garrido, S. Amsellem, S. Fishelson, D. Bouscary, F. Valensi, B. Varet, E. Solary, and O. Hermine. 2001. Caspase activation is required for terminal erythroid differentiation. J. Exp. Med. 193:247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]