

Abstract
We report the characterization of the dominant-negative CLA4t allele of the budding yeast CLA4 gene, encoding a member of the p21-activated kinase (PAK) family of protein kinases, which, together with its homologue STE20, plays an essential role in promoting budding and cytokinesis. Overproduction of the Cla4t protein likely inhibits both endogenous Cla4 and Ste20 and causes a delay in the onset of anaphase that correlates with inactivation of Cdc20/anaphase-promoting complex (APC)–dependent proteolysis of both the cyclinB Clb2 and securin. Although the precise mechanism of APC inhibition by Cla4t remains to be elucidated, our results suggest that Cla4 and Ste20 may regulate the first wave of cyclinB proteolysis mediated by Cdc20/APC, which has been shown to be crucial for activation of the mitotic exit network (MEN). We show that the Cdk1-inhibitory kinase Swe1 is required for the Cla4t-dependent delay in cell cycle progression, suggesting that it might be required to prevent full Cdc20/APC and MEN activation. In addition, inhibition of PAK kinases by Cla4t prevents mitotic exit also by a Swe1-independent mechanism impinging directly on the MEN activator Tem1.
Keywords: cytokinesis; mitotic exit network; Cla4; spindle checkpoint; Swe1
Introduction
In eukaryotic cells, entry into mitosis is driven by both activation of cyclinB-dependent Cdks and ubiquitin-dependent proteolysis triggered by the ubiquitin–ligase anaphase-promoting complex (APC).* At the metaphase–anaphase transition, the APC targets the Esp1 separase inhibitor securin for degradation, leading in turn to cleavage of the bonds between sister chromatids (Peters, 2002). APC-dependent proteolysis is also required at the end of mitosis to promote mitotic exit and cytokinesis. In particular, degradation of B-type cyclins seems to be crucial for Cdk inactivation, leading to spindle disassembly, cytokinesis, and entry into a new round of DNA replication (Zachariae and Nasmyth, 1999). To display full ubiquitin–ligase activity, the APC must bind to the Cdc20 and Cdh1 accessory factors that are responsible for securin and cyclinB degradation, respectively (Peters, 2002).
As in other eukaryotic systems, the onset of anaphase in budding yeast requires activation of the Cdk Cdc28 bound to any of four mitotic B-type cyclins, Clb1–4. Although the role of Clb1–4/Cdc28 kinases during this cell cycle transition can be attributed primarily to their essential function in spindle assembly (Surana et al., 1991; Fitch et al., 1992), some evidence suggests that they also stimulate the binding between Cdc20 and APC, thereby allowing mitotic exit (Rudner et al., 2000; Rudner and Murray, 2000). In this way, at the onset of anaphase, Clb1–4/Cdc28 kinases sow the seeds for their own destruction.
Work in budding yeast has shown that the Cdc14 protein phosphatase is critical to inactivate cyclinB/Cdks at the end of mitosis by dephosphorylating and activating both Cdh1 and the cyclinB/Cdk inhibitor Sic1 (Visintin et al., 1998). Cdc14 must be released from the nucleolus in order to be active during mitotic exit, and this requires a signal transduction cascade called mitotic exit network (MEN), involving the Tem1 GTPase, its GTPase exchange factor Lte1, several protein kinases and associated factors (Cdc5, Cdc15, Mob1/Dbf2, Dbf20), and a scaffold protein (Nud1; Bardin and Amon, 2001; Geymonat et al., 2002). Recently, an early anaphase release of Cdc14 from the nucleolus has been shown to be transiently driven by the Polo kinase Cdc5 and separase independently of the MEN, and to contribute to full MEN activation, thus generating a positive feedback loop (Pereira et al., 2002; Stegmeier et al., 2002; Yoshida et al., 2002).
Although Cdc20/APC reaches its maximal activity at the onset of anaphase, it is also necessary for inactivation of cyclinB/Cdks at the end of mitosis in different ways: (1) by triggering securin degradation, which in turn promotes separase activation and Cdc14 early release from the nucleolus; (2) by promoting degradation of the S-phase cyclin Clb5, whose associated Cdk activity prevents Cdc14 activation (Shirayama et al., 1999); and (3) by inducing directly a first wave of cyclinB degradation during anaphase (Baumer et al., 2000; Yeong et al., 2000), which is important for subsequent Cdh1 and MEN activation. Furthermore, a recent report has challenged the current view on mitotic exit regulation by highlighting the essential role of Cdc20/APC-driven degradation of the mitotic cyclin Clb2 (Wasch and Cross, 2002).
To ensure faithful chromosome transmission and genomic stability, progression through mitosis is tightly regulated by surveillance mechanisms (called checkpoints) that prevent or delay specific cell cycle transitions in response to different kinds of injuries, thus allowing correction of errors (Hartwell and Weinert, 1989). In particular, the metaphase to anaphase transition can be delayed by checkpoint mechanisms that respond to DNA damage (Weinert et al., 1994), spindle disassembly/misorientation (Hoyt, 2000; Gorbsky, 2001; Wassmann and Benezra, 2001), or faulty cytoskeletal organization (Lew, 2000). In budding yeast, the spindle assembly checkpoint is divided in two different branches, one monitoring kinetochore/microtubule attachment and/or kinetochore tension, and the other sensing spindle mispositioning. The kinetochore checkpoint, which involves Mad1, -2, and -3, Bub1 and -3, Mps1, Ndc10, and Ipl1, leads to inhibition of Cdc20/APC, thereby preventing both the anaphase onset and exit from mitosis (for review see Musacchio and Hardwick, 2002). Conversely, the spindle position checkpoint inhibits the Tem1 GTPase at the top of the MEN through the Bub2/Bfa1 two-component GTPase-activating protein (GAP), and therefore controls primarily mitotic exit and cytokinesis (Hoyt, 2000). Tem1 activation requires either inactivation of the GAP by the Cdc5 Polo kinase (Hu et al., 2001) or exposure to the GTPase exchange factor Lte1 (Shirayama et al., 1994). Strikingly, Tem1 resides at SPBs, whereas Lte1 is asymmetrically localized at the bud cortex, thus coupling spindle pole and nuclear migration in the bud with mitotic exit (Bardin et al., 2000; Pereira et al., 2000). Finally, the morphogenesis checkpoint delays the G2/M transition, specifically in response to perturbations in the actin cytoskeleton or in septin deposition, and depends on the Swe1-mediated phosphorylation and inactivation of Cdc28 (Lew, 2000).
Whether there are crosstalks between different checkpoints and how all the outputs of checkpoint controls are integrated in order to ensure the right progression of mitotic events are critical points still under investigation. In this paper, we present data that link cytokinesis, the spindle checkpoint, and the morphogenesis checkpoint by describing a dominant-negative CLA4 allele (CLA4t) that likely inhibits both endogenous Cla4 and Ste20. These two proteins share overlapping essential functions in controlling budding and cytokinesis (Cvrckova et al., 1995; Holly and Blumer, 1999; Weiss et al., 2000), and are members of the p21-activated kinase (PAK) family of protein kinases, which are effectors of the Cdc42 GTPase, playing a crucial role in regulating cell polarity in all eukaryotic cells (Johnson, 1999). From our data, PAK kinases appear to contribute to promote mitotic exit by regulating the MEN in two different ways; by allowing the first burst of cyclinB/Cdk inactivation that occurs at the onset of anaphase, and more directly, by establishing the conditions for Tem1 activation.
Results
High copy number of a truncated CLA4 gene can suppress the benomyl sensitivity of spindle checkpoint mutants
To gain insights into the Bub2/Bfa1 signal transduction cascade, we screened a budding yeast genomic library for high dosage suppressors of the sensitivity of a bub2Δ mutant to the microtubule-depolymerizing drug benomyl (see Materials and methods). Among the isolated clones, we found a plasmid carrying the whole promoter region and 92% of the CLA4 coding sequence (CLA4t). The Cla4t protein lacks 67 amino acids at its COOH-terminal region, where the essential kinase domain resides, but retains unaltered pleckstrin homology and PAK domains, the latter being involved in the association with Cdc42 (Benton et al., 1997; Fig. 1 A). Subcloning of both full-length and truncated CLA4 in the YEp13 2μ vector revealed that only high copy number CLA4t can suppress the benomyl sensitivity of bub2Δ, although to a lesser extent than wild-type BUB2 (Fig. 1 B). Moreover, 2μ CLA4t could also suppress the benomyl sensitivity of other spindle checkpoint mutants, like mad2Δ and bfa1Δ (Fig. 1 C), but not that of the β-tubulin mutant tub2–405 (unpublished data), suggesting that the suppression mechanism might involve differences in cell cycle progression rather than in microtubule stability.
Figure 1.
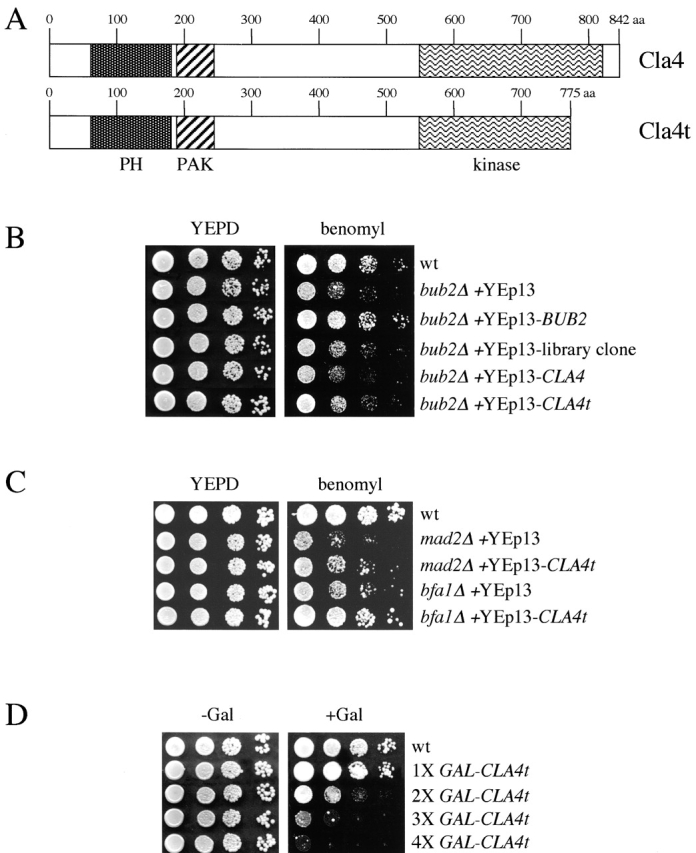
Effects of CLA4t overexpression on benomyl sensitivity and cell growth. (A) Schematic representation of wild-type Cla4 and its truncated Cla4t variant. PH, pleckstrin homology domain; PAK, p21-activated kinase family domain. (B and C) Serial dilutions of wild-type (W303), bub2Δ (ySP1071), mad2Δ (ySP1070), and bfa1Δ (ySP1243) strains carrying the indicated 2μ plasmids were spotted on YEPD plates with or without benomyl and incubated for two days at 25°C. (D) Serial dilutions of wild-type (W303) and isogenic strains carrying the indicated numbers of integrated GAL-CLAt fusions were spotted on YEPD (−Gal, GAL1 promoter off) and YEPRG (+Gal, GAL1 promoter on) plates and incubated for two days at 30°C.
Expression of CLA4t suppresses spindle checkpoint defects
To better control the expression of CLA4t, we cloned it under the galactose-inducible GAL1–10 promoter, and integrated the GAL-CLA4t fusion at the URA3 chromosomal locus. Galactose-induced expression of GAL-CLA4t had a strong dose effect on growth, with 4X GAL-CLA4t integrated copies almost totally inhibiting cells' ability to form colonies, whereas a single copy had little effect (Fig. 1 D). Therefore, we asked whether expression of GAL-CLA4t in bub2Δ, mad2Δ, and mad2Δ bub2Δ strains could suppress the checkpoint defects of these mutants. Synchronized G1 cultures of wild-type and the above mutants, either lacking or carrying a single copy of GAL-CLA4t, were released in the presence of galactose and the microtubule-depolymerizing drug nocodazole, to follow their kinetics of cell cycle progression by FACS® analysis (Fig. 2 A). As expected, in these conditions bub2Δ, mad2Δ, and mad2Δ bub2Δ mutants, unlike the wild-type strain, accumulated cells with DNA contents higher than 2C and mad2 bub2 double mutants re-replicated their genome much faster than either single mutant, as previously shown (Alexandru et al., 1999; Fesquet et al., 1999; Fraschini et al., 1999; Li, 1999). Strikingly, expression of GAL-CLA4t prevented bub2Δ and mad2Δ cells from entering a new round of DNA replication and restrained also a fraction of mad2Δ bub2Δ cells from doing so.
Figure 2.
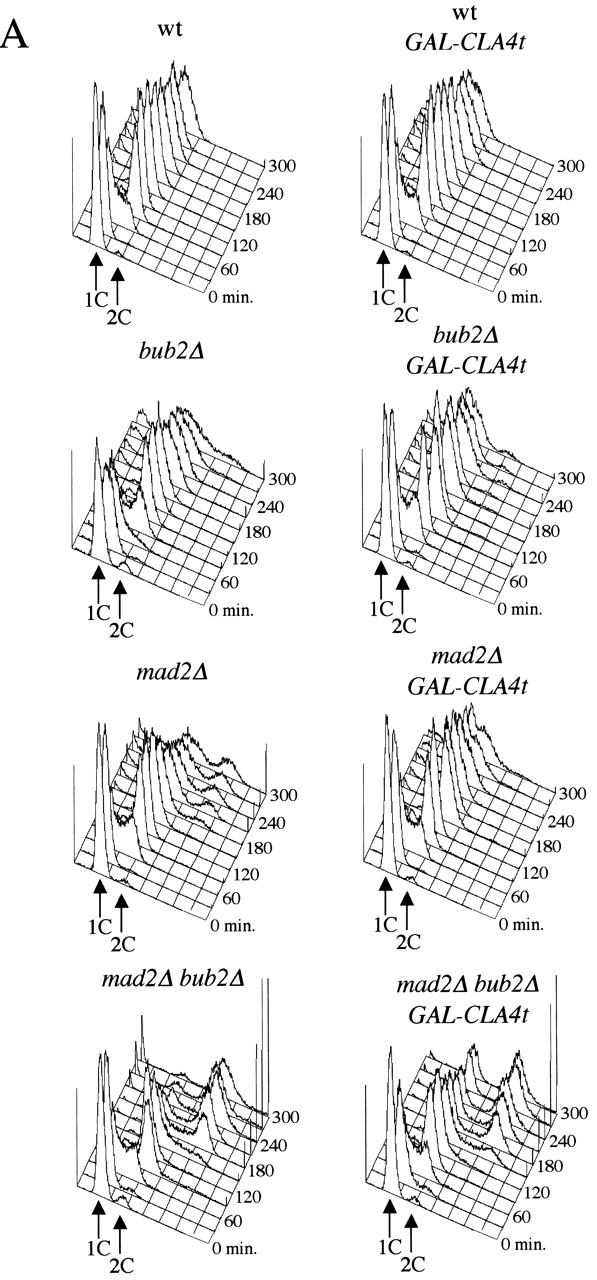

GAL-CLA4t expression suppresses the spindle checkpoint defects of mad2 and bub2 mutants. (A) Cultures of wild-type, bub2Δ, mad2Δ, and mad2Δ bub2Δ mutants, either lacking (W303, ySP3138, ySP1070, and ySP1086) or carrying one integrated copy of GAL-CLA4t (ySP2622, ySP2626, ySP2752, and ySP3068) were arrested in G1 by α-factor treatment and then released into YEPRG medium containing nocodazole (t = 0). (B) The same strains and procedure as in A were used, but 10 μg/ml α-factor was re-added to all cultures at t = 100' after release (at least 90% of budded cells) to prevent them from entering a new cell cycle. At the indicated times cells were collected for FACS® analysis of DNA contents (A) and for Western blot analysis of Clb2 and Sic1 protein levels (B). Swi6 was used as loading control. Cyc, cycling cells.
Because re-replication in the presence of nocodazole depends on inactivation of cyclinB-dependent Cdks by both APC-mediated proteolysis of cyclins and accumulation of Cdk inhibitors, we analyzed the levels of the main mitotic cyclinB (Clb2) and the Clb/Cdk inhibitor Sic1 in an experiment similar to the one above (Fig. 2 B). Although bub2Δ, mad2Δ, and mad2Δ bub2Δ cells underwent Clb2 proteolysis in the presence of nocodazole as described previously (Alexandru et al., 1999; Fraschini et al., 1999), expression of GAL-CLA4t in these mutants protected a fraction of Clb2 from degradation. In addition, Cla4t strongly reduced re-accumulation of Sic1 in bub2Δ cells, but only slightly (if at all) in mad2Δ and mad2Δ bub2Δ cells. This suggests that in the latter mutants, the phosphatase Cdc14, which promotes Sic1 transcription and activation (Visintin et al., 1998), might be active at least for what concerns accumulation of Sic1.
CLA4t inhibits PAK kinases and causes Swe1-dependent cell cycle delay
To gain insights into the molecular mechanism by which Cla4t might prevent inactivation of Clb/Cdk1 kinases in spindle checkpoint mutants, we investigated its effects when four copies of GAL-CLA4t were expressed in otherwise wild-type cells, where they almost completely inhibit cell proliferation (Fig. 1 D). Synchronized G1 wild-type cells, either lacking or carrying four integrated GAL-CLA4t copies (4X GAL-CLA4t), were released in the presence of galactose and analyzed at different time points for DNA contents, kinetics of nuclear division, and cell morphology. 4X GAL-CLA4t cells arrested with 2C DNA contents (Fig. 3 A) and undivided nuclei (Fig. 3, B and C), suggesting that not only mitotic exit, but also the onset of anaphase, failed to take place. Indeed, the securin Pds1 was stabilized in these cells (unpublished data), suggesting that Cdc20/APC could be inactive. In addition, cells displayed a dramatic cytokinetic defect due to a failure to form a proper bud neck (Fig. 3 C). This peanut-shaped cell phenotype closely resembled that observed after simultaneous inactivation of CLA4 and STE20 (Cvrckova et al., 1995), suggesting that these two redundant PAK kinases might be inactive in galactose-induced 4X GAL-CLA4t cells. In agreement with this hypothesis, a reduced fraction of these cells was able to assemble or maintain a proper septin ring at the bud neck (Fig. 3 E), as shown by localization of the Cdc3 septin fused to the GFP (Cdc3-GFP), similarly to what was shown for cla4 ste20 double mutants (Cvrckova et al., 1995; Holly and Blumer, 1999). In addition, overexpression of GAL-CLA4t disrupted localization of both myc-tagged Cla4 at the bud tip and myc-tagged Ste20 on polarized cell surface projections of pheromone-treated cells (Fig. 3 F), despite that it did not affect distribution of cortical actin (unpublished data; see Fig. 6 C). High levels of Cla4t also reduced the ability of endogenous Cla4 to phosphorylate myelin-basic protein in vitro (Fig. 3 D), suggesting that they could compromise Cla4 kinase activity in vivo. Finally, deletion of either CLA4 or STE20 reduced, but did not abolish, the ability of bub2Δ cells to re-replicate in the presence of nocodazole (Fig. 3 G).
Figure 3.


CLA4t overexpression inhibits endogenous Cla4 and Ste20 PAK kinases and causes a Swe1-dependent G2/M arrest. (A–C) Wild-type (W303), 4X GAL-CLA4t (ySP2625) and swe1Δ 4X GAL-CLA4t (ySP2711) cells were grown in YEPR, arrested in G1 with α-factor and then released in YEPRG medium (t = 0). Samples were collected at the indicated times for FACS® analysis of DNA contents (A) and to follow kinetics of nuclear division (B). Photographs were taken at t = 5 h after release (C). Bar, 5 μm. (D) wild-type (ySP3086, lanes 3–6) and 4X GAL-CLA4t cells (ySP3088, lanes 7–10), expressing myc-tagged Cla4 (Cla4myc18), as well as an untagged strain (W303, lanes 1 and 2) were grown in YEPR (lanes 1, 3, and 7) and then shifted to YEPRG for 2 h (lanes 4 and 8) and 3 h (lanes 5 and 9), or to YEPRG with nocodazole for 2.5 h (lanes 2, 6, and 10). Anti-myc immunoprecipitates from the corresponding cell extracts were subjected to Western blot analysis with anti-myc antibodies (IP) and to kinase assays using myelin basic protein (MBP) as substrate. (E) Wild-type (W303), 1X GAL-CLA4t (ySP2622) and 4X GAL-CLA4t (ySP2625) cells carrying a pRS316 plasmid with a GFP-CDC3 fusion (Vallen et al., 2000) were grown in YEPR and then shifted to YEPRG medium. After 3 h, GFP-Cdc3 was scored in unbudded and budded cells (n = 500) to monitor the presence of a septin ring. (F) Left; wild-type (ySP3086) and 4X GAL-CLA4t (ySP3088) cells, expressing Cla4myc18, were grown in YEPR, arrested in G1 with α-factor, and released in YEPRG (t = 0). Samples were collected every 10 min for 2 h for FACS® analysis of DNA contents (not depicted) and immunostaining of Cla4myc18. Photographs were taken at t = 50', when wild-type cells reached a peak of small budded cells with cortical Cla4myc18 (30% of total cells). The fraction of 4X GAL-CLA4t cells with cortical Cla4myc18 staining remained below 1% throughout the time course. Right; 4X GAL-CLA4t cells expressing myc-tagged Ste20 (Ste20myc18, ySP3091) were grown in YEPR, arrested in G1 with α-factor, and then shifted to either YEPD (glu) or YEPRG (gal) medium containing α-factor for 2 h to maintain the G1 arrest. The fraction of cells displaying polarized localization of Ste20 was 55% in YEPD and 3% in YEPRG (n = 100). Bar, 5 μm. (G) Wild-type (W303), cla4Δ (ySP3076), ste20Δ (ySP3078), bub2Δ (ySP3138), bub2Δ cla4Δ (ySP3186), and bub2Δ ste20Δ (ySP3198) cells were grown in YEPD, shifted to YEPD containing nocodazole (t = 0), and collected at the indicated times for FACS® analysis of DNA contents. The percentage of re-replicating cells at each time point was calculated as described in Materials and methods. (H) Wild-type (ySP3157) and 4X GAL-CLA4t (ySP3202) cells expressing myc-tagged Hsl1 were grown in YEPR, arrested in G1 with α-factor, and released in YEPRG. Samples were collected at different times for immunostaining of Hsl1myc18. Photographs were taken 90' after release. Bar, 5 μm.
Figure 6.
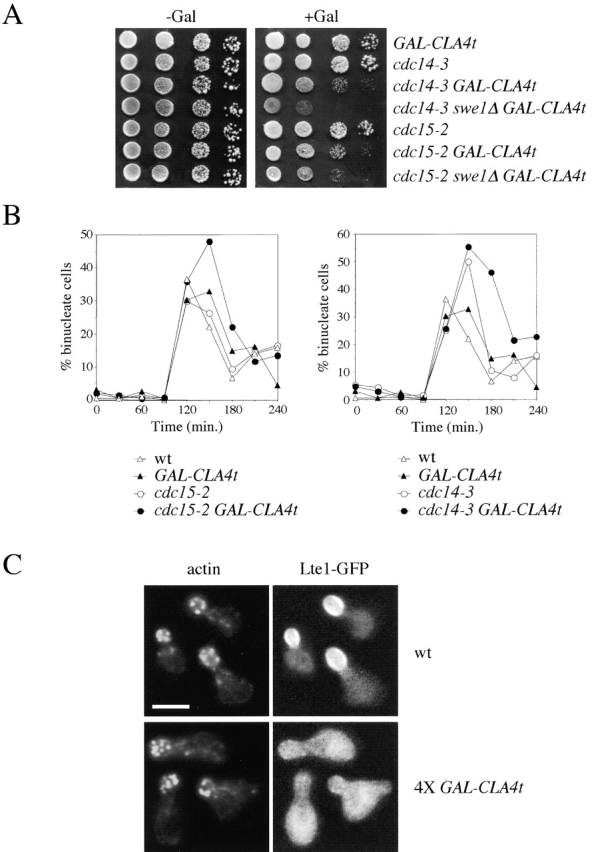
GAL-CLA4t expression delays MEN activation and prevents proper localization of Lte1 at the bud cortex. (A) Serial dilutions of 1X GAL-CLA4t (ySP2622), cdc14–3 (ySP284), cdc14–3 1X GAL-CLA4t (ySP3028), cdc14–3 swe1Δ 1X GAL-CLA4t (ySP3047), cdc15–2 (ySP51), cdc15–2 1X GAL-CLA4t (ySP3022), and cdc15–2 swe1Δ 1X GAL-CLA4t (ySP3023) strains were spotted on either YEPD (−Gal) or YEPRG (+Gal) plates and incubated for 2 d at 26°C. (B) Wild-type, cdc15–2, and cdc14–3 cells either lacking (W303, ySP51, and ySP284) or carrying (ySP2622, ySP3022, and ySP3028) one copy of GAL-CLA4t were grown in YEPR, arrested in G1 with α-factor, and released in YEPRG (t = 0). Samples were collected every 30' for FACS® analysis of the DNA contents (not depicted) and to score nuclear division. (C) Wild-type (ySP3222), 1X GAL-CLA4t (ySP3220, not depicted), and 4X GAL-CLA4t (ySP3221) cells were treated as in B. Samples were collected every 15' for 150' for FACS® analysis of the DNA contents (not depicted) and to score localization of GFP-Lte1 and actin by rhodamine-phalloidin. Photographs were taken 75' after release. Bar, 5 μm.
We also noticed that the Hsl1 protein kinase, responsible for targeting the Swe1 kinase to degradation (McMillan et al., 1999), was localized at the bud neck in virtually all wild-type budded cells from the onset of budding until late anaphase, but it was homogeneously distributed throughout the cells of the 4X GAL-CLA4t strain (Fig. 3 H) and might thus be inactive. Because deletion of CLA4 was shown to activate the morphogenesis checkpoint, resulting in a G2/M delay due to the Swe1-dependent phosphorylation of Cdc28 (Longtine et al., 2000; Weiss et al., 2000; Mitchell and Sprague, 2001), we asked whether the cell cycle delay caused by overproduced Cla4t could also depend on SWE1. Indeed, deletion of SWE1 allowed synchronized 4X GAL-CLA4t cells to undergo nuclear division with nearly wild-type kinetics in the presence of galactose (Fig. 3, B and C), suggesting that Swe1 is responsible for preventing anaphase in these cells. Conversely, lack of Swe1 did not rescue their cytokinetic defects (Fig. 3 C) and was only partially able to drive them out of mitosis and into a new round of DNA replication, as shown by the appearance of cells with 4C DNA contents (Fig. 3 A). Thus, Swe1 restrains mitotic exit in Cla4t-overproducing cells, but additional mechanisms are also involved.
Cla4t ability to suppress spindle checkpoint defects partially depends on Swe1
The data above indicate that Cla4t high levels delay the metaphase–anaphase transition in a Swe1-dependent manner, suggesting that Cla4t and Swe1 could prevent the onset of anaphase by inhibiting more directly the activity of Cdc20/APC. Therefore, we investigated whether expression of one copy of GAL-CLA4t, which has only mild effects on cell proliferation (Fig. 1 D) but suppresses spindle checkpoint defects (Fig. 2), might affect Cdc20/APC-dependent proteolysis of Pds1 and Clb2 during an unperturbed cell cycle. As shown in Fig. 4 A, Pds1 was stabilized in Cla4t expressing cells, and this correlated with a slight nuclear division delay (Fig. 4 B). Cla4t also protected a fraction of Clb2 from degradation, whereas no major effects were found on the levels of Sic1 accumulation. Thus, Cdc20/APC, which triggers both Pds1 and Clb2 degradation, might not be fully active in Cla4t expressing cells. Remarkably, deletion of SWE1 accelerated proteolysis of both Pds1 and Clb2 in these cells, although not to wild-type levels (Fig. 4 A), and restored normal kinetics of nuclear division (Fig. 4 B), suggesting that Swe1 is required for Cla4t to delay the onset of anaphase. The CDC28 Y19F allele, encoding an unphosphorylatable Cdc28 protein, had an intermediate effect because it reduced Pds1 levels but did not rescue the delay in either nuclear division or Clb2 degradation. However, we could not exclude that Swe1 exerts its inhibitory function through Cdc28 phosphorylation because our results could be biased by the fact that the CDC28 Y19F mutant displays, by itself, reduced Cdc20/APC activity (Rudner et al., 2000).
Figure 4.

Expression of GAL-CLA4t delays degradation of Cdc20/APC substrates with a mechanism partially dependent on Swe1. Wild-type (ySP1969), 1X GAL-CLA4t (ySP3073), swe1Δ 1X GAL-CLA4t (ySP3098), and CDC28 Y19F 1X GAL-CLA4t (ySP3074) cells expressing myc-tagged securin (Pds1myc18) were grown in YEPR, arrested in G1 with α-factor, and released in YEPRG (t = 0). Samples were collected at the indicated times for Western blot analysis (A) to score nuclear division (B) and for FACS® analysis of DNA contents (not depicted). 10 μg/ml α-factor was added back to the cultures at t = 110'.
Next, we asked whether deletion of SWE1 could also revert the Cla4t-dependent suppression of spindle checkpoint defects. For this purpose, we analyzed the ability of swe1Δ mad2Δ or swe1Δ bub2Δ strains carrying a single copy of GAL-CLA4t to support Clb2 degradation and Sic1 accumulation, as well as to re-replicate DNA, on nocodazole treatment. In a first experiment GAL-CLA4t, mad2Δ, mad2Δ GAL-CLA4t, mad2Δ swe1Δ GAL-CLA4t, and mad2Δ CDC28 Y19F GAL-CLA4t pheromone-treated G1 cells were released into the cell cycle in the presence of both galactose and nocodazole. As shown in Fig. 2, expression of CLA4t prevented a fraction of Clb2 from being degraded, slightly delayed Sic1 accumulation and prevented most mad2Δ cells from rebudding (Fig. 5 A) and re-replicating (unpublished data). Deletion of SWE1 in these conditions antagonized the effects of Cla4t, in that it accelerated kinetics of Clb2 degradation and Sic1 accumulation of mad2Δ GAL-CLA4t cells, although not to the levels observed for mad2Δ cells (Fig. 5 A). Furthermore, mad2Δ swe1Δ GAL-CLA4t rebudded and re-replicated only poorly in these conditions, suggesting that Cla4t prevents Clb2 degradation and re-replication in mad2Δ cells both by activating Swe1 and through an additional mechanism. Again, we found that the CDC28 Y19F allele had no major effect on either Clb2 and Sic1 protein levels (Fig. 5 A) or re-replication efficiency of mad2Δ GAL-CLA4t cells (unpublished data). Because Mad2 and Bub2 activate the spindle checkpoint via different pathways, we then performed the same analysis on bub2Δ cells. As shown in Fig. 5 B, knocking out Swe1 function in bub2Δ GAL-CLA4t cells restored a checkpoint defect similar to that of bub2Δ cells, in terms of Clb2 degradation, Sic1 re-accumulation, and kinetics of rebudding, erasing completely the effects of Cla4t. Thus, Swe1 can cooperate with either the Mad2- or the Bub2-dependent pathways in preventing exit from mitosis. Moreover, the different effects of Cla4t on the ability of mad2Δ and bub2Δ cells to enter a new cell cycle on checkpoint activation suggest that Cla4t can inhibit mitotic exit in at least two different ways, one dependent and another independent on Swe1; mad2Δ cells would be susceptible to both inhibitory mechanisms, whereas the lack of BUB2 antagonizes the Swe1-independent mechanism.
Figure 5.


Swe1 is required for Cla4t-dependent stabilization of Clb2 in nocodazole-treated spindle checkpoint mutants. (A) 1X GAL-CLA4t (ySP2622), mad2Δ (ySP1070), mad2Δ 1X GAL-CLA4t (ySP2752), mad2Δ swe1Δ 1X GAL-CLA4t (ySP3145), and mad2Δ CDC28 Y19F 1X GAL-CLA4t (ySP3148) cells (A) or 1X GAL-CLA4t (2622), bub2Δ (ySP3138), bub2Δ 1X GAL-CLA4t (ySP2626), and bubΔ swe1Δ 1X GAL-CLA4t (ySP2726) cells (B) were grown in YEPR, arrested in G1 with α-factor, and released in YEPRG (t = 0). Samples were collected at the indicated times for Western blot analysis of Clb2 and Sic1 and for FACS® analysis of the DNA contents (not depicted). 10 μg/ml α-factor was added back to the cultures at t = 120'. Swi6 was used as loading control. Cyc, cycling cells.
PAK kinases contribute to proper MEN activation
Because Cla4t prevents mitotic exit when heavily overproduced in wild-type cells or when cell cycle progression is partially compromised by spindle checkpoint activation, we wondered whether it could affect MEN activation. Indeed, the temperature-sensitive men mutants cdc15–2 and cdc14–3 were more sensitive than wild type to expression of a single copy of GAL-CLA4t at the permissive temperature (Fig. 6 A). Moreover, deletion of SWE1 did not rescue, but eventually accentuated, the lethality of cdc15–2 GAL-CLA4t and cdc14–3 GAL-CLA4t cells on galactose-containing plates, suggesting that Cla4t might directly affect MEN activation independently of Swe1. In the attempt to correlate with specific cell cycle defects the poor tolerance of cdc15–2 and cdc14–3 mutants to moderately high levels of Cla4t, we arrested in G1 wild-type, cdc15–2, and cdc14–3 cells, either carrying or lacking one copy of GAL-CLA4t, and followed the appearance and disappearance of binucleate anaphase cells on release from the G1 block in the presence of galactose at 25°C. Although Cla4t did not seem to affect the kinetics of nuclear division, comparison of the kinetics of disappearance of binucleate cells indicated that it delayed cell division by ∼30 min in both cdc15–2 and cdc14–3 cells (Fig. 6 B) independently of Swe1 (unpublished data), suggesting that PAK kinases contribute to MEN activation under these conditions. This is not achieved by promoting Bfa1 phosphorylation, which in turn correlates with inhibition of Bfa1/Bub2 GAP activity (Hu et al., 2001) because we found no major differences in the kinetics of Bfa1 phosphorylation between wild-type and swe1Δ GAL-CLA4t cells (unpublished data). We then verified whether high levels of Cla4t might displace and therefore inactivate Lte1, as wild-type Cla4 has been recently implicated in the correct localization of the Tem1 activator Lte1 at the bud cortex (Hofken and Schiebel, 2002; Seshan et al., 2002). Indeed, overexpression of CLA4t in 4X GAL-CLA4t cells, where we assume Cla4 and Ste20 are completely inactive, abolished polarized Lte1 but not actin distribution at the bud cortex (Fig. 6 C). Again, deletion of SWE1 did not reestablish proper Lte1 localization (unpublished data). Expression of a single copy of GAL-CLA4t slightly decreased the percentage of small- and medium-size budded cells with polarized Lte1 (70% in small/medium budded GAL-CLA4t cells versus 90% of wild-type cells). Thus, moderately high levels of Cla4t, although not completely inhibiting endogenous Cla4 and Ste20, might be sufficient to delay Tem1 activation at the end of mitosis in cells whose MEN activity is partially compromised. If partial Tem1 inactivation due to PAK kinases inhibition were the only reason for the mitotic exit delay caused by Cla4t in the absence of Swe1, one would predict that Tem1 activation could be rescued by eliminating its GAP Bfa1/Bub2, thereby counteracting the effect of Lte1 displacement from the bud cortex. To test this notion, we analyzed the effects of BUB2 deletion on cell cycle progression of swe1Δ 4X GAL-CLA4t, which we previously showed to be only partially able to exit mitosis (Fig. 3 A). Deletion of BUB2 by its own had negligible effects on the cell cycle block of galactose-induced 4X GAL-CLA4t cells, that arrested with 2C DNA contents (Fig. 7 A), undivided nuclei and metaphase spindles (Fig. 7 B). Strikingly, bub2Δ swe1Δ 4X GAL-CLA4t cells exited mitosis much faster than swe1Δ 4X GAL-CLA4t cells, entering a new round of DNA replication (Fig. 7 A) and disassembling their anaphase spindles with nearly wild-type kinetics, thus accumulating as multinucleate cells (Fig. 7 B). We obtained similar results by analyzing the effects of SWE1 and BUB2 deletion in the temperature-sensitive cla4–75 ste20 double mutant (Cvrckova et al., 1995), although Bub2 seemed to have a predominant role over Swe1 in preventing mitotic exit at 37°C (unpublished data). Therefore, PAK kinases appear to control MEN activation by at least two different mechanisms; one operating at the metaphase to anaphase transition and that can be inhibited by Swe1, and the other impinging more directly on Tem1 activation.
Figure 7.

Bub2 and Swe1 act synergistically in regulating mitotic exit. 4X GAL-CLA4t (ySP2625), bub2Δ 4X GAL-CLA4t (ySP2630), swe1Δ 4X GAL-CLA4t (ySP2711), and bub2Δ swe1Δ 4X GAL-CLA4t (ySP2728) cells were grown in YEPR, arrested in G1 with α-factor, and released in YEPRG medium (t = 0). Samples were collected at the indicated times for FACS® analysis of DNA contents (A) and to follow kinetics of spindle assembly/disassembly by α-tubulin immunostaining (B). Photographs were taken 3 h after release. Bar, 5 μm.
Discussion
Inhibition of PAK kinases by Cla4t
We have shown that overexpression of the truncated dominant-negative CLA4t allele prevents cell cycle progression of nocodazole-treated spindle checkpoint mutants and causes a mitotic arrest very reminiscent of the one reported for cla4 ste20 double mutants (Cvrckova et al., 1995). In fact, cells have duplicated chromosomes, but fail to undergo nuclear division and mitotic exit, and display a characteristic peanut-shaped morphology due to their inability to form a proper bud neck constriction in preparation to cytokinesis. This correlates with a slight defect in assembling the septin ring, but presumably other as yet unidentified cytokinetic processes are also impaired. Importantly, polar localization of both endogenous Cla4 and Ste20 is disrupted in CLA4t-overexpressing cells, and the in vitro kinase activity of immunoprecipitated endogenous Cla4 is reduced under the same conditions. The CLA4t gene product, which lacks part of the COOH-terminal kinase domain, but still contains protein–protein interaction motifs in the NH2-terminal part, might directly bind to endogenous Cla4 and Ste20 and therefore inhibit them. In fact, mammalian PAK kinases have been shown to dimerize and autoinhibit in trans, with the NH2-terminal portion of one subunit inhibiting the catalytic domain of the other (Parrini et al., 2002). Although immunoprecipitation assays did not reveal physical interactions between wild-type Cla4 and either itself or Ste20 (unpublished data), we cannot exclude that interaction is labile and stabilized when Cla4t acts as a partner in the dimers. Cla4t might also act by sequestering Cdc42 in an inactive form, resulting in the lack of activation of Cdc42 targets other than Cla4 and Ste20. If this were the case, we would expect to alleviate the effects of Cla4t overproduction by replacing wild-type CDC42 with the cdc42 V36A allele, which impairs more efficiently interaction of Cdc42 with Cla4 than with other effectors (Gladfelter et al., 2001), and should therefore reduce the capacity of Cla4t to titrate Cdc42 out, allowing it to activate other targets. Conversely, we found that not only did the cdc42 V36A allele not restore the ability to progress through the cell cycle in bub2Δ GAL-CLA4t cells treated with nocodazole, but also decreased by its own the ability of bub2Δ cells to re-replicate under these conditions (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200209097/DC1). In addition, concomitant overexpression of CDC42 from the GAL1 promoter does not counteract the toxicity caused by high levels of Cla4t (unpublished data). It is also worth noting that polarized actin distribution, which is affected in cells lacking either functional Cdc42 (Adams et al., 1990) or Cdc42 effectors other than Cla4 and Ste20, such as Gic1 and Gic2 (Brown et al., 1997; Chen et al., 1997), is not disrupted by either overexpression of Cla4t (Fig. 6 C) or inactivation of both Cla4 and Ste20 (Cvrckova et al., 1995). Therefore, we propose that even if Cla4t can sequester Cdc42, its effects on cell cycle progression might be at least partially attributed to inhibition of wild-type Cla4 and Ste20. Consistently, we show that deletion of either CLA4 or STE20 can reduce the fraction of bub2Δ cells that re-replicate in the presence of nocodazole (Fig. 3 G). Therefore, we think it is reasonable to assume that endogenous Cla4 and Ste20 are inactive in cells overproducing Cla4t, although we have not formally proven it.
PAK kinases and Swe1 regulate both the onset of anaphase and mitotic exit
In agreement with previous data (Longtine et al., 2000; Weiss et al., 2000; Mitchell and Sprague, 2001), we have shown that inactivation of Cla4 and Ste20 by Cla4t induces a Swe1-dependent G2/M delay, whose extent depends on Cla4t levels. This is likely the consequence of morphogenesis checkpoint activation (Lew and Reed, 1995) caused by septin ring defects and mislocalization of Hsl1 (Fig. 3, E and H), which normally targets Swe1 to degradation (McMillan et al., 1999; Longtine et al., 2000). Swe1 inactivates Clb1–4/Cdks by inhibitory phosphorylation of their catalytic subunit Cdc28 on tyrosine 19 (Booher et al., 1993). How do Clb1–4/Cdc28 kinases bring about the onset of anaphase? One process they definitely regulate is spindle assembly because inactivation of all mitotic cyclin genes CLB1–4, as well as overexpression of SWE1 or the presence of a CDC28 allele mimicking constitutive tyrosine 19 phosphorylation, inhibit separation of the duplicated SPBs and bipolar spindle assembly (Surana et al., 1991; Fitch et al., 1992; Lim et al., 1996). However, this is probably not the only anaphase mechanism controlled by Clb1–4/Cdks. In fact, we have shown that Swe1 is responsible for stabilizing the securin Pds1 on moderate CLA4t overexpression, when cells undergo normal cycles of spindle assembly/disassembly. In addition, Swe1 protects a fraction of Clb2 from degradation in spindle checkpoint mutants treated with nocodazole, i.e., in conditions where a spindle is not formed anyway. Both observations might be explained if Swe1 had a role in controlling Cdc20/APC activity. In fact, Cdc20/APC not only drives Pds1 (Visintin et al., 1997; Lim et al., 1998) and a first wave of Clb2 degradation (Baumer et al., 2000; Yeong et al., 2000; Wasch and Cross, 2002), but Cdc28 kinase activity is also necessary for full Cdc20/APC activation (Rudner et al., 2000; Rudner and Murray, 2000). Therefore, Swe1, by phosphorylating Cdc28, might prevent proper Cdc20/APC-dependent degradation of both Pds1 and Clb2, which is a prerequisite for full MEN activation at the end of mitosis (Cohen-Fix and Koshland, 1999; Tinker-Kulberg and Morgan, 1999; Yeong et al., 2000; Wasch and Cross, 2002). This hypothesis (depicted in Fig. 8) is consistent with our finding that Cla4t can delay mitotic exit of men mutants at permissive temperature and of nocodazole-treated spindle checkpoint mutants through a mechanism partially dependent on Swe1. Despite that the only known target of Swe1 thus far is Cdc28, we observed only a minor restoration of Clb2 degradation in cells expressing CLA4t in the presence of the CDC28 Y19F allele, which relieves the inhibitory function of Swe1. Thus, either Swe1 phosphorylates targets other than Cdc28, or Swe1-dependent inhibition of Clb2 proteolysis might still be mediated by Cdc28 phosphorylation, but we cannot properly see it because unphosphorylatable Cdc28 displays reduced kinase levels and Cdc20/APC activity (Rudner et al., 2000). Because a CDC28 T18VY19F allele impairs the physical association between Cdc20 and APC in a cdc15 telophase arrest (Rudner et al., 2000), we also investigated whether the CDC28 Y19F allele or overexpression of either SWE1 or CLA4t might similarly affect Cdc20/APC interaction. We found no major differences with a wild-type control (unpublished data), suggesting that other mechanisms, like phosphorylation of APC subunits, might influence the activity of the complex. A reduced Cdc20/APC activity could also explain why expression of a single copy of GAL-CLA4t, which barely delays the onset of anaphase in wild-type cells, can almost completely prevent mad2Δ and bub2Δ mutants from overriding a checkpoint arrest. In fact, Cla4t-dependent inhibition of Cdc20/APC would counteract its activation due to the lack of Mad2, thus reinforcing MEN inhibition operated by Bub2/Bfa1 (Fig. 8). Less intuitive is how reduced Cdc20/APC activity could prevent bub2Δ and bfa1Δ cells from exiting mitosis in the presence of nocodazole because Cdc20 should be kept inactive by Mad2 and Mad3 (Hwang et al., 1998; Fraschini et al., 2001; Hoyt, 2001). Remarkably, we have found that the ability of bub2Δ cells to separate sister chromatids and re-replicate in nocodazole requires Cdc20 function (Fig. S2), indicating that there might exist a pool of Cdc20 refractory to inhibition by Mad2 and Mad3.
Figure 8.

A model for the role of PAK kinases in controlling mitotic progression. See text for details.
Despite that the deletion of SWE1 accelerates Clb2 degradation and mitotic exit in cells expressing GAL-CLA4t, it does not rescue wild-type kinetics of mitotic exit either on heavy overproduction of Cla4t or in nocodazole-treated mad2Δ cells moderately overproducing Cla4t, suggesting that inhibition of PAK kinases by Cla4t might prevent MEN activation also by Swe1-independent means. We find that overexpression of CLA4t prevents Lte1 cortical localization, thus down-regulating the MEN, consistent with the recent finding that Cla4 phosphorylates and targets Lte1 to the bud cortex, thereby allowing timely activation of Tem1 (Hofken and Schiebel, 2002; Seshan et al., 2002). However, this cannot be the only level of MEN regulation exerted by PAK kinases, as LTE1 is not essential at the temperatures we performed our experiments (Adames et al., 2001). Indeed, deletion of BUB2, that allows Tem1 activation in the absence of Lte1, although not sufficient by its own to drive CLA4t overexpressing cells out of mitosis, greatly stimulates mitotic exit of swe1Δ GAL-CLA4t cells, indicating that SWE1 deletion and TEM1 activation act synergistically to fully activate the MEN, as proposed in Fig. 8. Interestingly, Hofken and Schiebel (2002) have reported a role for Ste20 in MEN activation unrelated to Lte1 association with the bud cortex. An intriguing hypothesis that would match our data is that Ste20 is implicated in down-regulating Swe1, thereby allowing full activation of Cdc20/APC at the onset of anaphase and the early release of Cdc14 from the nucleolus.
Based on our data, the PAK kinases Cla4 and Ste20 might be considered new members of the MEN. Remarkably, the Schizosaccharomyces pombe septation initiation network, which has similar organization to the MEN (Bardin and Amon, 2001; McCollum and Gould, 2001), includes the Sid1 PAK kinase (Guertin et al., 2000), and overexpression of the Sid1-interacting factor Cdc14 (unrelated to its Saccharomyces cerevisiae namesake) causes a G2 arrest that likely depends on the Swe1-like kinase Wee1 (Fankhauser and Simanis, 1993).
Interdependency between mitotic exit and cytokinesis
In budding yeast, the site of cell division is already established at the G1/S transition, concomitantly with bud emergence. During this process, the actin cytoskeleton undergoes profound rearrangements, so that actin patches get concentrated at the tip of the bud and actin cables orient toward it. At this cell cycle stage, the septin ring is also formed (Cooper and Kiehart, 1996; Longtine et al., 1996) and a contractile actomyosin ring starts assembling (Bi et al., 1998).
A dependency of cytokinesis on mitotic exit and inactivation of cyclinB/Cdks has long been established both in yeast and in other eukaryotic systems (Field et al., 1999; Surana et al., 2002). In addition, the requirement of Cdc20/APC-dependent proteolysis for MEN activation dictates a hierarchical order of events, where nuclear division occurs before mitotic exit, which is in turn a prerequisite for cytokinesis. Our data lead to the unforeseen conclusion that cytokinetic events can influence the kinetics of mitotic exit by allowing full activation of Cdc20/APC and by promoting cortical localization of Lte1 (Fig. 8). In summary, to exit mitosis, two conditions must be satisfied: (1) Cdc20/APC must be fully active to promote Pds1 and the first wave of Clb1–4 degradation. In fact, Pds1 inhibits the early release of Cdc14 from the nucleolus (Stegmeier et al., 2002; Yoshida et al., 2002), which together with the reduction in Clb/Cdks contributes to activation of Cdc15, and in turn, Cdc14 itself (Jaspersen and Morgan, 2000). Moreover, PAK and Clb1–4/Cdc28 kinases, as well as extinction of checkpoint signals, are necessary for complete activation of Cdc20/APC; and (2) nuclear division must take place along the mother-bud axis, thus allowing Tem1, which is localized at SPBs, to access Lte1 constrained in the bud (Bardin et al., 2000; Pereira et al., 2000) with a mechanism dependent on Cla4 (Hofken and Schiebel, 2002; Seshan et al., 2002; this work). Alternatively, inhibition of Bub2/Bfa1 by Cdc5 is sufficient to allow Tem1 activation (Adames et al., 2001; Hu et al., 2001). Such an intricate network of controls would guarantee the correct order of mitotic events and the dependency of mitotic exit on both nuclear division and cytokinesis. This latter feature might play a particularly relevant role in organisms like budding yeast, which specify the site of cytokinesis early in the cell cycle, before spindle assembly and orientation. Interestingly, a checkpoint control involving both Wee1 and the septation initiation network monitors proper accomplishment of early cytokinetic events in fission yeast (Liu et al., 2000), suggesting that it might be a common feature in eukaryotic cells.
Materials and methods
Strains, media, and reagents
All yeast strains (Table I) were derivatives of or were back-crossed at least three times to W303 (ade2–1, trp1–1, leu2–3,112, his3–11,15, ura3, ssd1), except for ySP3220, ySP3221, and ySP3222, which are congenic with each other. Cells were grown in YEP medium (1% yeast extract, 2% bactopeptone, 50 mg/l adenine) supplemented with 2% glucose (YEPD), 2% raffinose (YEPR), or 2% raffinose and 1% galactose (YEPRG). Unless otherwise stated, α-factor, nocodazole, and benomyl were used at 2 μg/ml, 15 μg/ml, and 12.5 μg/ml, respectively. Synchronization experiments were performed at 25°C, and unless otherwise stated, galactose was added half an hour before release from α-factor.
Table I. Table of strains.
| Name | Relevant genotype |
|---|---|
| ySP51 | MATa, cdc15-2 |
| ySP284 | MATa, cdc14-3 |
| ySP1070 | MATa, mad2::TRP1 |
| ySP1071 | MATa, bub2::HIS3 |
| ySP1086 | MATa, mad2::TRP1, bub2::HIS3 |
| ySP1243 | MATa, bfa1::KlTRP1 |
| ySP1969 | MATa, pds1::PDS1-myc18::LEU2 |
| ySP2457 | MATa, cdc20::MET3::HA3-CDC20::TRP1, his3::HIS3::tetR-GFP, ura3::URA3::336XtetO |
| ySP2622 | MATa, ura3::1X URA3::GAL1-CLA4t |
| ySP2623 | MATa, ura3::2X URA3::GAL1-CLA4t |
| ySP2624 | MATa, ura3::3X URA3::GAL1-CLA4t |
| ySP2625 | MATa, ura3::4X URA3::GAL1-CLA4t |
| ySP2626 | MATa, bub2::HIS3, ura3::1X URA3::GAL1-CLA4t |
| ySP2630 | MATa, bub2::HIS3, ura3::4X URA3::GAL1-CLA4t |
| ySP2631 | MATa, cdc20::MET3::HA3-CDC20::TRP1, bub2::HIS3, his3::HIS3::tetR-GFP, ura3::URA3::336XtetO |
| ySP2634 | MATa, bub2::HIS3, his3::HIS3::tetR-GFP, ura3::URA3::336XtetO |
| ySP2711 | MATa, swe1::LEU2, ura3::4X URA3::GAL1-CLA4t |
| ySP2726 | MATa, bub2::HIS3, swe1::LEU2, ura3::1X URA3::GAL1-CLA4t |
| ySP2728 | MATa, bub2::HIS3, swe1::LEU2, ura3::4X URA3::GAL1-CLA4t |
| ySP2752 | MATa, mad2::TRP1, ura3::1X URA3::GAL1-CLA4t |
| ySP3022 | MATa, cdc15-2, ura3::1X URA3::GAL1-CLA4t |
| ySP3023 | MATa, cdc15-2, swe1::LEU2, ura3::1X URA3::GAL1-CLA4t |
| ySP3028 | MATa, cdc14-3, ura3::1X URA3::GAL1-CLA4t |
| ySP3047 | MATa, cdc14-3, swe1::LEU2, ura3::1X URA3::GAL1-CLA4t |
| ySP3068 | MATa, mad2::TRP1, bub2::HIS3, ura3::1X URA3::GAL1-CLA4t |
| ySP3073 | MATa, ura3::1X URA3::GAL1-CLA4t, pds1::PDS1-myc18::LEU2 |
| ySP3074 | MATa, cdc28::CDC28 Y19F , ura3::1X URA3::GAL1-CLA4t, pds1::PDS1-myc18::LEU2 |
| ySP3076 | MATa, cla4::kanMX4 |
| ySP3078 | MATa, ste20::kanMX4 |
| ySP3086 | MATa, cla4::CLA4-myc18::KlTRP1 |
| ySP3088 | MATa, cla4::CLA4-myc18::KlTRP1, ura3::4X URA3::GAL1-CLA4t |
| ySP3091 | MATa, ste20::STE20-myc18::KlTRP1, ura3::4X URA3::GAL1-CLA4t |
| ySP3098 | MATa, swe1::LEU2, ura3::1X URA3::GAL1-CLA4t, pds1::PDS1-myc18::LEU2 |
| ySP3138 | MATa, bub2::HIS3 |
| ySP3145 | MATa, mad2::TRP1, swe1::LEU2, ura3::1X URA3::GAL1-CLA4t |
| ySP3148 | MATa, mad2::TRP1, cdc28::CDC28 Y19F , ura3::1X URA3::GAL1-CLA4t |
| ySP3157 | MATa, hsl1::HSL1-myc18::KlTRP1 |
| ySP3186 | MATa, bub2::HIS3, cla4::kanMX4 |
| ySP3198 | MATa, bub2::HIS3, ste20::kanMX4 |
| ySP3202 | MATa, ura3::4X URA3::GAL1-CLA4t, hsl1::HSL1-myc18::KlTRP1 |
| ySP3220 | MATa, ura3::1X URA3::GAL1-CLA4t, kanMX6::GAL1-GFP-LTE1 |
| ySP3221 | MATa, ura3::4X URA3::GAL1-CLA4t, kanMX6::GAL1-GFP-LTE1 |
| ySP3222 | MATa, kanMX6::GAL1-GFP-LTE1 |
Screen for high copy number suppressors of bub2Δ
A leu2 bub2Δ mutant (ySP1071) was transformed with a genomic library constructed in the LEU2 YEp13 2μ plasmid (from K. Nasmyth, Institute of Molecular Pathology, Vienna, Austria). 26,000 Leu+ transformants were replica-plated twice on YEPD plates containing benomyl 20 μg/ml. Plasmids were recovered from benomyl-resistant clones and used to transform again the original bub2Δ strain. Sequencing of both junctions of the inserts of 48 plasmids that confirmed suppression allowed to group them into 23 different classes. We then transformed with one plasmids for each class the tub2–405 mutant (Huffaker et al., 1988), whose benomyl sensitivity is due to defective β-tubulin. Plasmids increasing the benomyl resistance of tub2–405 cells were discarded, thus leaving eight plasmids specifically rescuing the benomyl sensitivity of bub2Δ cells.
Plasmid constructions and genetic manipulations
Standard techniques were used for genetic manipulations (Sherman, 1991; Maniatis et al., 1992). Wild-type and truncated CLA4 were amplified by PCR from genomic DNA and subcloned in the BamHI site of Yep13, to generate pSP150 and pSP164, respectively. To clone CLA4t under the GAL1–10 promoter (plasmid pSP176) a BglII PCR product containing the CLA4t coding region was cloned in the BamHI site of a GAL1–10-bearing YIplac211 vector. pSP176 integration was directed to the URA3 locus by ApaI digestion. Copy number of the integrated plasmid was verified by Southern analysis. CLA4 and STE20 chromosomal deletions were generated by one-step gene replacement (Wach et al., 1994). CLA4, STE20, HSL1, and BFA1 were tagged immediately before the stop codon by one-step gene tagging (Knop et al., 1999). The GAL-GFP-LTE1 construct (Pereira et al., 2002) is a gift from E. Schiebel (The Paterson Institute for Cancer Research, Manchester, UK). The cdc42 V36A , swe1::LEU2, and tub2-405 mutants were provided by D. Lew (Duke University Medical Center, Durham, NC), M.L. Agostoni Carbone (Dipartimento di Genetica, Milano, Italy), and T. Huffaker (Cornell University, Ithaca, NY), respectively.
Immunoprecipitations, kinase assays, and Western blot analysis
Cla4-myc18 was immunoprecipitated from 1 mg of total extract by protein A-sepharose beads cross-linked to anti-myc antibodies, and kinase assays were performed as described previously (Benton et al., 1997). For Western blot analysis, protein extracts were prepared according to Surana et al. (1993). Proteins transferred to Protran® membranes (Schleicher and Schuell) were probed with 9E10 mAb for myc-tagged Pds1, and with pAbs against Clb2, Sic1, and Swi6. Anti-Clb2 and anti-Sic1 antibodies were provided by W. Zachariae (Max Planck Institute of Molecular Cell Biology, Dresden, Germany) and M. Tyers (Department of Molecular and Medical Genetics, Toronto, Canada), respectively. Secondary antibodies were purchased from Amersham Biosciences, and proteins were detected by an ECL system according to the manufacturer.
Other techniques
Flow cytometric DNA quantitation was determined according to Fraschini et al. (1999) on a Becton Dickinson FACScan™. Due to a dramatic cytokinetic defect caused by CLA4 deletion in our genetic background, it was hard to distinguish on the FACS® histograms the fraction of cla4Δ cells that displayed DNA contents higher than 2C due to the ability to re-replicate in the presence of nocodazole from the fraction of multinucleate cells resulting from a cytokinesis failure. Therefore, the values plotted in Fig. 3 G were obtained by subtracting at each time point the percentage of microscopically scored cell clusters from the percentage of cells with DNA contents higher than 2C measured with CELLQuest™ software. Nuclear division was scored with a fluorescent microscope on cells stained with propidium iodide. In situ immunofluorescence was performed according to Fraschini et al. (1999). Immunostaining of α-tubulin was performed with the YOL34 mAb (Serotec) followed by indirect immunofluorescence using rhodamine-conjugated anti–rat Ab (1:100; Pierce Chemical Co.). Immunostaining of Cla4myc18, Ste20myc18, and Hsl1myc18 was detected by incubation with the 9E10 mAb followed by indirect immunofluorescence using CY3-conjugated goat anti–mouse Ab (1:500; Amersham Biosciences). Detection of Cdc3-GFP and Lte1-GFP was performed on ethanol-fixed cells, on wash with water and sonication. Cells were similarly fixed to stain actin with 20 U/ml rhodamine-phalloidin (Sigma-Aldrich). Digital images were taken with a CCD camera and software (CoolSNAP; Photometrics).
Online supplemental material
Supplemental figures correspond to Figs. S1 and S2. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200209097/DC1.
Acknowledgments
We are grateful to E. Schiebel for useful discussions and for communicating results before publication. Thanks to M.L. Agostoni Carbone, T. Huffaker, D. Lew, K. Nasmyth, E. Schiebel, M. Tyers, and W. Zachariae for providing strains, plasmids and reagents; to M.P. Longhese, A. Musacchio, and U. Surana for critical reading of the manuscript.
This work has been supported by grants from Associazione Italiana Ricerca sul Cancro and Centro Nazionale delle Richerche Agenzia 2000 to S. Piatti, and Cofinanziamento 2000 Ministero dell'Università delle Ricerca Scientifica e Tecnologica-Università di Milano-Bicocca and Consorzio Interuniversitario Biotecnologie to G. Lucchini.
E. Chiroli and R. Fraschini contributed equally to this paper.
The online version of this article includes supplemental material.
Footnotes
Abbreviations used in this paper: APC, anaphase-promoting complex; GAP, GTPase-activating protein; MEN, mitotic exit network; PAK, p21-activated kinase.
References
- Adames, N.R., J.R. Oberle, and J.A. Cooper. 2001. The surveillance mechanism of the spindle position checkpoint in yeast. J. Cell Biol. 153:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, A.E., D.I. Johnson, R.M. Longnecker, B.F. Sloat, and J.R. Pringle. 1990. CDC42 and CDC43, two additional genes involved in budding and the establishment of cell polarity in the yeast Saccharomyces cerevisiae. J. Cell Biol. 111:131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandru, G., W. Zachariae, A. Schleiffer, and K. Nasmyth. 1999. Sister chromatid separation and chromosome re-duplication are regulated by different mechanisms in response to spindle damage. EMBO J. 18:2707–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin, A.J., and A. Amon. 2001. Men and sin: what's the difference? Nat. Rev. Mol. Cell Biol. 2:815–826. [DOI] [PubMed] [Google Scholar]
- Bardin, A.J., R. Visintin, and A. Amon. 2000. A mechanism for coupling exit from mitosis to partitioning of the nucleus. Cell. 102:21–31. [DOI] [PubMed] [Google Scholar]
- Baumer, M., G.H. Braus, and S. Irniger. 2000. Two different modes of cyclin clb2 proteolysis during mitosis in Saccharomyces cerevisiae. FEBS Lett. 468:142–148. [DOI] [PubMed] [Google Scholar]
- Benton, B.K., A. Tinkelenberg, I. Gonzalez, and F.R. Cross. 1997. Cla4p, a Saccharomyces cerevisiae Cdc42p-activated kinase involved in cytokinesis, is activated at mitosis. Mol. Cell. Biol. 17:5067–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, E., P. Maddox, D.J. Lew, E.D. Salmon, J.N. McMillan, E. Yeh, and J.R. Pringle. 1998. Involvement of an actomyosin contractile ring in Saccharomyces cerevisiae cytokinesis. J. Cell Biol. 142:1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher, R.N., R.J. Deshaies, and M.W. Kirschner. 1993. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 12:3417–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J.L., M. Jaquenoud, M.P. Gulli, J. Chant, and M. Peter. 1997. Novel Cdc42-binding proteins Gic1 and Gic2 control cell polarity in yeast. Genes Dev. 11:2972–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, G.C., Y.J. Kim, and C.S. Chan. 1997. The Cdc42 GTPase-associated proteins Gic1 and Gic2 are required for polarized cell growth in Saccharomyces cerevisiae. Genes Dev. 11:2958–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Fix, O., and D. Koshland. 1999. Pds1p of budding yeast has dual roles: inhibition of anaphase initiation and regulation of mitotic exit. Genes Dev. 13:1950–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, J.A., and D.P. Kiehart. 1996. Septins may form a ubiquitous family of cytoskeletal filaments. J. Cell Biol. 134:1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvrckova, F., C. De Virgilio, E. Manser, J.R. Pringle, and K. Nasmyth. 1995. Ste20-like protein kinases are required for normal localization of cell growth and for cytokinesis in budding yeast. Genes Dev. 9:1817–1830. [DOI] [PubMed] [Google Scholar]
- Fankhauser, C., and V. Simanis. 1993. The Schizosaccharomyces pombe cdc14 gene is required for septum formation and can also inhibit nuclear division. Mol. Biol. Cell. 4:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesquet, D., P.J. Fitzpatrick, A.L. Johnson, K.M. Kramer, J.H. Toyn, and L.H. Johnston. 1999. A Bub2p-dependent spindle checkpoint pathway regulates the Dbf2p kinase in budding yeast. EMBO J. 18:2424–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field, C., R. Li, and K. Oegema. 1999. Cytokinesis in eukaryotes: a mechanistic comparison. Curr. Opin. Cell Biol. 11:68–80. [DOI] [PubMed] [Google Scholar]
- Fitch, I., C. Dahmann, U. Surana, A. Amon, K. Nasmyth, L. Goetsch, B. Byers, and B. Futcher. 1992. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell. 3:805–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraschini, R., E. Formenti, G. Lucchini, and S. Piatti. 1999. Budding yeast Bub2 is localized at spindle pole bodies and activates the mitotic checkpoint via a different pathway from Mad2. J. Cell Biol. 145:979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraschini, R., A. Beretta, L. Sironi, A. Musacchio, G. Lucchini, and S. Piatti. 2001. Bub3 interaction with Mad2, Mad3 and Cdc20 is mediated by WD40 repeats and does not require intact kinetochores. EMBO J. 20:6648–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geymonat, M., S. Jensen, and L. Johnston. 2002. Mitotic exit: the Cdc14 double cross. Curr. Biol. 12:R482–R484. [DOI] [PubMed] [Google Scholar]
- Gladfelter, A.S., J.J. Moskow, T.R. Zyla, and D.J. Lew. 2001. Isolation and characterization of effector-loop mutants of CDC42 in yeast. Mol. Biol. Cell. 12:1239–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbsky, G.J. 2001. The mitotic spindle checkpoint. Curr. Biol. 11:R1001–R1004. [DOI] [PubMed] [Google Scholar]
- Guertin, D.A., L. Chang, F. Irshad, K.L. Gould, and D. McCollum. 2000. The role of the sid1p kinase and cdc14p in regulating the onset of cytokinesis in fission yeast. EMBO J. 19:1803–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell, L.H., and T.A. Weinert. 1989. Checkpoints: controls that ensure the order of cell cycle events. Science. 246:629–634. [DOI] [PubMed] [Google Scholar]
- Hofken, T., and E. Schiebel. 2002. A role for cell polarity proteins in mitotic exit. EMBO J. 21:4851–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holly, S.P., and K.J. Blumer. 1999. PAK-family kinases regulate cell and actin polarization throughout the cell cycle of Saccharomyces cerevisiae. J. Cell Biol. 147:845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt, M.A. 2000. Exit from mitosis: spindle pole power. Cell. 102:267–270. [DOI] [PubMed] [Google Scholar]
- Hoyt, M.A. 2001. A new view of the spindle checkpoint. J. Cell Biol. 154:909–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, F., Y. Wang, D. Liu, Y. Li, J. Qin, and S.J. Elledge. 2001. Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell. 107:655–665. [DOI] [PubMed] [Google Scholar]
- Huffaker, T.C., J.H. Thomas, and D. Botstein. 1988. Diverse effects of beta-tubulin mutations on microtubule formation and function. J. Cell Biol. 106:1997–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, L.H., L.F. Lau, D.L. Smith, C.A. Mistrot, K.G. Hardwick, E.S. Hwang, A. Amon, and A.W. Murray. 1998. Budding yeast Cdc20: a target of the spindle checkpoint. Science. 279:1041–1044. [DOI] [PubMed] [Google Scholar]
- Jaspersen, S.L., and D.O. Morgan. 2000. Cdc14 activates Cdc15 to promote mitotic exit in budding yeast. Curr. Biol. 10:615–618. [DOI] [PubMed] [Google Scholar]
- Johnson, D.I. 1999. Cdc42: An essential Rho-type GTPase controlling eukaryotic cell polarity. Microbiol. Mol. Biol. Rev. 63:54–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop, M., K. Siegers, G. Pereira, W. Zachariae, B. Winsor, K. Nasmyth, and E. Schiebel. 1999. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 15:963–972. [DOI] [PubMed] [Google Scholar]
- Lew, D.J. 2000. Cell-cycle checkpoints that ensure coordination between nuclear and cytoplasmic events in Saccharomyces cerevisiae. Curr. Opin. Genet. Dev. 10:47–53. [DOI] [PubMed] [Google Scholar]
- Lew, D.J., and S.I. Reed. 1995. A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J. Cell Biol. 129:739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R. 1999. Bifurcation of the mitotic checkpoint pathway in budding yeast. Proc. Natl. Acad. Sci. USA. 96:4989–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, H.H., P.Y. Goh, and U. Surana. 1996. Spindle pole body separation in Saccharomyces cerevisiae requires dephosphorylation of the tyrosine 19 residue of Cdc28. Mol. Cell. Biol. 16:6385–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, H.H., P.Y. Goh, and U. Surana. 1998. Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr. Biol. 8:231–234. [DOI] [PubMed] [Google Scholar]
- Liu, J., H. Wang, and M.K. Balasubramanian. 2000. A checkpoint that monitors cytokinesis in Schizosaccharomyces pombe. J. Cell Sci. 113:1223–1230. [DOI] [PubMed] [Google Scholar]
- Longtine, M.S., D.J. DeMarini, M.L. Valencik, O.S. Al-Awar, H. Fares, C. De Virgilio, and J.R. Pringle. 1996. The septins: roles in cytokinesis and other processes. Curr. Opin. Cell Biol. 8:106–119. [DOI] [PubMed] [Google Scholar]
- Longtine, M.S., C.L. Theesfeld, J.N. McMillan, E. Weaver, J.R. Pringle, and D.J. Lew. 2000. Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae. Mol. Cell. Biol. 20:4049–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis, T., E.F. Fritsch, and J. Sambrook. 1992. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- McCollum, D., and K.L. Gould. 2001. Timing is everything: regulation of mitotic exit and cytokinesis by the MEN and SIN. Trends Cell Biol. 11:89–95. [DOI] [PubMed] [Google Scholar]
- McMillan, J.N., M.S. Longtine, R.A. Sia, C.L. Theesfeld, E.S. Bardes, J.R. Pringle, and D.J. Lew. 1999. The morphogenesis checkpoint in Saccharomyces cerevisiae: cell cycle control of Swe1p degradation by Hsl1p and Hsl7p. Mol. Cell. Biol. 19:6929–6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, D.A., and G.F. Sprague, Jr. 2001. The phosphotyrosyl phosphatase activator, Ncs1p (Rrd1p), functions with Cla4p to regulate the G(2)/M transition in Saccharomyces cerevisiae. Mol. Cell. Biol. 21:488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio, A., and K.G. Hardwick. 2002. The spindle checkpoint: structural insights into dynamic signalling. Nat. Rev. Mol. Cell Biol. 3:731–741. [DOI] [PubMed] [Google Scholar]
- Parrini, M.C., M. Lei, S.C. Harrison, and B.J. Mayer. 2002. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol. Cell. 9:73–83. [DOI] [PubMed] [Google Scholar]
- Pereira, G., T. Hofken, J. Grindlay, C. Manson, and E. Schiebel. 2000. The Bub2p spindle checkpoint links nuclear migration with mitotic exit. Mol. Cell. 6:1–10. [PubMed] [Google Scholar]
- Pereira, G., C. Manson, J. Grindlay, and E. Schiebel. 2002. Regulation of the Bfa1p-Bub2p complex at spindle pole bodies by the cell cycle phosphatase Cdc14p. J. Cell Biol. 157:367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, J.M. 2002. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell. 9:931–943. [DOI] [PubMed] [Google Scholar]
- Rudner, A.D., and A.W. Murray. 2000. Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol. 149:1377–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner, A.D., K.G. Hardwick, and A.W. Murray. 2000. Cdc28 activates exit from mitosis in budding yeast. J. Cell Biol. 149:1361–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshan, A., A.J. Bardin, and A. Amon. 2002. Control of lte1 localization by cell polarity determinants and Cdc14. Curr. Biol. 12:2098–2110. [DOI] [PubMed] [Google Scholar]
- Sherman, F. 1991. Getting started with yeast. Methods Enzymol. 194:3–21. [DOI] [PubMed] [Google Scholar]
- Shirayama, M., Y. Matsui, K. Tanaka, and A. Toh-e. 1994. Isolation of a CDC25 family gene, MSI2/LTE1, as a multicopy suppressor of ira1. Yeast. 10:451–461. [DOI] [PubMed] [Google Scholar]
- Shirayama, M., A. Toth, M. Galova, and K. Nasmyth. 1999. APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature. 402:203–207 (see comments). [DOI] [PubMed] [Google Scholar]
- Stegmeier, F., R. Visintin, and A. Amon. 2002. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell. 108:207–220. [DOI] [PubMed] [Google Scholar]
- Surana, U., A. Amon, C. Dowzer, J. McGrew, B. Byers, and K. Nasmyth. 1993. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J. 12:1969–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surana, U., H. Robitsch, C. Price, T. Schuster, I. Fitch, A.B. Futcher, and K. Nasmyth. 1991. The role of CDC28 and cyclins during mitosis in the budding yeast S. cerevisiae. Cell. 65:145–161. [DOI] [PubMed] [Google Scholar]
- Surana, U., F.M. Yeong, and H.H. Lim. 2002. MEN, destruction and separation: mechanistic links between mitotic exit and cytokinesis in budding yeast. Bioessays. 24:659–666. [DOI] [PubMed] [Google Scholar]
- Tinker-Kulberg, R.L., and D.O. Morgan. 1999. Pds1 and Esp1 control both anaphase and mitotic exit in normal cells and after DNA damage. Genes Dev. 13:1936–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallen, E.A., J. Caviston, and E. Bi. 2000. Roles of Hof1p, Bni1p, Bnr1p, and myo1p in cytokinesis in Saccharomyces cerevisiae. Mol. Biol. Cell. 11:593–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin, R., S. Prinz, and A. Amon. 1997. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 278:460–463. [DOI] [PubMed] [Google Scholar]
- Visintin, R., K. Craig, E.S. Hwang, S. Prinz, M. Tyers, and A. Amon. 1998. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk- dependent phosphorylation. Mol. Cell. 2:709–718. [DOI] [PubMed] [Google Scholar]
- Wach, A., A. Brachat, R. Pohlmann, and P. Philippsen. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 10:1793–1808. [DOI] [PubMed] [Google Scholar]
- Wasch, R., and F.R. Cross. 2002. APC-dependent proteolysis of the mitotic cyclin Clb2 is essential for mitotic exit. Nature. 418:556–562. [DOI] [PubMed] [Google Scholar]
- Wassmann, K., and R. Benezra. 2001. Mitotic checkpoints: from yeast to cancer. Curr. Opin. Genet. Dev. 11:83–90. [DOI] [PubMed] [Google Scholar]
- Weinert, T.A., G.L. Kiser, and L.H. Hartwell. 1994. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 8:652–665. [DOI] [PubMed] [Google Scholar]
- Weiss, E.L., A.C. Bishop, K.M. Shokat, and D.G. Drubin. 2000. Chemical genetic analysis of the budding-yeast p21-activated kinase Cla4p. Nat. Cell Biol. 2:677–685. [DOI] [PubMed] [Google Scholar]
- Yeong, F.M., H.H. Lim, C.G. Padmashree, and U. Surana. 2000. Exit from mitosis in budding yeast: biphasic inactivation of the Cdc28- Clb2 mitotic kinase and the role of Cdc20. Mol. Cell. 5:501–511. [DOI] [PubMed] [Google Scholar]
- Yoshida, S., K. Asakawa, and A. Toh-e. 2002. Mitotic exit network controls the localization of Cdc14 to the spindle pole body in Saccharomyces cerevisiae. Curr Biol. 12:944–950. [DOI] [PubMed] [Google Scholar]
- Zachariae, W., and K. Nasmyth. 1999. Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev. 13:2039–2058. [DOI] [PubMed] [Google Scholar]