

Abstract
cAMP controls many cellular processes mainly through the activation of protein kinase A (PKA). However, more recently PKA-independent pathways have been established through the exchange protein directly activated by cAMP (Epac), a guanine nucleotide exchange factor for the small GTPases Rap1 and Rap2. In this report, we show that cAMP can induce integrin-mediated cell adhesion through Epac and Rap1. Indeed, when Ovcar3 cells were treated with cAMP, cells adhered more rapidly to fibronectin. This cAMP effect was insensitive to the PKA inhibitor H-89. A similar increase was observed when the cells were transfected with Epac. Both the cAMP effect and the Epac effect on cell adhesion were abolished by the expression of Rap1–GTPase-activating protein, indicating the involvement of Rap1 in the signaling pathway. Importantly, a recently characterized cAMP analogue, 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate, which specifically activates Epac but not PKA, induced Rap-dependent cell adhesion. Finally, we demonstrate that external stimuli of cAMP signaling, i.e., isoproterenol, which activates the Gαs-coupled β2-adrenergic receptor can induce integrin-mediated cell adhesion through the Epac-Rap1 pathway. From these results we conclude that cAMP mediates receptor-induced integrin-mediated cell adhesion to fibronectin through the Epac-Rap1 signaling pathway.
Keywords: integrins; cyclic nucleotides; GTPases; guanine nucleotide exchange factor; cell adhesion
Introduction
cAMP is a common second messenger controlling many cellular processes. Protein kinase A (PKA)* is a general receptor for cAMP, resulting in the phosphorylation of a large variety of cellular targets. Specificity is regulated by A kinase anchoring proteins that target PKA to specific regions in the cell. A few years ago we discovered an additional cAMP target, exchange protein directly activated by cAMP (Epac)1. This protein and its close relative Epac2 contain cAMP-binding domains very similar to the cAMP-binding domains in the regulatory subunit of PKA and are exchange factors of the small GTPases Rap1 and Rap2 (de Rooij et al., 1998, 2000; Kawasaki et al., 1998).
Rap1 is a GTPase of the Ras superfamily, which functions as a molecular “switch,” cycling between inactive GDP- and active GTP-bound forms. Specific guanine nucleotide exchange factors are the “on switches,” and GTPase-activating proteins (GAPs) are the “off switches” (for review see Bos et al., 2001). Rap1 was initially identified in a screen for proteins that can suppress the transformed phenotype of fibroblasts transformed by oncogenic K-Ras (Kitayama et al., 1989), providing a model in which Rap1 functions as an antagonist of Ras signaling mainly by trapping Ras effectors (Raf-1) in an inactive complex. However, from numerous reports accumulated so far it is evident that Rap1 signaling is important in itself and independently of Ras regulates several important cellular processes (Bos et al., 2001). One of the most consistent findings is the involvement of Rap1 in integrin-mediated cell adhesion (Caron et al., 2000; Katagiri et al., 2000; Reedquist et al., 2000; Arai et al., 2001; Ohba et al., 2001; de Bruyn et al., 2002; Sebzda et al., 2002). Integrins are heterodimeric cell adhesion molecules consisting of one of several different α chains and one of at least five different β chains. One of the first indications was that in 32D cells granulocyte colony stimulating factor-induced cell adhesion could be abolished by the introduction of Spa1, a GAP for Rap proteins (Tsukamoto et al., 1999). This finding was followed by three independent observations showing a role for Rap1 in the inside-out signaling to integrins. First, in Jurkat cells introduction of Rap1 induced integrin αLβ2 (LFA1)-mediated adhesion to the intercellular adhesion molecule. Importantly, adhesion induced by ligation of the T cell receptor was inhibited by introduction of an interfering mutant of Rap1 (Katagiri et al., 2000). Second, also in Jurkat cells ligation of the adhesion molecule CD-31 induced activation of αLβ2, which was inhibited by blocking Rap1 signaling (Reedquist et al., 2000). Finally, in a macrophage cell line, complement-mediated phagocytosis, which requires activated αMβ2, was abolished by inhibition of Rap1 signaling (Caron et al., 2000). Other studies reached the same conclusion for integrins with a β1 chain, i.e., α5β1 (Arai et al., 2001) and for integrins with a β3 chain, i.e., αIIbβ3 (Bertoni et al., 2002). Recently, it was shown that in mice expressing active Rap1 in their T cell compartment both the thymocytes and mature T cells exhibited increased integrin-mediated cell adhesion. In addition, these cells showed enhanced T cell receptor–mediated responses (Sebzda et al., 2002). From the above results we hypothesized that cAMP or signals that raise cAMP levels may regulate integrin-mediated cell adhesion through Epac and Rap1. We have tested this model and found that indeed cAMP was able to induce integrin-mediated cell adhesion to fibronectin.
It has been reported previously that PKA was a key part in a signaling pathway activated by mAb 12G10, an antibody that can activate β1 integrins and induce integrin-mediated cell–cell and cell–substrate adhesion in human fibrosarcoma cells (Whittard and Akiyama, 2001). To distinguish which of the two independent cAMP signaling pathways, mediated by either PKA or Epac, is involved in integrin regulation in Ovcar3 cells, we used a novel analogue of cAMP, 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8CPT-2Me-cAMP) that specifically targets Epac and not PKA. We demonstrate that an Epac-Rap1 pathway mediates this effect independently of PKA. We also implicate physiological consequences of intracellular increases in cAMP in vivo by demonstrating that agonist stimulation of the β2-adrenergic receptor is linked to increased cell adhesion via Rap1.
Results and discussion
To investigate whether cAMP could induce integrin-mediated cell adhesion, we used ovarian carcinoma cells (Ovcar3), since these cells express the β1 integrin chain in association with α chains 1–6 and αv, with the α5β1 and αvβ3 integrins mediating binding to fibronectin (Cannistra et al., 1995; Buczek-Thomas et al., 1998). Cytomegalovirus-luciferase–transfected cells were detached with trypsin and allowed to reexpress cell surface markers. The cells were seeded onto fibronectin-coated multiwell plates in the presence or absence of 8-Br-cAMP, and the amount of cells that adhered after a certain period of time was quantified. We observed that 8-Br-cAMP augmented cell adhesion and activated Rap1 in a concentration-dependent manner to fibronectin (EC50, ∼0.2–0.5 mM) (Fig. 1, A and B). Rap1 was activated rapidly and remained active for at least 3 h. 8-Br-cAMP–induced adhesion was also observed using a different promoter (thymidine kinase [TK]–luciferase) driving luciferase expression and a direct method of measuring adhesion by counting cells (unpublished data). Cell adhesion induced by cAMP was insensitive to the PKA inhibitor H-89 when cells were pretreated for a short time just before adhesion (Fig. 1 C, Short). It has been reported that detachment of cells rapidly and transiently activates PKA, one of the well-established targets of cAMP (Howe and Juliano, 2000), raising the possibility that if a potential PKA substrate with a sustained phosphorylation profile was involved, addition of H-89 at a later time (post-PKA activation) may falsely imply a PKA-independent mechanism. However, when cells were treated with H-89 before trypsinization and throughout the recovery period, we found that cAMP-induced adhesion was not blocked (Fig. 1 C, Long), indicating that indeed PKA was not involved. As a control for H-89 activity, we measured cAMP-induced phosphorylation of the direct PKA target CREB (Gonzalez and Montminy, 1989) and ERK, which is also PKA-dependent (Fig. 1 D). Activation of Rap1, which is independent of PKA (de Rooij et al., 1998; Kawasaki et al., 1998; Enserink et al., 2002) was measured also. From these results we conclude that in Ovcar3 cells cAMP can induce cell adhesion to fibronectin independently of PKA.
Figure 1.

cAMP induces cell adhesion to fibronectin in a PKA-independent manner. (A) Treatment with 8-Br-cAMP induces adhesion to fibronectin. Ovcar3 cells were transiently transfected with CMV-luciferase plasmid, and cells adhering to fibronectin (2 μg/ml) in the presence of increasing concentrations of 8-Br-cAMP were quantified as described in Materials and methods. (B) 8-Br-cAMP induces Rap1 activation. (Top) Ovcar3 cells were treated with increasing concentrations 8-Br-cAMP for 15 min. Cells were lysed, and equal amounts of cell lysate were analyzed for activation of Rap1 (top blot) and CREB (bottom blot). Total levels of Rap1 in cell lysates are shown (middle blot). (Bottom) Ovcar3 cells were treated with 1 mM 8-Br-cAMP for the indicated times. Cells were lysed as above and analyzed for activation of Rap1 (top blot) and CREB (bottom blot). Total Rap1 levels are shown (middle blot). (C) 8-Br-cAMP– induced adhesion is independent of PKA. Ovcar3 cells transiently transfected with CMV-luciferase plasmid were either preincubated at 37°C for 30 min with the PKA inhibitor H-89 (10 μM) 30 min before seeding onto the wells (Short), or H-89 was added 30 min before trypsinization and during the recovery period (Long) and seeded onto wells with or without 8-Br-cAMP. Cells were allowed to adhere for 1 h, and nonadherent cells were removed. The percentage of adherent cells was quantified and plotted relative to unstimulated cells (range from 2–10%). The plot shown is representative of two (long pretreatment) and five (short pretreatment) experiments each in triplicate. Error bars represent SD. (D) Activation of CREB and ERK but not Rap1 is blocked by H-89. Ovcar3 cells were pretreated with either H-89 or carrier for 30 min followed by stimulation with 8-Br-cAMP for 15 min. Cells were lysed, and equal amounts of cell lysates were incubated with precoupled GST-RalGDS-RBD, and activation of Rap1 was analyzed by immunoblotting using a Rap1 antibody. Phosphorylation of CREB and ERK was assayed by Western blotting using phospho-specific antibodies.
Our finding that the induction of integrin-mediated cell adhesion by cAMP is independent of PKA suggested that Epac-Rap1 might be mediating this effect. To further test this idea, Ovcar3 cells were transiently transfected with Epac1. This resulted in an increase in basal adhesion to fibronectin, which was further increased by stimulation with 8-Br-cAMP (Fig. 2 A), suggesting that Epac mediates cAMP-induced cell adhesion. This observation was further strengthened by the introduction of Rap1GAPII, an inhibitor of Rap1 (Mochizuki et al., 1999), which attenuated Epac-induced cell adhesion (Fig. 2 A). These results show that ectopic expression of Epac is sufficient to induce Rap1-dependent cell adhesion to fibronectin, which can be enhanced by additional stimulation with cAMP. It should be noted that although Rap1GAPII is more effective on Rap1 than on Rap2, we cannot exclude a role for Rap2 in this process.
Figure 2.
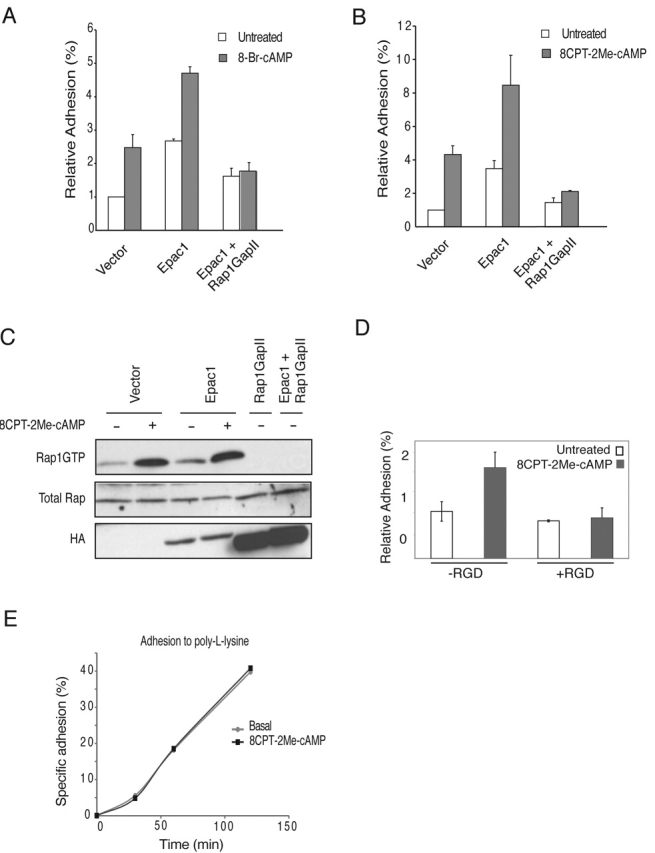
Overexpression of Epac1 increases cAMP-induced cell adhesion, which is Rap1GAPII sensitive. (A) 8-Br-cAMP-Epac1–induced cell adhesion is blocked by Rap1GAPII. Ovcar3 cells were transiently transfected with TK-luciferase plasmid and either mock DNA (vector), HA-tagged Epac1, or HA-tagged Rap1GAPII where indicated. Cells were stimulated with 8-Br-cAMP, and adhesion of cells to fibronectin was quantified. The percentage of adherent cells was plotted relative to unstimulated, mock-transfected cells. Representative data performed in triplicate are shown, and error bars represent SD. The experiments were repeated at least four times with identical results. (B) 8CPT-2Me-cAMP-Epac1–induced cell adhesion is blocked by Rap1GAPII. Ovcar3 cell were transiently transfected as above. Cells were stimulated with 8CPT-2Me-cAMP, and adhesion of cells to fibronectin was quantified. The percentage of adherent cells was plotted relative to unstimulated, mock-transfected cells. Representative data performed in triplicate are shown, and error bars represent SD. The experiments were repeated at least four times with identical results. (C) 8CPT-2Me-cAMP-Epac1–induced Rap1 activation is blocked by Rap1GAPII. Cells were treated with 50 μM 8CPT-2Me-cAMP for 15 min, lysed, and GTP-bound Rap1 levels were determined as described in Materials and methods (top). Rap1 protein levels were equal (middle), and expression of transfected proteins was confirmed with an anti-HA antibody (bottom). (D) A β1-integrin–blocking peptide containing the RGD sequence present in fibronectin inhibits 8CPT-2Me-cAMP–induced cell adhesion. Ovcar3 cells were pretreated for 20 min with RGD peptide (100 μM) where indicated and seeded in wells with or without 8CPT-2Me-cAMP. Cells were allowed to adhere for 1 h, and nonadherent cells were removed. The percentage of adherent cells was measured and plotted relative to unstimulated cells. Representative data from two experiments performed in triplicate are shown with error bars representing SD. (E) 8CPT-2Me-cAMP does not increase the rate of cell adhesion to poly- l-lysine. Ovcar3 cells were transfected with CMV-luciferase and seeded onto poly-l-lysine–coated plates. At various time points, nonadherent cells were removed and adherent cells were quantified. A representative experiment in triplicate is shown.
To formally exclude the possibility that cAMP and Epac are on parallel pathways, both of which would be required for the induction of cell adhesion, we used a newly characterized analogue of cAMP, 8CPT-2Me-cAMP, which specifically activates Epac but not PKA even at high concentrations (Enserink et al., 2002). As observed with 8-Br-cAMP, stimulation of Epac1-transfected cells with 8CPT-2Me-cAMP further increased cell adhesion to fibronectin (Fig. 2 B) and raised Rap1GTP levels (Fig. 2 C). Expression of Rap1GAPII inhibited adhesion of cells to fibronectin and completely abolished Rap1GTP levels (Fig. 2, B and C), indicating that Rap1 is critically involved in cAMP-induced cell adhesion.
We next investigated whether activation of endogenous Epac is sufficient to induce adhesion to fibronectin. Ovcar3 cells were treated with 8CPT-2Me-cAMP to activate endogenous Epac, which is abundantly expressed in ovary tissue (Kawasaki et al., 1998). Indeed, 8CPT-2Me-cAMP significantly induced cell adhesion to fibronectin (Fig. 2 D). To investigate whether cAMP-induced cell adhesion is indeed mediated by integrins, we pretreated Ovcar3 cells with the β1-integrin–binding arginine, glycine, aspartic acid (RGD) peptide. Peptides containing the RGD amino acid sequence motif bind to β1 integrins and have been shown to block fibronectin binding in ovarian carcinoma cells (Buczek-Thomas et al., 1998). As expected, 8CPT-2Me-cAMP–induced attachment to fibronectin was abolished (Fig. 2 D). 8CPT-2Me-cAMP did not increase the integrin-independent adhesion of Ovcar3 cells to poly-l-lysine (Fig. 2 E). From these results we conclude that activation of endogenous Epac induces integrin-mediated cell adhesion to fibronectin.
8CPT-2Me-cAMP enhanced cell adhesion to fibronectin and induced Rap1 activation at comparable concentrations (EC50, ∼30 μM) (Fig. 3 A). In a time-course analysis, we noted that increased adhesion was already observed after 30 min, which correlated with a rapid and sustained Rap1 activation (Fig. 3 B). As expected, the induction of adhesion and activation of Rap1 were insensitive to the PKA inhibitor H-89 (Fig. 3 C). However, even low levels of Rap1GAPII completely inhibited cAMP-induced adhesion of Ovcar3 cells to fibronectin (Fig. 3 D, left plot). Furthermore, the Rap1-inhibitory proteins Rap1GAPI and Ras-binding domain (RBD) of Ral guanine nucleotide dissociation stimulator (RalGDS) (Reedquist et al., 2000) also inhibited adhesion to fibronectin (Fig. 3 D, left plot). Transfection of cells with Rap1GAPs or RBD of RalGDS did not affect luciferase expression (Fig. 3 D, right plot).
Figure 3.
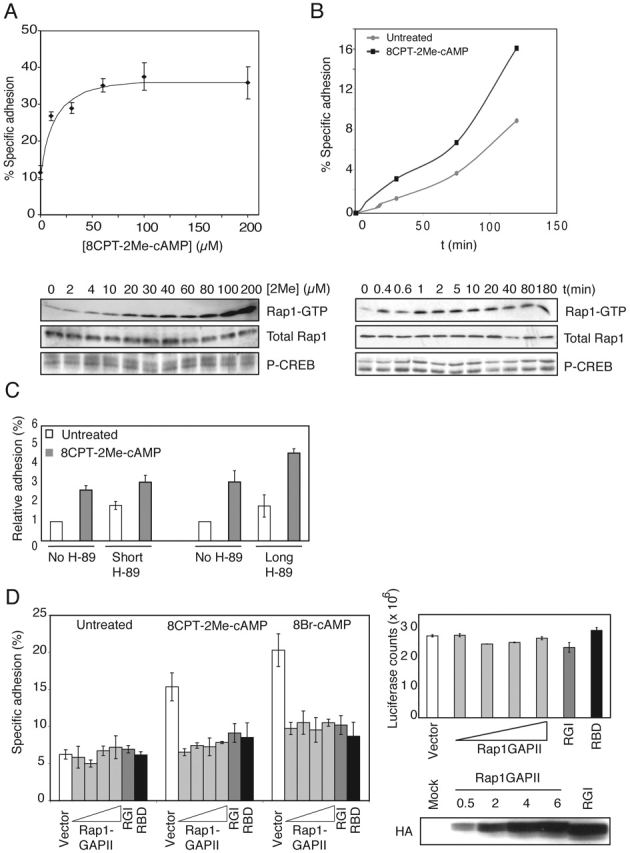
8CPT-2Me-cAMP induces cell adhesion via Epac and Rap1. (A) 8CPT-2Me-cAMP stimulates cell adhesion. (Top) Ovcar3 cells were transiently transfected with CMV- luciferase plasmid and treated with increasing concentrations of 8CPT-2Me-cAMP. Cells adhering to fibronectin (2 μg/ml) were quantified as described in Materials and methods. (Bottom) Ovcar3 cells were treated with increasing concentrations of 8CPT-2Me-cAMP for 15 min, and cells were lysed and analyzed for activation of Rap1 (top blot) and CREB (bottom blot). Total Rap1 levels are shown (middle blot). (B) 8CPT-2Me-cAMP increases the rate of cell adhesion. (Top) Ovcar3 cells were transfected with TK-luciferase and seeded onto fibronectin-coated plates. At various time points, nonadherent cells were removed, and adherent cells were quantified. (Bottom) cells were treated with 60 μM 8CPT-2Me-cAMP for the indicated times. Cells were lysed, and equal amounts of cell lysates were analyzed for activation of Rap1 (top blot) and CREB (bottom blot). Total levels of Rap1 in cell lysates are shown (middle blot). (C) Ovcar3 cells were pretreated with H-89 as described in the legend to Fig. 1 C and seeded onto wells in the absence or presence of 8CPT-2Me-cAMP (100 μM). Cells were allowed to adhere for 1 h, and nonadherent cells were removed. The percentage of adherent cells was quantified and plotted relative to unstimulated cells (range from 2–10%). The plot shown is representative of two (long pretreatment) and five (short pretreatment) experiments, each in triplicate. Error bars represent SD. (D) cAMP-induced adhesion to fibronectin is blocked by inhibitors of Rap1. (Left) Ovcar3 cells were transiently transfected with CMV-luciferase and either mock DNA, increasing concentrations of HA-Rap1GAP II (0.5, 1, 2, or 6 μg, respectively), HA-Rap1GAPI (6 μg), or HA-RBD of RalGDS (6 μg), respectively. Cells were treated with 8-Br-cAMP or 8CPT-2Me-cAMP, and adhesion to fibronectin (5 μg/ml) was determined and plotted relative to unstimulated, mock-transfected cells. Representative data from experiments performed in triplicate are shown with error bars representing SD. The experiments were repeated (Rap1GAPII, at least four times; Rap1GAPI and RBD, twice) with identical results. (Top right) Luciferase counts of total input cells per well in the above experiment are shown with error bars representing SD of triplicates. (Bottom left panel) Expression of HA-Rap1GAPs in the above experiment is shown.
Our observations that cAMP analogs could induce adhesion of Ovcar3 cells to fibronectin prompted us to test whether cAMP-elevating receptors could also mirror the same effect, thereby linking an in vivo cAMP signaling system to integrin activation. The β2-adrenergic receptor (β2-AR) couples to Gαs type of heterotrimeric G proteins, resulting in elevation of intracellular cAMP levels and subsequent activation of PKA and Epac1-Rap1 signaling cascades (Marinissen and Gutkind, 2001; Neves et al., 2002). Ovcar3 cells endogenously express the β2-AR and stimulation with isoproterenol, a ligand for the β2-AR receptor, significantly increased adhesion to fibronectin (Fig. 4 A). Treatment with isoproterenol also induced both activation of Rap1 and phosphorylation of CREB (Fig. 4 B) (EC50 for Rap1 activation and adhesion, ∼0.05 μM). Isoproterenol-induced adhesion was insensitive to short pretreatments with H-89 (Fig. 4 C, Short) but was partially inhibited when exposed very early to H-89 (Fig. 4 C, Long). Therefore, we looked at activation of Rap1 under similar conditions. We observed that after early (Fig. 4 C, Long) pretreatment with H-89, isoproterenol-induced Rap1 activation was clearly inhibited, whereas 8CPT-2Me-cAMP–induced Rap1 activation was not (Fig. 4 C, bottom). Since both 8-Br-cAMP–induced and 8CPT-2Me-cAMP–induced adhesion were not blocked by H-89 (Fig. 1 C and Fig. 3 C), the effect of very early treatment of H-89 on β2-AR signaling could likely be attributed to slow recovery and expression of the β2-AR on the cell surface. This possibility is consistent with the observation that PKA is involved in vesicle fusion (Morgan et al., 1993). Transient transfection of Ovcar3 cells with the β2-AR receptor further enhanced the isoproterenol-induced adhesion to fibronectin, which was sensitive to the Rap1-inactivating protein, Rap1GAPII (Fig. 4 D), showing a critical involvement of Rap1.
Figure 4.

Stimulation of the β2-AR with isoproterenol induces cell adhesion. (A) Isoproterenol induces adhesion to fibronectin. Ovcar3 cells transiently transfected with CMV- luciferase plasmid were treated with increasing concentrations of the β2-AR agonist isoproterenol, and cells adhering to fibronectin (2 μg/ml) were quantified as described in Materials and methods. (B) Isoproterenol induces activation of Rap1 and CREB. (Top) Ovcar3 cells were treated with increasing concentrations of isoproterenol for 5 min. Cells were lysed, and equal amounts of cell lysate were analyzed for activation of Rap1 (top) and CREB (bottom). Total levels of Rap1 in cell lysates are shown (middle blot). (Bottom) Cells were treated with 10 μM of isoproterenol for the indicated times. Cells were lysed, and equal amounts of cell lysate were analyzed for activation of Rap1 (top blot) and CREB (bottom blot). Total levels of Rap1 in cell lysates are shown (middle blot). (C) Isoproterenol-induced adhesion to fibronectin is independent of PKA. (Top) Ovcar3 cells were pretreated with H-89 as described in the legend to Fig. 1 C and seeded onto wells in the absence or presence of isoproterenol (100 μM). Cells were allowed to adhere for 1 h, and nonadherent cells were removed. The percentage of adherent cells was quantified and plotted relative to unstimulated cells (range from 2–10%). The plot shown is representative of two (long pretreatment) and four (short pretreatment) experiments, each in triplicate. Error bars represent SD. (Bottom) Cells were pretreated with either DMSO or H-89 for 30 min before trypsinization and during the recovery period (DMSO and long H-89 treatment, respectively) or during the last 30 min of recovery (short H-89 treatment). Then cells were stimulated with either 50 μM 8CPT-2Me-cAMP for 10 min or isoproterenol for 2 min, respectively. Cells were centrifuged, cell pellets were lysed, and equal amounts of cell lysate were incubated with precoupled GST-RalGDS-RBD, and activation of Rap1 was analyzed on Western blot using a Rap1 antibody. (D) Isoproterenol-induced adhesion to fibronectin is inhibited by Rap1GAPII. Ovcar3 cells were transfected with either mock DNA (Vector) or HA-Rap1GapII alone or in combination with a β2-AR expression vector where indicated. Adhesion of cells to fibronectin in the absence or presence of isoproterenol was quantified. The percentage of adherent cells was plotted relative to unstimulated, mock-transfected cells (range 5–15%). Summarizing data of four (for the left half of the plot) and two (for the right half of the plot) independent experiments performed in triplicate are shown with error bars representing SD.
Our results demonstrate a clear connection between cell surface receptors that induce cAMP, cAMP signaling, and integrin-mediated cell adhesion and show that this pathway is independent of PKA but mediated by the cAMP target Epac and the small GTPase Rap1. This conclusion is based on the observations that isoproterenol, which raises cAMP levels through activation of endogenous β2-AR, is able to induce integrin-mediated cell adhesion to fibronectin in a Rap1-dependent, PKA-independent manner. Furthermore, importantly, a cAMP analogue that specifically activates Epac but not PKA is also able to induce cell adhesion. However, our results do not entirely exclude a role for PKA in this process. Both Rap1 and Rap1GAP are substrates for PKA (Bos et al., 2001), and thus PKA may modulate the effect of the Epac-Rap1 signaling pathway on the adhesion process.
This novel function of cAMP was found in Ovcar3, an ovarian carcinoma cell line that expresses the fibronectin-binding integrin α5β1 and in NIH3T3 cells stably transfected with Epac1 (unpublished data). However, the effect may be more general and may include different cell types expressing Epac. Epac is particularly highly expressed in ovary, thyroid, kidney, adrenal gland, and brain (de Rooij et al., 1998; Kawasaki et al., 1998), and it is expected that the cAMP-Epac pathway leading to integrin activation may operate particularly in these tissues. In addition, Rap1 is implicated in the activation of a variety of integrins, including αLβ2 and αIIbβ3, and thus the effect may not be restricted to α5β1 integrins. The regulation of integrin-mediated adhesion plays an important role in many cellular processes including cell migration, cell division, and reactions to mechanical stress, and cAMP may impinge on these processes by activation of the Epac-Rap1 pathway. In ovarian cancer cells, for instance, functional integrins and molecular events that regulate them are important for invasion into the sub-mesothelial ECM (Cannistra et al., 1995; Buczek-Thomas et al., 1998; Strobel and Cannistra, 1999; for review see Brakebusch et al., 2002).
How Rap1 regulates integrin-mediated cell adhesion remains elusive. Integrin activity is regulated through various mechanisms, including cell surface expression (change of number), redistribution at the cell surface (change of avidity), and conformational changes (change of affinity) (Stewart and Hogg, 1996; Porter and Hogg, 1998; van Kooyk and Figdor, 2000; Brakebusch et al., 2002; Sebzda et al., 2002). Studies using activation-specific antibodies show that Rap1 regulates both avidity and affinity but not cell surface expression. For instance, in Jurkat cells Rap1 inhibits both the clustering and the increased affinity of αLβ2 (Katagiri et al., 2000; Reedquist et al., 2000; Sebzda et al., 2002), whereas in megakaryocytes Rap1 increases the affinity of αIIbβ3 (Bertoni et al., 2002). In addition, Rap1 is required for the direct activation of integrins by integrin-activating antibodies or manganese ions (de Bruyn et al., 2002). Apparently, Rap1 modulates a process before integrin activation, for instance, the recruitment of an essential cofactor. Interestingly, it has been reported recently that Rap1 is essential in the formation of adherens junctions, though it is less clear whether the process involves integrin-mediated signaling (Knox and Brown, 2002).
Materials and methods
Cells, plasmids, and transfections
NIH-OVCAR3 (Ovcar3) cells were maintained at 37°C in RPMI 1640 supplemented with 10% heat-inactivated (30 min at 56°C) FBS and 0.05% glutamine in the presence of penicillin and streptomycin. Hemagglutinin (HA)-tagged constructs of Epac1 and Rap1GapII in the PMT2HA expression vector have been described previously (de Rooij et al., 1998; de Bruyn et al., 2002). Transient transfection of Ovcar3 cells was performed using the FuGENE 6 transfection reagent (Roche Diagnostics Corporation) according to the manufacturer's procedures using 6 μg total DNA including either a TK-luciferase plasmid (1 μg) or CMV-luciferase plasmid (0.2 μg) as indicated. Cells were serum starved at least 16 h before stimulation.
Reagents
Western blotting of protein samples was performed using polyvinylidene difluoride membranes. Antibodies against dually phosphorylated p42/44MAPK and phosphorylated CREB (directed against phosphorylated Ser133) were obtained from Cell Signaling, and antibodies against K-Rev/Rap1 and polyclonal anti-HA were obtained from Santa Cruz Biotechnology, Inc. The following inhibitor and stimuli were used at the indicated concentrations: RGD peptide (100 μM) and H-89 (10 μM), obtained from Biomol Research Laboratories Inc. (Plymouth Meeting). Isoproterenol (10 μM, unless indicated otherwise) was obtained from Sigma-Aldrich and 8-Br-cAMP (1 mM, unless indicated otherwise) and 8CPT-2Me-cAMP (100 μM, unless indicated otherwise) was obtained from Biolog Life Science Institute.
Adhesion assay
24-well plates were coated overnight with fibronectin (Sigma-Aldrich; 1–5 μg/ml as indicated) in sodium bicarbonate buffer (Sigma-Aldrich). Poly-l-lysine was coated for 2 h at RT (0.1% wt/vol in water), washed twice with water, and dried overnight. Plates were washed in TSM buffer (20 mM Tris-HCl, pH 8, 150 mM NaCl, 1 mM CaCl2, 2 mM MgCl2) and blocked for 30–45 min at 37°C with 1% BSA/TSM. Transiently transfected Ovcar3 cells, serum starved 16 h before the adhesion assay, were detached by trypsinization. Cells were centrifuged at 1,500 rpm for 5 min and resuspended in serum-free RPMI containing 25 mM Hepes, 0.5% BSA, and 1 g/L glucose to allow recovery of cell surface markers at 37°C for 1.5–2 h with gentle rotation in suspension. Cells were centrifuged, counted, and resuspended at 3 × 105 cells/ml in serum-free RPMI with 0.5% BSA. The experiment was performed in triplicates, and to each well 150 μl of cells was added to 150 μl of medium with or without stimulus. In studies with H-89 (10 μM), cells were either preincubated at 37°C for 30 min with the inhibitor before seeding the wells (short pretreatment), or H-89 was added before trypsinization, during the recovery period, and before seeding wells (long pretreatment). Cells were allowed to adhere for 1 h at 37°C, and nonadherent cells were removed by gently washing plates three times with warmed 0.5% BSA/TSM. Adherent cells were lysed in luciferase lysis buffer (15% glycerol, 25 mM Tris-phosphate, pH 7.8, 1% Triton X-100, 8 mM MgCl2, 1 mM DTT) at 4°C for 30 min, and units of luciferase activity were quantified with addition of equal volume of luciferase assay buffer (25 mM Tris-phosphate, pH 7.8, 8 mM MgCl2, 1 mM DTT, 1 mM ATP, pH 7, 1 mM luciferin) using a luminometer (Lumat LB9507; Berthold Technologies). Unseeded cells (150 μl) were lysed separately to determine luciferase counts in the total input cells. Specific adhesion (%) was determined (counts in cells bound/counts in total input × 100) and plotted either directly or relative to the basal adhesion of HA vector–transfected cells. Error bars represent average deviation among experiments, and where representative experiments are depicted error bars represent average SD within each experiment. The expression of transfected constructs was confirmed by immunoblotting of total cell lysates.
Rap1 activation assay and phosphorylation of ERK and CREB
Rap1 activation assays were performed as described previously (Franke et al., 1997; van Triest et al., 2001). Briefly, adherent cells (unless stated otherwise) were serum starved overnight, treated, and lysed in 750 μl lysis buffer (10% glycerol, 1% Nonidet P-40, 50 mM Tris-Cl, pH 7.5, 200 mM NaCl, 2 mM MgCl2, 1 μM leupeptin, 0.1 μM aprotinin, 5 mM NaF, 1 mM NaVO3). Lysates were clarified by centrifugation, and 500 μl of lysate was incubated with GST-tagged RBD of RalGDS precoupled to glutathione beads to specifically pull down the GTP-bound forms of Rap1. Samples were incubated for 1 h at 4°C while tumbling. Beads were washed four times in lysis buffer, and remaining fluid was removed with an insulin syringe. Proteins were eluted with Laemmli sample buffer and analyzed by SDS-PAGE and Western blotting using Rap1 antibodies (Santa Cruz Biotechnology, Inc.). To 100 μl of clarified lysate 25 μl 5× Laemmli sample buffer was added, and phosphorylation of ERKs was analyzed by Western blotting using the phospho-specific antibody against p42/44MAPK. Phosphorylation of CREB was analyzed by Western blotting using a phospho-specific antibody directed against phosphorylated Ser133.
Acknowledgments
We thank J. Das and M.C. Verhoeven for technical assistance and our colleagues for discussions and for critically reading the manuscript.
S. Rangarajan was supported by a grant from the Human Frontiers Science Program, J.M. Enserink and H.B. Kuiperij by a grant from the Council of Earth and Life Sciences of the Netherlands Organization for Scientific Research, and L.S. Price by a grant from the Dutch Cancer Society.
S. Rangarajan and J.M. Enserink contributed equally to this work.
Footnotes
Abbreviations used in this paper: CMV, cytomegalovirus; 8CPT-2Me-cAMP, 8-(4-chloro-phenylthio)-2'-O-methyladenosine-3′,5′-cyclic monophosphate; Epac, exchange protein directly activated by cAMP; GAP, GTPase-activating protein; PKA, protein kinase A; RalGDS, Ral guanine nucleotide dissociation stimulator; RBD, ras-binding domain; RGD, arginine, glycine, aspartic acid; TK, thymidine kinase.
References
- Arai, A., Y. Nosaka, E. Kanda, K. Yamamoto, N. Miyasaka, and O. Miura. 2001. Rap1 is activated by erythropoietin or interleukin-3 and is involved in regulation of β1 integrin-mediated hematopoietic cell adhesion. J. Biol. Chem. 276:10453–10462. [DOI] [PubMed] [Google Scholar]
- Bertoni, A., S. Tadokoro, K. Eto, N. Pampori, L.V. Parise, G.C. White, and S.J. Shattil. 2002. Relationships between Rap1b, affinity modulation of integrin α IIbβ 3, and the actin cytoskeleton. J. Biol. Chem. 277:25715–25721. [DOI] [PubMed] [Google Scholar]
- Bos, J.L., J. de Rooij, and K.A. Reedquist. 2001. Rap1 signalling: adhering to new models. Nat. Rev. Mol. Cell Biol. 2:369–377. [DOI] [PubMed] [Google Scholar]
- Brakebusch, C., D. Bouvard, F. Stanchi, T. Sakai, and R. Fassler. 2002. Integrins in invasive growth. J. Clin. Invest. 109:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczek-Thomas, J.A., N. Chen, and T. Hasan. 1998. Integrin-mediated adhesion and signalling in ovarian cancer cells. Cell. Signal. 10:55–63. [DOI] [PubMed] [Google Scholar]
- Cannistra, S.A., C. Ottensmeier, J. Niloff, B. Orta, and J. DiCarlo. 1995. Expression and function of β1 and αVβ3 integrins in ovarian cancer. Gynecol. Oncol. 58:216–225. [DOI] [PubMed] [Google Scholar]
- Caron, E., A.J. Self, and A. Hall. 2000. The GTPase Rap1 controls functional activation of macrophage integrin alphaMbeta2 by LPS and other inflammatory mediators. Curr. Biol. 10:974–978. [DOI] [PubMed] [Google Scholar]
- de Bruyn, K.M., S. Rangarajan, K.A. Reedquist, C.G. Figdor, and J.L. Bos. 2002. The small GTPase Rap1 is required for Mn2+- and antibody-induced LFA-1– and VLA-4–mediated cell adhesion. J. Biol. Chem. 277:29468–29476. [DOI] [PubMed] [Google Scholar]
- de Rooij, J., F.J. Zwartkruis, M.H. Verheijen, R.H. Cool, S.M. Nijman, A. Wittinghofer, and J.L. Bos. 1998. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 396:474–477. [DOI] [PubMed] [Google Scholar]
- de Rooij, J., H. Rehmann, M. van Triest, R.H. Cool, A. Wittinghofer, and J.L. Bos. 2000. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J. Biol. Chem. 275:20829–20836. [DOI] [PubMed] [Google Scholar]
- Enserink, J.M., A.E. Christensen, J. de Rooij, M. van Triest, F. Schwede, H.G. Genieser, S.O. Doskeland, J.L. Blank, and J.L. Bos. 2002. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 4:901–906. [DOI] [PubMed] [Google Scholar]
- Franke, B., J.W. Akkerman, and J.L. Bos. 1997. Rapid Ca2+-mediated activation of Rap1 in human platelets. EMBO J. 16:252–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, G.A., and M.R. Montminy. 1989. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 59:675–680. [DOI] [PubMed] [Google Scholar]
- Howe, A.K., and R.L. Juliano. 2000. Regulation of anchorage-dependent signal transduction by protein kinase A and p21-activated kinase. Nat. Cell Biol. 2:593–600. [DOI] [PubMed] [Google Scholar]
- Katagiri, K., M. Hattori, N. Minato, S. Irie, K. Takatsu, and T. Kinashi. 2000. Rap1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol. Cell. Biol. 20:1956–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki, H., G.M. Springett, N. Mochizuki, S. Toki, M. Nakaya, M. Matsuda, D.E. Housman, and A.M. Graybiel. 1998. A family of cAMP-binding proteins that directly activate Rap1. Science. 282:2275–2279. [DOI] [PubMed] [Google Scholar]
- Kitayama, H., Y. Sugimoto, T. Matsuzaki, Y. Ikawa, and M. Noda. 1989. A ras-related gene with transformation suppressor activity. Cell. 56:77–84. [DOI] [PubMed] [Google Scholar]
- Knox, A.L., and N.H. Brown. 2002. Rap1 GTPase regulation of adherens junction positioning and cell adhesion. Science. 295:1285–1288. [DOI] [PubMed] [Google Scholar]
- Marinissen, M.J., and J.S. Gutkind. 2001. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol. Sci. 22:368–376. [DOI] [PubMed] [Google Scholar]
- Mochizuki, N., Y. Ohba, E. Kiyokawa, T. Kurata, T. Murakami, T. Ozaki, A. Kitabatake, K. Nagashima, and M. Matsuda. 1999. Activation of the ERK/MAPK pathway by an isoform of Rap1GAP associated with G α(i). Nature. 400:891–894. [DOI] [PubMed] [Google Scholar]
- Morgan, A., M. Wilkinson, and R.D. Burgoyne. 1993. Identification of Exo2 as the catalytic subunit of protein kinase A reveals a role for cyclic AMP in Ca(2+)-dependent exocytosis in chromaffin cells. EMBO J. 12:3747–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves, S.R., P.T. Ram, and R. Iyengar. 2002. G protein pathways. Science. 296:1636–1639. [DOI] [PubMed] [Google Scholar]
- Ohba, Y., K. Ikuta, A. Ogura, J. Matsuda, N. Mochizuki, K. Nagashima, K. Kurokawa, B.J. Mayer, K. Maki, J. Miyazaki, and M. Matsuda. 2001. Requirement for C3G-dependent Rap1 activation for cell adhesion and embryogenesis. EMBO J. 20:3333–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, J.C., and N. Hogg. 1998. Integrins take partners: cross-talk between integrins and other membrane receptors. Trends Cell Biol. 8:390–396. [DOI] [PubMed] [Google Scholar]
- Reedquist, K.A., E. Ross, E.A. Koop, R.M. Wolthuis, F.J. Zwartkruis, Y. van Kooyk, M. Salmon, C.D. Buckley, and J.L. Bos. 2000. The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J. Cell Biol. 148:1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebzda, E., M. Bracke, T. Tugal, N. Hogg, and D.A. Cantrell. 2002. Rap1A positively regulates T cells via integrin activation rather than inhibiting lymphocyte signaling. Nat. Immunol. 3:251–258. [DOI] [PubMed] [Google Scholar]
- Stewart, M., and N. Hogg. 1996. Regulation of leukocyte integrin function: affinity vs. avidity. J. Cell. Biochem. 61:554–561. [DOI] [PubMed] [Google Scholar]
- Strobel, T., and S.A. Cannistra. 1999. β1-integrins partly mediate binding of ovarian cancer cells to peritoneal mesothelium in vitro. Gynecol. Oncol. 73:362–367. [DOI] [PubMed] [Google Scholar]
- Tsukamoto, N., M. Hattori, H. Yang, J.L. Bos, and N. Minato. 1999. Rap1 GTPase-activating protein SPA-1 negatively regulates cell adhesion. J. Biol. Chem. 274:18463–18469. [DOI] [PubMed] [Google Scholar]
- van Kooyk, Y., and C.G. Figdor. 2000. Avidity regulation of integrins: the driving force in leukocyte adhesion. Curr. Opin. Cell Biol. 12:542–547. [DOI] [PubMed] [Google Scholar]
- van Triest, M., J. de Rooij, and J.L. Bos. 2001. Measurement of GTP-bound Ras-like GTPases by activation-specific probes. Methods Enzymol. 333:343–348. [DOI] [PubMed] [Google Scholar]
- Whittard, J.D., and S.K. Akiyama. 2001. Positive regulation of cell-cell and cell-substrate adhesion by protein kinase A. J. Cell Sci. 114:3265–3272. [DOI] [PubMed] [Google Scholar]