

Abstract
Activation of PKC depends on the availability of DAG, a signaling lipid that is tightly and dynamically regulated. DAG kinase (DGK) terminates DAG signaling by converting it to phosphatidic acid. Here, we demonstrate that DGKζ inhibits PKCα activity and that DGK activity is required for this inhibition. We also show that DGKζ directly interacts with PKCα in a signaling complex and that the binding site in DGKζ is located within the catalytic domain. Because PKCα can phosphorylate the myristoylated alanine-rich C-kinase substrate (MARCKS) motif of DGKζ, we tested whether this modification could affect their interaction. Phosphorylation of this motif significantly attenuated coimmunoprecipitation of DGKζ and PKCα and abolished their colocalization in cells, indicating that it negatively regulates binding. Expression of a phosphorylation-mimicking DGKζ mutant that was unable to bind PKCα did not inhibit PKCα activity. Together, our results suggest that DGKζ spatially regulates PKCα activity by attenuating local accumulation of signaling DAG. This regulation is impaired by PKCα-mediated DGKζ phosphorylation.
Keywords: diacylglycerol; diacylglycerol kinase; protein kinase C; spatial regulation; phosphorylation
Introduction
DAG is a lipid second messenger that transiently accumulates in cells stimulated by growth factors and other agonists (Ghosh et al., 1997; Hodgkin et al., 1998). The best-characterized role of DAG is as an allosteric activator of PKC (Nishizuka, 1992). The binding of DAG to the C1 domain in PKC induces an active conformation, allowing the PKC to phosphorylate its substrates (Newton, 2001). PKCs regulate a broad array of cell functions, such as growth, differentiation, apoptosis, and cytoskeletal reorganization (Nishizuka, 1995; Black, 2000; Dempsey et al., 2000). The mammalian PKC family comprises 10 isoforms divided into three groups: conventional, novel, and atypical (Newton, 1997). By selectively phosphorylating substrates, each PKC isoform likely mediates a unique set of cellular functions. This selectivity is dictated in part by the subcellular targeting of each isoform (Jaken, 1996; Newton, 2001). PKC function is compartmentalized through interactions with a number of proteins, such as scaffold proteins, its substrates, and proteins that regulate its activity (Pawson and Scott, 1997; Jaken and Parker, 2000; Mackay and Mochly-Rosen, 2001). This compartmentalization allows precise spatiotemporal activation of PKC, which influences subsequent signaling events. Because PKC activity depends on the availability of DAG, the accumulation of DAG in PKC compartments must be precisely regulated.
DAG kinases (DGKs)* are critical regulators of DAG signaling. DGKs metabolize DAG by phosphorylating it to generate phosphatidic acid (PA). To date, nine mammalian DGK isoforms have been cloned and divided into five classes based on common structural motifs (Topham and Prescott, 1999; van Blitterswijk and Houssa, 1999; Kanoh et al., 2002). Their structural diversity, together with their different subcellular localization (Topham and Prescott, 1999), suggests that each DGK isoform may regulate distinct DAG signaling events. DGKζ, a type IV DGK, contains a unique region homologous to the phosphorylation site domain (PSD) of the myristoylated alanine-rich C-kinase substrate (MARCKS) protein, a prominent substrate for PKC in cells (Blackshear, 1993; Bunting et al., 1996). This MARCKS motif is the predominant nuclear localization signal of DGKζ, and its phosphorylation by PKC isoforms reduces nuclear localization of DGKζ, which alters nuclear DAG accumulation (Topham et al., 1998). Thus, cells regulate the concentration of PKC-activating nuclear DAG by controlling nuclear localization of DGKζ. Interestingly, overexpression of wild-type DGKζ leads to decreased levels of nuclear DAG (Topham et al., 1998). This, combined with the fact that several DAG pools have been found in distinct, spatially separated compartments within the cell (Wakelam, 1998; D'Santos et al., 1999), suggests that the regulation of DAG signaling is achieved locally.
PKCα is strongly activated by DAG, and its regulation is likely spatially controlled (Wagner et al., 2000; Newton, 2001). We considered the possibility that DGKζ, by metabolizing signaling DAG, spatially regulates PKCα activity. We demonstrate here that DGKζ associates with PKCα and inhibits its activity in a signaling complex. This association was abolished when the MARCKS motif of DGKζ was phosphorylated by PKCα. Dissociation of the complex, in turn, attenuated the inhibition of PKCα activity; a phosphorylation-mimicking DGKζ mutant that could not bind to PKCα did not inhibit PKCα activity. Together, these data suggest that PKCα facilitates its own activation by phosphorylating DGKζ. This sequence may allow transient or even prolonged activation of PKCα in stimulated cells while inhibiting its activity in the basal state.
Results
DGKζ regulates PKCα activity
We previously demonstrated that DGKζ regulates the activity of Ras guanyl nucleotide–releasing protein (RasGRP) (Topham and Prescott, 2001), an enzyme that is allosterically activated by DAG. Because DAG is a crucial component of PKC activation, we considered the possibility that DGKζ could also regulate the enzymatic activity of some PKC isoforms. PKCα was an ideal candidate because the pattern of tissue and cellular expression closely parallels that of DGKζ (unpublished data). To test this possibility, we cotransfected PKCα along with wild-type DGKζ or a mutant, catalytically inactive DGKζ (ΔATP) into HEK293 cells and then measured PKC activity in the cell lysates. Lysates from PKCα-transfected cells demonstrated ∼10-fold higher PKC activity than the endogenous PKC activity, demonstrating that the majority of PKC activity that we measured was from transfected PKCα (Fig. 1). We found that simultaneous expression of DGKζ reduced PKCα activity by ∼50% (P < 0.001), whereas expression of a similar amount of inactive DGKζ had no significant effect on PKCα activity. Thus, DGK activity was required for PKCα inhibition. PA, the product of the DGKζ reaction, did not affect PKCα activity (unpublished data), indicating that its accumulation was not responsible for the inhibition. Supporting our contention that DGKζ regulates PKCα activity by converting DAG to PA, we found that DGKζ did not inhibit PKCα activity in the presence of PMA, a DAG analogue that cannot be metabolized by DGKζ (unpublished data). Demonstrating that the inhibition was selective to specific PKC isoforms, expression of DGKζ did not affect PKCδ activity under the same experimental conditions (unpublished data).
Figure 1.
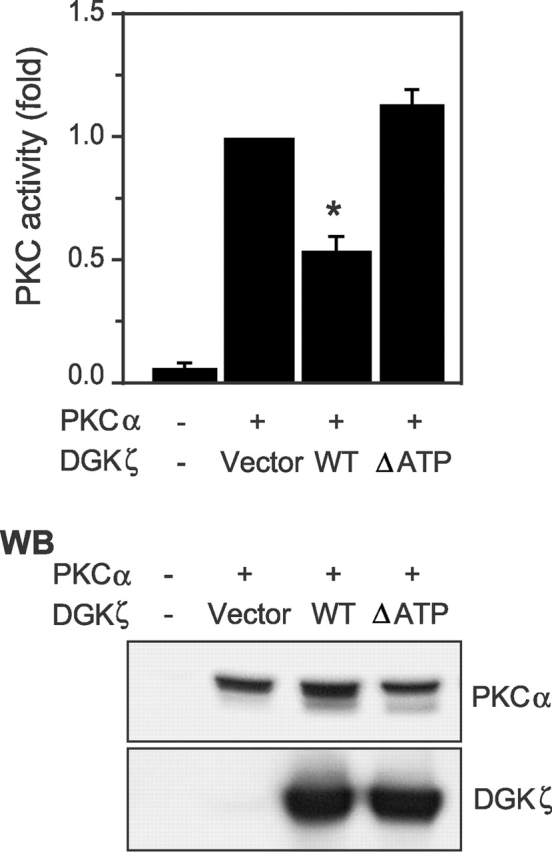
DGKζ inhibits PKCα activity in cells. HEK293 cells were transfected with PKCα along with a control vector, wild-type (WT) DGKζ, or inactive DGKζ (ΔATP). After 48 h, the cells were lysed, and PKC activity in the cell lysates was measured. Data are expressed as the mean ± SEM of four independent experiments. An asterisk indicates P < 0.001 compared with corresponding control value. Expression of PKCα and DGKζ in the cell lysates of a typical experiment is shown in the bottom panel.
PKCα and DGKζ physically associate in the cell
Evidence from several laboratories suggests that DGK function is spatially discrete rather than widespread and random, suggesting that DGKs specifically regulate a pool of DAG and the proteins activated by that DAG (van der Bend et al., 1994; Topham and Prescott, 2001; Kanoh et al., 2002). To specifically regulate a DAG-activated protein, a DGK isoform must closely associate with it. Thus, we hypothesized that DGKζ may interact with PKCα in cells. To test this possibility, we cotransfected HEK293 cells with PKCα and DGKζ–FLAG and then immunoprecipitated DGKζ with anti-FLAG antibody. We assayed for coprecipitation of PKCα by immunoblotting (Fig. 2 A). In these experiments, we found that PKCα clearly coimmunoprecipitated with DGKζ, whereas no PKCα was detected when we used a nonimmune antibody for the immunoprecipitation. Catalytically inactive DGKζ (ΔATP) still efficiently coimmunoprecipitated with PKCα. This result, combined with the fact that ΔATP did not inhibit PKCα activity (Fig. 1), confirms that association with PKCα is not sufficient for DGKζ to inhibit its activity. Demonstrating the specificity of this interaction, we could not detect coimmunoprecipitation of DGKζ and PKCδ under the same experimental conditions (unpublished data). Coimmunoprecipitation cannot distinguish direct from indirect protein–protein interactions. To test whether DGKζ and PKCα can directly bind to each other, we examined their interaction in vitro by incubating purified DGKζ–FLAG immobilized on anti-FLAG beads with recombinant PKCα. After precipitating the complexes, we observed by immunoblotting that PKCα specifically bound to DGKζ but did not bind to control beads (Fig. 2 B). Together, these data demonstrate that DGKζ and PKCα interact in vivo and that they likely bind to each other directly.
Figure 2.

DGKζ and PKCα associate with a signaling complex. (A) Lysates from HEK293 cells transiently transfected with PKCα and vector, FLAG-tagged DGKζ (WT or ΔATP), were immunoprecipitated using anti-FLAG or a control antibody (mouse IgG), and then the immunoprecipitates were subjected to immunoblot analysis with anti-PKCα. The blot was then stripped and reprobed to detect DGKζ. Expression of PKCα and DGKζ in the cell lysates is also shown. (B) Purified PKCα was incubated with purified DGKζ–FLAG bound to anti–FLAG-M2 agarose affinity gel or with affinity gel alone. The beads were washed, and proteins bound to the beads were immunoblotted with anti-PKCα. Input represents 15% of the initial recombinant PKCα used in this experiment. (C) Rat brain extracts were immunoprecipitated with anti-DGKζ or a control antibody (rabbit IgG), followed by immunoblotting with anti-PKCα. The blot was then stripped and reprobed to detect DGKζ. Expression of PKCα and DGKζ in the rat brain extracts is also shown. (D) Endogenous DGKζ in A172 cell lysates was immunoprecipitated using anti-DGKζ. Normal rabbit IgG was used as a control. The precipitates were subjected to immunoblot analysis with anti-PKCα. The blot was then stripped and reprobed to detect DGKζ. Expression of PKCα and DGKζ in the A172 cell lysate is shown in the bottom panel.
Because the above experiments examined the association of the proteins when they were abundant, which may not truly reflect their normal cellular stoichiometry, we tested whether endogenous DGKζ and PKCα could interact in rat brain extracts. Consistent with our initial results, endogenous PKCα coimmunoprecipitated with DGKζ from rat brain extracts (Fig. 2 C). We could not detect PKCα in immunoprecipitates using nonimmune IgG, demonstrating the specificity of their association. And, consistent with our results from transfected cells, we did not observe coprecipitation of endogenous DGKζ and PKCδ in rat brain extracts (unpublished data). We also tested for binding of endogenous DGKζ and PKCα in A172 cells, a glioblastoma cell line known to express both proteins (Xiao et al., 1994; Topham et al., 1998). Again, we observed a specific interaction (Fig. 2 D). Together, these data demonstrate that endogenous PKCα and DGKζ associate with the same signaling complex in vivo, and that they appear to bind directly to each other.
A portion of the catalytic domain of DGKζ is sufficient to bind PKCα
To map the domain in DGKζ that binds PKCα, we examined a series of DGKζ deletion mutants to determine which ones could associate with PKCα. We cotransfected each construct along with PKCα and then immunoprecipitated the FLAG-tagged DGKζ mutant with anti-FLAG antibodies and assessed coprecipitation of PKCα by immunoblotting. We found that a region (BD) near the COOH terminus of the catalytic domain was necessary for PKCα to coprecipitate with DGKζ (Fig. 3 A, lane 5). This region (BD), other than being part of the catalytic domain, lacks any identifiable protein motifs. Importantly, we also noted in these experiments that two different mutants (L and ΔM) lacking the MARCKS motif could still bind PKCα (Fig. 3 A, lanes 6 and 7). This binding in the absence of the MARCKS motif (the site of PKCα phosphorylation; Topham et al., 1998) demonstrates that their association was not simply a result of DGKζ being a substrate for PKCα.
Figure 3.
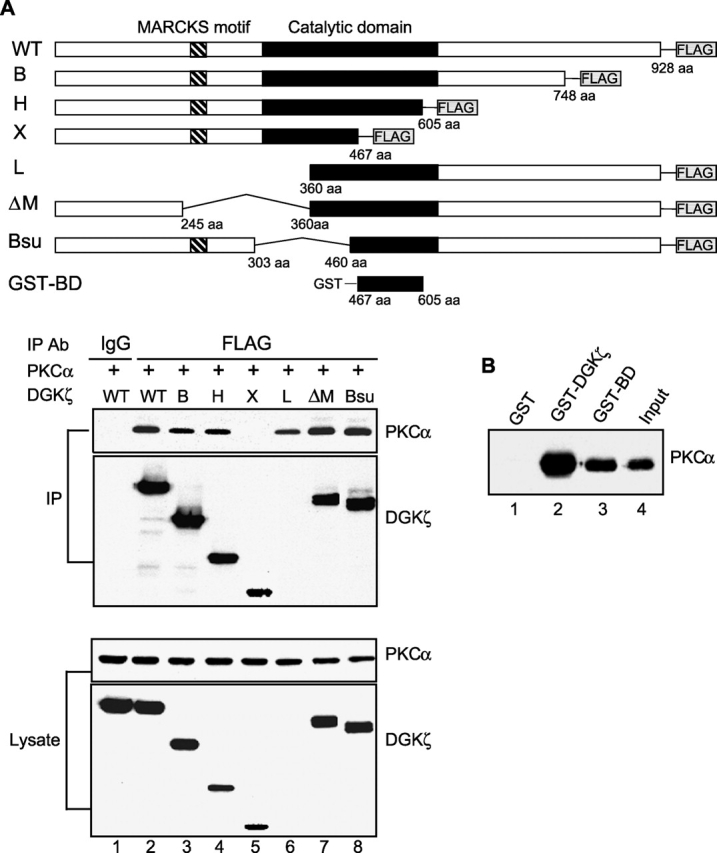
A portion of the catalytic domain of DGKζ is sufficient to bind PKCα. (A) PKCα was transfected into HEK293 cells along with wild-type (WT) DGKζ or deletion mutants of DGKζ (B, H, X, L, ΔM, and Bsu) containing FLAG epitope tags at their COOH termini. DGKζ proteins in the cell lysates were immunoprecipitated with anti-FLAG or a control antibody (mouse IgG), and coimmunoprecipitation of PKCα was detected by immunoblotting. The blot was then stripped and reprobed with anti-DGKζ. Because the DGKζ antibody we used was the NH2-terminal anti-peptide rabbit antibody, we could not detect the NH2 terminus deletion DGKζ mutant L (lane 6). However, we detected DGKζ L protein in the same blot using anti-FLAG antibody (not depicted). Expression of PKCα and DGKζ in the cell lysates is also shown. (B) Purified recombinant PKCα was incubated with the glutathione-sepharose–bound GST (lane 1) or GST fusion proteins that contain either full-length DGKζ (GST–DGKζ, lane 2) or a portion of the catalytic domain of DGKζ (GST–BD, lane 3). The beads were collected by centrifugation, and then the proteins bound to beads were subjected to immunoblot analysis with anti-PKCα. Input represents 5% of initial recombinant PKCα used in this experiment.
To determine whether the BD region of DGKζ was sufficient to interact with PKCα, we incubated recombinant PKCα with GST fusion proteins containing full-length DGKζ (GST–DGKζ), the putative binding domain (GST–BD), or control GST protein. We assessed coprecipitation of PKCα by immunoblotting and found that PKCα coprecipitated with both the GST–DGKζ and GST–BD fusion proteins, but not with the GST control protein (Fig. 3 B). Taken together, our results demonstrate that a region in the COOH terminus of the catalytic domain of DGKζ is both necessary and sufficient to bind to PKCα.
Activation of PKCα impairs its association with DGKζ
Because activated PKCα regulates the subcellular localization of DGKζ (Topham et al., 1998), we wondered if the association of DGKζ and PKCα was similarly dependent on the activation state of PKCα. To assess this possibility, we examined whether phorbol esters, potent activators of PKCα, affected coprecipitation of DGKζ and PKCα. Initially, we monitored the binding between DGKζ and PKCα when both proteins were overexpressed in HEK293 cells and observed that treating the cells with PMA significantly reduced coprecipitation of PKCα and DGKζ (Fig. 4 A). A PKC inhibitor abolished this reduction, indicating that PKC activity was required to attenuate their interaction. To examine this more directly, we tested whether PMA inhibited in vitro binding of recombinant PKCα and purified DGKζ. As demonstrated in Fig. 4 B, the recombinant PKCα could specifically bind to purified DGKζ (lane 2), but PMA dramatically attenuated their association (lane 1). To test this in vivo, we used confocal microscopy to examine whether endogenous DGKζ and PKCα colocalized in NIH 3T3 cells and whether PMA had any effect. We found that the proteins colocalized in the basal state but not after addition of PMA (Fig. 5). Thus, activation of PKCα inhibits its association with DGKζ.
Figure 4.

Activation of PKCα impairs its association with DGKζ. (A) HEK293 cells transfected with PKCα and DGKζ–FLAG were stimulated with PMA or vehicle for 30 min. DGKζ in the cell lysates was immunoprecipitated by anti-FLAG, and coimmunoprecipitation of PKCα was detected by immunoblotting. To inhibit PKC activity, cells were treated with Gö 6983 for 10 min before PMA stimulation. The blot was then stripped and reprobed to detect DGKζ. Expression of DGKζ and PKCα in the cell lysates is also shown. (B) Purified recombinant PKCα was incubated with purified DGKζ–FLAG bound to anti–FLAG-M2 agarose affinity gel or with affinity gel alone in PKC assay buffer (containing phosphatase inhibitors) in the presence or absence of PMA. After 2 h, the beads were washed, and proteins bound to beads were immunoblotted to detect PKCα. Input represents 5% of the initial recombinant PKCα. (C) A172 cells, treated with either 50 ng/ml of PDGF or vehicle for 30 min, were lysed, and then endogenous PKCα proteins were immunoprecipitated with anti-PKCα or normal rabbit IgG as a control. The precipitates were then used for DGK activity assays. To inhibit PKC activity, the cells were treated with Gö 6983 before PDGF stimulation. Data are expressed as the mean ± SEM of three independent experiments. An asterisk indicates P < 0.01 compared with corresponding control value. (D) PKCα was immunoprecipitated from control or PDGF-treated A172 cells as described in the legend to C, and then the precipitates were used for immunoblotting with a DGKζ antibody to detect coimmunoprecipitation of DGKζ. The blot was then stripped and reprobed to detect PKCα. Expression of DGKζ and PKCα in the cell lysates is shown in the bottom.
Figure 5.

DGKζ and PKCα colocalize in the basal state but not after stimulation. Control NIH 3T3 cells or cells treated with PMA (90 nM) for 30 min were immunostained to detect DGKζ (red) and PKCα (green). Confocal images were then obtained to assay for colocalization of the proteins. Bar, 10 μm.
To further examine this, we used A172 cells to test the effects of a physiologic agonist, PDGF, on the interaction of endogenous DGKζ and PKCα. Control A172 cells or those treated with PDGF were lysed, and then endogenous PKCα was immunoprecipitated. We then measured DGK activity in PKCα immunoprecipitates (Fig. 4 C). Under the experimental conditions, PDGF, which stimulates PKCα through PLCγ (Valius and Kazlauskas, 1993), activated endogenous PKCα (unpublished data). We found that PKCα immunoprecipitates had 2.1 times more DGK activity than control (nonimmune IgG) immunoprecipitates. Stimulation with PDGF almost completely removed DGK activity from PKCα immunoprecipitates, but not in the presence of a PKC inhibitor. Thus, activation of PKCα by PDGF impaired its association with DGK in A172 cells. Supporting our contention that the associated DGK was DGKζ, we found by immunoblotting that PDGF significantly attenuated coprecipitation of DGKζ and PKCα, and this was blocked by a PKCα inhibitor (Fig. 4 D). These data demonstrate that the association between endogenous DGKζ and PKCα depends on the activation state of PKCα.
Phosphorylation of the MARCKS motif causes DGKζ and PKCα to dissociate
Because PKCα can phosphorylate the MARCKS motif of DGKζ (Topham et al., 1998), we considered the possibility that this phosphorylation regulated the association of DGKζ and PKCα. Initially, we investigated whether the MARCKS motif was essential for regulation of their association. To test this, we cotransfected HEK293 cells with PKCα and the MARCKS motif deletion mutant (ΔM) and then examined the effect of PMA on the coprecipitation of PKCα and ΔM. As shown in Fig. 6 A, treating cells with PMA did not significantly affect coprecipitation of PKCα and ΔM (lane 4) but markedly reduced the binding between PKCα and wild-type DGKζ (lane 2), indicating that the MARCKS motif was necessary for the binding regulation. Under these experimental conditions, PKCα phosphorylated wild-type DGKζ on the MARCKS motif upon PMA stimulation (unpublished data). Thus, these data suggested that phosphorylation of the MARCKS motif inhibited association of DGKζ and PKCα. To further test this possibility, we cotransfected HEK293 cells with PKCα and either wild-type DGKζ or a mutant of DGKζ in which serine residues in the MARCKS motif were changed to aspartates (DGKζ S/D) to mimic phosphorylation of these residues (Swierczynski and Blackshear, 1995; Topham et al., 1998). We compared the ability of these DGKζ proteins to coimmunoprecipitate PKCα. As shown in Fig. 6 B, DGKζ S/D did not interact with PKCα (lane 3), whereas wild-type DGKζ (lane 2) and a second mutant of DGKζ in which the same serines in the MARCKS motif were changed to asparagines (DGKζ S/N) (lane 4) efficiently coimmunoprecipitated with PKCα. These results demonstrated that phosphorylation of the MARCKS motif by PKCα inhibited the interaction between DGKζ and PKCα. To verify this observation, we examined the effect of PMA on association of PKCα and DGKζ S/N. We expected that activation of PKCα by PMA would not cause their dissociation because the S/N mutation prevents phosphorylation in the MARCKS motif (unpublished data). Indeed, we found that PMA did not significantly affect coprecipitation of PKCα and DGKζ S/N (Fig. 6 C).
Figure 6.

Phosphorylation of the MARCKS motif induces the dissociation between DGKζ and PKCα. (A) HEK293 cells transfected with PKCα and either wild-type (WT) DGKζ or a MARCKS deletion mutant (ΔM). After 48 h, the cells were stimulated with PMA or vehicle for 30 min. DGKζ in the cell lysates was immunoprecipitated by anti-FLAG, and coimmunoprecipitation of PKCα was detected by immunoblotting. The blot was then stripped and reprobed to detect DGKζ. Expression of PKCα and DGKζ in the cell lysates is also shown. (B) PKCα was transfected into HEK293 cells along with wild-type (WT) DGKζ, DGKζ S/D, or DGKζ S/N. DGKζ proteins in the cell lysates were immunoprecipitated with anti-FLAG or a control antibody (mouse IgG), and coimmunoprecipitation of PKCα was detected by immunoblotting. The blot was then stripped and reprobed to detect DGKζ. Expression of PKCα and DGKζ in the cell lysates is also shown. (C) HEK293 cells transfected with PKCα and either wild-type (WT) DGKζ or DGKζ S/N were stimulated with PMA or vehicle for 30 min. DGKζ in the cell lysates was immunoprecipitated by anti-FLAG, and coimmunoprecipitation of PKCα was detected by immunoblotting. To inhibit PKC activity, cells were treated with Gö 6983 for 10 min before PMA stimulation. The blot was then stripped and reprobed to detect DGKζ. Expression of DGKζ and PKCα in the cell lysates is also shown.
Phosphorylation-mimicking DGKζ does not inhibit PKCα activity
Our data indicate the organization of a regulated signaling complex in which DGKζ binds to and locally inhibits PKCα activity. To extend our observation that phosphorylation of DGKζ caused PKCα and DGKζ to dissociate, consequently relieving PKCα inhibition, we tested whether a mutant DGKζ that could not bind to PKCα could still affect inhibition of PKCα. We transfected HEK293 cells with wild-type DGKζ, a mutant DGKζ (S/D) that could not bind efficiently to PKCα, or another mutant DGKζ (S/N) that could still bind to PKCα. After immunoprecipitation of endogenous PKCα, we compared its activity in the immunoprecipitates. We found in these experiments that expression of wild-type DGKζ significantly reduced endogenous PKCα activity, consistent with our previous results (Fig. 1), whereas expression of DGKζ S/D did not inhibit endogenous PKCα activity (Fig. 7, top). Protein expression levels of these DGKζ were similar in these experiments (Fig. 7, bottom). The lack of inhibition by DGKζ S/D was not caused by loss of its DGK activity, because it demonstrated a high DGK activity level in the cell lysates (Fig. 7, middle). A control mutant DGKζ S/N also efficiently inhibited endogenous PKCα in cells, demonstrating that negative charge (i.e., phosphorylation) within the MARCKS motif, which causes dissociation of DGKζ and PKCα, is necessary to relieve inhibition of PKCα. Consistent with this, we found that wild-type DGKζ did not inhibit PKCα activity when PMA was included in the PKC activity reaction (unpublished data). Taken together, our data demonstrate that DGKζ associates with PKCα to negatively regulate its protein kinase activity. But, PKCα can relieve this inhibition by phosphorylating DGKζ, thus causing dissociation of the two proteins.
Figure 7.
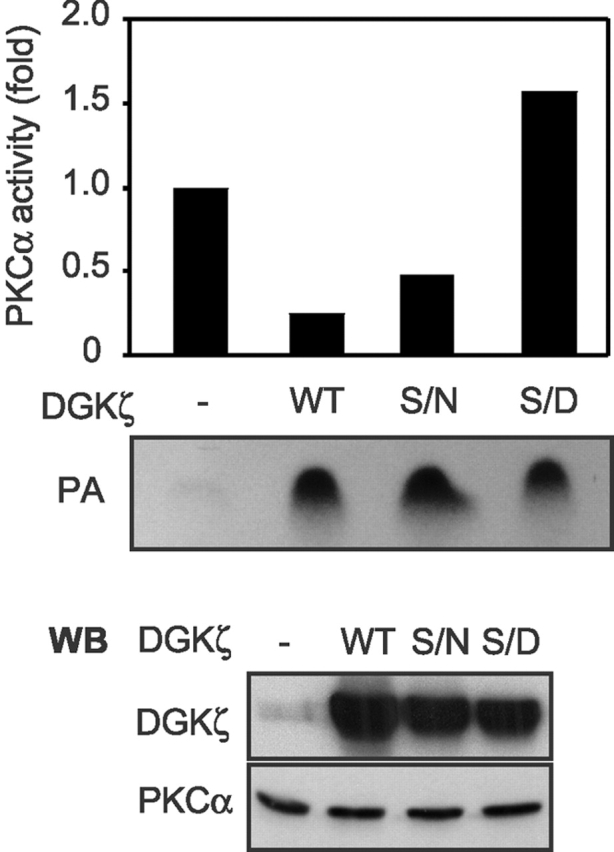
Phosphorylation of the MARCKS motif of DGKζ abolishes its inhibitory effect on PKCα activity in cells. HEK293 cells were transfected with a control vector, wild-type (WT) DGKζ, DGKζ S/N, or DGKζ S/D. After 48 h, the cells were lysed, and then endogenous PKCα was immunoprecipitated. The immunoprecipitates were used for in vitro PKC kinase assays. The DGK activity in the cell lysates was also determined using an in vitro DGK kinase assay. The top panel shows PKCα activity, and the DGK activity in the cell lysate is shown below. The bottom panel shows the protein levels of DGKζ and PKCα in the cell lysates. The data are representative of results observed from three independent experiments.
Discussion
The results presented here demonstrate that DGKζ acts to regulate the activity of PKCα. This regulation is accomplished by DGKζ binding to PKCα and metabolizing local DAG that would otherwise activate the PKCα enzyme, thus inhibiting PKCα activity. Indeed, we observed that endogenous DGKζ coimmunoprecipitated with endogenous PKCα in A172 cells and rat brain extracts and that the proteins colocalized in NIH 3T3 cells. In addition, we found that DGKζ directly interacted with PKCα. Another finding confirmed that the regulation of PKCα requires an association between DGKζ and PKCα; the phosphorylation-mimicking DGKζ (DGKζ S/D) could not bind to PKCα and inhibit its activity. Lack of PKCα inhibition by DGKζ S/D likely resulted from its inability to access, and consequently remove, the local signaling molecule, DAG. Demonstrating that this inhibition was specific for the PKCα isoform, we observed that DGKζ did not inhibit the activity of PKCδ, another DAG-dependent PKC isoform that did not associate with DGKζ. Thus, changes mediated by DGKζ in the concentration of signaling DAG occur locally and may not affect the function of PKCs found elsewhere within the cell. Taken together, these data strongly indicate that both the target (PKCα) and the attenuator (DGKζ) of DAG signaling are spatially organized in a signaling complex that is able to fine tune DAG signaling.
From immunoprecipitation experiments of endogenous DGKζ and PKCα, we estimated, using densitometry, that ∼10–20% of cellular DGKζ coimmunoprecipitated with ∼10–20% of cellular PKCα (Fig. 4 D; unpublished data). Although rough estimates, they correlate well with the amount of overlap that we observed using confocal microscopy and indicate that this regulation affects a subset of cellular PKCα. This is not surprising, given the numerous biologic functions of PKCα and the importance of its spatial regulation, and it suggests that DGKζ regulates PKCα in some, but not all, of its intracellular compartments. Because of the diverse functions of PKCα, the lack of specific PKCα or DGKζ inhibitors, and the absence of direct in vivo PKCα activity assays, we were unable to determine a distinct physiologic consequence of this regulation. However, we previously demonstrated a functional association between DGKζ and PKCα in the nucleus, indicating that DGKζ may regulate signaling mediated by nuclear PKCα (Topham et al., 1998). In NIH 3T3 cells, though, we found no evidence that these proteins colocalized in the nucleus (Fig. 5). However, another DGKζ splice variant that is not present in NIH 3T3 cells predominantly localizes in the nucleus (unpublished data), suggesting that it might regulate nuclear PKCα activity in cells where it is expressed.
PKC is not the only protein allosterically activated by DAG; several other proteins, including RasGRP, the chimaerins, Unc-13, and protein kinase D (Hurley et al., 1997; Kazanietz, 2002), have C1 domains and can bind and are activated by DAG. We have previously demonstrated (Topham and Prescott, 2001) that RasGRP associated with DGKζ and that DGKζ regulated the activation status of RasGRP by metabolizing local DAG. Additionally, Miller et al. (1999) and Nurrish et al. (1999) found that a DGK in Caenorhabditis elegans, an ortholog of mammalian DGKθ, negatively regulated synaptic transmission by metabolizing DAG that would otherwise activate Unc-13, a protein that is involved in neurotransmitter secretion. Thus, it appears that regulation of DAG signaling is frequently spatially restricted and is achieved through association of DAG target proteins and DGKs. This may be a common mechanism to regulate the amplitude or duration of signaling events. Indeed, Tasken et al. (2001) and Dodge et al. (2001) demonstrated that phosphodiesterase, which metabolizes cAMP, associated with PKA, a cAMP-dependent protein kinase, in cells. In this signaling complex, phosphodiesterase could tightly control cAMP levels to regulate the activity of PKA and consequently the phosphorylation state of proteins regulated by PKA. Also, Divecha et al. (2000) observed that phosphatidylinositol 4-phosphate 5 kinase interacted with phospholipase D in a signaling complex. It has been shown that the products of the two enzymes each stimulate the opposite enzyme (Jenkins et al., 1994; Exton, 2000). Thus, they proposed a mutual positive regulation, leading to a rapid high local increase in both products, PA and phosphatidylinositol 4,5-bisphosphate (PIP2), which may be important in a series of cellular functions. These observations, along with ours, support a paradigm where regulation of second messengers, such as DAG, cAMP, and PIP2, is spatially controlled within an assembled signaling complex.
Increasing evidence suggests that signaling proteins are organized into localized compartments where regulation of signaling events can be precisely controlled (Hunter, 2000). Targeting of protein kinases to the close proximity of their substrates ensures that they phosphorylate only the proper targets and prevents inappropriate phosphorylation events (Pawson and Nash, 2000; Smith and Scott, 2002). PKCs can directly bind to many substrates, such as adducin, GAP43, STICK72, MARCKS, and MARCKS-related proteins (Dekker and Parker, 1997; Jaken and Parker, 2000). The association of DGKζ and PKCα, combined with the fact that PKCα can phosphorylate DGKζ, suggests that DGKζ is a physiologic substrate for PKCα. Interestingly, we showed that the interaction between DGKζ and PKCα was abolished when PKCα phosphorylated DGKζ. Jaken's group (Chapline et al., 1993; Dong et al., 1995) similarly demonstrated that adducin bound to PKC and that phosphorylation of adducin reduced their interaction. Because protein phosphorylation often leads to dramatic conformational changes, phosphorylation of DGKζ likely changes its structure, resulting in its dissociation from PKCα. Supporting this idea, Bubb et al. (1999) observed a dramatic conformational change when a peptide corresponding to the PSD of the MARCKS protein was phosphorylated. This structural change may have resulted from the new negatively charged phosphates transiently interacting with positively charged amino acids in the PSD. The new structure was more compact and caused obliteration of an actin binding site in the MARCKS protein. The MARCKS motif of DGKζ is homologous to the PSD of the MARCKS protein (Bunting et al., 1996), so it would not be surprising if phosphorylation of the MARCKS motif in DGKζ caused similar conformational changes, resulting in dissociation of DGKζ and PKCα. A functional consequence of this phosphorylation may be to prevent phosphorylated DGKζ from accessing and removing local DAG that activates PKCα.
Our data support a model (Fig. 8) where in the basal state, when DAG levels in the cell are low, DGKζ associates with and regulates PKCα. This allows DGKζ to metabolize local DAG and prevent PKCα activation. Upon stimulation, when PKCα activity is required, local DAG levels increase transiently and overcome the ability of DGKζ to remove DAG. Consequently, PKCα becomes activated by DAG and then phosphorylates DGKζ, which causes dissociation of PKCα and DGKζ. This sequence results in a transient increase in PKCα activity, allowing it to phosphorylate other substrates. Presumably, PKCα activity is eventually attenuated by a variety of mechanisms, including inactivation of PLCs, dephosphorylation of PKCα or its proteolytic degradation, and reassociation with DGKζ. The duration of PKCα activation likely depends on a variety of circumstances, and its regulation is probably complex. Some cellular responses, such as proliferation and differentiation, require sustained activation of PKC, whereas other responses require it to be activated only transiently (Nishizuka, 1995; Black, 2000). For example, Balciunaite et al. (2000) demonstrated in HepG2 cells that PDGF stimulated PKC activity at two distinct times, within 10 min after PDGF treatment and then for a longer duration, between 5 and 19 h. The late phase of PKC activity was required for the PDGF-dependent transition from G0 into S phase. Aihara et al. (1991) found that sustained activation of PKCα is essential for differentiation of HL-60 cells to macrophages. Prolonged activation of PKCα may occur by regulation of DGKζ protein levels, as we have observed that expression of DGKζ significantly decreases during differentiation of HL-60 cells (unpublished data). We have previously described a functional correlation of DGKζ and PKCs in the nucleus (Topham et al., 1998), and Shirai et al. (2000) observed that DGKγ and PKCγ showed spatially similar, but temporally different, translocation after purinergic receptor activation in living cells. They suggested that the time lag between the translocation of DGKγ and PKCγ may regulate the duration of PKCγ activation. Thus, regulation of conventional PKC isoforms by DGKs may be a common theme.
Figure 8.

Model for the spatial regulation between DGKζ and PKCα. In the basal condition, DGKζ phosphorylates DAG, thereby preventing it from activating PKCα. Upon stimulation, local DAG levels increase and overcome the ability of DGKζ to phosphorylate DAG. High levels of DAG activate PKCα, which then phosphorylates DGKζ, causing the two proteins to dissociate. This favors transient or even prolonged activation of PKCα.
In conclusion, we identified a mechanism where DGKζ, by metabolizing local DAG, negatively regulates PKCα activity. In turn, by phosphorylating DGKζ, PKCα removes this inhibition, allowing its own activation. This mechanism provides low basal PKCα activity but allows for transient, or even prolonged, PKCα activation, depending on the cellular context.
Materials and methods
Materials
PMA, PDGF, phosphatase inhibitor cocktail, and anti–FLAG-M2 agarose affinity gel were obtained from Sigma-Aldrich. Rabbit polyclonal PKCα antibody, Gö 6983, and human recombinant PKCα (prepared from Sf9 insect cells) were purchased from Calbiochem. [γ-32P]ATP (6,000 Ci/mmol) was purchased from Amersham Biosciences. Normal rabbit IgG, normal mouse IgG–coupled agarose, and protein A/G agarose were from Santa Cruz Biotechnology, Inc. Glutathione–sepharose 4B was obtained from Amersham Biosciences. PKC assay kit was purchased from Upstate Biotechnology. LipofectAMINE and all cell culture reagents were obtained from Invitrogen.
Expression plasmids
Wild-type DGKζ was cloned into pcDNA1/Amp, and a FLAG epitope tag was placed at the COOH terminus, as described previously (Topham and Prescott, 2001). Generation of the progressive COOH-terminal deletions of DGKζ constructs (B, H, and X) has been published previously (Topham and Prescott, 2001). The NH2-terminal deletion DGKζ L was generated by BamH1 digestion of the DGKζ–FLAG plasmid followed by religation. For the DGKζ Bsu construct, DGKζ–FLAG plasmid was digested with Bsu36I and then religated. The catalytically inactive DGKζ mutant (ΔATP), MARCKS motif deletion (ΔM), and MARCKS motif mutants of DGKζ (S/N and S/D), in which all four serines in the MARCKS motif were altered, were generated as described previously (Topham et al., 1998). pGEX-5X plasmid containing wild-type DGKζ was a gift from Sarah Leibowitz (Rockefeller University, New York, NY). GST–BD was generated by cloning a PCR fragment comprising nucleotides 1400–1906 of human DGKζ into pGEX-5X. PKCα and PKCδ were subcloned into pcDNA3/Amp plasmid (Invitrogen).
Cell culture and transfection
HEK293 and NIH 3T3 cells were maintained in DME containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. HEK293 cells were transiently transfected as previously described (Bunting et al., 1996). After 48 h, the cells were stimulated with 90 nM PMA or vehicle for 30 min. Where indicated, Gö 6983 (500 nM) was added for 10 min before PMA stimulation. A172 cells were cultured as previously described (Topham et al., 1998).
Preparation of rat brain extracts
Rat brain was homogenized by Dounce homogenizer in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 10 μg/ml leupeptin, and 10 μg/ml aprotinin) containing 1 mM DTT and then centrifuged at 100,000 g for 30 min. Cleared lysates were used as brain extracts.
Immunoprecipitation and immunoblotting
FLAG-tagged DGKζ was transfected along with PKCα into HEK293 cells. After 48 h, the cells were harvested in lysis buffer containing phosphatase inhibitor cocktail (Sigma-Aldrich), allowed to lyse for 30 min on ice, and then centrifuged to remove debris. 1 ml of cleared lysate (3 mg of total protein) was incubated with 25 μl monoclonal anti–FLAG-M2 agarose affinity gel or normal mouse IgG coupled to agarose beads for 2 h. After centrifugation for 5 s at 10,600 g, the beads were washed with 500 μl wash buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl) three times. Immunoprecipitates were used for SDS-PAGE. Polyclonal anti-PKCα (Calboichem) was used to immunoblot for PKCα, and DGKζ was detected with a previously described polyclonal DGKζ antibody (Topham et al., 1998). To immunoprecipitate endogenous DGKζ or PKCα, A172 cell lysates and rat brain extracts were precleared with normal rabbit IgG together with protein A/G agarose for 30 min at 4°C, and then the supernatants were incubated with anti-DGKζ or anti-PKCα (Santa Cruz Biotechnology, Inc.) overnight at 4°C. The immunocomplexes were collected using protein A/G agarose, washed three times with wash buffer, and then used for immunoblotting as described above.
Immunofluorescence and confocal microscopy
NIH 3T3 cells grown on glass coverslips were rinsed in PBS, fixed with 4% formaldehyde in PBS for 20 min, permeabilized with 0.1% Triton X-100 in PBS for 10 min, and blocked with PBS containing 5% BSA for 1 h. The cells were then incubated for 1 h with 1:50 rabbit anti-DGKζ antibody and 1:100 mouse anti-PKCα antibody (Transduction Laboratories), followed by Oregon green–conjugated anti–mouse and Texas red–conjugated anti–rabbit IgG (Molecular Probes). After being washed with PBS, the immunofluorescently stained cells were imaged using a confocal microscope (Bio-Rad Laboratories).
In vitro binding assay
DGKζ–FLAG was expressed in HEK293 cells and immunoprecipitated with anti–FLAG-M2 agarose affinity gel. The immunoprecipitates were mixed with purified recombinant PKCα (0.7 μg) and incubated for 2 h at 4°C. Then, the beads were washed with wash buffer three times and analyzed for PKCα binding by immunoblotting. GST–DGKζ, GST–BD, and GST proteins were expressed in bacterial strain BL21 and purified by incubation with glutathione–sepharose 4B according to the manufacturer's instructions. The glutathione–sepharose 4B–bound GST fusion proteins were incubated with purified recombinant PKCα (0.7 μg) for 2 h at 4°C. The beads were washed three times with wash buffer and then used for immunoblotting as described above.
Kinase activity assay
PKCα or PKCδ was transfected along with a control vector, DGKζ–FLAG, or ΔATP–FLAG into HEK293 cells. After 48 h, the cells were harvested in 200 μl of lysis buffer containing phosphatase inhibitor cocktail. PKC activity in the cell lysate was determined by a PKC assay kit (Upstate Biotechnology) according to the manufacturer's instructions. To measure endogenous PKCα activity, PKCα proteins from lysates of HEK293 cells transfected with different DGKζ constructs were immunoprecipitated by polyclonal PKCα antibody (Santa Cruz Biotechnology, Inc.). The PKCα immunoprecipitates were collected using protein A/G agarose and washed three times with wash buffer and once in PKC assay buffer (20 mM MOPS, pH 7.2, 25 mM β–glycerol phosphate, 1 mM sodium orthovanadate, 1 mM DTT, 1 mM CaCl2). The immunoprecipitate was used for PKC activity assays. The DGK kinase assay was performed as previously described using lysates of HEK293 cells transfected with DGKζ (Bunting et al., 1996). 1,2-dioleoyl-sn-glycerol was used as the substrate. The reaction was performed for 10 min in the presence of [γ-32P]ATP. Lipids were extracted and separated by TLC, and PA was visualized by autoradiography.
Acknowledgments
We thank Drs. Debra Regier and Mark Wade for many helpful discussions. We are grateful to Debra Regier, Ellen Wilson, and Diana Lim for preparation of the manuscript.
This work was supported by the Huntsman Cancer Foundation.
Footnotes
Abbreviations used in this paper: DGK, DAG kinase; MARCKS, myristoylated alanine-rich C-kinase substrate; PA, phosphatidic acid; PSD, phosphorylation site domain; RasGRP, Ras guanyl nucleotide–releasing protein.
References
- Aihara, H., Y. Asaoka, K. Yoshida, and Y. Nishizuka. 1991. Sustained activation of protein kinase C is essential to HL-60 cell differentiation to macrophage. Proc. Natl. Acad. Sci. USA. 88:11062–11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciunaite, E., S. Jones, A. Toker, and A. Kazlauskas. 2000. PDGF initiates two distinct phases of protein kinase C activity that make unequal contributions to the G0 to S transition. Curr. Biol. 10:261–267. [DOI] [PubMed] [Google Scholar]
- Black, J.D. 2000. Protein kinase C-mediated regulation of the cell cycle. Front. Biosci. 5:D406–D423. [DOI] [PubMed] [Google Scholar]
- Blackshear, P.J. 1993. The MARCKS family of cellular protein kinase C substrates. J. Biol. Chem. 268:1501–1504. [PubMed] [Google Scholar]
- Bubb, M.R., R.H. Lenox, and A.S. Edison. 1999. Phosphorylation-dependent conformational changes induce a switch in the actin-binding function of MARCKS. J. Biol. Chem. 274:36472–36478. [DOI] [PubMed] [Google Scholar]
- Bunting, M., W. Tang, G.A. Zimmerman, T.M. McIntyre, and S.M. Prescott. 1996. Molecular cloning and characterization of a novel human diacylglycerol kinase ζ. J. Biol. Chem. 271:10230–10236. [PubMed] [Google Scholar]
- Chapline, C., K. Ramsay, T. Klauck, and S. Jaken. 1993. Interaction cloning of protein kinase C substrates. J. Biol. Chem. 268:6858–6861. [PubMed] [Google Scholar]
- Dekker, L.V., and P.J. Parker. 1997. Regulated binding of the protein kinase C substrate GAP-43 to the V0/C2 region of protein kinase C-Δ. J. Biol. Chem. 272:12747–12753. [DOI] [PubMed] [Google Scholar]
- Dempsey, E.C., A.C. Newton, D. Mochly-Rosen, A.P. Fields, M.E. Reyland, P.A. Insel, and R.O. Messing. 2000. Protein kinase C isozymes and the regulation of diverse cell responses. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L429–L438. [DOI] [PubMed] [Google Scholar]
- Divecha, N., M. Roefs, J.R. Halstead, S. D'Andrea, M. Fernandez-Borga, L. Oomen, K.M. Saqib, M.J. Wakelam, and C. D'Santos. 2000. Interaction of the type Iα PIPkinase with phospholipase D: a role for the local generation of phosphatidylinositol 4, 5-bisphosphate in the regulation of PLD2 activity. EMBO J. 19:5440–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge, K.L., S. Khouangsathiene, M.S. Kapiloff, R. Mouton, E.V. Hill, M.D. Houslay, L.K. Langeberg, and J.D. Scott. 2001. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 20:1921–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, L., C. Chapline, B. Mousseau, L. Fowler, K. Ramsay, J.L. Stevens, and S. Jaken. 1995. 35H, a sequence isolated as a protein kinase C binding protein, is a novel member of the adducin family. J. Biol. Chem. 270:25534–25540. [DOI] [PubMed] [Google Scholar]
- D'Santos, C.S., J.H. Clarke, R.F. Irvine, and N. Divecha. 1999. Nuclei contain two differentially regulated pools of diacylglycerol. Curr. Biol. 9:437–440. [DOI] [PubMed] [Google Scholar]
- Exton, J.H. 2000. Phospholipase D. Ann. NY Acad. Sci. 905:61–68. [DOI] [PubMed] [Google Scholar]
- Ghosh, S., J.C. Strum, and R.M. Bell. 1997. Lipid biochemistry: functions of glycerolipids and sphingolipids in cellular signaling. FASEB J. 11:45–50. [DOI] [PubMed] [Google Scholar]
- Hodgkin, M.N., T.R. Pettitt, A. Martin, R.H. Michell, A.J. Pemberton, and M.J. Wakelam. 1998. Diacylglycerols and phosphatidates: which molecular species are intracellular messangers? Trends Biochem. Sci. 23:200–204. [DOI] [PubMed] [Google Scholar]
- Hunter, T. 2000. Signaling–2000 and beyond. Cell. 100:113–127. [DOI] [PubMed] [Google Scholar]
- Hurley, J.H., A.C. Newton, P.J. Parker, P.M. Blumberg, and Y. Nishizuka. 1997. Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 6:477–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaken, S. 1996. Protein kinase C isozymes and substrates. Curr. Opin. Cell Biol. 8:168–173. [DOI] [PubMed] [Google Scholar]
- Jaken, S., and P.J. Parker. 2000. Protein kinase C binding partners. Bioessays. 22:245–254. [DOI] [PubMed] [Google Scholar]
- Jenkins, G.H., P.L. Fisette, and R.A. Anderson. 1994. Type I phosphatidylinositol 4-phosphate 5-kinase isoforms are specifically stimulated by phosphatidic acid. J. Biol. Chem. 269:11547–11554. [PubMed] [Google Scholar]
- Kanoh, H., K. Yamada, and F. Sakane. 2002. Diacylglycerol kinases: emerging downstream regulators in cell signaling systems. J. Biochem. (Tokyo). 131:629–633. [DOI] [PubMed] [Google Scholar]
- Kazanietz, M.G. 2002. Novel “nonkinase” phorbol ester receptors: the C1 domain connection. Mol. Pharmacol. 61:759–767. [DOI] [PubMed] [Google Scholar]
- Mackay, K., and D. Mochly-Rosen. 2001. Localization, anchoring, and functions of protein kinase C isozymes in the heart. J. Mol. Cell. Cardiol. 33:1301–1307. [DOI] [PubMed] [Google Scholar]
- Miller, K.G., M.D. Emerson, and J.B. Rand. 1999. Goα and diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans. Neuron. 24:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton, A.C. 1997. Regulation of protein kinase C. Curr. Opin. Cell Biol. 9:161–167. [DOI] [PubMed] [Google Scholar]
- Newton, A.C. 2001. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 101:2353–2364. [DOI] [PubMed] [Google Scholar]
- Nishizuka, Y. 1992. Intracellular signalling by hydrolysis of phospholipids and activation of protein kinase C. Science. 258:607–614. [DOI] [PubMed] [Google Scholar]
- Nishizuka, Y. 1995. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 9:484–496. [PubMed] [Google Scholar]
- Nurrish, S., L. Segalat, and J.M. Kaplan. 1999. Serotonin inhibition of synaptic transmission: Gαo decreases the abundance of UNC-13 at release sites. Neuron. 24:231–242. [DOI] [PubMed] [Google Scholar]
- Pawson, T., and P. Nash. 2000. Protein-protein interactions define specificity in signal transduction. Genes Dev. 14:1027–1047. [PubMed] [Google Scholar]
- Pawson, T., and J.D. Scott. 1997. Signaling through scaffold, anchoring, and adaptor proteins. Science. 278:2075–2080. [DOI] [PubMed] [Google Scholar]
- Shirai, Y., S. Segawa, M. Kuriyama, K. Goto, N. Sakai, and N. Saito. 2000. Subtype-specific translocation of diacylglycerol kinase α and γ and its correlation with protein kinase C. J. Biol. Chem. 275:24760–24766. [DOI] [PubMed] [Google Scholar]
- Smith, F.D., and J.D. Scott. 2002. Signaling complexes: junctions on the intracellular information super highway. Curr. Biol. 12:R32–R40. [DOI] [PubMed] [Google Scholar]
- Swierczynski, S.L., and P.J. Blackshear. 1995. Membrane association of the myristoylated alanine-rich C kinase substrate (MARCKS) protein. Mutational analysis provides evidence for complex interactions. J. Biol. Chem. 270:13436–13445. [DOI] [PubMed] [Google Scholar]
- Tasken, K.A., P. Collas, W.A. Kemmner, O. Witczak, M. Conti, and K. Tasken. 2001. Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J. Biol. Chem. 276:21999–22002. [DOI] [PubMed] [Google Scholar]
- Topham, M.K., and S.M. Prescott. 1999. Mammalian diacylglycerol kinases, a family of lipid kinases with signaling functions. J. Biol. Chem. 274:11447–11450. [DOI] [PubMed] [Google Scholar]
- Topham, M.K., and S.M. Prescott. 2001. Diacylglycerol kinase ζ regulates Ras activation by a novel mechanism. J. Cell Biol. 152:1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham, M.K., M. Bunting, G.A. Zimmerman, T.M. McIntyre, P.J. Blackshear, and S.M. Prescott. 1998. Protein kinase C regulates the nuclear localization of diacylglycerol kinase-ζ. Nature. 394:697–700. [DOI] [PubMed] [Google Scholar]
- Valius, M., and A. Kazlauskas. 1993. Phospholipase C-γ1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor's mitogenic signal. Cell. 73:321–334. [DOI] [PubMed] [Google Scholar]
- van Blitterswijk, W.J., and B. Houssa. 1999. Diacylglycerol kinases in signal transduction. Chem. Phys. Lipids. 98:95–108. [DOI] [PubMed] [Google Scholar]
- van der Bend, R.L., J. de Widt, H. Hilkmann, and W.J. van Blitterswijk. 1994. Diacylglycerol kinase in receptor-stimulated cells converts its substrate in a topologically restricted manner. J. Biol. Chem. 269:4098–4102. [PubMed] [Google Scholar]
- Wagner, S., C. Harteneck, F. Hucho, and K. Buchner. 2000. Analysis of the subcellular distribution of protein kinase Cα using PKC-GFP fusion proteins. Exp. Cell Res. 258:204–214. [DOI] [PubMed] [Google Scholar]
- Wakelam, M.J.O. 1998. Diacylglycerol–when is it an intracellular messenger? Biochim. Biophys. Acta. 1436:117–126. [DOI] [PubMed] [Google Scholar]
- Xiao, H., D.A. Goldthwait, and T. Mapstone. 1994. The identification of four protein kinase C isoforms in human glioblastoma cell lines: PKC α, γ, ε and ζ. J. Neurosurg. 81:734–740. [DOI] [PubMed] [Google Scholar]