

Abstract
Mitochondrial distribution and morphology depend on MDM33, a Saccharomyces cerevisiae gene encoding a novel protein of the mitochondrial inner membrane. Cells lacking Mdm33 contain ring-shaped, mostly interconnected mitochondria, which are able to form large hollow spheres. On the ultrastructural level, these aberrant organelles display extremely elongated stretches of outer and inner membranes enclosing a very narrow matrix space. Dilated parts of Δmdm33 mitochondria contain well-developed cristae. Overexpression of Mdm33 leads to growth arrest, aggregation of mitochondria, and generation of aberrant inner membrane structures, including septa, inner membrane fragments, and loss of inner membrane cristae. The MDM33 gene is required for the formation of net-like mitochondria in mutants lacking components of the outer membrane fission machinery, and mitochondrial fusion is required for the formation of extended ring-like mitochondria in cells lacking the MDM33 gene. The Mdm33 protein assembles into an oligomeric complex in the inner membrane where it performs homotypic protein–protein interactions. Our results indicate that Mdm33 plays a distinct role in the mitochondrial inner membrane to control mitochondrial morphology. We propose that Mdm33 is involved in fission of the mitochondrial inner membrane.
Keywords: membrane fission; mitochondria; mitochondrial dynamics; organelle morphology; Saccharomyces cerevisiae
Introduction
Mitochondria perform essential metabolic functions in virtually every eukaryotic cell (Scheffler, 2001). Each cell type contains mitochondria in characteristic copy number, distribution, and shape, often reflecting the energy requirements of the cell. As mitochondria cannot be synthesized de novo, they must be inherited upon cell division. Mitochondrial inheritance and maintenance of normal mitochondrial morphology depend on active transport of the organelles along cytoskeletal tracks and balanced membrane fusion and fission events (Bereiter-Hahn, 1990; Warren and Wickner, 1996; Yaffe, 1999; Griparic and van der Bliek, 2001; Westermann, 2002). Recent work has shown that mitochondrial dynamics plays an important role in cellular function and cell differentiation processes. For example, mitochondrial fusion is required for sperm development in Drosophila (Hales and Fuller, 1997). Mitochondrial fission is crucial for embryonic development in nematodes (Labrousse et al., 1999) and is involved in apoptosis in mammalian cells (Frank et al., 2001). Moreover, exchange of mitochondrial content by frequent fusion and fission is thought to be a defense mechanism against cellular aging (Ono et al., 2001).
Baker's yeast, Saccharomyces cerevisiae, turned out to be a powerful model organism to study the molecular machinery mediating mitochondrial distribution and morphology (Hermann and Shaw, 1998; Jensen et al., 2000; Boldogh et al., 2001; Shaw and Nunnari, 2002). Yeast mitochondria form a branched tubular network located immediately below the cell cortex (Hoffmann and Avers, 1973; Egner et al., 2002). The continuity of the mitochondrial reticulum is maintained by continuous membrane fusion and fission events (Nunnari et al., 1997; Bleazard et al., 1999; Sesaki and Jensen, 1999). Two proteins of the outer membrane, Fzo1 and Ugo1, were found to be involved in the fusion of mitochondria. Yeast mutants lacking either one of these proteins exhibit fragmented mitochondria because fusion is blocked in the presence of ongoing fission. As a secondary consequence of aberrant mitochondrial morphology, fzo1 and ugo1 mutants fail to inherit mitochondrial DNA (mtDNA),* resulting in a respiratory-deficient growth phenotype (Hermann et al., 1998; Rapaport et al., 1998; Sesaki and Jensen, 2001).
Division of the mitochondrial outer membrane in yeast depends on the dynamin-related GTPase Dnm1, which assembles on the surface of mitochondrial tubules at sites of organelle constriction and fission (Otsuga et al., 1998; Bleazard et al., 1999; Sesaki and Jensen, 1999). Dnm1 cooperates with the soluble protein Mdv1 (Fekkes et al., 2000; Tieu and Nunnari, 2000; Cerveny et al., 2001; Tieu et al., 2002) and the integral outer membrane protein Fis1 (Mozdy et al., 2000). Mutants lacking either one of these proteins exhibit long tubular mitochondria or closed planar nets of interconnected mitochondria, a phenotype indicative of a strong defect in mitochondrial division. Inheritance of mtDNA is not affected in these mutants. Mitochondrial fragmentation and the respiratory growth defect of mutants defective in fusion can be suppressed by deletion of components of the outer membrane division machinery (Bleazard et al., 1999; Sesaki and Jensen, 1999; Fekkes et al., 2000; Mozdy et al., 2000; Tieu and Nunnari, 2000; Cerveny et al., 2001; Sesaki and Jensen, 2001).
Mitochondria are complex organelles bounded by two membranes. Thus, mechanisms must exist that coordinate fusion and fission events of the outer and inner membranes. The fusion machinery of the outer membrane is in contact with as yet unknown factors of the inner membrane, and this connection appears to be critical to coordinate fusion of four mitochondrial membranes (Fritz et al., 2001). A second mitochondrial dynamin-related protein, Mgm1, is located in the intermembrane space. In contrast to Dnm1, it is not clear whether Mgm1 is involved in membrane fission events. However, it has been suggested that Mgm1 is involved in remodeling events of the mitochondrial inner membrane (Wong et al., 2000). Other proteins that might be involved in fusion and fission of the inner membrane have not been described.
We recently conducted a systematic genome-wide screen to identify novel yeast genes important for mitochondrial distribution and morphology, MDM. One of the mutants isolated in this screen, mdm33, exhibits giant ring-like mitochondrial structures, a phenotype that has never been described before for any other yeast mutant (Dimmer et al., 2002). Here, we report on the functional characterization of Mdm33 and discuss a possible role of this protein in fission of the mitochondrial inner membrane.
Results
MDM33 encodes a novel 54-kD protein
The MDM33 gene (systematic name YDR393W) encodes a protein of 456 amino acid residues (∼54 kD). The Mdm33 protein shares 19% amino acid identity with the predicted protein CaShe9 of Candida albicans (Andaluz et al., 2001) and 18% with SPAC823.13Cp of Schizosaccharomyces pombe (GenBank/EMBL/DDBJ accession no. CAB90158) (for an alignment see Fig. 1 A). These three proteins have a similar domain structure. Hydropathy analysis (Hofmann and Stoffel, 1993) predicts two transmembrane segments, one after about two thirds of the protein and another one at the extreme COOH terminus (Fig. 1 A). The COILS algorithm (Lupas et al., 1991) predicts a high propensity of the protein to form coiled coils (Fig. 1 B).
Figure 1.
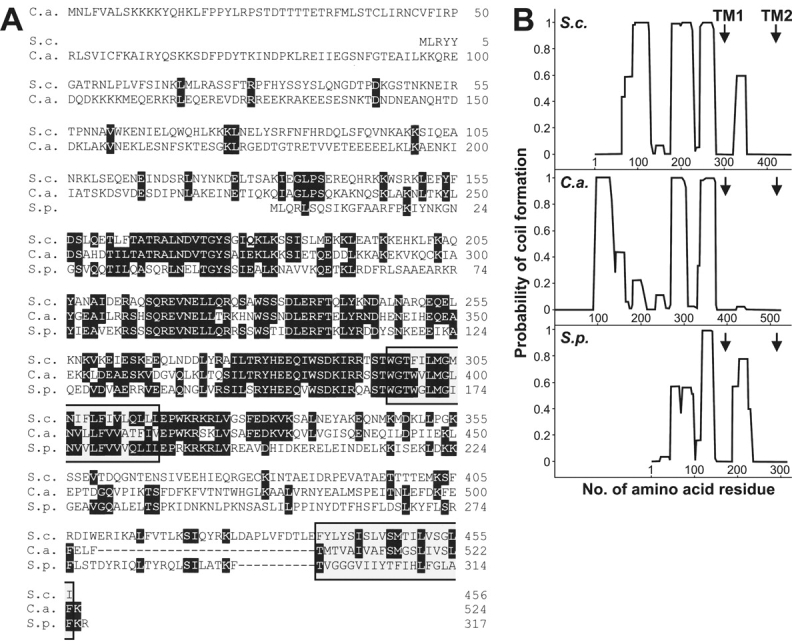
Mdm33 is a novel protein with predicted transmembrane segments and coiled-coil structures. (A) Comparison of the predicted amino acid sequences of Mdm33-related proteins. The amino acid sequences of Mdm33 of Saccharomyces cerevisiae (S.c.), CaShe9 of Candida albicans (C.a.), and SPAC823.13C of Schizosaccharomyces pombe (S.p.) were aligned using the DNAMAN software (Lynnon BioSoft). Amino acids that are identical in all three proteins are in black boxes, and gaps introduced to maximize the alignment are indicated by dashes. The positions of two predicted transmembrane segments (Hofmann and Stoffel, 1993), which are present in all three homologues, are indicated by gray boxes. (B) Plots of coiled-coil probabilities (Lupas et al., 1991) for the protein sequences shown in A. The window size used was 21. The positions of predicted transmembrane segments (TM1 and TM2) are indicated by arrows.
Mdm33 is located in the mitochondrial inner membrane
The NH2 terminus of Mdm33 resembles classical mitochondrial presequences (Hartl et al., 1989); it is rich in positively charged residues, lacks acidic amino acid residues, and has a high content of hydroxylated residues (Fig. 1 A). Possible cleavage sites for the matrix processing peptidase (Hartl et al., 1989) are after residues 24 and 30. To examine whether the NH2 terminus of Mdm33 functions as a presequence in vivo, we fused the first 41 amino acid residues to GFP and expressed the chimeric protein in yeast. Specific mitochondrial staining was observed by fluorescence microscopy (Fig. 2 A), suggesting that Mdm33 is targeted to mitochondria.
Figure 2.
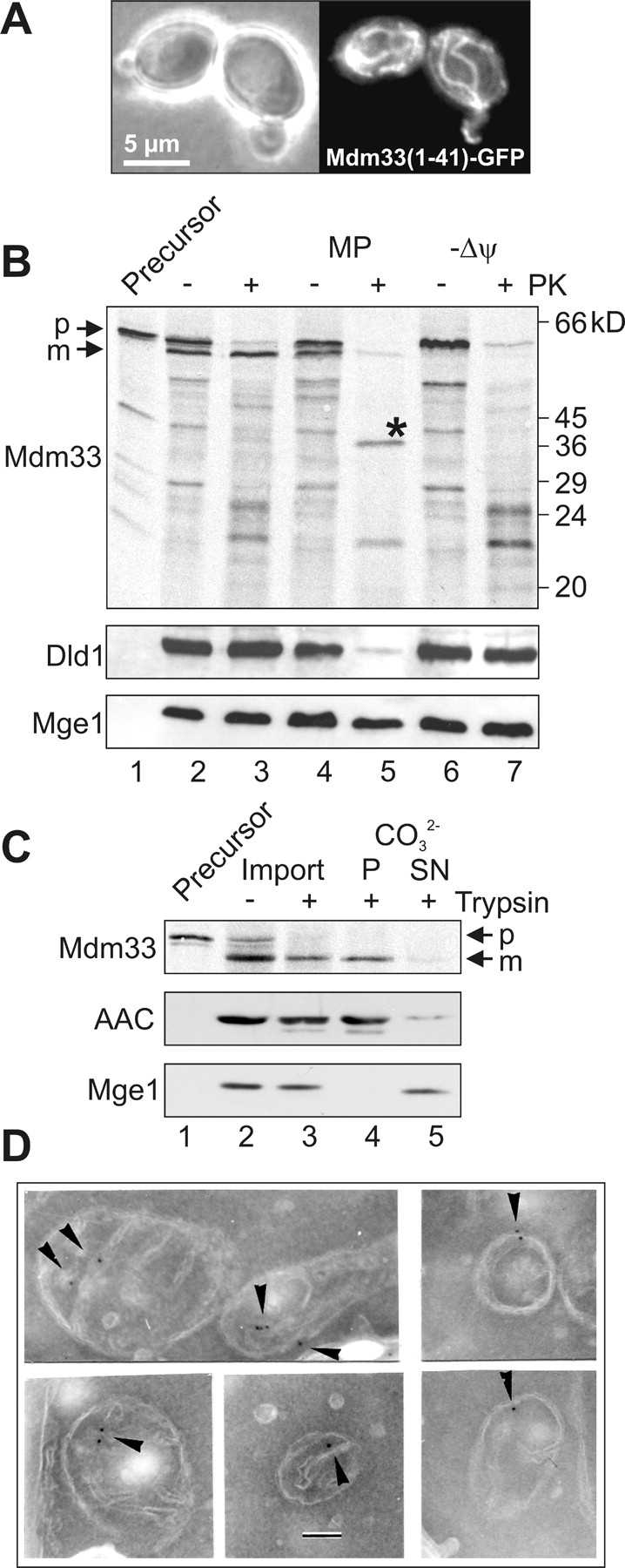
Mdm33 is a protein of the mitochondrial inner membrane. (A) Mitochondrial targeting of Mdm33(1–41)–GFP. Wild-type yeast cells expressing a chimeric protein consisting of the NH2-terminal 41 amino acid residues of Mdm33 fused to GFP (Mdm33[1–41]–GFP) were grown to log phase in YPGal (yeast extract-peptone-galactose) medium and examined by phase contrast (left panel) and fluorescence (right panel) microscopy. (B) Import in vitro of Mdm33. Mdm33 was synthesized in reticulocyte lysate in the presence of [35S]methionine (Precursor; lane 1) and imported into isolated mitochondria. The amount of precursor loaded in lane 1 corresponds to 10% of the amount used for each import reaction. For import in vitro, the protein was incubated with isolated mitochondria, and the organelles were reisolated by centrifugation. Equal aliquots were either left untreated on ice (lane 2), treated with proteinase K (PK; lane 3), converted to mitoplasts (MP) by hypotonic swelling in the absence (lane 4) or presence (lane 5) of PK, or import was performed in the presence of valinomycin to dissipate the membrane potential (Δψ) without (lane 6) and with (lane 7) subsequent treatment with PK. The asterisk indicates a specific fragment of imported Mdm33 that is generated only upon protease treatment of mitoplasts. The intactness of the mitochondrial membranes was controlled by immunoblotting using antisera against Dld1, an integral protein of the inner membrane that exposes its major part to the intermembrane space, and Mge1, a soluble matrix protein. p, precursor; m, mature form of Mdm33. (C) Carbonate extraction of imported Mdm33. Mdm33 was imported into mitochondria as in B. Aliquots were either left untreated on ice (lane 2) or treated with trypsin (lane 3). Then, protease treatment was stopped, and trypsin-treated samples were extracted with carbonate (CO3 2-) and separated into pellet (P; lane 4) and supernatant (SN; lane 5) fractions. All samples were precipitated with TCA and analyzed by SDS-PAGE and autoradiography. The fractionation into soluble and membrane proteins was controlled by immunoblotting using antisera against the ADP/ATP carrier (AAC), an integral protein of the inner membrane, and Mge1. p, precursor; m, mature form of Mdm33. (D) Immunocytochemistry of GFP–Mdm33. Yeast cells expressing a chimeric protein consisting of Mdm33 and GFP, inserted between the presequence and the mature part of Mdm33, were analyzed by immunoelectron microscopy using antibodies against GFP. Arrowheads point to inner membrane regions containing GFP–Mdm33. Bar, 100 nm.
We tested the import of Mdm33 into isolated mitochondria. Upon translation in vitro in the presence of [35S]methionine, SDS-PAGE, and autoradiography, a prominent band with an apparent molecular mass of ∼60 kD was observed (Fig. 2 B, lane 1). The relatively slow running behavior of Mdm33 in SDS-PAGE might be explained by its rather basic nature (the isoelectric point is 9.36). Upon incubation with isolated mitochondria, Mdm33 was processed to a slightly smaller form (Fig. 2 B, lane 2) and translocated to a protease-protected location (Fig. 2 B, lane 3) in a membrane potential–dependent manner (Fig. 2 B, lanes 6 and 7).
When the outer membrane was opened by osmotic swelling of mitochondria after import, a specific fragment was generated by protease treatment of mitoplasts (Fig. 2 B, lane 5). The apparent molecular mass of this fragment corresponds well to the predicted size of a segment ranging from the processing site to the end of the first predicted transmembrane domain (34 kD), suggesting that this part is protected by the inner membrane in mitoplasts. The fragment was not generated in the absence of a membrane potential (Fig. 2 B, lane 7), indicating that it was a derivative of imported Mdm33. As the imported protein was resistant to alkaline extraction (Fig. 2 C), it is an integral part of the membrane. We conclude that Mdm33 is located in the mitochondrial inner membrane with its NH2-terminal part in the matrix and its COOH-terminal part exposed to the intermembrane space.
To further investigate the intracellular localization, we expressed epitope-tagged versions of Mdm33 in a Δmdm33 background. A tagged version carrying 13 copies of the myc epitope fused to the COOH terminus of Mdm33 was exclusively detected in mitochondria upon subfractionation of cells (unpublished data). However, this construct was unable to rescue the mitochondrial morphology defect of the deletion mutant, suggesting that the COOH-terminal end of Mdm33 is important for function of the protein. Next, we constructed a chimeric protein carrying a GFP moiety inserted between the mitochondrial presequence and the NH2-terminal end of mature Mdm33. This construct, GFP–Mdm33, rescued the mitochondrial defect of the deletion mutant (unpublished data). Immunoelectron microscopy demonstrated an association of GFP–Mdm33 with the inner membrane, both at the cristae and the inner boundary membrane, the part of the inner membrane that faces the outer membrane (Fig. 2 D). The fact that a fraction of GFP–Mdm33 was found in cristae suggests that Mdm33 does not form stable contacts to the outer membrane.
Δmdm33 cells harbor aberrant mitochondria
We examined the phenotype of the Δmdm33 mutant by various methods. Cells harboring mitochondrial matrix–targeted GFP (mtGFP) were grown to mid-logarithmic growth phase in liquid cultures containing different carbon sources and examined by fluorescence microscopy. Under all conditions, the majority of Δmdm33 cells harbored either large mitochondrial ring-like structures (Fig. 3 B) or two to four smaller mitochondrial rings that were often interconnected (Fig. 3 C). The remainder of the cells displayed either single elongated structures or aggregated or fragmented mitochondria. Branched tubular networks resembling wild-type cells (Fig. 3 A) could never be observed in Δmdm33 cells. A quantification of mitochondrial morphology of Δmdm33 cells and its isogenic wild type is presented in Table I.
Figure 3.

Δmdm33 cells exhibit grossly altered mitochondrial morphology. (A–C) Mitochondrial morphology. Wild-type (WT) and Δmdm33 cells expressing mtGFP were grown to log phase in YPD medium and examined by phase contrast (left) and fluorescence (right) microscopy. (B) A representative Δmdm33 cell with a large ring-like mitochondrion. (C) A representative Δmdm33 cell with small interconnected ring-like mitochondria. (D) Double staining of mitochondria and cytosol. Δmdm33 cells coexpressing mtGFP and cRFP were grown to log phase in YPD medium and examined by confocal fluorescence microscopy. Left, bright field image taken in the transmission mode; right, merged confocal green and red fluorescence images. (E) Mitochondrial fusion. Δmdm33 cells of opposite mating types containing mitochondria preloaded with mtGFP or mtRFP were mated and analyzed by fluorescence microscopy. The distribution of mtGFP and mtRFP in a representative zygote containing a medial bud (asterisk) is shown. (F and I) Morphology of the ER. Wild-type and Δmdm33 cells expressing ER-targeted GFP were grown to log phase in glucose-containing synthetic minimal medium lacking methionine and examined by phase contrast and fluorescence microscopy. (G and J) Vacuolar morphology. Wild-type and Δmdm33 cells were grown to log phase in YPD medium, stained with 7-amino-4-chloromethylcoumarin, l-arginine amide to label vacuoles and examined by phase contrast and fluorescence microscopy. (H and K) Structure of the actin cytoskeleton. Wild-type and Δmdm33 cells were grown to log phase in YPD medium, fixed, stained with rhodamine-phalloidin to visualize filamentous actin, and examined by phase contrast and fluorescence microscopy.
Table I.
Quantification of mitochondrial morphology in Δmdm33 cells
| Strain/medium | Mitochondrial morphology (% of cells) | No. of cells scored | ||||
|---|---|---|---|---|---|---|
| Wild type–like | Large rings | Small rings | Elongated/tubules | Aggregated/fragmented | ||
| MDM33/YPD | 97 | − | − | 1 | 2 | 394 |
| MDM33/YPGal | 99 | − | − | − | 1 | 281 |
| MDM33/YPG | 89 | − | − | − | 11 | 109 |
| Δmdm33/YPD | − | 41 | 39 | 13 | 7 | 433 |
| Δmdm33/YPGal | − | 40 | 41 | 10 | 9 | 242 |
| Δmgm33/YPG | − | 52 | 17 | 5 | 26 | 104 |
Confocal fluorescence microscopy of Δmdm33 cells coexpressing mtGFP and cytosolic red fluorescent protein (cRFP) suggested that the interior of mitochondrial rings contained cytosolic proteins (Fig. 3 D). Similarly, cross sections of Δmdm33 cells analyzed by electron microscopy did not provide evidence for an association of mitochondrial rings with other organelles (Fig. 6, compare D–F). Thus, the generation of mitochondrial rings in Δmdm33 cells does not depend on a stable interaction of mitochondria with other cellular membranes. Upon mating of Δmdm33 cells harboring different fluorescent matrix markers (Nunnari et al., 1997), the fluorescent labels immediately intermixed in zygotes, indicating that Mdm33 is not required for mitochondrial fusion (Fig. 3 E). Deletion of the MDM33 gene did not affect the structure of other organelles, such as the ER (Fig. 3 I), the vacuole (Fig. 3 J), or the actin cytoskeleton (Fig. 3 K). We conclude that Mdm33 is specifically required for maintenance of mitochondrial shape.
Figure 6.
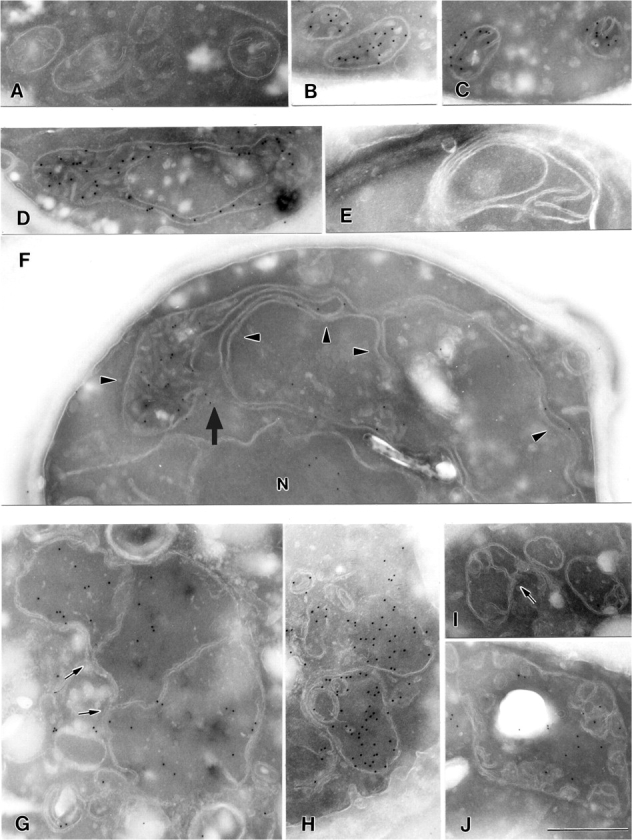
Ultrastructure of mitochondria in Mdm33-deficient and Mdm33-overexpressing cells. (A-C) Cross sections of mitochondria of wild-type cells. (D-F) Cross sections of mitochondria of Δmdm33 cells. N, nucleus. Arrowheads point to extended interconnected mitochondrial structures; the arrow points to a putative intramitochondrial fusion site. (G–J) Cross sections of mitochondria of Mdm33-overexpressing cells. Induction of GAL–MDM33 was as described for Fig. 5 B. Arrows point to inner membrane septa. (B, C, D, F, G, H and J) Immunogold labeling of mtGFP to identify the mitochondrial matrix compartment. It should be noted that the mitochondrial outer membrane appears relatively faint due to its low protein content. Bar, 500 nm.
Aberrant Δmdm33 mitochondria form hollow spheres, interconnected sheets, and rings
To examine the three-dimensional organization of Δmdm33 mitochondria, cells expressing mtGFP were analyzed by confocal fluorescence microscopy. A representative Δmdm33 cell containing a giant spherical organelle is depicted in Fig. 4. Large mitochondrial rings were observed in several consecutive focal planes (Fig. 4, A–K). Ring-shaped structures similar to those seen in the x/y planes were observed also in sections corresponding to z/y (Fig. 4 L) and z/x planes (Fig. 4 M). Apparently, this mitochondrion formed a large hollow sphere of ∼4 μm diameter. A view onto a surface-rendered three-dimensional representation of this organelle is depicted in Fig. 4 O. Similar structures were observed in many other Δmdm33 cells. Often these organelles appeared to be closed, sometimes they contained openings of varying sizes, or they formed sheets typically located at one side of the cell (see supplemental figures, Figs. S1–S6, available at http://www.jcb.org/cgi/content/full/jcb.200211113/DC1).
Figure 4.

Mitochondrial sphere in a Δmdm33 mutant cell. Δmdm33 cells expressing mtGFP were grown to log phase in YPGal medium. A representative cell was analyzed by confocal fluorescence microscopy. (A–K) Single optical planes (x/y); distance between serial planes is 285 nm. (L) Section corresponding to a z/y plane. (M) Section corresponding to a z/x plane. (N) Bright field image taken in the transmission mode. (O) Surface-rendered three-dimensional representation. This image has been turned 90° counter clockwise when compared with panels A–K.
Overexpression of Mdm33 leads to growth arrest and aggregation of mitochondria
The MDM33 gene (alternate name SHE9; sensitive to high expression) was previously found in a screen for genes that cause growth arrest when highly overexpressed (Espinet et al., 1995). We asked whether this growth arrest is concomitant with changes of mitochondrial morphology. The MDM33 coding sequence was placed under control of the strong inducible GAL1 promoter in a multicopy plasmid and transformed into wild-type and Δmdm33 yeast strains. Induction of the GAL1 promoter by change of the carbon source from glucose to galactose resulted in a severe reduction of growth in both strains (Fig. 5 A). The generation time in minimal medium containing galactose as carbon source was 3 h for both the wild-type and Δmdm33 strains carrying the empty vector. It increased to ∼10 h for the strains overexpressing Mdm33 from the GAL1 promoter. Mitochondria were highly aggregated and/or fragmented in these cells (Fig. 5 B; Table II). A similar aggregation behavior was observed upon GAL1-induced overexpression of an Mdm33 fusion protein carrying a GFP moiety fused to its COOH terminus, Mdm33–GFP. Fluorescence microscopy indicated that mitochondrial targeting was not affected under these conditions (unpublished data). From the comparison of fluorescent signals of GAL1-overexpressed Mdm33–GFP and nonoverexpressed GFP–Mdm33, we estimate that GAL1-driven overexpression was at least 100-fold (unpublished data). We conclude that overexpression of Mdm33 severely affects mitochondrial morphology.
Figure 5.
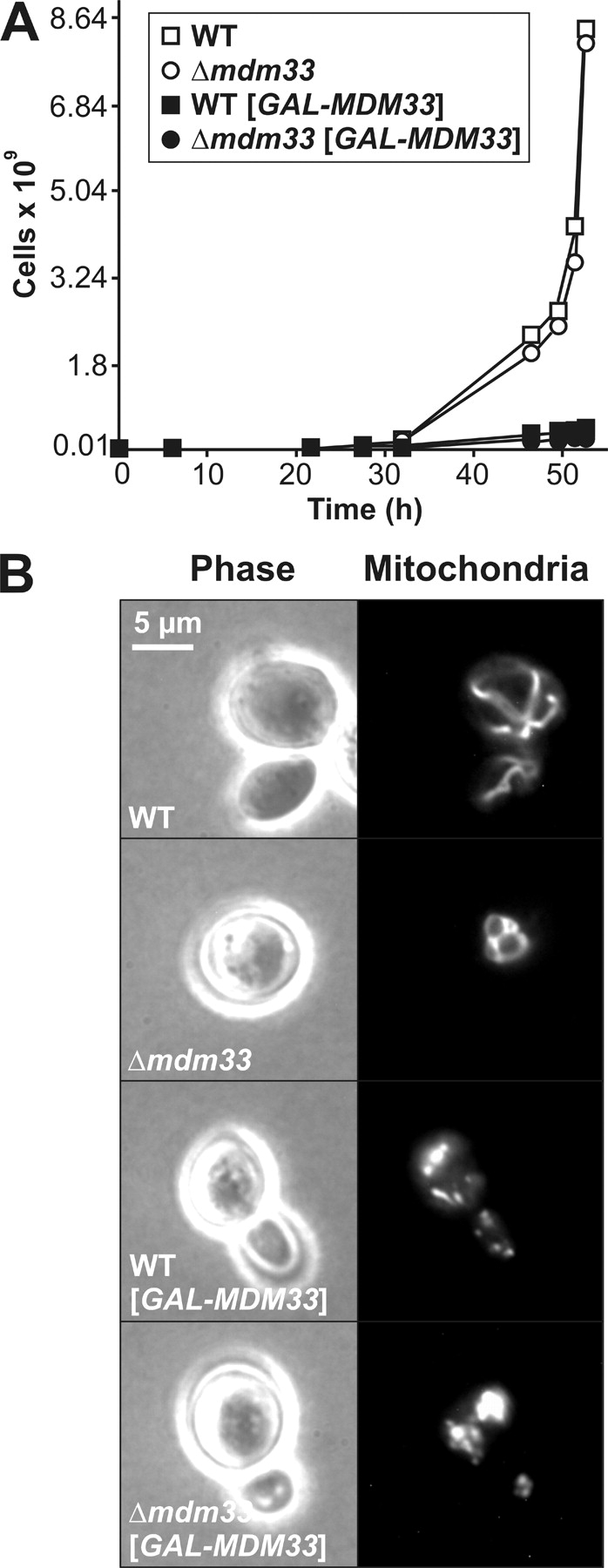
Overexpression of Mdm33 induces growth arrest and aggregation of mitochondria. (A) Wild-type (WT) and Δmdm33 cells harboring empty vectors (open symbols) or multicopy plasmids with the MDM33 coding sequence under control of the GAL1 promoter (filled symbols; GAL–MDM33) were grown in galactose-containing synthetic complete medium lacking histidine to select for the overexpressing plasmid. At the indicated time points, the OD578 was measured, cultures were diluted when the OD exceeded 1, and growth was plotted as total number of cells. (B) Wild-type and Δmdm33 cells harboring empty vectors or multicopy plasmids with the MDM33 coding sequence under control of the GAL1 promoter (GAL–MDM33) were grown overnight in galactose-containing synthetic complete medium lacking histidine and examined by phase contrast (left) and fluorescence (right) microscopy. Mitochondria were visualized with mtGFP.
Table II. Quantification of mitochondrial morphology in Mdm33-overexpressing cells.
| Strain | Mitochondrial morphology (% of cells) | No. of cells scored | |||
|---|---|---|---|---|---|
| Wild type–like | Rings | Elongated/tubules | Aggregated/fragmented | ||
| MDM33 | 98 | − | − | 2 | 104 |
| Δmdm33 | − | 84 | 10 | 6 | 101 |
| MDM33[GAL–MDM33] | 16 | − | 5 | 79 | 109 |
| Δmdm33[GAL–MDM33] | 2 | 11 | 5 | 82 | 102 |
Mdm33 affects the internal structure of mitochondria
To investigate the role of Mdm33 in the organization of the mitochondrial membranes, wild-type cells (Fig. 6, A–C), Δmdm33 mutant cells (Fig. 6, D–F), and Mdm33-overexpressing cells (Fig. 6, G–J) were examined by electron microscopy. In some experiments, mtGFP was labeled with immunogold to unequivocally identify the mitochondrial matrix compartment. Sections of Δmdm33 cells often showed extremely elongated mitochondria that extended through half of the cell (Fig. 6 F). These organelles displayed very long stretches of mitochondrial double membranes that enclosed a very narrow matrix space (see immunogold label in Fig. 6, D and F). Typically, these structures were dilated at the ends where the inner membrane formed cristae (Fig. 6 F). It is conceivable that these mitochondria are similarly extended in the third dimension perpendicular to the cross section, and that they correspond to the large rings or elongated structures seen in the fluorescence microscope (see above).
Some images showed extended mitochondrial structures in close proximity originating from the same organelle. As Δmdm33 mitochondria are not defective in membrane fusion, these structures are able to fuse with each other (the arrow in Fig. 6 F points to a putative fusion site). Such fusion events would form mitochondrial rings (Fig. 6, D and E) that eventually might close also in the third dimension and form the hollow structures that were observed by confocal fluorescence microscopy (see above). We propose that Δmdm33 cells contain abnormally extended mitochondria that may form hollow spheres by fusion of mitochondrial membranes of the same organelle.
We examined the internal structure of mitochondria in cells overexpressing Mdm33 (Fig. 6, G–J). The most striking phenotype was the formation of inner membrane septa, membranous partitions separating the inner compartment into distinct chambers. Apparently, these partitions consisted of two parallel membranes in direct continuity with the inner membrane (Figs. 6, G and I, arrows). Furthermore, vesicular structures accumulated within the organelles, some of which appeared to be disconnected from the inner boundary membrane (Fig. 6, H and J). Some organelles were seen that were largely devoid of cristae (Fig. 6 H). All mitochondria were still bounded by a double membrane. We consider it likely that overexpression of Mdm33 initially results in the formation of inner membrane septa. With ongoing septation of the mitochondrial interior, smaller inner membrane fragments are generated that eventually become disconnected from the inner boundary membrane, leaving a largely cristae-free space. Our results demonstrate an important role of Mdm33 in maintenance of the structure of the mitochondrial inner membrane.
Epistatic relationships of Δmdm33, Δfis1, and Δfzo1 mutations
Establishment and maintenance of a tubular mitochondrial network depends on antagonistic fission and fusion events (Nunnari et al., 1997). Block of mitochondrial fission by deletion of any one of the DNM1, MDV1, or FIS1 genes leads to the formation of long tubular mitochondria or closed mitochondrial nets (Otsuga et al., 1998; Bleazard et al., 1999; Sesaki and Jensen, 1999; Fekkes et al., 2000; Mozdy et al., 2000; Tieu and Nunnari, 2000; Cerveny et al., 2001), whereas block of mitochondrial fusion by deletion of either one of the FZO1 or UGO1 genes leads to highly fragmented mitochondria (Hermann et al., 1998; Rapaport et al., 1998; Sesaki and Jensen, 2001). To investigate whether Mdm33 is involved in these processes, we constructed Δmdm33/Δfis1 and Δmdm33/Δfzo1 double mutants by genetic crosses and examined their phenotypes.
The Δmdm33/Δfis1 double mutant was viable on nonfermentable carbon sources (unpublished data). Mitochondrial morphology was very similar to the Δmdm33 parent, and clearly different from Δfis1. Most of the Δmdm33/Δfis1 cells displayed mitochondrial ring structures, whereas the elongated net-like mitochondria of Δfis1 cells (Mozdy et al., 2000) could never be found in the double mutant (Table III). Thus, the Δmdm33 mitochondrial morphology defect is epistatic to the Δfis1 mitochondrial morphology defect. Similarly, overexpression of Mdm33 in Δfis1 cells induced the same growth arrest and mitochondrial aggregation phenotypes that were observed in a FIS1 wild-type background (unpublished data). These results demonstrate that Mdm33 acts upstream of Fis1.
Table III.
Quantification of mitochondrial morphology in Δmdm33/Δfis1 cells
| Strain | Mitochondrial morphology (% of cells) | No. of cells scored | |||||
|---|---|---|---|---|---|---|---|
| Wild type–like | Large rings | Small rings | Elongated/tubules | Elongated net-like | Aggregated/fragmented | ||
| MDM33/FIS1 | 95 | − | − | − | − | 5 | 114 |
| MDM33/Δfis1 | − | − | 1 | 9 | 84 | 6 | 108 |
| Δmdm33/FIS1 | − | 29 | 41 | 17 | − | 13 | 115 |
| Δmdm33/Δfis1 | − | 17 | 44 | 22 | − | 17 | 115 |
The Δmdm33/Δfzo1 double mutant is inviable on nonfermentable carbon sources (unpublished data), as is the Δfzo1 single mutant (Hermann et al., 1998; Rapaport et al., 1998). Mitochondrial morphology was indiscernible from the Δfzo1 parent. Most of the Δmdm33/Δfzo1 cells displayed highly fragmented mitochondria, whereas the characteristic ring-shaped mitochondria of Δmdm33 cells could not be found in the double mutant (Table IV). Thus, the Δfzo1 mitochondrial morphology defect is epistatic to the Δmdm33 mitochondrial morphology defect, implying that Fzo1 is required to form the extended mitochondrial structures seen in the Δmdm33 mutant. This genetic finding is consistent with our morphological observations, which suggest the formation of mitochondrial rings and spheres by self-fusion of the organelles in Δmdm33 cells.
Table IV.
Quantification of mitochondrial morphology in Δmdm33/Δfzo1 cells
| Strain | Mitochondrial morphology (% of cells) | No. of cells scored | |||||
|---|---|---|---|---|---|---|---|
| Wild type–like | Large rings | Small rings | Elongated/tubules | Fragmented | Aggregated | ||
| MDM33/FZO1 | 98 | − | − | − | 2 | − | 146 |
| MDM33/Δfzo1 | − | − | − | − | 83 | 17 | 103 |
| Δmdm33/FZO1 | − | 43 | 43 | 13 | 1 | − | 126 |
| Δmdm33/Δfzo1 | − | − | <1 | − | 78 | 22 | 218 |
Mdm33 is part of a high molecular weight complex with homo-oligomeric interactions
The function of Mdm33 in mitochondrial morphogenesis may involve protein–protein interactions. In particular, the predicted coiled-coil domains could be responsible for formation of homo- and/or hetero-oligomers. We investigated whether Mdm33 assembles into a high molecular weight complex. In vitro–translated Mdm33 protein was imported into mitochondria. After completion of import, mitochondria were solubilized with detergent, and protein complexes were analyzed by gel filtration chromatography. Mdm33 was eluted from the column in fractions corresponding to a molecular weight of ∼300 kD (Fig. 7 A), indicating assembly into an oligomeric complex.
Figure 7.
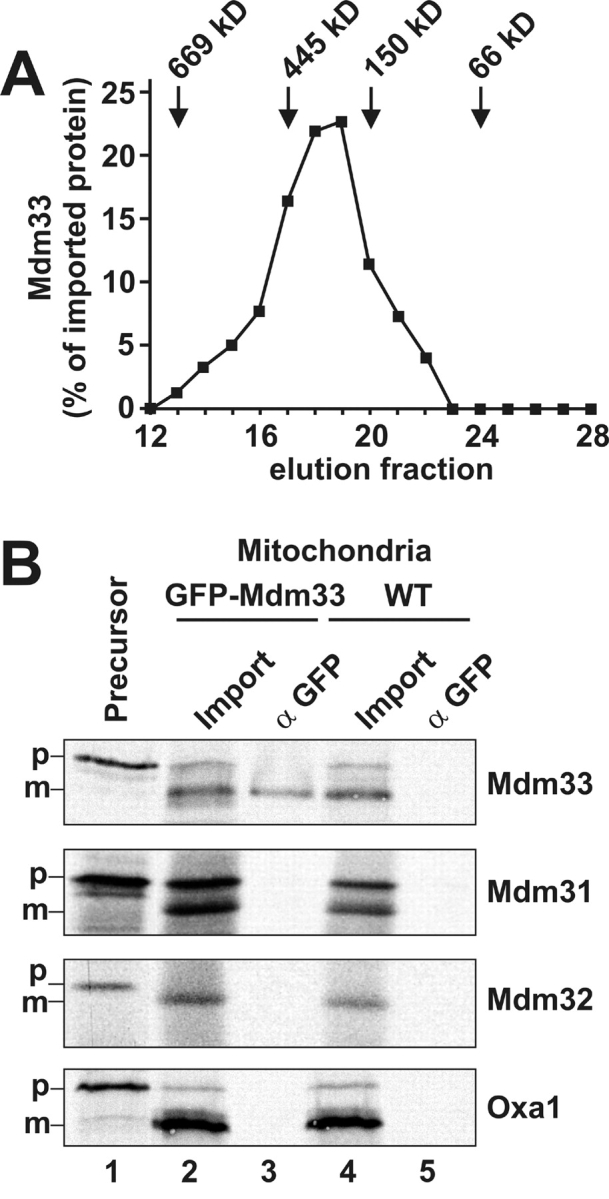
Mdm33 assembles into higher molecular complexes. (A) Gel filtration of Mdm33. Mdm33 protein synthesized in vitro was imported into wild-type mitochondria as in Fig. 2 B. After completion of import, mitochondria (1 mg) were washed, reisolated, and solubilized in 300 μl buffer containing 0.5% Triton X-100. Nonsolubilized and aggregated material was separated by centrifugation, and the supernatant was loaded on a superose-6 column. After chromatography, the collected fractions were analyzed by SDS-PAGE, blotting to nitrocellulose, and autoradiography. Recovered Mdm33 was quantified by densitometry. Marker proteins thyroglobulin, apoferritin, alcohol dehydrogenase, and bovine serum albumin were used to calibrate the column. (B) Coimmunoprecipitation of Mdm33. Mdm33 translated in vitro was imported into mitochondria harboring GFP–Mdm33 or nontagged Mdm33 (WT). Mitochondria were washed, reisolated, lysed with Triton X-100, and subjected to coimmunoprecipitation with antibodies directed against GFP. In control reactions, two other putative inner membrane proteins involved in mitochondrial morphogenesis, Mdm31 and Mdm32, and a nonrelated inner membrane protein, Oxa1, were imported. Lane 1, [35S]methionine-labeled precursor proteins, corresponding to 10% of the amounts added to the import reactions. Lane 2, import reactions using GFP–Mdm33 mitochondria, corresponding to 10% of the amounts used for the coimmunoprecipitations. Lane 3, coimmunoprecipitations from GFP–Mdm33 mitochondria using anti-GFP antiserum. Lane 4, import reactions using wild-type mitochondria. Lane 5, coimmunoprecipitations from wild-type mitochondria using anti-GFP antiserum. p, precursor forms; m, mature forms of imported proteins.
To investigate protein–protein interactions involving Mdm33, we performed coimmunoprecipitation experiments. In vitro–translated precursor proteins were imported into wild-type mitochondria or mitochondria harboring GFP–Mdm33. After completion of import, mitochondria were lysed and subjected to coimmunoprecipitation with antibodies against GFP. We tested the interactions of GFP–Mdm33 with imported Mdm33, Mdm31, Mdm32, and Oxa1. Mdm31 and Mdm32 are two predicted inner membrane proteins that were shown to be essential for mitochondrial distribution and morphology. Mutants lacking these proteins, however, do not exhibit the mitochondrial ring structures seen in Δmdm33 mutants (Dimmer et al., 2002). Oxa1 is an inner membrane protein not related to mitochondrial morphogenesis and served as a negative control. Imported Mdm33 was specifically coprecipitated with GFP–Mdm33, indicating that Mdm33 performs homotypic protein–protein interactions (Fig. 7 B, lane 3). Mdm33 was not precipitated from mitochondria lacking the GFP tag (Fig. 7 B, lane 5), and no interactions of GFP–Mdm33 with Mdm31, Mdm32, and Oxa1 were found (Fig. 7 B).
Stable interactions of Mdm33 with outer membrane proteins known to be involved in mitochondrial morphogenesis appear unlikely, because Mdm33 is present both in the inner boundary membrane and in cristae (Fig. 2 D). If Mdm33 would be in contact to outer membrane proteins, it would be expected to be located exclusively in parts of the inner membrane that face the outer membrane. Moreover, genetic evidence suggests that Mdm33 function is not coupled to that of the outer membrane proteins Fis1 or Fzo1 (see above). Yet, we cannot exclude that Mdm33 interacts with other known or unknown proteins. However, the observed homotypic interactions of Mdm33 might explain why mdm33 was the only mutant displaying giant ring-shaped mitochondria in a comprehensive genome-wide screen for mitochondrial morphology mutants (Dimmer et al., 2002). Thus, no other known MDM component would be a likely interaction partner of Mdm33 based on its location, function, or phenotype. We suggest that Mdm33 performs homo-oligomeric interactions.
Discussion
Our studies establish an important function of Mdm33 in the regulation of mitochondrial morphology. Several lines of evidence point to a possible role of Mdm33 in fission of the inner membrane. First, the formation of ring-like and spherical mitochondria is unique for the Δmdm33 mutant. However, the phenotype of the Δmdm33 mutant bears some similarities to other yeast mutants known to be affected in mitochondrial fission, such as Δdnm1, Δmdv1, and Δfis1 (Otsuga et al., 1998; Fekkes et al., 2000; Mozdy et al., 2000; Tieu and Nunnari, 2000; Cerveny et al., 2001). Mutant cells contain only one or a few individual mitochondria per cell; the aberrant mitochondrial structures often remain interconnected and are located at only one side of the cell; and, in contrast to many other mitochondrial morphology mutants, mutants defective in mitochondrial fission do not lose mtDNA and do not acquire a respiratory-deficient growth phenotype.
Second, deletion of the MDM33 gene leads to the formation of large organelles that typically contain long stretches of outer and inner membranes enclosing a very narrow matrix space. These extremely extended structures can form only in the absence of frequent organellar division events. As mitochondrial fusion is not compromised in the Δmdm33 strain, elongated organelles likely undergo self-fusion when two ends of the same organelle approach one another. Such a self-fusion event initially would form a ring structure, and this is indeed observed by electron microscopy. The enclosed spheres seen by confocal microscopy are probably generated when two-dimensional rings subsequently close also in the third dimension.
Third, overexpression of Mdm33 results in septation and vesiculation of the inner membrane and disappearance of inner membrane cristae. This phenotype can be best explained by greatly enhanced inner membrane fission activity in the presence of excess Mdm33. In particular, the appearance of septated mitochondria induced by Mdm33 overexpression bears striking similarity to dividing mitochondria in various tissues described in the literature (e.g., Tandler et al., 1969; Larsen, 1970; Tandler and Hoppel, 1972; Wakabayashi et al., 1974; Duncan and Greenaway, 1981; for review see Griparic and van der Bliek, 2001). Partitioning of mitochondria is likely due to enhanced fission activity of the inner membrane in the absence of division of the outer membrane.
Fourth, Mdm33 is an integral protein of the inner membrane and has extensive coiled-coil domains exposed to the matrix side. This topology and the observed homotypic protein–protein interactions are in agreement with the following possible mode of action. Mdm33 protein complexes on apposing inner membranes would interact with each other from the matrix side, possibly by forming α-helical bundles via Mdm33 coiled coils. Formation of α-helical bundles would then mediate constriction and/or fission of the inner membrane from the matrix side.
Maintenance of normal mitochondrial morphology by balanced membrane fission and fusion depends on several distinct, yet interdependent, processes. Based on the results reported herein and in previous studies by others, we propose a hypothetical cycle of sequential reactions of inner membrane fission, outer membrane fission, and mitochondrial fusion (Fig. 8). The first step of the cycle is inner membrane fission. Division of the mitochondrial outer and inner membranes can be uncoupled. Mitochondria of worms expressing dominant interfering mutants of DRP-1, the Caenorhabditis elegans homologue of yeast Dnm1, had swollen mitochondria with irregular shapes and sizes. Intriguingly, these mitochondria contained separated matrix compartments that were still connected by thin tubules of outer membrane. Thus, inner membrane fission persists, even when outer membrane fission is blocked by loss of DRP-1 function (Labrousse et al., 1999). An independent machinery appears to mediate fission of the mitochondrial inner membrane. Mdm33 is likely to play an important role to control this process in yeast.
Figure 8.

Hypothetical cycle of epistatic relationships between components with a role in mitochondrial dynamics. Arrows leaving the cycle indicate the mitochondrial phenotypes of the deletion mutants. Each gene deletion is epistatic to the following one, i.e., double mutants obtained by genetic crosses display the phenotype of the mutant that is preceding in the cycle. See text for further details. IM, inner membrane; OM, outer membrane; WT, wild type.
The next step of the cycle is fission of the outer membrane by Dnm1 and its cofactors, Fis1 and Mdv1. Membrane pinching by dynamin-like proteins requires a considerable constriction of the organelle before a dynamin ring can assemble (Labrousse et al., 1999). In the case of mitochondria, constriction of the outer membrane may be facilitated by severing of the inner membrane. Consistent with this idea, the Δmdm33 mutation was found to be epistatic to Δfis. Thus, the outer membrane fission machinery is not required for the formation of extended mitochondria in Δmdm33 cells. Conversely, Mdm33 is required to form the net structures in Δfis1 mutants. Thus, Mdm33 acts upstream of the outer membrane fission machinery.
The cycle is completed by mitochondrial fusion. In yeast, mutations leading to deficiency in outer membrane fission are epistatic to mutations leading to deficiency in fusion, i.e., mitochondrial fragmentation and loss of mtDNA in fusion mutants is blocked by mutation of components of the outer membrane fission machinery (Bleazard et al., 1999; Sesaki and Jensen, 1999, 2001; Fekkes et al., 2000; Mozdy et al., 2000; Tieu and Nunnari, 2000; Cerveny et al., 2001). On the other hand, mitochondrial fusion is a prerequisite for the formation of extended Δmdm33 mitochondria, as we found that Δfzo1 is epistatic to Δmdm33. Why does deletion of the MDM33 gene not prevent fragmentation of mitochondria in Δfzo1 cells, similar to deletion of genes encoding components of the outer membrane fission machinery? Interestingly, Sesaki and Jensen (1999) observed that mitochondrial fragments persisted in ∼40% of the cells when cells were first disrupted for FZO1 and subsequently for DNM1. This suggests that once the equilibrium of fission and fusion has been shifted in the direction of fission, it is difficult to rebuild larger mitochondrial structures without fusion activity. In case of the Δmdm33/Δfzo1 strain, the action of Dnm1 and its cofactors might well be sufficient to induce and maintain fragmentation of mitochondria, even in the absence of Mdm33.
Abnormal ring-shaped or cup-shaped mitochondria in various animal and human tissues have been described. These include, for example, parathyroid gland cells of senile dogs (Setoguti, 1977), renal oncocytes in rats (Krech et al., 1981), avian retinal pigment epithelium cells under continuous light (Lauber, 1982), Purkinje cells of the rat cerebrellar cortex after prolonged alcohol consumption (Tavares and Paula-Barbosa, 1983), and human salivary tumor cells (Kataoka et al., 1991). It will be interesting to see in the future whether there is a link between defects of mitochondrial division and various pathological conditions, and whether proteins similar to Mdm33 are involved.
Materials and methods
Plasmid constructions
Standard methods were used for cloning procedures (Sambrook et al., 1989). PCR was performed using Pfu polymerase (Stratagene) according to the manufacturer's instructions. The plasmid for expression of GFP fused to the presequence of Mdm33 in yeast, pYES-Mdm33(1–41)–GFP, was generated by PCR amplification of the first 123 bp of the MDM33 gene from genomic yeast DNA using the primers 5′-GGG AAG CTT ATG TTG AGA TAC TAT GGG GCG-3′ and 5′-GGG GGA TCC ACC GTT CTG TAG AGA ATA TGA AC-3′ and cloning into the HindIII/BamHI sites of vector pYES-mtGFP (Westermann and Neupert, 2000). The plasmid for in vitro transcription of MDM33, pMM106-1, was generated by PCR amplification of the MDM33 coding region from genomic yeast DNA using the primers 5′-AAA CCA TGG TGA GAT ACT ATG GGG CG and 5′-TTT AAG CTT TCA TAT CAG CCC GGA TAC C-3′ and ligation into the pGEM-T vector (Promega) according to the manufacturer's instructions. The plasmid for in vitro transcription of MDM31, pKSD31.1, was generated by PCR amplification of the MDM31 coding region from genomic yeast DNA using the primers 5′-AAA GAG CTC ACC ATG TCC CTT TTT ACC AGG C-3′ and 5′-AAA AAG CTT TCA CGC GAT AGC ACC GAG AC-3′ and ligation into pGEM-T. The plasmid for in vitro transcription of MDM32, pKSD32.1, was generated by PCR amplification of the MDM32 coding region from genomic yeast DNA using the primers 5′-AAA GAG CTC ACC ATG CAT ATT AAA GTT GAC AGT AG-3′ and 5′-AAA AAG CTT TCA AAC CAT GGC ACC AAA CC-3′ and ligation into the pPCR-script Amp SK(+) vector (Stratagene) according to the manufacturer's instructions. For construction of the plasmid expressing GFP–Mdm33 under control of the MDM33 regulatory sequences, pMM112, the following three DNA fragments were PCR amplified and successively cloned into vector pRS316 (Sikorski and Hieter, 1989): (1) MDM33 promoter and Mdm33 presequence encoding region amplified from genomic yeast DNA using the primers 5′-AAA GAG CTC AAT TTC ATC ATG GAG GTG CAT G-3′ and 5′-TTT GGA TCC AGA ATA TGA ACT ATA ATG AAA TGG-3′, cloned with SacI/BamHI; (2) yeast enhanced GFP encoding region amplified from pUG34 (GenBank/EMBL/DDBJ accession no. AF298784) using primers 5′-AAA GGA TCC ATG TCT AAA GGT GAA GAA TTA TTC-3′ and 5′-TTT AAG CTT TTT GTA CAA TTC ATC CAT ACC AT-3′, cloned with BamHI/HindIII; and (3) Mdm33 mature part encoding region and MDM33 terminator amplified from genomic yeast DNA using primers 5′-AAA AAG CTT TTT CAT TAT AGT TCA TAT TCT CTA C-3′ and 5′-AAA CTC GAG ACA CAC TGA GAA TCT GAG CC-3′, cloned with HindIII/XhoI. The plasmid for expression of cRFP, pVT102U-cRFP, was generated by cloning a BamHI/XhoI fragment of pRS424-ADH1+PrF 0ATP9–RFP (Mozdy et al., 2000) encoding the red fluorescent protein, DsRed, into vector pVT102-U (Vernet et al., 1987). The plasmid for expression of Mdm33 from the GAL1 promoter in yeast, pYX223-MDM33, was generated by cloning an NcoI/HindIII fragment of pMM106-1 into vector pYX223 (R&D Systems).
Yeast strain constructions
Standard methods were used for growth and manipulation of yeast strains (Sherman et al., 1986; Gietz et al., 1992). Growth of yeast was always at 30°C. All strains used for microscopy and genetic studies were isogenic to BY4741, BY4742, and BY4743 (Brachmann et al., 1998). Haploid deletion strains Δmdm33, Δfis1, and Δfzo1 were obtained from EUROSCARF (Frankfurt, Germany). Double mutants Δmdm33/Δfis1 and Δmdm33/Δfzo1 were generated by mating of haploid strains, sporulation, and tetrad dissection. Genotypes of haploid progeny were determined by PCR. For overexpression of Mdm33, yeast strains were transformed with pYX223-MDM33 and grown on galactose-containing medium lacking histidine to select for the overexpressing plasmid. For expression of GFP–Mdm33 in yeast, pMM112 was transformed into Δmdm33.
For visualization of mitochondria, yeast strains were transformed with plasmid pVT100U-mtGFP, pYX113-mtGFP, pYX122-mtGFP, pYX142-mtGFP (Westermann and Neupert, 2000), or pRS416-GAL1+PrF 0ATP9–RFP (Mozdy et al., 2000). For visualization of the ER, yeast strains were transformed with pWP1055 (Prinz et al., 2000), and transformants were grown in the absence of methionine to induce the MET25 promoter for expression of ER-targeted GFP. For visualization of the cytosol, yeast strains were transformed with pVT102U-cRFP.
Import of radiolabeled proteins into isolated mitochondria
Mitochondria of strain W303 were isolated by differential centrifugation as previously described (Diekert et al., 2001). Translation of proteins in vitro in the presence of [35S]methionine and import into isolated mitochondria were performed essentially as previously described (Ryan et al., 2001), with the exception that an energy-regenerating system was added, and 1 μM valinomycin was used to dissipate the mitochondrial membrane potential (Westermann et al., 1995). Hypotonic swelling, protease treatment of mitochondria (Diekert et al., 2001), and carbonate extraction of imported proteins (Fritz et al., 2001) were performed as previously described. Samples were analyzed by SDS-PAGE, blotting to nitrocellulose, and autoradiography.
Microscopy
Yeast cultures were grown in liquid YPD medium (yeast extract, peptone, dextrose) to mid-logarithmic growth phase, if not indicated otherwise. Epifluorescence microscopy was according to standard procedures (Westermann and Neupert, 2000). Quantification of mitochondrial morphology defects was performed without prior reference to strain identity. Confocal images were taken with a Leica TCS SP2 beam scanning confocal microscope equipped with a 1.4 NA oil immersion lens (Leica 100X; Planapo). For imaging, living cells were embedded in 1% low melting point agarose.
Electron microscopy and immunocytochemistry were performed as previously described (Kärgel et al., 1996). In brief, yeast cells were harvested by centrifugation and fixed for 1 h with 4% formaldehyde and 0.5% glutaraldehyde under culture conditions (pH and temperature were kept constant), cryoprotected by a mixture of 25% polyvinylpyrrolidone (PVP K15; M r 10,000; Fluka) and 1,6 M sucrose for 3 h, and frozen in liquid nitrogen. Ultrathin cryosections were prepared with glass knifes and transferred to formvar-carbon–coated copper grids using the cryoprotectant mixture. Labeling with anti-GFP IgG primary antibodies and secondary antibody–gold complexes (10 nm; Dianova) was performed as described (Kärgel et al., 1996). Finally, the sections were stained and stabilized by a freshly prepared mixture of 3% tungstosilicic acid hydrate (Fluka) and 2.5% polyvinyl alcohol (M r 10,000; Sigma-Aldrich).
Assay of mitochondrial fusion in vivo
Mitochondrial fusion was examined in vivo essentially as previously described (Nunnari et al., 1997; Mozdy et al., 2000) with some minor modifications. Cells of opposite mating types harboring plasmids encoding mtGFP or mtRFP under control of the GAL1 promoter were precultured in synthetic raffinose-containing medium under selection for the plasmids. Then, cells were grown to log phase in synthetic medium containing raffinose and galactose to induce expression of the fluorescent proteins. Then, yeast cells were incubated for 2 h in YPD medium (pH adjusted to 3.5) to shut off expression of the fluorescent proteins and to synchronize the cell cycle. Cultures were mixed, cells were transferred to nitrocellulose, placed for 3 h on YPD plates (pH 4.5) to allow mating, resuspended in water, and analyzed by fluorescence microscopy.
Miscellaneous
Gel filtration (Rapaport et al., 1998) and coimmunoprecipitation (Herrmann et al., 2001) were performed according to published procedures. Staining of the actin cytoskeleton with rhodamine-phalloidin (Molecular Probes) was performed as previously described (Amberg, 1998). Staining of the vacuole with 7-amino-4-chloromethylcoumarin, l-arginine amide (Molecular Probes) was performed according to the manufacturer's instructions.
Online supplemental material
The supplemental figures (Figs. S1–S6) are available online at http://www.jcb.org/cgi/content/full/jcb.200211113/DC1. Mitochondrial morphology of wild-type and Δmdm33 cells expressing mtGFP was analyzed by confocal fluorescence microscopy as in Fig. 4.
Supplemental Material
Acknowledgments
We thank Margit Vogel for expert technical assistance, Stefan Hell for continuous support and discussions concerning advanced optical microscopy, Hannes Herrmann for critical comments on the manuscript, William Prinz, Janet Shaw, and Johannes Hegemann for providing plasmids, and William Wickner for providing antiserum against GFP.
This work was supported by the Deutsche Forschungsgemeinschaft through grants WE 2174/2-3 and SFB 413/B3.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: cRFP, cytosolic red fluorescent protein; mtDNA, mitochondrial DNA; mtGFP, mitochondrial matrix–targeted GFP.
References
- Amberg, D.C. 1998. Three-dimensional imaging of the yeast actin cytoskeleton through the budding cell cycle. Mol. Biol. Cell. 9:3259–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andaluz, E., J.J. Coque, R. Cueva, and G. Larriba. 2001. Sequencing of a 4.3 kbp region of chromosome 2 of Candida albicans reveals the presence of homologues of SHE9 from Saccharomyces cerevisiae and of bacterial phosphatidylinositol-phospholipase C. Yeast. 18:711–721. [DOI] [PubMed] [Google Scholar]
- Bereiter-Hahn, J. 1990. Behavior of mitochondria in the living cell. Int. Rev. Cytol. 122:1–63. [DOI] [PubMed] [Google Scholar]
- Bleazard, W., J.M. McCaffery, E.J. King, S. Bale, A. Mozdy, Q. Tieu, J. Nunnari, and J.M. Shaw. 1999. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldogh, I.R., H.-C. Yang, and L.A. Pon. 2001. Mitochondrial inheritance in budding yeast. Traffic. 2:368–374. [DOI] [PubMed] [Google Scholar]
- Brachmann, C.B., A. Davies, G.J. Cost, E. Caputo, J. Li, P. Hieter, and J.D. Boeke. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14:115–132. [DOI] [PubMed] [Google Scholar]
- Cerveny, K.L., J.M. McCaffery, and R.E. Jensen. 2001. Division of mitochondria requires a novel DNM1-interacting protein, Net2p. Mol. Biol. Cell. 12:309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekert, K., A.I. de Kroon, G. Kispal, and R. Lill. 2001. Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae. Methods Cell Biol. 65:37–51. [DOI] [PubMed] [Google Scholar]
- Dimmer, K.S., S. Fritz, F. Fuchs, M. Messerschmitt, N. Weinbach, W. Neupert, and B. Westermann. 2002. Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol. Biol. Cell. 13:847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan, C.J., and H.C. Greenaway. 1981. The induction of septation and subdivision in muscle mitochondria. Comp. Biochem. Physiol. 69A:329–331. [Google Scholar]
- Egner, A., S. Jakobs, and S.W. Hell. 2002. Fast 100 nm resolution 3D-microscope reveals structural plasticity of mitochondria in live yeast. Proc. Natl. Acad. Sci. USA. 99:3370–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinet, C., M.A. de la Torre, M. Aldea, and E. Herrero. 1995. An efficient method to isolate yeast genes causing overexpression-mediated growth arrest. Yeast. 11:25–32. [DOI] [PubMed] [Google Scholar]
- Fekkes, P., K.A. Shepard, and M.P. Yaffe. 2000. Gag3p, an outer membrane protein required for fission of mitochondrial tubules. J. Cell Biol. 151:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, S., B. Gaume, E.S. Bergmann-Leitner, W.W. Leitner, E.G. Robert, F. Catez, C.L. Smith, and R.J. Youle. 2001. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell. 1:515–525. [DOI] [PubMed] [Google Scholar]
- Fritz, S., D. Rapaport, E. Klanner, W. Neupert, and B. Westermann. 2001. Connection of the mitochondrial outer and inner membranes by Fzo1 is critical for organellar fusion. J. Cell Biol. 152:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz, D., A.S. Jean, R.A. Woods, and R.H. Schiestl. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20:1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic, L., and A.M. van der Bliek. 2001. The many shapes of mitochondrial membranes. Traffic. 2:235–244. [DOI] [PubMed] [Google Scholar]
- Hales, K.G., and M.T. Fuller. 1997. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 90:121–129. [DOI] [PubMed] [Google Scholar]
- Hartl, F.-U., N. Pfanner, D.W. Nicholson, and W. Neupert. 1989. Mitochondrial protein import. Biochim. Biophys. Acta. 988:1–45. [DOI] [PubMed] [Google Scholar]
- Hermann, G.J., and J.M. Shaw. 1998. Mitochondrial dynamics in yeast. Annu. Rev. Cell Dev. Biol. 14:265–303. [DOI] [PubMed] [Google Scholar]
- Hermann, G.J., J.W. Thatcher, J.P. Mills, K.G. Hales, M.T. Fuller, J. Nunnari, and J.M. Shaw. 1998. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J. Cell Biol. 143:359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann, J.M., B. Westermann, and W. Neupert. 2001. Analysis of protein-protein interactions in mitochondria by coimmunoprecipitation and chemical cross-linking. Methods Cell Biol. 65:217–230. [DOI] [PubMed] [Google Scholar]
- Hoffmann, H.-P., and C.J. Avers. 1973. Mitochondrion of yeast: ultrastructural evidence for one giant, branched organelle per cell. Science. 181:749–750. [DOI] [PubMed] [Google Scholar]
- Hofmann, K., and W. Stoffel. 1993. TMbase - a database of membrane spanning protein segments. Biol. Chem. Hoppe-Seyler. 347:166. [Google Scholar]
- Jensen, R.E., A.E. Aiken Hobbs, K.L. Cerveny, and H. Sesaki. 2000. Yeast mitochondrial dynamics: fusion, division, segregation, and shape. Microsc. Res. Tech. 51:573–583. [DOI] [PubMed] [Google Scholar]
- Kärgel, E., R. Menzel, H. Honeck, F. Vogel, A. Böhmer, and W.-H. Schunck. 1996. Candida maltosa NADPH-cytochrome P450 reductase: cloning of a full-length cDNA, heterologous expression in Saccharomyces cerevisiae and function of the N-terminal region for membrane anchoring and proliferation of the endoplasmic reticulum. Yeast. 12:333–348. [DOI] [PubMed] [Google Scholar]
- Kataoka, R., Y. Hyo, T. Hoshiya, H. Miyahara, and T. Matsunaga. 1991. Ultrastructural study of mitochondria in oncocytes. Ultrastruct. Pathol. 15:231–239. [DOI] [PubMed] [Google Scholar]
- Krech, R., H. Zerban, and P. Bannasch. 1981. Mitochondrial anomalies in renal oncocytes induced in rat by N-nitrosomorpholine. Eur. J. Cell Biol. 25:331–339. [PubMed] [Google Scholar]
- Labrousse, A.M., M.D. Zappaterra, D.A. Rube, and A.M. van der Bliek. 1999. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell. 4:815–826. [DOI] [PubMed] [Google Scholar]
- Larsen, W.J. 1970. Genesis of mitochondria in insect fat body. J. Cell Biol. 47:373–383. [PMC free article] [PubMed] [Google Scholar]
- Lauber, J.K. 1982. Retinal pigment epithelium: ring mitochondria and lesions induced by continuous light. Curr. Eye Res. 2:855–862. [DOI] [PubMed] [Google Scholar]
- Lupas, A., M. Van Dyke, and J. Stock. 1991. Predicting coiled coils from protein sequences. Science. 252:1162–1164. [DOI] [PubMed] [Google Scholar]
- Mozdy, A., J.M. McCaffery, and J.M. Shaw. 2000. Dnm1p GTPase-mediated mitochondrial fusion is a multistep process requiring the novel integral membrane component Fis1p. J. Cell Biol. 151:367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari, J., W.F. Marshall, A. Straight, A. Murray, J.W. Sedat, and P. Walter. 1997. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell. 8:1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, T., K. Isobe, K. Nakada, and J.I. Hayashi. 2001. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 28:272–275. [DOI] [PubMed] [Google Scholar]
- Otsuga, D., B.R. Keegan, E. Brisch, J.W. Thatcher, G.J. Hermann, W. Bleazard, and J.M. Shaw. 1998. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J. Cell Biol. 143:333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz, W.A., L. Grzyb, M. Veenhuis, J.A. Kahana, P.A. Silver, and T.A. Rapoport. 2000. Mutants affecting the structure of the cortical endoplasmic reticulum in Saccharomyces cerevisiae. J. Cell Biol. 150:461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport, D., M. Brunner, W. Neupert, and B. Westermann. 1998. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J. Biol. Chem. 273:20150–20155. [DOI] [PubMed] [Google Scholar]
- Ryan, M.T., W. Voos, and N. Pfanner. 2001. Assaying protein import into mitochondria. Methods Cell Biol. 65:189–215. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 1659 pp.
- Scheffler, I.E. 2001. A century of mitochondrial research: achievements and perspectives. Mitochondrion. 1:3–31. [DOI] [PubMed] [Google Scholar]
- Sesaki, H., and R.E. Jensen. 1999. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J. Cell Biol. 147:699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki, H., and R.E. Jensen. 2001. UGO1 encodes an outer membrane protein required for mitochondrial fusion. J. Cell Biol. 152:1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoguti, T. 1977. Electron microscopic studies of the parathyroid gland of senile dogs. Am. J. Anat. 148:65–83. [DOI] [PubMed] [Google Scholar]
- Shaw, J.M., and J. Nunnari. 2002. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 12:178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F., G.R. Fink, and J. Hicks. 1986. Methods in Yeast Genetics: A Laboratory Course. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 198 pp.
- Sikorski, R.S., and P. Hieter. 1989. A system of shuttle vectors and host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandler, B., and C.L. Hoppel. 1972. Possible division of cardiac mitochondria. Anat. Rec. 173:309–323. [DOI] [PubMed] [Google Scholar]
- Tandler, B., R.A. Erlandson, A.L. Smith, and E.L. Wynder. 1969. Riboflavin and mouse hepatic cell structure and function. II. Division of mitochondria during recovery from simple deficiency. J. Cell Biol. 41:477–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares, M.A., and M.M. Paula-Barbosa. 1983. Mitochondrial changes in rat Purkinje cells after prolonged alcohol consumption. A morphologic assessment. J. Submicrosc. Cytol. 15:713–720. [PubMed] [Google Scholar]
- Tieu, Q., and J. Nunnari. 2000. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J. Cell Biol. 151:353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu, Q., V. Okreglak, K. Naylor, and J. Nunnari. 2002. The WD repeat protein, Mdv1p, functions as a molecular adaptor by interacting with Dnm1p and Fis1p during mitochondrial fission. J. Cell Biol. 158:445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernet, T., D. Dignard, and D.Y. Thomas. 1987. A family of yeast expression vectors containing the phage f1 intergenic region. Gene. 52:225–233. [DOI] [PubMed] [Google Scholar]
- Wakabayashi, T., M. Asano, and C. Kurono. 1974. Some aspects of mitochondria having a “septum.” J. Electron Microsc. (Tokyo). 23:247–254. [PubMed] [Google Scholar]
- Warren, G., and W. Wickner. 1996. Organelle inheritance. Cell. 84:395–400. [DOI] [PubMed] [Google Scholar]
- Westermann, B. 2002. Merging mitochondria matters. Cellular role and molecular machinery of mitochondrial fusion. EMBO Rep. 3:527–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann, B., and W. Neupert. 2000. Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast. 16:1421–1427. [DOI] [PubMed] [Google Scholar]
- Westermann, B., C. Prip-Buus, W. Neupert, and E. Schwarz. 1995. The role of the GrpE homologue, Mge1p, in mediating protein import and folding in mitochondria. EMBO J. 14:3452–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, E.D., J.A. Wagner, S.W. Gorsich, J.M. McCaffery, J.M. Shaw, and J. Nunnari. 2000. The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion-competent mitochondria. J. Cell Biol. 151:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe, M.P. 1999. The machinery of mitochondrial inheritance and behavior. Science. 283:1493–1497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}