

Abstract
We report the development and characterization of an in vitro system for the formation of filopodia-like bundles. Beads coated with actin-related protein 2/3 (Arp2/3)–activating proteins can induce two distinct types of actin organization in cytoplasmic extracts: (1) comet tails or clouds displaying a dendritic array of actin filaments and (2) stars with filament bundles radiating from the bead. Actin filaments in these bundles, like those in filopodia, are long, unbranched, aligned, uniformly polar, and grow at the barbed end. Like filopodia, star bundles are enriched in fascin and lack Arp2/3 complex and capping protein. Transition from dendritic to bundled organization was induced by depletion of capping protein, and add-back of this protein restored the dendritic mode. Depletion experiments demonstrated that star formation is dependent on Arp2/3 complex. This poses the paradox of how Arp2/3 complex can be involved in the formation of both branched (lamellipodia-like) and unbranched (filopodia-like) actin structures. Using purified proteins, we showed that a small number of components are sufficient for the assembly of filopodia-like bundles: Wiskott-Aldrich syndrome protein (WASP)–coated beads, actin, Arp2/3 complex, and fascin. We propose a model for filopodial formation in which actin filaments of a preexisting dendritic network are elongated by inhibition of capping and subsequently cross-linked into bundles by fascin.
Keywords: filopodia; actin; Arp2/3; capping protein; fascin
Introduction
Lamellipodia and filopodia are the two major types of protrusive organelles in crawling cells. Multiple lines of evidence indicate that lamellipodial protrusion occurs by a dendritic nucleation/array treadmilling model (Mullins et al., 1998; Borisy and Svitkina, 2000). In this model, members of the Wiskott-Aldrich syndrome protein (WASP)* family activate the actin-related protein 2/3 (Arp2/3) complex and nucleate the formation of actin filaments on preexisting filaments, which function as coactivators (Higgs and Pollard, 2001). Repeated dendritic nucleation generates a branched array of filaments, as found at the leading edge of cells (Svitkina et al., 1997; Svitkina and Borisy, 1999) or in comet tails (Cameron et al., 2001). Capping protein functions to cap excessive barbed ends (Cooper and Schafer, 2000), thus channeling actin polymerization close to the membrane.
Whereas lamellipodia seem designed for protrusion over a smooth surface, filopodia seem designed for exploring the extracellular matrix and surfaces of other cells. Filopodia, in contrast to the branched network of lamellipodia, contain an unbranched bundle of actin filaments that are aligned axially, packed tightly together, and of uniform polarity (Small et al., 1978; Lewis and Bridgman, 1992). Unlike lamellipodia, the Arp2/3 complex is excluded from filopodia (Svitkina and Borisy, 1999), and filaments in filopodia are relatively long and do not turn over rapidly (Mallavarapu and Mitchison, 1999). The roots of filopodia extend well into the cell lamellipodium. As proposed by the filament treadmilling model (Small et al., 1994), filaments elongate with their barbed ends oriented toward the leading edge, pushing the membrane and at the same time continuously depolymerizing from the pointed ends.
A major question in understanding filopodial formation is how they are initiated. One member of the WASP family, N-WASP, facilitates Cdc42-induced filopodia formation in cells (Miki et al., 1998), suggesting that Arp2/3-mediated nucleation of actin filaments plays a role in generating parallel bundles. How might Arp2/3 complex be involved in the formation of unbranched actin structures? One possibility is that the nucleation and branching activities of Arp2/3 complex can be separated, resulting in the production of dendritic or parallel actin structures, depending on the way in which it is activated. Another possibility is that the dendritic array initially produced by Arp2/3-mediated nucleation is subsequently transformed into parallel bundles of actin filaments.
Valuable insights into the mechanism of lamellipodial formation and protrusion have been obtained using bacterial- and bead-based in vitro motility systems (Theriot et al., 1994; Loisel et al., 1999; Cameron et al., 2000). Filopodial formation is less understood, one reason being that a similar in vitro approach is lacking. In this study, we report the development of in vitro systems for producing filopodia-like bundles, one of which employs cytoplasmic extracts and another that reconstitutes filopodia-like bundles from purified proteins. Using these systems, we provide evidence that filopodia-like bundles are formed by reorganization of the dendritic array.
Results
Formation of bead-associated actin bundles in extracts
Because filopodia are especially abundant in neuronal cell growth cones, we reasoned that brain cytoplasmic extracts might be a good source of filopodia-promoting factors, even though such extracts have previously been demonstrated to support comet tail motility (Laurent et al., 1999; Yarar et al., 1999). WASP-coated beads were introduced into rat brain extracts along with rhodamine–actin. Strikingly different structures were found associated with the beads depending on their position on the coverslip. In the center of the coverslip, beads were associated with a cloud of actin filaments or a typical comet tail (Fig. 1 A). In contrast, at the edge of the coverslip, straight actin bundles radiated from a bead in a star-like configuration (Fig. 1, A and B). These stars represented 84 ± 10% (n = 1,030) of all bead-associated actin structures at the edges of the coverslips (outermost third of the coverslip radius). No stars were found in the center of the coverslips (innermost third of the radius). In the transition zone between the center and edge of the coverslip, we observed large actin clouds and chimeras, structures intermediate between tails and stars (Fig. 1 A).
Figure 1.

Different actin structures are assembled on the beads. Actin assembly was assayed on Arp2/3 activator-coated beads in brain extract supplemented with rhodamine-labeled actin. (A) The pattern of actin assembly depends on the location of the bead on the coverslip. In the center of the coverslip, beads induce the formation of tails. Halfway between the center and edge of the coverslip, actin clouds and chimeras are formed. At the edge of the coverslip, star-like structures are associated with the beads. Beads are shown in yellow. Bar, 5 μm. (B) Stars are the dominant actin structure at the edge of the sample. Low magnification view of a field at the edge of a coverslip. Bar, 50 μm. (C) Percentage of stars increases with extract dilution. 1, center of the coverslip; 2, transitional zone; 3, edge of the coverslip. (D) Percentage of stars produced at the edge of the coverslip in undiluted rat brain extract by beads coated with different Arp2/3-activating proteins.
To determine whether star formation somehow resulted from special conditions at the coverslip edge, we altered the geometry of sample preparation. In the previous experiment, a 1-μl drop of assay mix was placed on a glass slide, and a coverslip was applied such that the drop spread outward from the coverslip's center. Here, the coverslip was applied such that one edge contacted the drop of assay mix, which was then forced to spread toward the coverslip's center. With this design, stars were not observed at the initial contacting edge of the coverslip (n = 527), whereas in the center of the coverslip, stars were abundant (83 ± 10%, n = 675), and in the transition zone, they represented 2 ± 2% (n = 745) of all structures. Thus, star formation was not a result of proximity to the coverslip edge. Rather, star formation correlated with distance of spreading across the glass surface.
One possible explanation for the formation of stars instead of comet tails was depletion of some protein(s) by adsorption to the glass during sample spreading. We tested this idea by blocking and preadsorption experiments. Pretreatment of the coverslip with 1% BSA blocked star formation, whereas other actin structures (tails and clouds) did form (unpublished data), suggesting that protein adsorption to glass plays a critical role in star formation. Preadsorption was performed by mixing extract with ground glass to simulate protein depletion during sample spreading, followed by centrifugation to collect the unadsorbed fraction. Glass-depleted extracts supported star formation throughout the entire coverslip, not only at the edges. Further, glass-depleted extracts also supported the formation of stars on WASP-coated beads in plastic tubes. If absorption to glass reduced the concentration of some star-inhibiting factor(s), then simple dilution of the extract might be sufficient to induce stars. Indeed, as shown in Fig. 1 C, when extracts were diluted fivefold with buffer, stars formed across the entire coverslip. The percentage of stars at the center of the coverslip increased from 0% (n = 1603), for undiluted extracts, to 73 ± 6% (n = 542), for extracts diluted fivefold. Thus, the formation of stars could be induced by lowering the concentration of some factor(s) in the extract. Star formation was not limited to rat brain extracts. Extracts of Xenopus oocytes and rat embryo fibroblasts (REFs) also supported star formation (unpublished data), but required greater dilution than rat brain extracts. 10% Xenopus oocyte extracts and 50% REF extracts were comparable to full-strength brain extracts in their ability to produce stars.
Star formation in extracts is an Arp2/3-dependent process
The formation of bundles in association with WASP-coated beads suggested the involvement of the Arp2/3 complex. To investigate the role of Arp2/3 complex in star assembly, it was depleted from brain extracts by GST–verprolin-homology/connecting/acidic domain of WASP (VCA) sepharose beads. At least 90% depletion was achieved, as assayed by immunoblotting (Fig. 2 A). In control and mock-depleted extracts, stars were present throughout the entire coverslip, 96% (n = 126) and 90% (n = 138), respectively (Fig. 2, B and C). In Arp2/3-depleted extracts (Fig. 2 D), actin assembly around the beads was completely abolished (0%, n = 213). Only spontaneously polymerized filaments could be observed in the background. Add-back of pure Arp2/3 complex to the depleted brain extracts restored star formation (Fig. 2 E) (84%, n = 170), although stars were slightly smaller than in control samples. Based on our immunoblotting experiments, the concentration of Arp2/3 complex necessary to rescue star formation (0.5 μM) was similar to that calculated to be present in glass-depleted extracts (0.45 μM). Lower concentrations of added Arp2/3 complex induced the formation of branched filaments on the background or actin clouds on the beads. We conclude that star formation is mediated by the Arp2/3 complex.
Figure 2.
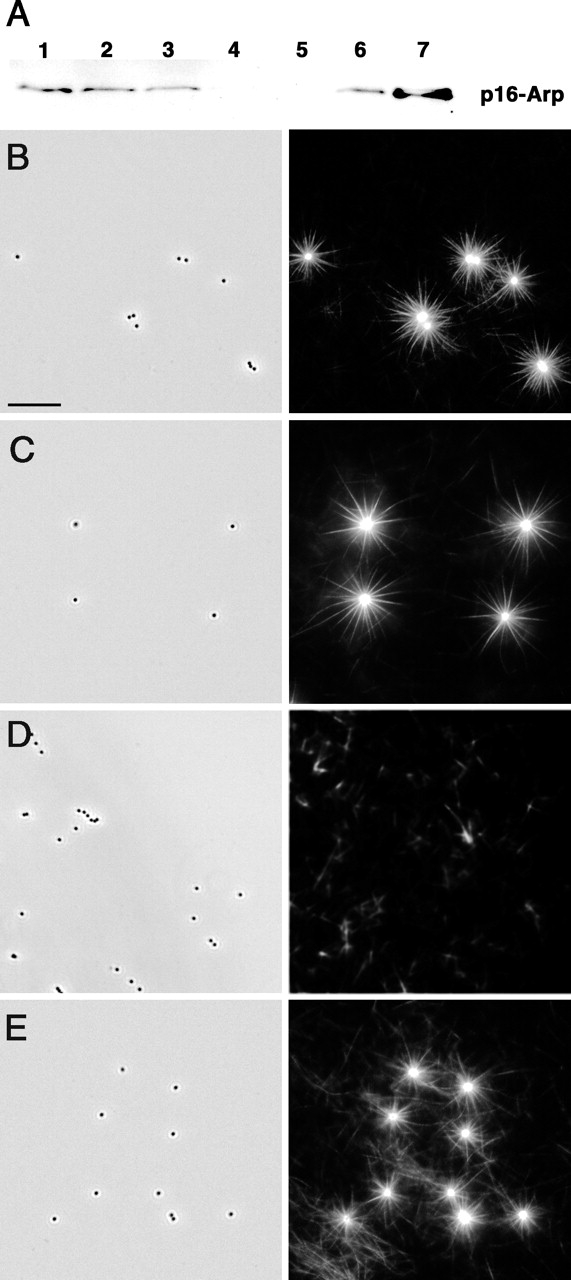
Arp2/3 complex is essential for star formation. (A) Arp2/3 complex depletion in brain extract. Glutathione-Sepharose or glutathione-Sepharose–coupled GST–VCA beads were incubated with 40 μl of brain extract. Arp2/3 depletion was monitored by immunoblotting using anti-p16 polyclonal antibody. Lane 1, 10 μl of untreated extract; lane 2, 10 μl of glass-depleted extract; lane 3, 10 μl of mock-depleted extract; lane 4, 10 μl of Arp2/3-depleted extract; lane 5, Arp2/3 associated with glutathione-Sepharose beads; lane 6, Arp2/3 associated with GST–VCA beads; lane 7, pure Arp2/3 from bovine brain, 4 μg. (B–E) Star assembly in Arp2/3-depleted brain extract. (B) Control, glass-depleted extract. (C) Mock-depleted extract. (D) Arp2/3-depleted extract; stars do not form. (E) Arp2/3-depleted extract rescued by add-back of 0.64 μM pure Arp2/3 complex; star formation is restored. Left panels, phase contrast. Individual 0.5-μm beads are visible. Right panels, fluorescence. Bright stars are evident on a background of faint individual filaments in B, C, and E. Only faint filaments are seen in D. Bar, 10 μm.
We next assayed whether star formation would be supported by different Arp2/3 activators. Beads were coated with either the bacterial protein ActA or cellular proteins WASP or Scar1. All activator-coated beads induced star formation in rat brain extracts, whereas beads coated with BSA did not (Fig. 1 D). COOH-terminal domains of WASP and Scar (pVCA) proteins, which were sufficient for Arp2/3 activation (Higgs and Pollard, 2001), also induced stars in extracts. We also tested whether star formation depended on beads or could also occur on bacteria. Stars assembled on Listeria expressing ActA and on Escherichia coli expressing the Shigella protein IcsA (unpublished data), indicating that the formation of stars was not restricted to coated synthetic beads. Because Shigella protein IcsA is known to recruit N-WASP from the extracts (Egile et al., 1999), this result also suggests that N-WASP supports star formation. Thus, star formation required active Arp2/3 complex but did not depend on a specific Arp2/3 activator.
Stars display both lamellipodial and filopodial types of actin organization
The radial bundles comprising stars bear a superficial similarity to filopodia in cells. To test whether these two kinds of structures have a deeper similarity, we analyzed the kinetics of star formation, their structural organization, sites of actin incorporation, and protein composition. Star development was observed by time-lapse microscopy. Initially, a diffuse actin cloud was formed around the bead (Fig. 3 A, 15 min). As time progressed, radial actin bundles appeared and began to elongate, with an average rate of 0.15 μm/min. Finally, we observed long stable actin bundles radiating from the bead-associated cloud. Some bundles in the course of star formation fused together by zippering in a proximal–distal direction, producing thicker bundles (Fig. 3 B). Zippering in a distal–proximal direction was also observed (unpublished data).
Figure 3.

Kinetics of star formation. (A) Time-lapse sequence of star assembly in rat brain extract. Bar, 5 μm. (B) Actin bundle zippering. Two bundles zipper together in a centrifugal direction. Time shown in min.
The structural organization of stars was examined by EM of platinum replicas (Fig. 4 A). Proximal to the bead, actin filaments formed a dendritic network, similar to that in lamellipodia. Many long unbranched filaments emanated from this network and, distal to the bead, gradually merged into bundles structurally similar to filopodia. Because one of the hallmarks of native filopodial bundles is uniform polarity of actin filaments, we performed myosin S1 decoration of actin filaments in stars to determine their polarity. The high density of filaments near the bead prevented determination of filament polarity in this region as well as in tight bundles. However, in the looser bundles distal to the bead (>1 μm), 93% (n = 429) of actin filaments had uniform polarity with their barbed ends pointing away from the bead (Fig. 4 B). Thus, stars display both lamellipodial (dendritic network) and filopodial (parallel bundle) types of actin organization, with the transition from one to the other occurring with distance away from the bead. The transition appeared as bundling of long filaments arising from the dendritic network.
Figure 4.

Structural organization of stars and polarity of actin assembly. (A) Actin filaments form a dendritic network around the bead and filament bundles away from the bead. Platinum replica EM. Bar, 0.5 μm. Overview of the star is shown in the left inset. Examples of branched filaments are highlighted in yellow and enlarged in the small panels at right. (B) Actin filaments in bundles are oriented with their barbed end away from the bead. Polarity of actin filaments was determined by myosin S1 decoration and is indicated by arrowheads. The bead is on the left. Bar, 0.1 μm. (C) Actin assembly occurs at tips of bundles and around the bead. Pulse labeling of actin incorporation. Rhodamine–actin (red) marks sites of new actin incorporation. All actin filaments were labeled with FITC–phalloidin (green). Bar, 5 μm.
Sites of actin polymerization in stars were analyzed by pulse-labeling experiments. After allowing stars to form, the distribution of newly incorporated rhodamine-labeled actin was determined relative to total actin, which was labeled with fluorescein-conjugated phalloidin (Fig. 4 C). Two major sites of actin incorporation were found: near the bead and at the tips of actin bundles. We interpret bead-associated sites to represent the growth of branches nucleated by WASP-activated Arp2/3 complex. In contrast, we interpret the incorporation at tips of radial bundles to represent elongation from preexisting, uncapped barbed ends, because no branched filaments were observed by EM at the tips of bundles. Actin incorporation was sometimes seen along the bundle, distant from its tip, consistent with EM data showing that some filaments in the bundle are shorter than others. This may result from unequal elongation of filaments or zippering of bundles of unequal length. Thus, stars display two modes of actin polymerization: Arp2/3-mediated nucleation at the bead, similar to that in lamellipodia, and barbed-end elongation at the tips of bundles, like that in filopodia.
Lamellipodia and comet tails, on the one hand, and filopodia in cells, on the other hand, have distinct protein composition (Goldberg, 2001; Small et al., 2002). For example, the Arp2/3 complex and capping protein are present in lamellipodia and comet tails but have not been found in filopodia (Svitkina and Borisy, 1999; Svitkina et al., 2003), whereas fascin is enriched in filopodia and less abundant in lamellipodia (Kureishy et al., 2002). α-Actinin has been found in lamellipodia (Langanger et al., 1984) but only in the roots of filopodia (Svitkina et al., 2003). We determined the localization of these proteins in stars by immunofluorescence staining or by incorporation of the labeled protein (Fig. 5). Arp2/3 complex and capping protein were found in the dendritic network proximal to the bead but not in actin bundles. α-Actinin was clearly enriched around the bead but could be faintly detected in bundles when a high amount of exogenous protein was added. In contrast, fascin was strongly localized to actin bundles but was diminished in the network surrounding the bead. Thus, by structural, kinetic, and biochemical criteria, our data demonstrate that the proximal dendritic network and radial bundles of stars are similar to the actin organization of lamellipodia and filopodia, respectively.
Figure 5.

Localization of actin binding proteins in stars. Arp2/3 complex, capping protein, and α-actinin are enriched near the beads but not in bundles, whereas fascin is enriched in bundles but is not prominent at beads. Stars were labeled with rhodamine–actin (green) and FITC–α-actinin (red) during assembly. Immunostaining for Arp2/3 (p16 Arc), capping protein (β2), or fascin (red) was performed after fixation of stars. Bar, 5 μm.
Parallel bundle formation can be shifted to dendritic network formation by capping protein
The absorption and dilution experiments indicated that reduced levels of factor(s) in the extract are critical in shifting the balance from a dendritic organization toward parallel bundles of actin. Several considerations suggest that one likely candidate for this role is capping protein. First, it has been shown (DiNubile et al., 1995) that increasing the concentration of neutrophil extract, and thus concentration of added capping protein, inhibited the extent and rate of polymerization of actin on spectrin–actin seeds. Conversely, we interpret that in our system, star formation after dilution or glass depletion might be due to decreased capping protein concentration. Second, when bacterial motility was reconstituted from purified proteins, suboptimal concentrations of capping protein (35 nM) produced comet tails with a fishbone appearance (Pantaloni et al., 2000), similar to chimeras observed in our samples in the transitional zone (Fig. 1 A). Third, immunostaining for capping protein detected high levels of fluorescence on the coverslip (Fig. 3), suggesting that capping protein was adsorbed to the glass and thus depleted from the extract. Therefore, we investigated whether star formation was dependent on the concentration of capping protein in the extract.
Capping protein concentration was estimated in terms of a chicken capping protein standard by immunoblotting. Rat brain extracts contained 3.8 ng/μl (58 nM) and REF extracts contained 8 ng/μl (122 nM) capping protein (Fig. 6 A). The twofold higher concentration of capping protein in REF extracts was consistent with the twofold dilution of this extract required to produce stars. We were unable to estimate the concentration of capping protein in Xenopus oocyte extracts because of a lack of cross-reactivity of the antibodies used. The initial total protein concentration for all tested extracts was similar: brain extract, 16 mg/ml; REF, 21 mg/ml; and Xenopus oocyte, 24 mg/ml. Next, we examined how much capping protein was depleted from brain extract by its adsorption on ground glass. As assayed by immunoblotting, 35% of capping protein was depleted (Fig. 6 B), whereas actin was not depleted (0%) (Fig. 6 B), Arp2/3 was 18% depleted (Fig. 2 A), and fascin was only 8% depleted (unpublished data). These results demonstrate that glass absorbs motility proteins differentially and that capping protein is preferentially depleted.
Figure 6.

Formation of stars depends on the concentration of capping protein. (A) Capping protein concentration in rat brain and REF extracts (7.5 μl per lane) was determined by Western blotting. Purified CapZ was used as a standard at concentrations shown above the respective lanes. (B) Capping protein and actin concentration in control and glass-depleted brain extract. (C) Addition of capping protein inhibits star formation. Percentage of the indicated bead-associated structure (Y axis) is shown versus concentration of added capping protein (X axis) to the 50% diluted rat brain extract. (D) Addition of capping protein induces growth of clouds. Time-lapse sequence of actin assembly around a WASP-coated bead in 50% brain extract supplemented with 50 nM capping protein. Bar, 5 μm. (E) Dendritic organization of clouds formed after the addition of 50 nM capping protein to the 50% brain extract. Bar, 0.2 μm. Overview of the cloud is shown in the left panel. Bar, 1 μm. Examples of branched filaments are highlighted in boxes and enlarged in small right panels.
Next, we supplemented 50% diluted brain extract with increasing amounts of exogenous capping protein. Capping protein inhibited star formation and facilitated cloud formation in a concentration-dependent manner (Fig. 6 C). In a kinetic analysis using time-lapse observation, addition of 50 nM capping protein to the extract (which normally produced mostly stars) blocked bundle formation around beads but allowed for continuous growth of actin clouds up to 20 times the bead diameter (Fig. 6 D). EM analysis of such clouds showed that actin filaments were organized into an extended dendritic network (Fig. 6 E). Higher concentrations of added capping protein, as expected, inhibited the extensive growth of clouds (unpublished data). At 400 nM capping protein, the diameter of the cloud was reduced to approximately twice the bead diameter. These results show that parallel bundle formation can be shifted to extended dendritic network formation by an optimal level of capping protein. As a specificity control, other proteins known to participate in actin dynamics were added to glass-depleted and diluted brain extracts. Addition of actin (7.5 μM), Arp2/3 complex (0.05, 0.1, and 0.6 μM), profilin (1.0, 2.5, and 10 μM), cofilin (2.5, 5.0, and 10 μM), and α-actinin (0.15, 0.25, 0.35, and 0.5 μM) did not affect star formation, whereas addition of 50 nM capping protein blocked star formation but allowed growth of actin clouds. These results show that capping protein was specific in antagonizing star formation.
Reconstitution of filopodia-like bundles using pure proteins
We tried to define a minimal set of components necessary for star assembly. Because data obtained with extracts indicated that Arp2/3 complex was necessary and capping protein had to be depleted, initial reconstitution experiments were performed with WASP-coated beads, actin, and Arp2/3 complex. Concentrations of proteins were based on published data for reconstitution of comet tails (Loisel et al., 1999). Beads coated with WASP were put into physiological ionic strength buffer solution, pH 7, containing rhodamine-labeled actin (6.9 μM) and increasing amounts of Arp2/3 complex over the range 0.1–0.9 μM. At 0.7 μM Arp2/3 complex and above, actin polymerization at the bead surface resulted in clouds within 15 min (Fig. 7 A). Clouds were ∼3.2 mm in diameter (measured as full width at 1/e of maximum fluorescence) and were azimuthally homogeneous in intensity except for apparently stochastic fluctuations. Lower concentrations of Arp2/3 did not generate clouds or did so more slowly.
Figure 7.

Reconstitution of filopodia-like bundles from pure proteins. Samples of WASP-coated beads preincubated on ice with 6.9 μM rhodamine-labeled actin and 0.7 μM Arp2/3 complex were brought to RT to allow for actin assembly. (A) Actin clouds formed in the absence of fascin. (B) Addition of 3.1 μM fascin to the sample before incubation at RT produced stars. Low magnification panels (top row) show distinctive pattern of actin assembly under each condition. (C) EM of star bundles formed in the presence of fascin as in B. Bars: (top row) 50 μM; (bottom row) 5 μm; (C) 100 nm.
Because Arp2/3 alone was insufficient to induce stars, we then introduced a bundling protein. Fascin was selected to complement the reconstitution system because it is the major bundler in filopodia and in star bundles formed in extracts. Recombinant fascin was used in these experiments. Fascin was added to samples of WASP-coated beads preincubated with Arp2/3 complex (0.7 μM) and actin (6.9 μM) on ice for 20 min, and star formation was evaluated within 15 min incubation at RT. Increasing amounts of fascin promoted bundle formation both on beads and of filaments in the background. At 3.1 μM fascin, >95% of the beads showing actin polymerization displayed stars with straight, needle-like rays (Fig. 7 B). EM demonstrated that the star bundles formed in the presence of fascin were composed of long actin filaments that were packed tightly together (Fig. 7 C), similar to those of filopodia and bundles made in extracts. The actin cross-linking protein, α-actinin, also generated star-like structures, but they were qualitatively different from those induced by fascin. The bundles of α-actinin stars were wavy, not straight, and EM showed them to consist of loosely packed and cross-linked filaments (unpublished data), as previously reported for mixtures of α-actinin and actin (Jockusch and Isenberg, 1982). The reconstitution experiments demonstrate that in the absence of barbed-end capping, four components, WASP-coated beads, Arp2/3 complex, actin, and fascin, are sufficient for star formation.
Discussion
Formation of filopodia-like structures in vitro and initiation of filopodia in the cellular context (Svitkina et al., 2003) reveal remarkable similarities. A comparison of these two processes is presented in Fig. 8. The initial step, dendritic nucleation of actin filaments, is essentially the same in both cases. Nucleation is driven by activation of the Arp2/3 complex and results in the formation of new branches on preexisting filaments. Under conditions where capping activity is favored, filaments not specifically protected become terminated soon after nucleation, resulting in an overall dendritic organization, forming tails or clouds in the bead assay (Fig. 8, top left) and lamellipodia in cells (top right). In the in vitro system, using activator-coated beads, a reduced concentration of capping protein allows filaments to remain uncapped and continue elongation, followed by cross-linking into bundles to form stars (Fig. 8, bottom left). In the cellular context, where the cytoplasmic concentration of capping protein is presumably high, we postulate that filaments with barbed ends at the membrane are protected from capping, allowing them to elongate and be bundled to form filopodia (bottom right). Thus, we recognize three processes to be necessary for star or filopodia formation: nucleation, elongation, and bundling. These three processes and the molecules likely to be involved in them are discussed in turn.
Figure 8.

Model for the formation of filopodial bundles. We propose that filopodia are formed from a preexisting dendritic network by barbed-end elongation of actin filaments and their subsequent cross-linking into bundles. At normal levels of capping activity, clouds and tails are formed around the bead in the in vitro system (top left), and lamellipodia are formed in cells (top right). If the concentration of capping protein is lowered in the in vitro system, filaments elongate and become bundled by cross-linking proteins, e.g., fascin (bottom left). Two examples of cross-linking are presented. Thin bundles may further zipper into the thicker bundles (arrows). In the cell, some filament barbed ends at the membrane become protected from capping, perhaps by Ena/VASP proteins, so that they can elongate and be cross-linked to form bundles in filopodia (bottom right).
Nucleation
The Arp2/3 complex is thought to play a role in filopodia formation because one of its activators, N-WASP, induces filopodia in cells (Miki et al., 1998). Because Arp2/3 is absent from established filopodia (Svitkina and Borisy, 1999; Svitkina et al., 2003), one may infer that it likely participates in initiation, not in steady-state elongation of filopodia. The question then becomes precisely how the Arp2/3 complex is involved in the initiation process. We suggest that our in vitro system for producing filopodia-like bundles reflects the situation in vivo and provides insights into the mechanism. First, formation of filopodia-like bundles, as well as dendritic clouds, depended on the presence and activity of the Arp2/3 complex. These findings agree with the observations in vivo, that perturbation of Arp2/3 complex function inhibits the formation of both lamellipodia and filopodia (Machesky and Insall, 1998; Li et al., 2002). Localization of Arp2/3 in stars was also analogous to the in vivo situation; it was present in dendritic arrays at the base of bundles, but not in bundles per se.
One possibility for how the action of the Arp2/3 complex can be explained is that it promotes dendritic or filopodial initiation depending upon the specific Arp activator. This idea is consistent with findings that different members of the WASP family vary in their ability to activate the Arp2/3 complex (Zalevsky et al., 2001). However, we found no significant differences in the process of star formation when they were induced by a variety of Arp2/3 activators, including ActA, WASP, Scar, N-WASP, and the pVCA COOH-terminal domains of WASP and Scar. ActA was active both as endogenous bacterial protein and as a recombinant protein at the bead surface. Thus, our data do not support a model that Arp2/3 complex produces different arrays depending on the specific activator.
Our data are in agreement with a model in which the Arp2/3 complex, irrespective of its activator, produces a normal dendritic array, which subsequently becomes reorganized into bundles. Time-lapse observations showed that diffuse clouds with dendritic organization preceded the formation of bundles. Structural studies demonstrated a gradual transition from dendritic arrays around the bead to distal radial bundles. Both results suggest that a normal dendritic network serves as a precursor for bundles. These results are in close agreement with recent results elucidating the in vivo mechanism of filopodial initiation (Svitkina et al., 2003). We propose that the role of the Arp2/3 complex in the in vitro system is to supply barbed ends. The high local concentration of barbed ends created by activator-coated beads and the high concentration of Arp2/3 in solution were essential for bundle formation. A similar method may be used by cells if Arp2/3 activators (or molecules recruiting them) are not evenly distributed along the leading edge, but exist in clusters. A high local concentration of barbed ends could also be created by the clustering of some barbed end–binding molecules at the membrane. Possible candidates for such a role are members of the enabled/vasodilator-stimulated phosphoprotein (Ena/VASP) family (Bear et al., 2002; Svitkina et al., 2003) and formins (Pruyne et al., 2002), which have recently been shown to bind barbed ends but allow for filament elongation. Formins can also nucleate unbranched actin filaments (Pruyne et al., 2002; Sagot et al., 2002b) and thus are candidates for an alternative, Arp2/3-independent pathway of bundle initiation, similar to how actin cables in yeast are formed (Evangelista et al., 2002; Sagot et al., 2002a). However, this mechanism does not account for the N-WASP induction of filopodia.
Elongation
After filaments are nucleated, they have to elongate and encounter each other before they can form a bundle. A priori, elongation of filaments could be facilitated by increasing the concentration of an “elongation” factor or by decreasing the concentration of a “termination” factor. The main arguments in favor of the latter possibility are that star formation was induced in extracts by depletion of capping protein and was antagonized specifically by add-back of capping protein. A key point here is that add-back of capping protein to depleted extracts did not simply block all actin polymerization. Rather, it induced the formation of clouds as opposed to stars. Our interpretation is that cloud formation was the result of the termination of filament elongation shortly after branch nucleation under conditions when active Arp2/3 complex continuously nucleates new filaments. This process results in short filaments and a dense dendritic network. The fact that stars could be reconstituted in a pure protein system lacking barbed-end capping proteins but allowing for filament nucleation, elongation, and bundling is consistent with the interpretation that star formation was facilitated by decreasing a termination factor.
In the cellular context, the cytoplasmic concentration of capping protein is high (Huang et al., 1999). In lamellipodia where barbed ends are constantly being produced, the high concentration of capping protein can be understood as necessary to cap unproductive barbed ends. In filopodia where filaments elongate continuously, their barbed ends need to be protected from capping. Protection may be provided by Ena/VASP family proteins because they are present at the extreme leading edge, bind to barbed ends, and can antagonize capping in vitro and in vivo (Bear et al., 2002). Ena/VASP proteins are also enriched at the tips of filopodia (Lanier et al., 1999; Rottner et al., 1999) and become gradually accumulated at the tips of filopodial precursors during the filopodial initiation (Svitkina et al., 2003). It is attractive to speculate that the presence of Ena/VASP at the filopodial tips in the cellular context prevents filament termination and allows filopodial elongation. Consistent with this idea, our results showing that the level of capping protein controls the transition between two types of actin arrays in vitro match the observations that the level of Ena/VASP proteins performs analogous control in vivo, but in the opposite direction. That is, a low level of Ena/VASP induced short branched filaments, and an excess of Ena/VASP promoted the formation of long filaments (Bear et al., 2002).
In our in vitro system, the rates of bundle elongation in stars were ∼0.15 μm/min, similar to the reported rates of comet tail motility in undiluted brain extracts (0.2–0.3 μm/min) (Laurent et al., 1999; Yarar et al., 1999). Whereas in Xenopus oocyte extract or during leading edge protrusion in cells, the rates of actin array assembly are at least an order of magnitude higher (Mallavarapu and Mitchison, 1999). These observations indicate that additional factors may contribute to the overall performance of actin machinery, but balance between nucleation and elongation seems to be critical for the determination of supramolecular organization of the actin array.
Bundling
Bundling is necessary to allow long filaments to push efficiently without buckling under the cell surface. The leading candidate for filament bundling in filopodia is fascin. It shows the greatest enrichment in filopodial bundles in cells (Kureishy et al., 2002), where it significantly prevails over α-actinin (Svitkina et al., 2003), and it is essential for the maintenance of filopodia (Yamashiro et al., 1998; Adams et al., 1999; Cohan et al., 2001).
Consistent with in vivo data, we found that fascin was the major bundling protein present in stars assembled in cytoplasmic extracts. Fascin was also sufficient to form filopodia-like bundles in a reconstitution system in which fascin represented the only bundling protein. The straightness of the fascin-induced star bundles suggests that they were quite rigid, similar to filopodia. In contrast to fascin, the other actin filament cross-linker, α-actinin, was more abundant in the dendritic network surrounding beads and was absent from star bundles. Although α-actinin was also able to drive star assembly in a reconstitution system, the resulting star bundles had wavy rays and a dendritic, not parallel bundled, organization. Thus, α-actinin did not recapitulate filopodia formation in vitro. These results suggest that the two cross-linkers may be specialized with respect to the particular actin filament array that they stabilize. Segregation of fascin and α-actinin to bundles and the dendritic network, respectively, correlates with the biochemical properties of these two cross-linkers: fascin is a short cross-linker that makes tight parallel bundles (Yamashiro-Matsumura and Matsumura, 1985) and α-actinin is a longer molecule that, when combined with actin, makes loose bundles with both parallel and antiparallel filament orientation. The absence of α-actinin in filopodial bundles in more complex systems, such as extracts or cells, when both proteins are present may be explained as fascin outcompeting α-actinin because of a slightly higher affinity or, possibly, specific recruitment to areas of filopodial assembly.
In the course of star formation, we frequently observed zippering of radial bundles, an effect that superficially resembles fusion of filopodia during their normal dynamics in cells. However, distinctions between these two phenomena should be noted. In cells, the fusion site moves backward because of treadmilling and retrograde flow of the whole assembly, but no actual displacement of bundles, with respect to each other, occurs. In stars, we did not find independent indications of retrograde flow, suggesting that zippering in vitro actually brings distant bundles together. A likely reason for the difference between the two systems is that the movement of bundles in vivo is precluded by the more crowded conditions and cross-linking occurring in the cytoplasm, compared with extracts. Nevertheless, the similarity of the systems suggests that cross-linking molecules, presumably fascin, have a potential for zippering and may accomplish this function under permissive circumstances, for formation of bundles in vitro and for filopodia in vivo.
In summary, our results suggest that an Arp2/3-mediated pathway is compatible with filopodia formation, and that it is not necessary to postulate unusual properties of WASP family members to stimulate a nonbranching mode of actin filament formation. Filopodia-like structures can be accounted for as a transformation from a dendritic organization by a combination of elongation and bundling.
Materials and methods
Proteins
Actin was purified from rabbit muscle as previously described (Spudich and Watt, 1971). Rhodamine–actin was prepared by labeling actin with N-hydroxy succinimido-rhodamine (Molecular Probes) as previously described (Isambert et al., 1995) and stored at −80°C. Before use, labeled G-actin was recycled by polymerization for 2 h on ice in the presence of 50 mM KCl, 2 mM MgCl2, and 1 mM ATP, sedimentation at 100,000 g for 1.5 h at 4°C, resuspension in cold G buffer (2 mM Tris-Cl, 0.2 mM CaCl2, 0.2 mM ATP, and 0.5 mM DTT) to a final concentration of 2 mg/ml, and dialysis overnight against G buffer using microdialysis buttons (Hampton Research) and dialysis tubing (Pierce Chemical Co.).
DNA encoding WASP tagged at its NH2 terminus with both Met, Arg, Gly, Ser (MRGS) 6xHis and FLAG epitopes was amplified by PCR from a human WASP cDNA (a gift of Arie Abo, PPD Discovery, Menlo Park, CA) and subcloned into pFastBac1 (Life Technologies; Amersham Biosciences). DNA encoding human Scar1 was amplified by PCR and subcloned into the WASP-pFastbac1 vector in the place of the WASP DNA. Recombinant WASP and Scar1 proteins were expressed in Sf9 cells using the baculovirus system. Baculovirus strains were generated and used for infections according to the Bac-to-Bac baculovirus expression system (Life Technologies). After 72 h of infection, cells were harvested by centrifugation at 500 g for 10 min at 25°C, resuspended in lysis buffer (50 mM NaH2PO4, pH 8.0, 300 mM KCl) with protease inhibitors (1 mM PMSF and 10 μg/ml leupeptin, pepstatin, and chymostatin [LPC]), and frozen in liquid N2. To prepare the lysate, cells were thawed and centrifuged at 200,000 g for 15 min at 4°C.
To purify the recombinant proteins, Sf9 lysates were supplemented with 20 mM imidazole, incubated with Ni-NTA agarose (QIAGEN) resin for 45 min at 4°C, washed with 50 mM NaH2PO4, pH 8.0, 300 mM KCl, 20 mM imidazole, and eluted with 200 mM imidazole, 50 mM NaH2PO4, pH 8.0, 300 mM KCl, and protease inhibitors. Eluted proteins were further purified by gel filtration chromatography on a Superdex-200 column (Amersham Biosciences) equilibrated with 20 mM MOPS, pH 7.0, 100 mM KCl, 2 mM MgCl2, 5 mM EGTA, 1 mM EDTA, 0.2 mM ATP, 0.5 mM DTT, 10% vol/vol glycerol. Full-length recombinant WASP has constitutive ability to activate Arp2/3 complex (Higgs and Pollard, 2001).
DNA encoding human fascin was amplified by PCR and sublcloned into the pGEX-4T-3 vector (Amersham Biosciences) using BamH1/Xho I sites. Recombinant human fascin was prepared by a modification of the method of Ono et al. (1997). E. coli carrying the plasmid was grown at 37°C until the A600 reached 0.6. Protein expression was induced by adding 0.1 mM IPTG at 20°C for 4 h. Cells were harvested by centrifugation and extracted with B-PER in phosphate buffer (Pierce Chemical Co.) plus 1 mM PMSF and 1 mM DTT. The lysate was centrifuged at 20,000 g for 20 min, and the supernatant was mixed for 1 h at RT with 2 ml glutathione–Sepharose 4B (Amersham Biosciences) equilibrated with PBS plus 1 mM DTT. The glutathione-Sepharose was poured into a column and washed with 20 ml of PBS plus 1 mM DTT. 80 μl of thrombin (Amersham Biosciences) was added, and digestion was allowed to proceed overnight at 4°C. Flowthrough fractions were collected in 2 mM PMSF and concentrated by Centricon 10 (Amicon).
Arp2/3 was purified from bovine brain as described by Laurent et al. (1999). Recombinant chicken CapZ (Soeno et al., 1998; Palmgren et al., 2001) was provided by John Cooper (Washington University School of Medicine, St. Louis, MO). FITC-labeled α-actinin was provided by Marion Greaser (University of Wisconsin, Madison, WI). ActA protein (Cameron et al., 1999) was provided by Julie Theriot (Stanford University School of Medicine, Stanford, CA). Recombinant human cofilin and human profilin were purchased from Cytoskeleton, Inc.
Cytoplasmic extracts
Rat brain extract was prepared as previously described (Laurent et al., 1999). Metaphase Xenopus oocyte extract was prepared as previously described (Murray, 1991) and kept frozen at −80°C. Before use, it was centrifuged at 100,000 g for 1 h at 4°C, and the supernatant was used for experiments. Rat embryonic fibroblast extract was prepared as described by Saoudi et al. (1998).
Actin polymerization bead assay
Carboxylated polystyrene beads (Polysciences) were coated with ActA as previously described (Cameron et al., 1999). For coating beads with WASP/Scar proteins, we took 15 μl of 0.5 μM WASP or Scar proteins, mixed up with 15 μl of brain buffer (BB) (Laurent et al., 1999) or Xenopus buffer (Murray, 1991) and 0.5 μl of 0.5-μm carboxylated polystyrene beads. This mixture was incubated at RT for 1 h. Beads were washed twice with the appropriate buffer and resuspended in 10 μl of buffer. For longer storage, beads were supplemented with 50% glycerol and placed at −80°C.
Coated beads (0.5 μl) were introduced into 10 μl of cell extract supplemented with energy mix (15 mM creatine phosphate, 2 mM ATP, and 2 mM MgCl2) and 1.25 μM rhodamine-labeled actin. In experiments to evaluate the effect of capping protein concentration, the assay mix was supplemented with increasing amounts of capping protein. The assay mix was incubated on ice for 1 h before preparation for observation.
For reconstitution system, 0.5 μl of coated beads was introduced in 1× KME buffer (50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 10 mM imidazole, pH 7) supplemented with Arp2/3 complex. After incubation for 5 min at RT, 6.9 μM actin was added, and the mixture was incubated on ice for 20 min before the addition of fascin and allowing for actin assembly at RT.
Microscopy
For light microscopy, a 1-μl sample was removed and pressed tightly between a microscope slide and a 22-mm square glass to create a chamber ∼5 μm thick, which was then sealed with vaseline/lanolin/paraffin (at 1:1:1). Samples were incubated at RT for 15 min and then observed with a Nikon Eclipse inverted microscope equipped with phase contrast and epifluorescence optics. Time-lapse images were acquired with a back-thinned CCD camera (CH250; Photometrics) using METAMORPH (Universal Imaging Corp.) software. Fluorescence images were recorded every 5 min for 2 h.
For EM, samples were prepared as described by Cameron et al. (2001). After incubation for 2 h at RT in a humid environment, chambers were opened into solution containing 0.2% Triton X-100 and 2 μM phalloidin in BB. Although some stars were washed out during this procedure, some remained attached to the coverslip. Coverslips were washed with 2 μM phalloidin in BB, fixed with 2% glutaraldehyde, and processed for EM. Procedures for S1 decoration and EM were as previously described (Svitkina and Borisy, 1998). Polarity of filaments, determined in blind experiments by two independent observers, gave similar results.
Immunostaining
Rabbit polyclonal antibody to the Arc p16 subunit of the Arp2/3 complex was prepared against the RFRKVDVDEYDENKFVDEED peptide of the human sequence. By Western blotting, the affinity-purified antibody recognized in rat brain extract a doublet of closely spaced bands in the16-kD range, supposedly corresponding to two isoforms of Arc p16 (Millard et al., 2003). Polyclonal rabbit antibody against capping protein (R22) was provided by Dorothy A. Schafer (University of Virginia, Charlottesville, VA). Mouse monoclonal α-actinin antibody was from Sigma-Aldrich, and mouse monoclonal fascin antibody was from DakoCytomation. Immunostaining was performed in perfusion chambers. Solutions were applied on one side of the chamber with a pipet and withdrawn from the other side with filter paper. A 4-μl assay sample was sandwiched between a glass slide and a coverslip (22 × 22 mm) separated by two strips of teflon tape and sealed. Stars were allowed to form for 2 h at RT in humid conditions. 10 μl of BB containing 0.2% Triton X-100 and 2 μM phalloidin was perfused through the chamber, followed by 10 μl of 2 μM phalloidin in BB. For most immunostaining experiments, stars were fixed with 0.2% glutaraldehyde for 20 min at RT, washed with PBS, and quenched for 20 min with 2 mg/ml of NaBH4 in PBS supplemented with 0.1% Tween 20. For fascin staining, samples were fixed with methanol for 10 min at −20°C.
Pulse-labeling assay
Stars were allowed to form for 1 h in a perfusion chamber containing 4 μl of assay sample without labeled actin. Then 1 μl of 1.8 μM rhodamine–actin was added by diffusion from the edge. After 10 min, FITC–phalloidin was added in the same way to label all actin filaments. The chamber was sealed, and observations were made after 10 min of incubation at RT.
Glass depletion experiment
30 μl of brain extract was applied to 70 mg of ground glass coverslip in a mini filtration tube (0.65 μm; UltraFree-MC; Millipore), and the filtrate was collected by centrifugation twice for 1 min at 11,000 g.
Depletion of Arp2/3 complex
Arp2/3 complex was depleted from glass-depleted brain extract using a variation of the method described by Egile et al. (1999). 15 μl of glutathione-Sepharose–coupled GST–VCA beads was incubated with 40 μl of brain extract for 30 min at 4°C on a rotating wheel. Beads were pelleted at 10,000 g for 2 min. Mock depletion is performed by the same amount of glutathione-Sepharose beads. Depletion of the Arp2/3 complex was monitored by Western blotting of aliquots of extracts and beads. The add-back experiment was performed using the Arp2/3 complex purified from bovine brain. For the microscopy assay, we scored the percentage of stars assembled on all beads.
Quantification of capping protein in cell extracts by Western blotting
Different amounts of rat brain and rat embryonic fibroblast extracts were subjected to SDS-PAGE (4–20% polyacrylamide) and immunoblotting with anti-CapZ. The amount of capping protein present was evaluated by comparing the intensity of the bands of each sample with the chicken CapZ standard by densitometry using NIH image software.
Acknowledgments
We thank Drs. Julie Theriot, Dorothy Schafer, John Cooper, Marie-France Carlier, Josephine Adams, Marion Greaser, and Elena S. Nadezhdina for generous gifts of reagents and bacterial strains. We also thank Dr. Tom Keating and members of the Borisy laboratory for constructive discussions.
This work was supported by United States Army USAMRMC grant DAMD 17-00-1-0386 (D. Vignjevic), National Institutes of Health (NIH) grant GM 62431 (G.G. Borisy), and NIH Glue Grant on Cell Migration IU 54 GM 63126.
Footnotes
Abbreviations used in this paper: Arp2/3, actin-related protein 2/3; BB, brain buffer; Ena/VASP, enabled/vasodilator-stimulated phosphoprotein; REF, rat embryo fibroblast; VCA, verprolin-homology/connecting/acidic domain of WASP; WASP, Wiskott-Aldrich syndrome protein.
References
- Adams, J.C., J.D. Clelland, G.D. Collett, F. Matsumura, S. Yamashiro, and L. Zhang. 1999. Cell-matrix adhesions differentially regulate fascin phosphorylation. Mol. Biol. Cell. 10:4177–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear, J.E., T.M. Svitkina, M. Krause, D.A. Schafer, J.J. Loureiro, G.A. Strasser, I.V. Maly, O.Y. Chaga, J.A. Cooper, G.G. Borisy, and F.B. Gertler. 2002. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 109:509–521. [DOI] [PubMed] [Google Scholar]
- Borisy, G.G., and T.M. Svitkina. 2000. Actin machinery: pushing the envelope. Curr. Opin. Cell Biol. 12:104–112. [DOI] [PubMed] [Google Scholar]
- Cameron, L.A., M.J. Footer, A. van Oudenaarden, and J.A. Theriot. 1999. Motility of ActA protein-coated microspheres driven by actin polymerization. Proc. Natl. Acad. Sci. USA. 96:4908–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron, L.A., P.A. Giardini, F.S. Soo, and J.A. Theriot. 2000. Secrets of actin-based motility revealed by a bacterial pathogen. Nat. Rev. Mol. Cell Biol. 1:110–119. [DOI] [PubMed] [Google Scholar]
- Cameron, L.A., T.M. Svitkina, D. Vignjevic, J.A. Theriot, and G.G. Borisy. 2001. Dendritic organization of actin comet tails. Curr. Biol. 11:130–135. [DOI] [PubMed] [Google Scholar]
- Cohan, C.S., E.A. Welnhofer, L. Zhao, F. Matsumura, and S. Yamashiro. 2001. Role of the actin bundling protein fascin in growth cone morphogenesis: localization in filopodia and lamellipodia. Cell Motil. Cytoskeleton. 48:109–120. [DOI] [PubMed] [Google Scholar]
- Cooper, J.A., and D.A. Schafer. 2000. Control of actin assembly and disassembly at filament ends. Curr. Opin. Cell Biol. 12:97–103. [DOI] [PubMed] [Google Scholar]
- DiNubile, M.J., L. Cassimeris, M. Joyce, and S.H. Zigmond. 1995. Actin filament barbed-end capping activity in neutrophil lysates: the role of capping protein-β2. Mol. Biol. Cell. 6:1659–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egile, C., T.P. Loisel, V. Laurent, R. Li, D. Pantaloni, P.J. Sansonetti, and M.F. Carlier. 1999. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell Biol. 146:1319–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista, M., D. Pruyne, D.C. Amberg, C. Boone, and A. Bretscher. 2002. Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat. Cell Biol. 4:32–41. [DOI] [PubMed] [Google Scholar]
- Goldberg, M.B. 2001. Actin-based motility of intracellular microbial pathogens. Microbiol. Mol. Biol. Rev. 65:595–626 (table of contents). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs, H.N., and T.D. Pollard. 2001. Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu. Rev. Biochem. 70:649–676. [DOI] [PubMed] [Google Scholar]
- Huang, M., C. Yang, D.A. Schafer, J.A. Cooper, H.N. Higgs, and S.H. Zigmond. 1999. Cdc42-induced actin filaments are protected from capping protein. Curr. Biol. 9:979–982. [DOI] [PubMed] [Google Scholar]
- Isambert, H., P. Venier, A.C. Maggs, A. Fattoum, R. Kassab, D. Pantaloni, and M.F. Carlier. 1995. Flexibility of actin filaments derived from thermal fluctuations. Effect of bound nucleotide, phalloidin, and muscle regulatory proteins. J. Biol. Chem. 270:11437–11444. [DOI] [PubMed] [Google Scholar]
- Jockusch, B.M., and G. Isenberg. 1982. Vinculin and α-actinin: interaction with actin and effect on microfilament network formation. Cold Spring Harb. Symp. Quant. Biol. 46:613–623. [DOI] [PubMed] [Google Scholar]
- Kureishy, N., V. Sapountzi, S. Prag, N. Anilkumar, and J.C. Adams. 2002. Fascins, and their roles in cell structure and function. Bioessays. 24:350–361. [DOI] [PubMed] [Google Scholar]
- Langanger, G., J. de Mey, M. Moeremans, G. Daneels, M. de Brabander, and J.V. Small. 1984. Ultrastructural localization of α-actinin and filamin in cultured cells with the immunogold staining (IGS) method. J. Cell Biol. 99:1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier, L.M., M.A. Gates, W. Witke, A.S. Menzies, A.M. Wehman, J.D. Macklis, D. Kwiatkowski, P. Soriano, and F.B. Gertler. 1999. Mena is required for neurulation and commissure formation. Neuron. 22:313–325. [DOI] [PubMed] [Google Scholar]
- Laurent, V., T.P. Loisel, B. Harbeck, A. Wehman, L. Grobe, B.M. Jockusch, J. Wehland, F.B. Gertler, and M.F. Carlier. 1999. Role of proteins of the Ena/VASP family in actin-based motility of Listeria monocytogenes. J. Cell Biol. 144:1245–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, A.K., and P.C. Bridgman. 1992. Nerve growth cone lamellipodia contain two populations of actin filaments that differ in organization and polarity. J. Cell Biol. 119:1219–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., E.S. Kim, and E.L. Bearer. 2002. Arp2/3 complex is required for actin polymerization during platelet shape change. Blood. 99:4466–4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loisel, T.P., R. Boujemaa, D. Pantaloni, and M.F. Carlier. 1999. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 401:613–616 (see comments). [DOI] [PubMed] [Google Scholar]
- Machesky, L.M., and R.H. Insall. 1998. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 8:1347–1356. [DOI] [PubMed] [Google Scholar]
- Millard, T.H., B. Behrendt, S. Launay, K. Futterer, and L.M. Machesky. 2003. Identification and characterisation of a novel human isoform of Arp2/3 complex subunit p16-ARC/ARPC5. Cell Motil. Cytoskeleton. 54:81–90. [DOI] [PubMed] [Google Scholar]
- Mallavarapu, A., and T. Mitchison. 1999. Regulated actin cytoskeleton assembly at filopodium tips controls their extension and retraction. J. Cell Biol. 146:1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki, H., T. Sasaki, Y. Takai, and T. Takenawa. 1998. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature. 391:93–96. [DOI] [PubMed] [Google Scholar]
- Mullins, R.D., J.A. Heuser, and T.D. Pollard. 1998. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. USA. 95:6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, A.W. 1991. Cell cycle extracts. Methods Cell Biol. 36:581–605. [PubMed] [Google Scholar]
- Ono, S., Y. Yamakita, S. Yamashiro, P.T. Matsudaira, J.R. Gnarra, T. Obinata, and F. Matsumura. 1997. Identification of an actin binding region and a protein kinase C phosphorylation site on human fascin. J. Biol. Chem. 272:2527–2533. [DOI] [PubMed] [Google Scholar]
- Palmgren, S., P.J. Ojala, M.A. Wear, J.A. Cooper, and P. Lappalainen. 2001. Interactions with PIP2, ADP-actin monomers, and capping protein regulate the activity and localization of yeast twinfilin. J. Cell Biol. 155:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni, D., R. Boujemaa, D. Didry, P. Gounon, and M.F. Carlier. 2000. The Arp2/3 complex branches filament barbed ends: functional antagonism with capping proteins. Nat. Cell Biol. 2:385–391. [DOI] [PubMed] [Google Scholar]
- Pruyne, D., M. Evangelista, C. Yang, E. Bi, S. Zigmond, A. Bretscher, and C. Boone. 2002. Role of formins in actin assembly: nucleation and barbed-end association. Science. 297:612–615. [DOI] [PubMed] [Google Scholar]
- Rottner, K., B. Behrendt, J.V. Small, and J. Wehland. 1999. VASP dynamics during lamellipodia protrusion. Nat. Cell Biol. 1:321–322. [DOI] [PubMed] [Google Scholar]
- Sagot, I., S.K. Klee, and D. Pellman. 2002. a. Yeast formins regulate cell polarity by controlling the assembly of actin cables. Nat. Cell Biol. 4:42–50. [DOI] [PubMed] [Google Scholar]
- Sagot, I., A.A. Rodal, J. Moseley, B.L. Goode, and D. Pellman. 2002. b. An actin nucleation mechanism mediated by Bni1 and profilin. Nat. Cell Biol. 4:626–631. [DOI] [PubMed] [Google Scholar]
- Saoudi, Y., R. Fotedar, A. Abrieu, M. Doree, J. Wehland, R.L. Margolis, and D. Job. 1998. Stepwise reconstitution of interphase microtubule dynamics in permeabilized cells and comparison to dynamic mechanisms in intact cells. J. Cell Biol. 142:1519–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, J.V., G. Isenberg, and J.E. Celis. 1978. Polarity of actin at the leading edge of cultured cells. Nature. 272:638–639. [DOI] [PubMed] [Google Scholar]
- Small, J.V., M. Herzog, M. Haner, and U. Abei. 1994. Visualization of actin filaments in keratocyte lamellipodia: negative staining compared with freeze-drying. J. Struct. Biol. 113:135–141. [DOI] [PubMed] [Google Scholar]
- Small, J.V., T. Stradal, E. Vignal, and K. Rottner. 2002. The lamellipodium: where motility begins. Trends Cell Biol. 12:112–120. [DOI] [PubMed] [Google Scholar]
- Soeno, Y., H. Abe, S. Kimura, K. Maruyama, and T. Obinata. 1998. Generation of functional β-actinin (CapZ) in an E. coli expression system. J. Muscle Res. Cell Motil. 19:639–646. [DOI] [PubMed] [Google Scholar]
- Spudich, J.A., and S. Watt. 1971. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246:4866–4871. [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1998. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods Enzymol. 298:570–592. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1999. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina, T.M., A.B. Verkhovsky, K.M. McQuade, and G.G. Borisy. 1997. Analysis of the actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J. Cell Biol. 139:397–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina, T.M., Bulanova E.A., Chaga O.Y., Vignjevic D., Kojima S., Vasiliev J.M., and Borisy G.G. 2003. Mechanism of filopodia initiation by reorganization of a dendritic network. J. Cell Biol. 160:409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot, J.A., J. Rosenblatt, D.A. Portnoy, P.J. Goldschmidt-Clermont, and T.J. Mitchison. 1994. Involvement of profilin in the actin-based motility of L. monocytogenes in cells and in cell-free extracts. Cell. 76:505–517. [DOI] [PubMed] [Google Scholar]
- Yamashiro, S., Y. Yamakita, S. Ono, and F. Matsumura. 1998. Fascin, an actin-bundling protein, induces membrane protrusions and increases cell motility of epithelial cells. Mol. Biol. Cell. 9:993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro-Matsumura, S., and F. Matsumura. 1985. Purification and characterization of an F-actin-bundling 55-kilodalton protein from HeLa cells. J. Biol. Chem. 260:5087–5097. [PubMed] [Google Scholar]
- Yarar, D., W. To, A. Abo, and M.D. Welch. 1999. The Wiskott-Aldrich syndrome protein directs actin-based motility by stimulating actin nucleation with the Arp2/3 complex. Curr. Biol. 9:555–558. [DOI] [PubMed] [Google Scholar]
- Zalevsky, J., L. Lempert, H. Kranitz, and R.D. Mullins. 2001. Different WASP family proteins stimulate different Arp2/3 complex-dependent actin-nucleating activities. Curr. Biol. 11:1903–1913. [DOI] [PubMed] [Google Scholar]