

Abstract
We have demonstrated previously that adult human synovial membrane-derived mesenchymal stem cells (hSM-MSCs) have myogenic potential in vitro (De Bari, C., F. Dell'Accio, P. Tylzanowski, and F.P. Luyten. 2001. Arthritis Rheum. 44:1928–1942). In the present study, we have characterized their myogenic differentiation in a nude mouse model of skeletal muscle regeneration and provide proof of principle of their potential use for muscle repair in the mdx mouse model of Duchenne muscular dystrophy. When implanted into regenerating nude mouse muscle, hSM-MSCs contributed to myofibers and to long term persisting functional satellite cells. No nuclear fusion hybrids were observed between donor human cells and host mouse muscle cells. Myogenic differentiation proceeded through a molecular cascade resembling embryonic muscle development. Differentiation was sensitive to environmental cues, since hSM-MSCs injected into the bloodstream engrafted in several tissues, but acquired the muscle phenotype only within skeletal muscle. When administered into dystrophic muscles of immunosuppressed mdx mice, hSM-MSCs restored sarcolemmal expression of dystrophin, reduced central nucleation, and rescued the expression of mouse mechano growth factor.
Keywords: Duchenne muscular dystrophy; dystrophin; cell transplantation; muscle development; insulin-like growth factor I
Introduction
Skeletal muscle consists predominantly of syncytial fibers with peripheral, postmitotic myonuclei. Its intrinsic repair potential in adulthood relies on the persistence of a resident reserve population of undifferentiated mononuclear cells, termed satellite cells. In mature skeletal muscle, most satellite cells are quiescent and are activated in response to environmental cues such as injury to mediate postnatal muscle regeneration. After division, satellite cell progeny, termed myoblasts, undergo terminal differentiation and become incorporated into muscle fibers (Bischoff, 1994).
Myogenesis is regulated by a family of transcription factors (myogenic regulatory factors [MRFs]*), including MyoD, Myf5, myogenin, and MRF4 (Sabourin and Rudnicki, 2000). During embryonic development, MyoD and Myf5 are involved in the establishment of the skeletal muscle lineage (Rudnicki et al., 1993), whereas myogenin is required for terminal differentiation (Hasty et al., 1993; Nabeshima et al., 1993). During muscle repair, satellite cells recapitulate the MRF expression program manifested during embryonic development. Quiescent satellite cells do not express detectable levels of MRFs. After muscle injury, they proliferate and express Myf5 and MyoD (Cornelison and Wold, 1997; Cooper et al., 1999). Myogenin is expressed later and is associated with fusion and terminal differentiation (Smith et al., 1994; Yablonka-Reuveni and Rivera, 1994).
Duchenne muscular dystrophy (DMD), the most common lethal genetic disorder in children, is an X-linked recessive muscle disease characterized by the absence of dystrophin at the sarcolemma of muscle fibers (Hoffman et al., 1987). Dystrophin is associated with a large oligomeric complex of glycoproteins called dystrophin-associated proteins, which provide linkage to the extracellular membrane (Ervasti and Campbell, 1991). The absence of dystrophin results in destabilization of the extracellular membrane–sarcolemma-cytoskeleton architecture, making muscle fibers susceptible to contraction-associated mechanical stress and degeneration. In the first phase of the disease, new muscle fibers are formed by satellite cells. After depletion of the satellite cell pool in childhood, skeletal muscles degenerate progressively and irreversibly and are replaced by fibrotic tissue (Cossu and Mavilio, 2000). Like DMD patients, the mdx mouse lacks dystrophin in skeletal muscle fibers (Hoffman et al., 1987; Sicinski et al., 1989). However, the mdx mouse develops a mild dystrophic phenotype, probably because muscle regeneration by satellite cells is efficient for most of the animal's life span (Cossu and Mavilio, 2000). The lack of dystrophin is associated with secondary dysregulation of molecular pathways possibly involved in muscle regeneration. Mechano growth factor (MGF) is a splice variant of insulin-like growth factor-I expressed by skeletal myofibers and up-regulated in response to mechanical stimulation (Yang et al., 1996). MGF is an autocrine/paracrine growth factor that appears to play a role in local muscle maintenance and repair (Yang et al., 1996; Goldspink, 1999). It was reported that MGF mRNA is not detectable in dystrophic mdx muscles even when subjected to mechanical stimulation (Goldspink, 1999; Yang and Goldspink, 2002). This is likely to contribute to the muscle impairment in the mdx mouse.
Myoblasts represent the natural first choice in cellular therapeutics for skeletal muscle because of their intrinsic myogenic commitment (Grounds et al., 2002). However, myoblasts are recovered in low number from DMD muscle biopsies, are poorly expandable in vitro, and rapidly undergo senescence (Cossu and Mavilio, 2000). An alternative source of muscle progenitor cells is desirable. We have reported recently the isolation and characterization of mesenchymal stem cells (MSCs) from the synovial membrane (SM) of adult human donors (De Bari et al., 2001). Human synovial membrane-derived MSCs (hSM-MSCs) are easily expandable in culture, maintain a stable molecular profile, and retain multipotentiality in vitro over at least 10 passages (De Bari et al., 2001). In the present work, we have characterized the myogenic differentiation of hSM-MSCs in an in vivo model of skeletal muscle regeneration and provide evidence of their capacity to partially restore specific pathophysiologic features of the dystrophic muscle in the mdx mouse model of DMD.
Results
Contribution to myofibers
To study myogenic differentiation of hSM-MSCs in vivo, we adopted a well-defined mouse model of skeletal muscle regeneration. The model consists of injuring the tibialis anterior (TA) muscle by injecting cardiotoxin (CTX). 24 h later, culture-expanded hSM-MSCs were injected into the same TA muscle. Nude mice were chosen to avoid rejection of the human cells.
To localize the injected human cells, we chose to trace the nuclei, which preserve their individuality within syncytial fibers, by using in situ hybridization (ISH) for human Alu genomic repeats. At 4 wk after transplantation, 60% (109 out of 180) of the stained longitudinal serial sections throughout the mouse TA muscle contained human nuclei (87 ± 45 per section, mean ± SD, range 3–285) (Fig. 1, A and B). To investigate the integration of human cells into muscle fibers, we followed two strategies. First, we implanted into regenerating TA muscles hSM-MSCs transduced with an adenovirus containing the LacZ gene under the transcriptional control of the cytomegalovirus (CMV) promoter (AdCMV-LacZ). At 1 wk, bacterial β-galactosidase (β-gal) was detected in mononuclear cells but not in myofibers (unpublished data). At 3 wk, some myofibers displayed diffuse β-gal expression (Fig. 1 C), demonstrating incorporation of at least one transduced human cell for each β-gal–positive fiber. The second strategy consisted of staining sections of TA muscles for human β2-microglobulin (β2M). After 3 wk, some fibers displayed sarcolemmal expression of human β2M in the TA muscle injected with hSM-MSCs (Fig. 1 D). The detection of human β2M at the sarcolemma of muscle fibers represents indirect evidence that each human-β2M–positive fiber was contributed by at least one hSM-MSC. The muscle fibers contributed by human cells were mostly located in regenerating areas as suggested by the heterogeneous size of the myofibers and the central location of their myonuclei (Gillis, 1999).
Figure 1.
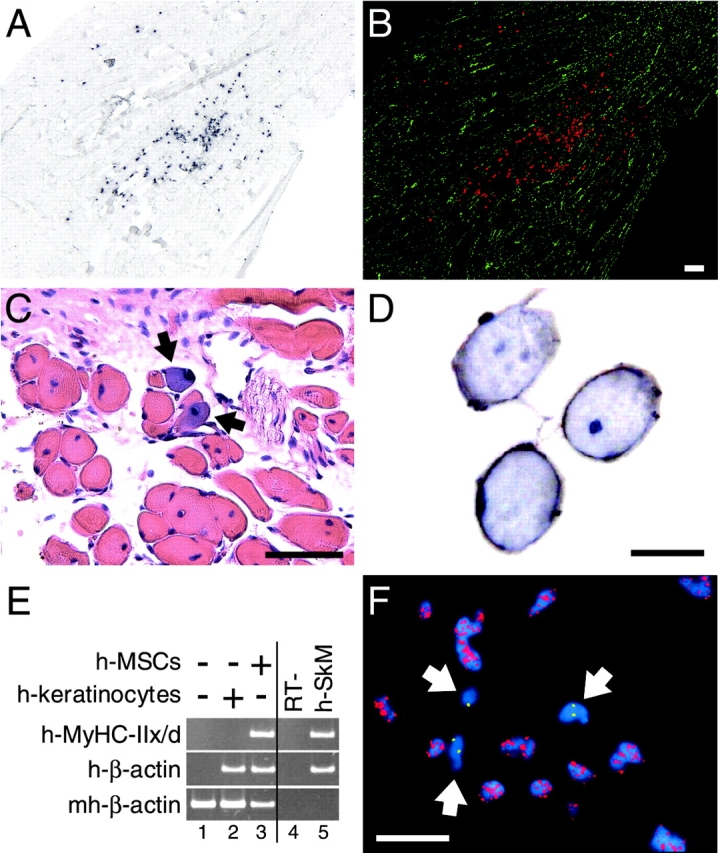
Contribution of hSM-MSCs to skeletal muscle regeneration in vivo. (A and B) Localization of the human nuclei within the mouse skeletal muscle. (A) ISH for human Alu repeats on a representative longitudinal cryosection from a nude mouse TA muscle at 4 wk after hSM-MSC transplantation. Human nuclei are stained black. In B, brightfield and fluorescence (DAPI counterstaining) images were given artificial colors and superimposed. The Alu-positive nuclei are shown in red, and the Alu-negative, DAPI-stained nuclei are shown in green. Bar, 200 μm. (C) X-gal staining with hematoxylin-eosin counterstaining at 3 wk after transplantation of hSM-MSCs transduced with AdCMV-LacZ. Arrows point to muscle fibers with diffuse β-gal expression. Bar, 50 μm. (D) Immunohistochemistry for human β2M (brown). Nuclei are counterstained with hematoxylin (blue). Bar, 20 μm. (E) SQ–RT-PCR for human MyHC-IIx/d. CTX-treated TA muscle was injected with either human keratinocytes (lane 2) or hSM-MSCs (lane 3); CTX-treated muscle injected with PBS was used as tissue-negative control (lane 1). Lane 4, RT-negative control of lane 3; lane 5, human skeletal muscle (h-SkM) as a positive control. cDNAs were equalized for the expression of human β-actin. At 3 wk after transplantation, human MyHC-IIx/d was detected only in the muscle injected with SM-MSCs. (F) Double genomic FISH on a cryosection from a TA muscle at 4 wk after hSM-MSC transplantation using a probe for human CEN18 (green) and a probe for mouse centromeric satellite DNA (red). Nuclei were counterstained with DAPI (blue). Arrows point to human nuclei. Bar, 50 μm.
To determine whether the human cells implanted in the mouse TA muscles acquired the skeletal muscle phenotype, we used RT-PCR with primers specific for human cDNAs. At 3 wk, we detected human myosin heavy chain type IIx/d (MyHC-IIx/d) in the TA muscles injected with hSM-MSCs. Under similar experimental conditions, we did not detect human muscle markers in the TA muscles injected with human keratinocytes, although human β-actin was detectable (Fig. 1 E).
Cell nuclear fusion has been suggested recently as a possible explanation for apparent stem cell plasticity (Terada et al., 2002; Ying et al., 2002). To investigate the occurrence of fusion hybrids between donor hSM-MSC nuclei and recipient mouse muscle nuclei, we performed double FISH using a probe for human centromere 18 (CEN18) and a probe for mouse centromeric satellite DNA on sections of TA muscles 4 wk after hSM-MSC transplantation (Fig. 1 F). Over 160 human nuclei counted in two longitudinal sections, 154 (96%) were distinct from mouse nuclei. The remaining six human nuclei were located in areas where mouse and human nuclei were clustered, making it impossible to distinguish between overlapping or fused nuclei (unpublished data). Together, these findings provide evidence that implanted hSM-MSCs can fuse with host myofibers and contribute their genetic information to the mosaic fibers generated. The detection of human MyHC-IIx/d suggests that some human cells acquired the skeletal muscle phenotype. Contribution to muscle regeneration was reproducible regardless of cell storage in liquid nitrogen or donor age within the ranges examined and was inherent to individual cell clones of multipotent SM-MSCs (Fig. S1 available at http://www.jcb.org/cgi/content/full/jcb.200212064/DC1).
SM-MSC differentiation recapitulates embryonic myogenesis
To test the hypothesis that the mature skeletal muscle phenotype of the human cells was acquired through a cascade of molecular events reminiscent of embryonic myogenesis, we analyzed expression of muscle differentiation genes at several time points by semiquantitative (SQ) RT-PCR using primers specific for human cDNAs. We observed an early peak of human Myf5 and proliferating cell nuclear antigen (PCNA) at 24 h after cell implantation. With the decline of Myf5, the expression of human myogenin increased, peaking at 8 d, and then decreased, whereas markers of mature muscle, such as MyHC, muscle creatine kinase, and dystrophin, progressively reached plateau levels (Fig. 2 A). These data indicate that muscle differentiation of hSM-MSCs, in this model, appears to recapitulate embryonic muscle formation. The proliferation, suggested by high levels of PCNA, paralleled an increase in the expression of human β-actin per TA muscle over time (Fig. 2 B). The expression of human β-actin was considered indicative of the number of human cells or, since some human cells fused with myofibers, human “cell equivalents” (CE) per TA muscle. Values are expressed as the total number of human CE per TA muscle. They were obtained by performing a regression analysis of the ratio of human β-actin to mouse/human β-actin using a standard curve generated by injecting from 2.5 million to 30 hSM-MSCs (unpublished data). The experimental values were within the linear range of the curve.
Figure 2.
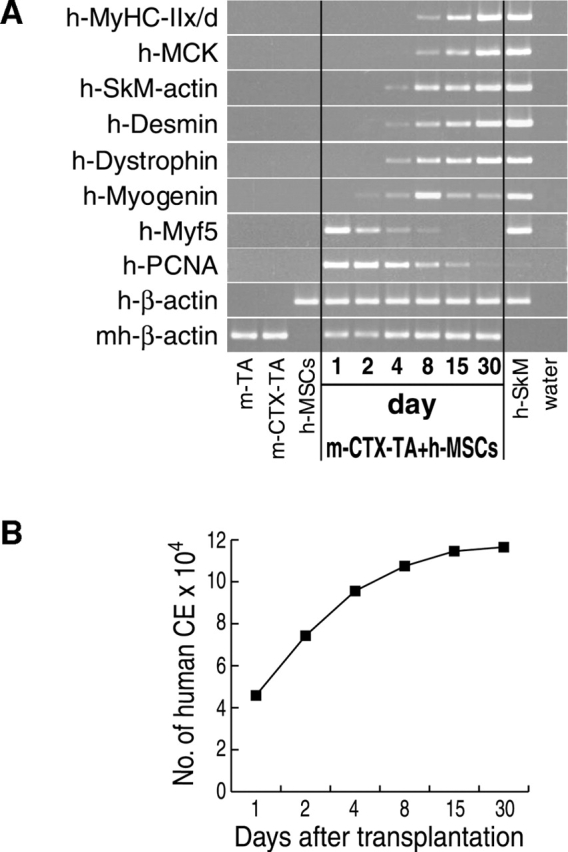
Muscle differentiation of hSM-MSCs recapitulates embryonic myogenesis. (A) hSM-MSCs were injected into regenerating (CTX-treated) TA muscles of nude mice. Mice were killed at different time points as indicated. The dissected muscles were subjected to SQ–RT-PCR analysis. TA muscles containing human cells were equalized for the expression of human β-actin. TA muscles with no human cells served as controls for species specificity of the primers for human cDNAs. For each time point, a PBS-injected regenerating (CTX-treated) mouse TA muscle was included for a negative control. m-TA, uninjected mouse TA muscle; m-CTX-TA, mouse CTX-treated TA muscle. (B) Number of human CE per TA muscle over time as determined by Q–RT-PCR (see Results for details).
Differentiation is sensitive to environmental cues
Systemic delivery is an attractive route of administration of cells to target tissue and organ systems. We tested whether hSM-MSCs preferentially home to damaged skeletal muscle and contribute to muscle regeneration when delivered systemically. We administered 5 × 106 hSM-MSCs in the tail vein of six mice, which had received a CTX injection into one TA muscle 24 h earlier. Two animals were killed at each time point examined, up to 6 mo. hSM-MSCs homed preferentially to the CTX-injured TA muscle. However, the number of human CE, initially not detected in the uninjured muscle, increased progressively over time in both TA muscles, remaining constantly higher in the CTX-injured muscle (Fig. 3 A). At 6 mo, we detected human MyHC-IIx/d in both CTX-treated and -untreated TA muscles (Fig. 3 B) and localized human nuclei by ISH for human Alu genomic repeats (Fig. 3 C). We do not report expression of human MyHC-IIx/d at 3 and 8 wk because at these time points the numbers of human CE in TA muscles were too low for a reliable estimation of its expression levels. At 6 mo, the CTX-induced muscle injury did not influence the expression of human MyHC-IIx/d normalized for human β-actin (Fig. 3 B), as in the experiments performed with local implantation of hSM-MSCs at 3 mo (Fig. S2 available at http://www.jcb.org/cgi/content/full/jcb.200212064/DC1). Although, as determined by RT-PCR for human β-actin and/or ISH for human Alu repeats, human cells were also found in other mouse tissues and organs such as lungs, we have been unable to detect expression of human MyHC-IIx/d and muscle creatine kinase in tissues other than skeletal muscle at any of the time points examined (Fig. 3 B). Together, these results suggest that hSM-MSCs engrafted into skeletal muscle after systemic administration, with preferential homing to the damaged muscle and long-term contribution to muscle regeneration. The expression of human MyHC-IIx/d was specific to skeletal muscle with no apparent heterotopic muscle formation, suggesting a context-sensitive differentiation response of the hSM-MSCs. We have never observed any adverse effect(s), such as ectopic tissue formation or tumor development, after injection in nude mice of hSM-MSCs even at high doses (2 × 107 cells) regardless of the site and the route of administration (Fig. S2).
Figure 3.

Differentiation is sensitive to environmental cues. (A) Preferential homing to the damaged skeletal muscle of systemically delivered hSM-MSCs. As determined by Q–RT-PCR, the number of human CE increased in parallel over time in both CTX-injured and uninjured host TA muscles, remaining constantly higher in the CTX-injured muscles. Two mice were killed at each time point examined with similar results. (B) SQ–RT-PCR analysis at the 6 mo time point. cDNAs were equalized for the expression of mouse/human β-actin. Human MyHC-IIx/d was detected in both the CTX-treated and -untreated TA muscles (lanes 2 and 3). Lungs (lane 4) expressed human β-actin at levels at least comparable to TA muscles, but MyHC-IIx/d was not detectable. Controls were mouse TA muscle (lane 1) and RT-negative control of lane 2 (lane 5). (C) ISH for human Alu repeats on a longitudinal section from a TA muscle at 6 mo after systemic administration of hSM-MSCs. Counterstaining with eosin. Bar, 50 μm.
Contribution to functional satellite cells
Satellite cells are mononucleated cells located between the sarcolemma and the basal lamina of the muscle fibers (Bischoff, 1994) and are considered the muscle-specific stem cells (LaBarge and Blau, 2002). To test the possibility that some of the implanted human cells contributed to the satellite cell compartment, we performed double immunostaining for laminin, which identifies the basal lamina, and human β2M, which labels the human cells, on sections of TA muscles 6 mo after human SM-MSC transplantation. We detected human-β2M–positive mononuclear cells residing between basal lamina and muscle fibers (Fig. 4 A). The staining for human β2M did not extend to the sarcolemma of the adjacent myofibers, indicating that the human cells had not fused with mouse muscle fibers. Given the absence of specific markers, EM is the most reliable way to identify satellite cells (Grounds et al., 2002). We analyzed sections of TA muscles of nude mice transplanted 6 mo earlier with hSM-MSCs by transmission electron microscopy (TEM), after staining for human β2M. Some mononuclear cells that fulfilled the ultrastructural criteria of satellite cells (Bischoff, 1994) displayed staining for human β2M that was associated with the plasma membrane (Fig. 4 B). These results demonstrate that a subpopulation of the implanted hSM-MSCs can persist for at least 6 mo as satellite cells.
Figure 4.

Contribution to functional satellite cells. (A) Double IF staining for laminin (red) and human β2M (green) at 6 mo after hSM-MSC transplantation into mouse TA muscle. The nuclei were revealed by DAPI staining (blue). A mononuclear cell staining positive for human β2M (arrowhead) is shown between the laminin-positive basal lamina and a distinct (β2M-negative) myofiber. The arrow points to another mononuclear cell of human origin lying outside of the basal lamina. Bar, 50 μm. (B) TEM of a hSM-MSC–derived satellite cell at 6 mo after hSM-MSC transplantation. The high magnification (bar, 1 μm) of a satellite cell shows a plasma membrane positive for human β2M (white arrows), separating the satellite cell from its adjacent β2M-negative myofiber, the continuous basal lamina (black arrow) surrounding the satellite cell and myofiber, and the heterochromatic appearance of the nucleus. Inset shows an inverted, magnified view of the staining for human β2M. (C) Satellite cell-like response of hSM-MSCs in vivo upon muscle injury. 6 mo after hSM-MSC bilateral implantation into regenerating TA muscles, CTX was injected into the right TA muscle and PBS into the contralateral muscle. Animals were killed 12 h later. SQ–RT-PCR analysis was performed after equalization of cDNAs for human β-actin. High levels of human Myf5 and human PCNA were observed in the CTX-treated TA muscle (lane 4) compared with the contralateral muscle (lane 5), indicating the presence of hSM-MSC–derived functional satellite cells. The following were controls: mouse TA muscle (lane 1); mouse TA muscle at 12 h after CTX treatment (lane 2); mouse CTX-treated TA muscle implanted with hSM-MSCs and harvested after 6 mo as external control (lane 3); and RT-negative control of lane 4 (lane 6). (D) Human mononuclear cells recovered from first recipient mice retain in vivo myogenic activity when transplanted into a second recipient. 6 mo after hSM-MSC transplantation, first recipient mice were killed, and cultures of primary myoblasts were established from the injected TA muscles. At 70% confluence, these cells were injected into regenerating TA muscles of second recipient mice, which were killed 1 mo later. Lane 1, control mouse TA muscle; lane 2, TA muscle from first mouse recipient; lane 3, first recipient primary myoblasts; lane 4, TA muscle from second mouse recipient; lane 5, RT-negative control of lane 4.
Quiescent (Myf5-negative) satellite cells respond to muscle injury with proliferation and expression of activation markers, such as Myf5 (Cornelison and Wold, 1997; Cooper et al., 1999). To test whether the engrafted hSM-MSCs contributed long term functional satellite cells, we injected CTX to injure the right TA muscles of three mice that 6 mo earlier had received hSM-MSCs bilaterally into regenerating TA muscles. Contralateral TA muscles received PBS. After 12 h, SQ–RT-PCR revealed high expression levels of human PCNA and Myf5 in the CTX-injured TA muscles compared with the CTX-untreated contralateral muscles (Fig. 4 C), indicating that human cells transplanted 6 mo earlier persisted within the regenerated muscle as quiescent (Myf5-negative) satellite cells and were capable of activation upon injury.
Satellite cells are known to be able to form myotubes under low serum conditions in vitro and to contribute to muscle repair when injected into a regenerating muscle in vivo (Seale and Rudnicki, 2000). To investigate whether the hSM-MSC–derived satellite cells displayed similar properties, we established cultures of primary myoblasts derived from satellite cells (Zammit and Beauchamp, 2001) from mouse TA muscles that had been injected with hSM-MSCs 6 mo earlier (first recipients). During in vitro expansion, human cell nuclei remained distinct from mouse cell nuclei with no apparent fusion as determined by double genomic FISH (unpublished data). As reported with primary myoblasts (Smith et al., 1994; Cornelison and Wold, 1997), in our cultures of first recipient primary myoblasts we detected human Myf5 but not terminal differentiation markers such as human MyHC-IIx/d (Fig. 4 D). Under low serum conditions, cells underwent terminal differentiation and formed multinucleated myotubes that expressed human MyHC (unpublished data). This property was not shared by hSM-MSCs before implantation in the first recipients (unpublished data). At ∼70% confluence, the first recipient primary myoblasts were implanted into regenerating TA muscles of second recipient mice. 1 mo later, we detected human MyHC-IIx/d (Fig. 4 D), indicating that recovered human cells contributed to muscle regeneration in second recipients.
Partial restoration of muscle pathology in mdx mice
To explore whether hSM-MSCs can correct the genetic muscle disorder of the mdx mouse, we transplanted hSM-MSCs into the right TA muscles of three immunosuppressed mdx mice. The left TA muscles were injected with PBS as internal controls. After 4 wk, mdx TA muscles injected with hSM-MSCs expressed human dystrophin and MyHC-IIx/d, whereas the contralateral PBS-injected TA muscles did not (Fig. 5 A). To localize human dystrophin protein, we performed an immunostaining using an antibody that does not react with mouse dystrophin (Huard et al., 1993). Therefore, putative revertant fibers expressing dystrophin protein (Lu et al., 2000) would not be detected (Braun et al., 2000). In hSM-MSC–injected mdx TA muscles, we observed clusters of myofibers with peripheral human-dystrophin immunoreactivity (Fig. 5 B). Human nuclei were detected in a parallel, nonconsecutive section in an area corresponding to the location of human-dystrophin–positive myofibers (Fig. 5 C).
Figure 5.

Restoration of dystrophin in mdx muscle fibers by hSM-MSCs. (A) SQ–RT-PCR for human dystrophin. cDNAs were equalized for mouse/human β-actin expression. In all mdx mice, TA muscles injected with hSM-MSCs expressed human dystrophin and human MyHC-IIx/d, whereas the contralateral muscles did not. (B) IF staining for human dystrophin (red) of a TA muscle transverse cryosection from an mdx mouse at 4 wk after hSM-MSC transplantation. Nuclei are shown in green. Clusters of muscle fibers display peripheral localization of the human dystrophin protein. Bar, 100 μm. (C) ISH for human Alu repeats on a parallel, nonconsecutive section, showing human nuclei in the squared area of (B). Counterstaining with eosin. Bar, 100 μm. (D) The percentage of CN human-dystrophin–positive fibers was significantly lower in the hSM-MSC–injected TA muscles than in the contralateral PBS-injected TA muscle fibers in three mdx mice (P < 0.05).
The percentage of centronucleated (CN) myofibers is indicative of previous cycles of degeneration and regeneration and is inversely correlated with the ability of dystrophin to protect muscle fibers from these cycles (Gillis, 1999). In hSM-MSC–injected mdx TA muscles, the percentage of CN human-dystrophin–positive fibers was significantly lower than the contralateral PBS-injected TA muscle fibers counted after hematoxylin-eosin staining (hSM-MSCs, 53.1% ± 5.2 versus PBS, 71.0% ± 1.6; P < 0.05) (Fig. 5 D). Together, these data indicate that transplantation of hSM-MSCs into dystrophic mdx muscle is associated with restoration of sarcolemmal expression of dystrophin and reduction of the central nucleation.
Partial rescue of mouse MGF in mdx muscle
Dysregulation of MGF expression has been described in mdx muscle. In particular, MGF was not detected by RT-PCR (Goldspink, 1999; Yang and Goldspink, 2002). Compared with PBS-injected contralateral TA muscles, we observed a reproducible partial restoration of mouse-specific MGF mRNA at 4 wk after hSM-MSC transplantation (Fig. 6 A). For comparison, we injected a plasmid DNA containing full-length human dystrophin (pCMV-dystrophin; Braun et al., 2000) into TA muscles of three immunosuppressed mdx mice. To increase the efficacy of in vivo transduction, electrotransfer (ET) was applied after plasmid DNA injection in additional three mice. After 4 wk, sarcolemmal expression of human dystrophin was detected by immunostaining in transverse sections from all pCMV-dystrophin–injected TA muscles (unpublished data). The maximal number of human-dystrophin–positive fibers per transverse section was 69.3 ± 10.3 in TA muscles injected with hSM-MSCs, 42.0 ± 7.9 in TA muscles injected with pCMV-dystrophin, and 275.0 ± 81.2 when ET was applied (Fig. 6 B). We observed no significant difference in the percentage of CN human-dystrophin–positive myofibers (hSM-MSCs, 53.1% ± 5.2, pCMV-dystrophin, 50.4% ± 4.9, pCMV-dystrophin ET, 47.3 ± 6.6) (Fig. 6 C). However, the expression levels of mouse MGF, as determined by quantitative (Q) RT-PCR, remained low in all pCMV-dystrophin–injected muscles, analogous to the PBS-injected or pCMV-LacZ–injected mdx muscles (Fig. 6 D). On longitudinal sections, dystrophin immunostaining was segmental in both SM-MSC–injected and pCMV-dystrophin–injected mdx TA muscles, extending over ∼100–700 μm (unpublished data), which reflects the dystrophin nuclear domain reported in previous studies (Gussoni et al., 1997; Vilquin et al., 2001).
Figure 6.
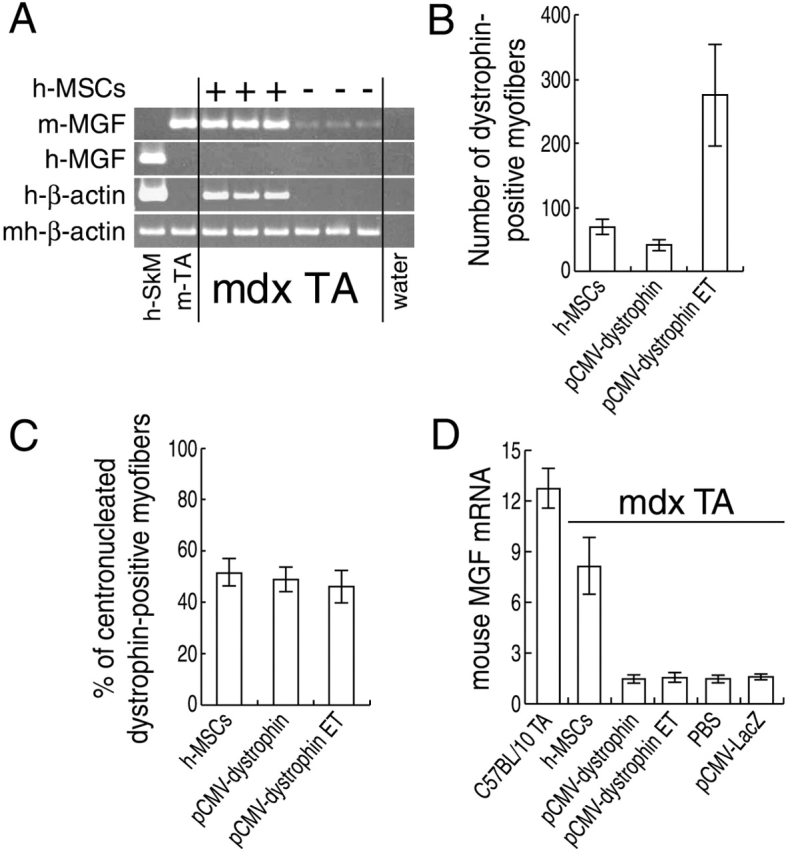
Rescue of mouse MGF in mdx muscle by hSM-MSCs. (A) SQ–RT-PCR for mouse MGF. cDNAs were equalized for mouse/human β-actin expression. In the first lane, human skeletal muscle was added for mouse primer specificity. Mouse MGF expression was reproducibly restored in the mdx TA muscles injected with hSM-MSCs to levels comparable to the TA muscle from a wild-type C57BL/10 mouse. (B) Maximal numbers of human-dystrophin–positive fibers in TA muscles of mdx mice injected with either hSM-MSCs or pCMV-dystrophin. In three mice, ET was applied after plasmid DNA injection. Animals were assayed for human-dystrophin expression at 4 wk after injections. For each treatment group, three TA muscles were serially cross sectioned and stained for human dystrophin. The sections containing the highest number of human- dystrophin–positive fibers were selected. Data are mean ± SD of maximal numbers of human-dystrophin–positive fibers per treatment group. (C) Percentage of CN human-dystrophin–positive fibers. We observed no significant difference comparing the three methods of dystrophin delivery. (D) Real-time Q–RT-PCR for mouse MGF normalized for mouse/human β-actin. The expression levels of mouse MGF in mdx TA muscles injected with hSM-MSCs were significantly higher (P < 0.05) than those found in mdx TA muscles injected with pCMV-dystrophin (with or without ET), with PBS, or with pCMV-LacZ. Normal TA muscles were from three age-matched C57BL/10 mice.
Discussion
We have described previously the isolation and characterization in vitro of MSCs from adult human SM (De Bari et al., 2001). In the present study, we report that hSM-MSCs can participate in skeletal muscle regeneration in vivo by long term persistence and contribution to both myofibers and functional satellite cells. In addition, we provide evidence that hSM-MSCs can partially restore muscle pathophysiology in mdx mice.
The identity of human SM-MSCs
A recent report (Gerhart et al., 2001) showed that MyoD-positive cells were present in many fetal chick organs and that these cells differentiated into skeletal muscle in culture. Whether similar committed myogenic cells are resident in postnatal tissues and organs in humans, and in particular in SM, remains to be determined. Nevertheless, we were unable to detect expression of MRFs in either human synovial tissue or SM-MSCs in vitro by RT-PCR.
Bone marrow (BM) is known to contain two types of stem cells, the hematopoietic stem cells and the MSCs, both displaying myogenic potential (Ferrari et al., 1998) and both circulating in the bloodstream (Kuznetsov et al., 2001; Wright et al., 2001). SM-MSCs are indeed similar to BM-MSCs in their behavior in vitro. Like BM-MSCs (Prockop, 1997; Pittenger et al., 1999), SM-MSCs rapidly adhere to plastic and can be expanded for several passages, preserving their molecular profile and multipotentiality (De Bari et al., 2001). These characteristics make MSCs, irrespective of their origin, distinct from hematopoietic stem cells (Prockop, 1997; Pittenger et al., 1999). Whether the SM-MSCs are derived from endogenous resident cells or originate from circulating MSCs (Kuznetsov et al., 2001) is debatable. Increasing evidence suggests that MSCs isolated from different tissues and organs would have different phenotypic and biological traits in vitro and in vivo (Kuznetsov et al., 2001; unpublished data). However, manipulations, such as tissue dissection, cell isolation, and subsequent culture expansion, can profoundly influence patterns of gene expression and differentiation potentials.
Recently, it has been demonstrated that postnatal BM contains primitive progenitors termed multipotent adult progenitor cells, which copurify with MSCs (Jiang et al., 2002). We cannot exclude that our hSM-MSCs also contain a subpopulation of these progenitors.
Muscle differentiation of hSM-MSCs recapitulates embryonic myogenesis
Expression by hSM-MSCs of muscle-specific genes does not appear to be the consequence of reprogramming of human nuclei secondary to fusion into host muscle fibers (Blau et al., 1985). First, some hSM-MSCs can differentiate in vitro to form MyHC-positive multinucleated myotubes (De Bari et al., 2001), demonstrating that fusion with muscle cells is not required for myogenesis. Second, we have documented immediate-early muscle-specific differentiation events happening and even extinguishing before fusion with myofibers could be proven. Although we cannot exclude sporadic early fusion with host muscle fibers, under our experimental conditions we did not detect muscle fibers contributed by human cells before 7 d. In contrast, we observed a peak of human Myf5 as early as 24 h after hSM-MSC transplantation. During embryonic development, Myf5 is necessary to restrict undifferentiated cells to myogenesis (Tajbakhsh et al., 1996). The subsequent decrease in Myf5 expression coincided with the peak of myogenin at 8 d followed by plateau levels of mature muscle markers. It is known that myogenin expression occurs during fusion and terminal differentiation (Hasty et al., 1993; Nabeshima et al., 1993; Smith et al., 1994; Yablonka-Reuveni and Rivera, 1994). Our findings suggest that hSM-MSCs undergo a multistep differentiation process, which comprises proliferation, commitment to the myogenic lineage, and eventual terminal maturation and fusion. These kinetics of muscle differentiation appear to be similar to those of BM cells, since it was reported that myoblasts injected into regenerating TA muscle fused into muscle fibers within 5 d, whereas BM cells required at least 2 wk for integration into muscle fibers (Ferrari et al., 1998).
hSM-MSCs can contribute to functional satellite cells
The number of satellite cells decreases with age, and this progressive decline is dramatically accelerated in DMD. The depletion of the satellite cell pool, with consequent irreversible muscle degeneration, is believed to be responsible for terminal muscle failure in DMD (Cossu and Mavilio, 2000). The ability of implanted stem cells to replenish the satellite cell compartment may ensure long term efficacy by restoring the regeneration potential necessary for muscle tissue homeostasis and repair. The persistence as quiescent satellite cells has been reported with muscle cells (Yao and Kurachi, 1993; Asakura et al., 2002; Qu-Petersen et al., 2002) and recently with BM cells (LaBarge and Blau, 2002). In the present study, we have demonstrated that a small population of the implanted hSM-MSCs persisted as functional satellite cells in muscle tissues for over 6 mo. Human mononuclear cells recovered from first recipient mice displayed in vitro phenotypic and functional properties of primary myoblasts and retained their myogenic capacity into second recipient mice. Since conversion of syncytial myofibers to mononucleated myoblasts has been described in mammals under specific conditions (Odelberg et al., 2000), we cannot exclude that recovered human mononuclear cells might have derived from disassembly and dedifferentiation of human myonuclei.
Recently, it has been demonstrated that adult skeletal muscle contains a population of stem cells distinct from satellite cells located outside of the muscle fiber basal lamina (Asakura et al., 2002). As shown in Fig. 4 A, we detected human mononucleated cells lying outside of the basal lamina. The phenotype and biology of these cells are currently under investigation.
Rescue of peculiar pathophysiologic features of dystrophic mdx muscle
The pathologic traits of the dystrophic mdx muscle include the absence of dystrophin protein and a high number of CN fibers (Gillis, 1999). Like dystrophin complementation via gene transfer, hSM-MSC transplantation resulted in restoration of dystrophin at the sarcolemma of muscle fibers and in a decrease of centronucleation in human-dystrophin–positive myofibers. In addition, hSM-MSCs rescued, at least in part, the capacity of mdx mouse muscle cells to produce MGF, a critical factor controlling local muscle maintenance and repair (Yang et al., 1996) not detectable in dystrophic mdx muscles (Goldspink, 1999; Yang and Goldspink, 2002).
Dystrophin, possibly involved in mechanotransduction mechanisms, may play a role in the regulation of MGF expression in muscle fibers in response to mechanical stimuli (Goldspink, 1999). The expression of a full-length human dystrophin by plasmid DNA delivery failed to rescue mouse MGF. However, the experimental protocols may explain, at least in part, this discrepancy. Cell implantation was localized, whereas plasmid injections were distributed along the muscle length. Possibly, in the first case a small number of fibers were expressing sufficient levels of dystrophin to recover mouse MGF expression. In the second case, a larger number of fibers were expressing lower amounts, not sufficient to reach the threshold needed for the correction of this specific disorder. Additionally, one must bear in mind that the correction of the molecular defect of DMD muscle with a functional dystrophin protein may not be curative for secondary events already established during the course of the hereditary disorder.
In conclusion, our data provide evidence that hSM-MSCs cannot only correct the initial molecular defect in mdx mice but also restore expression of mouse MGF, a molecule involved in maintenance and repair of skeletal muscle (Goldspink, 1999). These findings further support the potential of cellular therapeutics in patients with dystrophic muscle disorders.
Materials and methods
MSC isolation and culture
Random biopsies of SM (wet weight 10–50 mg) were obtained aseptically from the knee joints of human donors of various ages (mean 48 yr, range 18–83 yr) within 12 h postmortem. MSCs were isolated and expanded in monolayer on plastic in growth medium (DME [Life Technologies] containing 10% FBS [BioWhittaker] and antibiotics [Life Technologies]) as described previously (De Bari et al., 2001). At passage 3, aliquots of cells were cryopreserved in liquid nitrogen, thawed after variable times (range 3–36 mo), replated, and expanded. For all experiments, we used SM-MSCs between passages 3 and 10.
Adenovirus transduction
The replication-deficient recombinant AdCMV-LacZ and the empty backbone adenovirus were gifts from The Center for Transgene Technology and Gene Therapy (Leuven, Belgium). For transduction, cells were replated in growth medium after addition of the virus at 10 multiplicity of infection. The next day, the virus supernatant was removed, and the cells were washed with several changes of medium. 5 d later, cells were harvested for the in vivo myogenesis assay. The efficiency of transduction was ∼50%.
Animals and transplant procedure
Animal experimentation protocols were approved by the local ethics committee. 8-wk-old female NMRI nu−/− mice were used for the in vivo model of muscle regeneration. 25 μl of 10 μM CTX (Latoxan) were injected in the TA muscle. The day after, 5 × 105 hSM-MSCs suspended in 25 μl PBS were administered (single-point injection) into the same TA muscle. For systemic injections, 5 × 106 hSM-MSCs in 250 μl DME were infused into the bloodstream of a tail vein.
Dystrophin-deficient mdx mice (C57BL/10ScSn DMDmdx/J) were purchased from Jackson ImmunoResearch Laboratories. 2-mo-old mice were used for all experiments. Transplantation was performed by single-point injection of 106 hSM-MSCs in 50 μl PBS into the right TA muscle. The left TA muscle served as internal control receiving PBS. Recipient mice were immunosuppressed with FK506 (Fujisawa Pharmaceutical Co. Ltd.) administered intraperitoneally at the dose of 2.5 mg/kg per day (Kinoshita et al., 1994) from the day of transplantation until the animals were killed 4 wk after transplantation.
DNA injection and electric pulse delivery
pTG11025 plasmid containing full-length human dystrophin driven by the CMV promoter (Braun et al., 2000) was a gift from S. Braun (Transgene, Strasbourg, France). Six mdx mice were anesthetized, and 50 μg of plasmid DNA in 50 μl of 0.9% NaCl was injected percutaneously into the right TA muscle in 5–10 different sites (5–10 μl per site). Sham control injections were done with pCMV-LacZ or PBS. Immediately after DNA administration, transcutaneous electric pulses were applied to the hindlimbs of three mice as described previously (Vilquin et al., 2001). The animals were immunosuppressed with FK506 and killed 4 wk after plasmid DNA injection.
Myoblast isolation and transplantation
Primary myoblasts were isolated from TA muscles of six nude mice (14 mo of age) transplanted with hSM-MSCs 6 mo earlier as described previously (Ferrari et al., 1998). Dissociated single cells were plated on plastic Petri dishes and maintained for 2 h at 37°C in growth medium to allow attachment of fibroblasts. Nonadherent cells were collected and plated on gelatin-coated culture plates in DME containing 20% FBS and antibiotics. Differentiation to myotubes was induced by exposing confluent myoblast culture to DME containing 2% horse serum (Life Technologies) for 48 h. At 70% confluence, myoblasts were released with trypsin and implanted into regenerating TA muscles. Since the expanded myoblast population contained both mouse and human cells, we used 3 × 106 cells in 60 μl PBS per injection.
Tissue processing
Mice were killed by cervical dislocation, and TA muscles were excised. For total RNA extraction, TA muscles were homogenized in TRIzol (Life Technologies). For histology, histochemistry, and ISH, unless differently stated, they were either fixed overnight at 4°C in 10% neutral buffered formalin, embedded in paraffin, and sectioned at 5 μm, or cryoembedded and sectioned at 10 μm. For staining for human β2M, specimens were fixed with 2% glutaraldehyde in 0.05 M cacodylate buffer (pH 7.3) at 4°C for 60 min, embedded in paraffin, and sectioned at 7 μm. Sections were mounted on poly-l-lysine–coated glass slides and Thermanox coverslips (Electron Microscopy Sciences) for light microscopy and TEM, respectively. TA muscles from mdx mice were divided transversely into two equal parts, of which one was used for total RNA extraction and the other to make cryosections.
Histochemistry
Whole mount X-gal staining of TA muscles was performed overnight at 30°C according to the standard method. Muscles were then embedded in paraffin and sectioned at 7 μm to observe LacZ expression at the cellular level.
To perform immunostaining for human β2M, sections were deparaffinized and blocked with sheep anti–mouse Ig (Chemicon). Endogenous peroxidase was quenched with 0.5% H2O2 in methanol. Sections were incubated for 1 h with a mouse mAb specific to human β2M (BD Biosciences) diluted 1:50 in PBS. For light microscopy, immunoreactivity was detected using the peroxidase-based EnVisionTM System (Dako) using DAB as a chromogenic substrate and Mayer's hematoxylin to counterstain nuclei. For TEM, immunoreactivity was detected using a silver-enhanced preembedding colloidal gold immunohistochemistry system (Aurion). After immunolabeling, sections were postfixed in 2% osmium tetroxide, stained with 2% uranyl acetate in 10% acetone, dehydrated, and embedded on the plastic coverslip in araldite according to the “Pop-Off technique.” Ultrathin sections (0.2 μm) were mounted on 0.7% formvar-coated grids, stained with uranyl acetate and lead citrate, and examined with a Philips EM 208 transmission electron microscope.
For double immunofluorescence (IF) staining, cryostat sections were fixed with 4% PFA, blocked with sheep anti–mouse Ig, and incubated overnight at 4°C with 10 μg/ml rabbit antilaminin polyclonal antibody (InnoGenex) and mouse anti–human β2M mAb diluted 1:50. The secondary antibodies were Cy3-conjugated goat anti–rabbit IgG (Jackson ImmunoResearch Laboratories) and Cy2-conjugated goat anti–mouse IgG (H+L) (Kirkegaard & Perry Laboratories). To detect human dystrophin, we used the mAb NCL-DYS3 (Novocastra Laboratories) following the manufacturer's protocol, with Cy3-conjugated goat anti–mouse IgG (Jackson ImmunoResearch Laboratories) as secondary antibody. Slides were mounted with Mowiol containing DAPI. Tissue and Ig isotype negative controls were included.
In situ hybridization
ISH for human Alu genomic repeats was performed as described elsewhere (Dell'Accio et al., 2001). Slides were mounted with Mowiol containing DAPI. For FISH, mouse centromeric probe was generated by PCR using the following primers: forward, 5′-GGAAAATGATAAAAACCACACTG-3′ and reverse, 5′-TGTTTCTCATTGTAACTCATTGAT-3′. The human probe was generated from BAC DNA (RPCI-373M8) containing CEN18 sequences. Labeling of the human CEN18 DNA and the mouse centromeric PCR product with fluorescein-dUTP and lissamine-dUTP, respectively, was performed using the BioNick™ Labeling System kit (Life Technologies). Cryostat sections were treated with pepsin, fixed in 1% acid-free formaldehyde solution in PBS, washed with PBS, dehydrated, and air dried. Chromosomes were denatured by incubating the slides at 72°C in a 70% formamide/2× SSC solution and dehydrated through an ice-cold ethanol series. Probes were denatured in hybridization mixture (50% formamide, 2× SSC, 10% dextran-sulfate) for 5 min at 75°C and applied on the slides, which were incubated overnight at 37°C. The next day, slides were washed 1 min in 0.4× SSC/0.3% NP-40 at 73°C, 1 min in 2× SSC/0.1% NP-40 at RT, and 5 min in 4T (4× SSC, 0.05% Tween 20, pH 7.0) at RT. Slides were dehydrated and mounted with antifade medium (Vectashield; Vector Laboratories) containing DAPI.
RT-PCR
Total RNA was isolated using TRIzol. After DNase treatment, cDNAs were obtained by reverse transcription of 2 μg of total RNA (Thermoscript; Life Technologies) using oligo(dT)20 as primer. SQ-PCR was performed as described previously (De Bari et al., 2001). Gene expression of human cells within muscle tissues was evaluated using primers specific for human cDNAs. When mouse/human chimeric samples were equalized for the expression of human β-actin, control mouse samples with no human cells were normalized to the mouse/human chimeric sample of the series with the highest mouse/human β-actin. In the mouse/human chimeric samples, the molar ratio of human over mouse β-actin mRNA was <0.01 as determined by Q–RT-PCR. Therefore, in Figs. 1 E and 2 A, after normalization for human β-actin at 25 cycles, the 18 cycles performed for mouse/human β-actin were optimal to show that the mouse controls contained at least as much cDNA template as the most concentrated of the experimental mouse/human samples but not sufficient to reach detection levels of β-actin in the human reference samples. For MGF, the reverse primer was located within the insert that differentiates MGF from insulin-like growth factor-I (Yang et al., 1996) (sequence data available from GenBank/EMBL/DDBJ under accession no. U40870 for human cDNA, and NM_010512 for mouse cDNA). Sequencing of the PCR product confirmed the specificity of the primers for mouse MGF. Real-time Q-PCR was performed using the ABI PRISM 7700 detection system (Taqman; Applied Biosystems). The sequences of the primers and the expected sizes of the amplification products are listed in the Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200212064/DC1).
Statistics
An investigator, unaware of the treatment group, analyzed a minimum of 300 fibers per mdx TA muscle. Results are mean ± SD. Data were analyzed using the Student's t test for the comparison of the CN muscle fibers, and the one way repeated measures ANOVA followed by multiple comparisons with the Student-Newman-Keuls method to compare the values of MGF expression. P < 0.05 was significant.
Online supplemental material
Fig. S1 shows that the in vivo myogenic potential of hSM-MSCs was reproducible, regardless of cell storage in liquid nitrogen or donor age, within the ranges examined and was inherent to individual cell clones of multipotent SM-MSCs. Fig. S2 shows that CTX treatment was not required for muscle differentiation of hSM-MSCs and that differentiation of locally implanted hSM-MSCs to skeletal muscle was a tissue-specific event with no detectable heterotopic tissue formation. In Table S1, the sequences of the primers and the expected sizes of the amplification products are listed. All supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200212064/DC1.
Acknowledgments
We thank J.M. Gillis, M. Moos, J.T. Thomas, P. Carmeliet, and F. Wuytack for critical reading of this manuscript, and J.T. Vilquin for helpful discussion about in vivo gene transfer. We also thank the Association Belge contre les Maladies Neuromusculaires for providing the mdx mice. We are grateful to J. Peeters, J. Neys, and I. Derese for processing tissue blocks for histology.
This work was supported by Fonds voor Wetenschappelijk Onderzoek grant G.0192.99 and Innovatie door Wetenschap en Technologie grant N. 000259, and in part by the Association Française contre les Myopathies. J.M. Raymackers is a research fellow of the Fonds National de la Recherche Scientifique.
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: AdCMV-LacZ, adenovirus containing the LacZ gene under the CMV promoter; β-gal, β-galactosidase; β2M, β2-microglobulin; BM, bone marrow; CE, cell equivalents; CEN18, centromere 18; CMV, cytomegalovirus; CN, centronucleated; CTX, cardiotoxin; DMD, Duchenne muscular dystrophy; ET, electrotransfer; hSM-MSC, human synovial membrane-derived mesenchymal stem cell; IF, immunofluorescence; ISH, in situ hybridization; MGF, mechano growth factor; MRF, myogenic regulatory factor; MSC, mesenchymal stem cell; MyHC-IIx/d, myosin heavy chain type IIx/d; PCNA, proliferating cell nuclear antigen; Q, quantitative; pCMV-dystrophin, plasmid DNA containing human dystrophin under the CMV promoter; SM, synovial membrane; SQ, semiquantitative; TA, tibialis anterior; TEM, transmission electron microscopy.
References
- Asakura, A., P. Seale, A. Girgis-Gabardo, and M.A. Rudnicki. 2002. Myogenic specification of side population cells in skeletal muscle. J. Cell Biol. 159:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff, R. 1994. The satellite cell and muscle regeneration. Myogenesis. A.G. Engel and C. Franszini-Armstrong, editors. McGraw-Hill, New York. 97–118.
- Blau, H.M., G.K. Pavlath, E.C. Hardeman, C.P. Chiu, L. Silberstein, S.G. Webster, S.C. Miller, and C. Webster. 1985. Plasticity of the differentiated state. Science. 230:758–766. [DOI] [PubMed] [Google Scholar]
- Braun, S., C. Thioudellet, P. Rodriguez, D. Ali-Hadji, F. Perraud, N. Accart, J.M. Balloul, C. Halluard, B. Acres, B. Cavallini, and A. Pavirani. 2000. Immune rejection of human dystrophin following intramuscular injections of naked DNA in mdx mice. Gene Ther. 7:1447–1457. [DOI] [PubMed] [Google Scholar]
- Cooper, R.N., S. Tajbakhsh, V. Mouly, G. Cossu, M. Buckingham, and G.S. Butler-Browne. 1999. In vivo satellite cell activation via Myf5 and MyoD in regenerating mouse skeletal muscle. J. Cell Sci. 112:2895–2901. [DOI] [PubMed] [Google Scholar]
- Cornelison, D.D., and B.J. Wold. 1997. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev. Biol. 191:270–283. [DOI] [PubMed] [Google Scholar]
- Cossu, G., and F. Mavilio. 2000. Myogenic stem cells for the therapy of primary myopathies: wishful thinking or therapeutic perspective? J. Clin. Invest. 105:1669–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bari, C., F. Dell'Accio, P. Tylzanowski, and F.P. Luyten. 2001. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 44:1928–1942. [DOI] [PubMed] [Google Scholar]
- Dell'Accio, F., C. De Bari, and F.P. Luyten. 2001. Molecular markers predictive of the capacity of expanded human articular chondrocytes to form stable cartilage in vivo. Arthritis Rheum. 44:1608–1619. [DOI] [PubMed] [Google Scholar]
- Ervasti, J.M., and K.P. Campbell. 1991. Membrane organization of the dystrophin-glycoprotein complex. Cell. 66:1121–1131. [DOI] [PubMed] [Google Scholar]
- Ferrari, G., A.G. Cusella-De Angelis, M. Coletta, E. Paolucci, A. Stornaiuolo, G. Cossu, and F. Mavilio. 1998. Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 279:1528–1530. [DOI] [PubMed] [Google Scholar]
- Gerhart, J., B. Bast, C. Neely, S. Iem, P. Amegbe, R. Niewenhuis, S. Miklasz, P.F. Cheng, and M. George-Weinstein. 2001. MyoD-positive myoblasts are present in mature fetal organs lacking skeletal muscle. J. Cell Biol. 155:381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis, J.M. 1999. Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J. Muscle Res. Cell Motil. 20:605–625. [DOI] [PubMed] [Google Scholar]
- Goldspink, G. 1999. Changes in muscle mass and phenotype and the expression of autocrine and systemic growth factors by muscle in response to stretch and overload. J. Anat. 194:323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grounds, M.D., J.D. White, N. Rosenthal, and M.A. Bogoyevitch. 2002. The role of stem cells in skeletal and cardiac muscle repair. J. Histochem. Cytochem. 50:589–610. [DOI] [PubMed] [Google Scholar]
- Gussoni, E., H.M. Blau, and L.M. Kunkel. 1997. The fate of individual myoblasts after transplantation into muscles of DMD patients. Nat. Med. 3:970–977. [DOI] [PubMed] [Google Scholar]
- Hasty, P., A. Bradley, J.H. Morris, D.G. Edmondson, J.M. Venuti, E.N. Olson, and W.H. Klein. 1993. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 364:501–506. [DOI] [PubMed] [Google Scholar]
- Hoffman, E.P., R.H. Brown, Jr., and L.M. Kunkel. 1987. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 51:919–928. [DOI] [PubMed] [Google Scholar]
- Huard, J., G. Tremblay, S. Verreault, C. Labrecque, and J.P. Tremblay. 1993. Utilization of an antibody specific for human dystrophin to follow myoblast transplantation in nude mice. Cell Transplant. 2:113–118. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., B.N. Jahagirdar, R.L. Reinhardt, R.E. Schwartz, C.D. Keene, X.R. Ortiz-Gonzalez, M. Reyes, T. Lenvik, T. Lund, M. Blackstad, et al. 2002. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 418:41–49. [DOI] [PubMed] [Google Scholar]
- Kinoshita, I., J.T. Vilquin, B. Guerette, I. Asselin, R. Roy, and J.P. Tremblay. 1994. Very efficient myoblast allotransplantation in mice under FK506 immunosuppression. Muscle Nerve. 17:1407–1415. [DOI] [PubMed] [Google Scholar]
- Kuznetsov, S.A., M.H. Mankani, S. Gronthos, K. Satomura, P. Bianco, and P.G. Robey. 2001. Circulating skeletal stem cells. J. Cell Biol. 153:1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBarge, M.A., and H.M. Blau. 2002. Biological progression from adult bone marrow to mononucleate muscle stem cell to multinucleate muscle fiber in response to injury. Cell. 111:589–601. [DOI] [PubMed] [Google Scholar]
- Lu, Q.L., G.E. Morris, S.D. Wilton, T. Ly, O.V. Artem'yeva, P. Strong, and T.A. Partridge. 2000. Massive idiosyncratic exon skipping corrects the nonsense mutation in dystrophic mouse muscle and produces functional revertant fibers by clonal expansion. J. Cell Biol. 148:985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeshima, Y., K. Hanaoka, M. Hayasaka, E. Esumi, S. Li, I. Nonaka, and Y. Nabeshima. 1993. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature. 364:532–535. [DOI] [PubMed] [Google Scholar]
- Odelberg, S.J., A. Kollhoff, and M.T. Keating. 2000. Dedifferentiation of mammalian myotubes induced by msx1. Cell. 103:1099–1109. [DOI] [PubMed] [Google Scholar]
- Pittenger, M.F., A.M. Mackay, S.C. Beck, R.K. Jaiswal, R. Douglas, J.D. Mosca, M.A. Moorman, D.W. Simonetti, S. Craig, and D.R. Marshak. 1999. Multilineage potential of adult human mesenchymal stem cells. Science. 284:143–147. [DOI] [PubMed] [Google Scholar]
- Prockop, D.J. 1997. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 276:71–74. [DOI] [PubMed] [Google Scholar]
- Qu-Petersen, Z., B. Deasy, R. Jankowski, M. Ikezawa, J. Cummins, R. Pruchnic, J. Mytinger, B. Cao, C. Gates, A. Wernig, and J. Huard. 2002. Identification of a novel population of muscle stem cells in mice: potential for muscle regeneration. J. Cell Biol. 157:851–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki, M.A., P.N. Schnegelsberg, R.H. Stead, T. Braun, H.H. Arnold, and R. Jaenisch. 1993. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 75:1351–1359. [DOI] [PubMed] [Google Scholar]
- Sabourin, L.A., and M.A. Rudnicki. 2000. The molecular regulation of myogenesis. Clin. Genet. 57:16–25. [DOI] [PubMed] [Google Scholar]
- Seale, P., and M.A. Rudnicki. 2000. A new look at the origin, function, and “stem-cell” status of muscle satellite cells. Dev. Biol. 218:115–124. [DOI] [PubMed] [Google Scholar]
- Sicinski, P., Y. Geng, A.S. Ryder-Cook, E.A. Barnard, M.G. Darlison, and P.J. Barnard. 1989. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 244:1578–1580. [DOI] [PubMed] [Google Scholar]
- Smith, C.K., M.J. Janney, and R.E. Allen. 1994. Temporal expression of myogenic regulatory genes during activation, proliferation, and differentiation of rat skeletal muscle satellite cells. J. Cell. Physiol. 159:379–385. [DOI] [PubMed] [Google Scholar]
- Tajbakhsh, S., D. Rocancourt, and M. Buckingham. 1996. Muscle progenitor cells failing to respond to positional cues adopt non-myogenic fates in myf-5 null mice. Nature. 384:266–270. [DOI] [PubMed] [Google Scholar]
- Terada, N., T. Hamazaki, M. Oka, M. Hoki, D.M. Mastalerz, Y. Nakano, E.M. Meyer, L. Morel, B.E. Petersen, and E.W. Scott. 2002. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 416:542–545. [DOI] [PubMed] [Google Scholar]
- Vilquin, J.T., P.F. Kennel, M. Paturneau-Jouas, P. Chapdelaine, N. Boissel, P. Delaere, J.P. Tremblay, D. Scherman, M.Y. Fiszman, and K. Schwartz. 2001. Electrotransfer of naked DNA in the skeletal muscles of animal models of muscular dystrophies. Gene Ther. 8:1097–1107. [DOI] [PubMed] [Google Scholar]
- Wright, D.E., A.J. Wagers, A.P. Gulati, F.L. Johnson, and I.L. Weissman. 2001. Physiological migration of hematopoietic stem and progenitor cells. Science. 294:1933–1936. [DOI] [PubMed] [Google Scholar]
- Yablonka-Reuveni, Z., and A.J. Rivera. 1994. Temporal expression of regulatory and structural muscle proteins during myogenesis of satellite cells on isolated adult rat fibers. Dev. Biol. 164:588–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S., M. Alnaqeeb, H. Simpson, and G. Goldspink. 1996. Cloning and characterization of an IGF-1 isoform expressed in skeletal muscle subjected to stretch. J. Muscle Res. Cell Motil. 17:487–495. [DOI] [PubMed] [Google Scholar]
- Yang, S.Y., and G. Goldspink. 2002. Different roles of the IGF-I Ec peptide (MGF) and mature IGF-I in myoblast proliferation and differentiation. FEBS Lett. 522:156–160. [DOI] [PubMed] [Google Scholar]
- Yao, S.N., and K. Kurachi. 1993. Implanted myoblasts not only fuse with myofibers but also survive as muscle precursor cells. J. Cell Sci. 105:957–963. [DOI] [PubMed] [Google Scholar]
- Ying, Q.L., J. Nichols, E.P. Evans, and A.G. Smith. 2002. Changing potency by spontaneous fusion. Nature. 416:545–548. [DOI] [PubMed] [Google Scholar]
- Zammit, P., and J. Beauchamp. 2001. The skeletal muscle satellite cell: stem cell or son of stem cell? Differentiation. 68:193–204. [DOI] [PubMed] [Google Scholar]