

Abstract
Stress granules (SGs) are formed in the cytoplasm in response to various toxic agents, and are believed to play a critical role in the regulation of mRNA metabolism during stress. In SGs, mRNAs are stored in an abortive translation initiation complex that can be routed to either translation initiation or degradation. Here, we show that G3BP, a phosphorylation-dependent endoribonuclease that interacts with RasGAP, is recruited to SGs in cells exposed to arsenite. G3BP may thus determine the fate of mRNAs during cellular stress. Remarkably, SG assembly can be either dominantly induced by G3BP overexpression, or on the contrary, inhibited by expressing a central domain of G3BP. This region binds RasGAP and contains serine 149, whose dephosphorylation is induced by arsenite treatment. Critically, a phosphomimetic mutant (S149E) fails to oligomerize and to assemble SGs, whereas a nonphosphorylatable G3BP mutant (S149A) does both. These results suggest that G3BP is an effector of SG assembly, and that Ras signaling contributes to this process by regulating G3BP dephosphorylation.
Keywords: mRNA stability; protein phosphorylation; mammalian endoribonucleases; Ras signaling; RNA-binding protein
Introduction
Cells have evolved protective responses from stress. Different stresses, such as elevated temperature, oxidative conditions, or exposure to UV light trigger a similar response. This response is characterized by the induction of the heat-shock proteins, stress-induced transcription factors, and formation of stress granules (SGs;* Theodorakis and Morimoto, 1987; Petersen and Lindquist, 1988; Nover et al., 1983, 1989; Morimoto, 1998; Kedersha et al., 1999, 2000). In both plant and animal cells, SGs correspond to large, dynamic cytoplasmic structures at which untranslated mRNAs accumulate (Nover et al., 1989; Kedersha et al., 1999, 2002). There are few granules per cell, and they can easily be observed by light microscopy. In mammalian cells, assembly of SGs can be triggered by the phosphorylation of the translation initiation factor eIF2α (Kedersha et al., 1999), which effectively prevents formation of the eIF2–GTP–Met-tRNAi complex. This inhibits protein synthesis and leads to the accumulation of a 48S preinitiation complex (Kedersha et al., 1999, 2002). Consistently, SGs contain many components of this complex, including eukaryotic initiation factors eIF3, eIF4E, eIF4G, and small (but not large) ribosomal subunits, but lack eIF2 (Kedersha et al., 2002).
Assembly of SGs in response to the phosphorylation of eIF2α likely involves effector proteins, such as the related RNA-binding proteins TIA-1 and TIAR (Kedersha et al., 1999). These proteins possess three RNA recognition motifs (RRMs) at the NH2 terminus and a glutamine-rich, prion-related domain at the COOH terminus (Tian et al., 1991). The prion-related domain is responsible for self-aggregation of the protein, and therefore is thought to modulate SG assembly. Other RNA-binding proteins associated with SGs include the poly(A)+ binding protein I (PABP-I) and the mRNA stabilizing protein HuR (Kedersha et al., 2002). These and other major components of SGs (eIF3, eIF4E, and eIF4G) assemble and disassemble from SGs with similar kinetics, suggesting that they are coordinately recruited as a complex (Kedersha et al., 2000). Given the dynamic shuttling of SG-associated proteins, it is postulated that SGs may be sites of mRNA sorting at which the structure and composition of individual mRNPs could determine whether mRNAs are repacked into translationally-competent mRNPs or degraded (Kedersha et al., 2000, 2002).
Information regarding components of SGs involved in mRNA degradation is lacking. Therefore, we analyzed the intracellular localization of the endoribonuclease G3BP under stress conditions. G3BP is an evolutionarily conserved RNA-binding protein that was initially characterized through its interaction with a Ras-GTPase–activating protein (RasGAP p120; Parker et al., 1996). Importantly, the RNase activity of G3BP is positively regulated by phosphorylation at several sites, one of which, serine 149 (Ser 149) is dependent on RasGAP and negatively regulated by Ras signaling (Parker et al., 1996; Gallouzi et al., 1998; Tourrière et al., 2001; for review see Tourrière et al., 2002).
Results and discussion
P21ras influences G3BP recruitment to SGs
First, we analyzed the intracellular localization of endogenous G3BP by performing immunofluorescence with a specific antibody in Cos cells. As previously observed in untreated cells (Parker et al., 1996; Gallouzi et al., 1998), the protein is diffusely distributed throughout the cytoplasm (Fig. 1 A). However, when cells are stressed with either arsenite or high temperature, G3BP becomes localized in large cytoplasmic structures that resemble SGs (Fig. 1 A; unpublished data). One of the markers of SGs is poly(A)+ RNA, and double labeling of G3BP and oligo-dT probe demonstrated that the sites of G3BP enrichment are indeed SGs (Fig. 1 A). G3BP is an evolutionarily conserved protein, and homologues have been found in Drosophila, Caenorhabditis, and yeast (Pazman et al., 2000). To test whether the recruitment of G3BP to SGs is conserved across evolution, we fused the Drosophila protein to GFP and expressed it in Cos cells. Similar to human G3BP, Drosophila G3BP was efficiently recruited to SGs after arsenite treatment (Fig. 1 A and Fig. 2 A).
Figure 1.
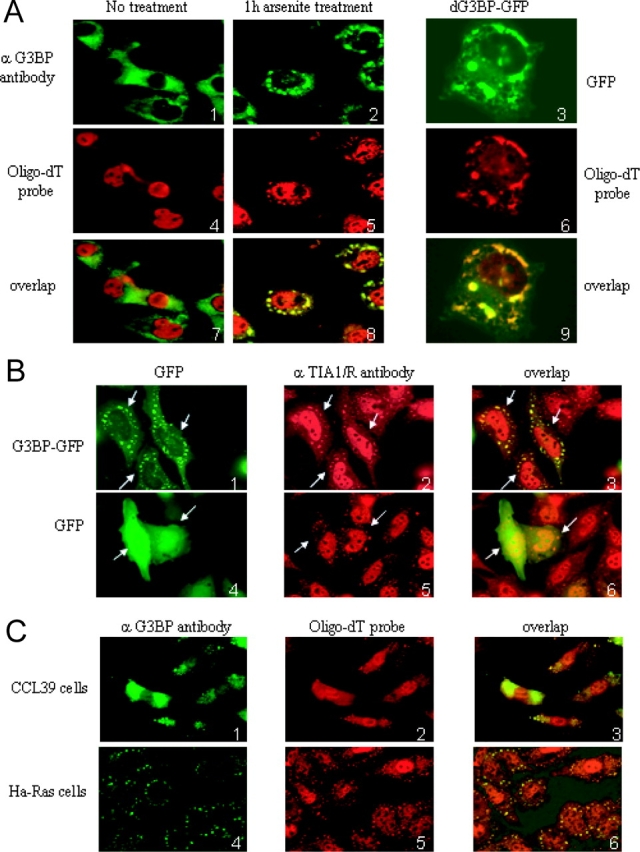
G3BP is recruited to SGs. (A) Fixed Cos cells were stained with an anti-G3BP antibody (1 and 2) or transfected with Drosophila G3BP-GFP (dG3BP-GFP, 3) fusion (green) and a fluorescent oligo-dT probe to reveal SGs (red). (B) Intracellular localization of G3BP-GFP fusion (green) in transfected HeLa cells treated with 0.5 mM arsenite for 1 h, which were fixed and stained with anti-TIA1/R antibody (red). Arrows indicate transfected cells expressing G3BP-GFP fusion. (C) Fixed CCL39 and Ha-Ras cells were treated with 1 mM arsenite for 20 min and stained with an anti-G3BP antibody (green) and a fluorescent oligo-dT probe to reveal SGs (red).
Figure 2.
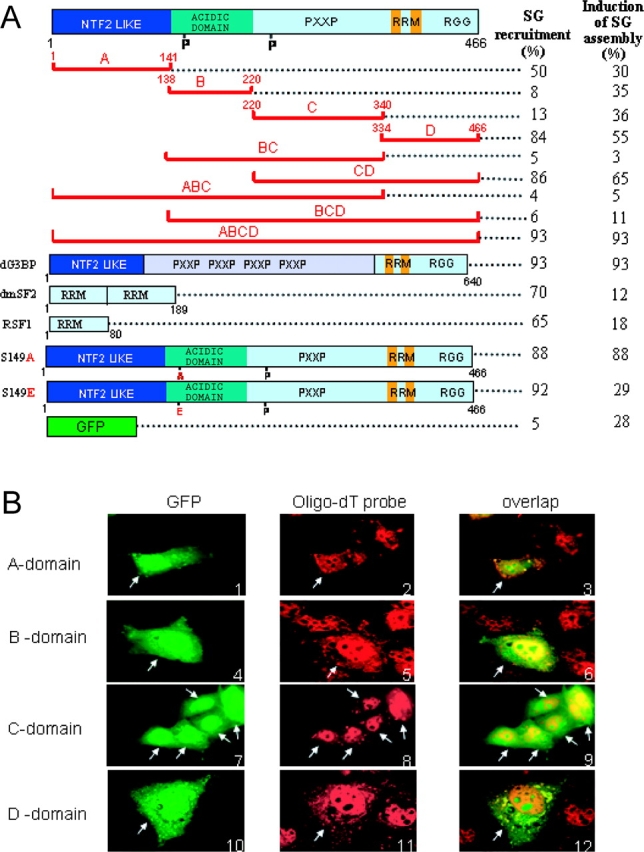
G3BP domain A and D can direct GFP fusion proteins to arsenite-induced SGs. (A) Efficiency of recruitment to SGs. Schematic representation of the GFP fusion proteins. G3BP domain A, B, C, and D, and phosphorylation mutants S149A and S149E (see text); G3BP from Drosophila (dG3BP); RRM of dmSF2 and RSF1. Numbers refer to the first and to the last residue of each region. Transfected Cos cells were scored (100 transfected cells averaged from two experiments) for the ability of each GFP fusion to either be recruited to SGs after arsenite treatment (left column), or dominantly induce SGs assembly without treatment (right column). In each case, hybridization with oligo-dT probe was included to positively identify SGs. (B) Intracellular localization of G3BP domains fused to GFP (green). SGs were visualized with fluorescent oligo-dT probes (red). Arrows indicate transfected cells expressing GFP fusions.
Previously, it has been shown that arsenite causes most of cytoplasmic TIA-1 and TIAR (TIA1/R) RNA-binding proteins to accumulate at SGs in human treated cells (Kedersha et al., 1999). Therefore, it was essential to determine whether G3BP and TIA1/R colocalized to the same SGs. To test this possibility, wild-type G3BP was fused to GFP and transfected into HeLa cells, while endogenous TIA1/R protein was detected with specific antibodies. Overlays of images obtained from transfected cells treated with arsenite show that GFP-G3BP colocalized with cytoplasmic (but not nuclear) TIA1/R, and all of the observed SGs contained both proteins (Fig. 1 B).
G3BP associates with RasGAP, and its RNase activity appears to be negatively regulated by p21ras (Gallouzi et al., 1998). Therefore, it was important to know whether p21ras would play a role in SG assembly and/or recruitment of G3BP to SGs. For this purpose, we determined the rate of recruitment of G3BP into SGs in a pair of cell lines, differing only by the expression of constitutively activated p21ras; the factor-dependent hamster lung fibroblasts CCL39, which are tightly regulated by growth factors; and CCL39 derivatives transformed with Ha-ras (Ras-Val12; Seuwen et al., 1988). Fig. 1 C shows that G3BP SGs assemble more rapidly in transformed CCL39 expressing constitutively active Ha-Ras compared with untransformed CCL39. Although 100% of Ha-Ras cells demonstrated SGs containing G3BP at 20 min of arsenite treatment, <50% of CCL39 cells contained SGs (Fig. 1 C). However, longer exposure to arsenite (1 h) leads to indistinguishable levels of SGs between the two cell lines (unpublished data). The results altogether indicate that G3BP is a stable component of SGs whose recruitment is influenced in a time-dependent fashion by p21ras.
The NTF2-like and the RNA-binding domains of G3BP mediate its recruitment to SGs
G3BP shows a modular organization in four domains, which will be termed ABCD, going from the NH2- to the COOH terminus of the protein (Fig. 2 A). Domain A is an NTF2-like domain, possibly mediating protein–protein interactions (Bullock et al., 1996; Kent et al., 1996); domain B is highly acidic, and contains the serum-dependent phosphorylation site Ser 149 (Gallouzi et al., 1998; Tourrière et al., 2001); domain C can potentially bind the SH3 domain of Ras-GAP; and domain D is the RNA-binding domain, which contains a classical RRM and an arginine glycine-rich box. To understand how G3BP is recruited to SGs, we fused each of its domains to GFP and expressed them in Cos cells in order to analyze their localization after treatment with arsenite. In all cases, double labeling with an oligo-dT probe was included to positively identify SGs. Domains A and D, containing the NTF2-like and RNA-binding domains, respectively, were recruited to SGs, albeit less efficiently for domain A (Fig. 2, A and B). In contrast, domains B and C showed the same behavior as GFP alone (Fig. 2, A and B), and were not recruited to SGs.
SGs contain high concentrations of RNA. Thus, recruitment of domain D to SGs could simply be mediated by its affinity for RNA. To test this hypothesis, we analyzed the localization of other RNA-binding domains. We chose the RRMs of two Drosophila proteins involved in splicing, RSF1, and dmSF2 because these were unlikely to have a functional partner in the cytoplasm of monkey cells. The RRM of RSF1 is normally present in both the cytoplasm and the nucleus, whereas the two RRMs of dmSF2 are nuclear (Allemand et al., 2001, 2002). Despite this, both RNA-binding domains became concentrated in SGs after arsenite treatment (Fig. 2 A), suggesting their ability to bind RNA may be sufficient to concentrate them in SGs. It is striking that even RNA-binding domains that are nuclear can become concentrated in SGs. These data suggest that many RNA-binding proteins will be found within SGs, even those that have a nuclear localization. A transient localization of nuclear protein to the cytoplasm could be important to adjust the nuclear metabolism of RNA to conditions of stress, and has in fact been observed for hnRNP A1 and HuR (Kedersha et al., 2000; van der Houven van Oordt et al., 2000). Alternatively, the RRMs could localize to SGs simply due to the movement of mRNAs to which they are attached.
Domain A is homologous to the Ran binding domain of NTF2 (Bullock et al., 1996; Kent et al., 1996). Thus, it was possible that recruitment of this domain to SGs was mediated through Ran or another small GTPase. To test this hypothesis, we analyzed whether G3BP could coimmunoprecipitate with Ran, RhoA, Rac1, and also eIF2α, which is involved in SG formation. Despite the efficient immunoprecipitation of G3BP, no interactions could be detected with any of these proteins, either before or after addition of arsenite (unpublished data). NTF2-like domains are also known to mediate interaction with themselves (Bullock et al., 1996; Kent et al., 1996). Thus, domain A could be recruited to SGs by dimerizing with the endogenous G3BP. To test this possibility, we transfected wild-type G3BP-GFP or a mutant lacking domain A, immunoprecipitated extracts with an antibody against GFP, and analyzed the bound proteins with an antibody against G3BP. G3BP could interact with itself, as shown by the ability of G3BP-GFP to immunoprecipitate endogenous G3BP. In contrast, G3BP-GFP lacking domain A could not interact with wild-type G3BP, suggesting that it indeed mediates protein multimerization (see Fig. 5 A).
Figure 5.

Phosphorylation of Ser 149 prevents G3BP self-association. (A) Whole-cell extracts prepared from Cos cells transfected with GFP, GFP-G3BP wild type, and phosphorylation mutants GFP-S149A, GFP-S149E, GFP-S232A, and GFP-S232E were immunoprecipitated with anti-GFP antibodies. Proteins were revealed by immunoblot analysis using anti-G3BP antibody. S, supernatant; IP, immunoprecipitation. (B) Glutaraldehyde cross-linking analysis of purified G3BP phosphorylation mutants S149A (lanes 1–6) and S149E (lanes 7–11). Proteins (0.2 μg each) were incubated for the indicated time with glutaraldehyde (G.A.), and were then analyzed by Western blotting with an anti-G3BP. Asterisks correspond to proteolytic fragments of G3BP-GFP that can be detected by anti-GFP antibodies, whereas the band at level of G3BP is only seen with antiG3BP antibody.
The acidic RasGAP binding domain of G3BP harbors an arsenite-regulated phosphorylation site and dominantly inhibits SG formation
Having determined the role of individual G3BP domains in SG recruitment, we went on to analyze the localization of various deletion mutants (Fig. 2 A, numbers in right panels). In cells subjected to arsenite stress, the CD mutant was recruited to SGs (in 86% of transfected cells). In contrast, the BC, ABC, and BCD forms of G3BP were diffusely distributed in the cytoplasm (Fig. 3 A). Most importantly, although poly(A)+ RNA and endogenous G3BP accumulated in SGs of untransfected cells, they remained diffusely distributed in cells transfected with BC, ABC, or BCD mutants (Fig. 3 A). This indicates that the BC domain is able to block the recruitment of poly(A)+ RNA to SGs, thereby acting as a transdominant inhibitor of SG assembly. Suppression of SG assembly by BC-containing mutants did not reflect overexpression of these mutants in transfected cells because SGs were absent from cells expressing low levels of these mutants (Fig. 3 A, 1–3). These results suggested that domain BC interacts with and eventually titrates a critical regulator of SG formation, and raised the possibility that G3BP itself was involved in SG assembly.
Figure 3.
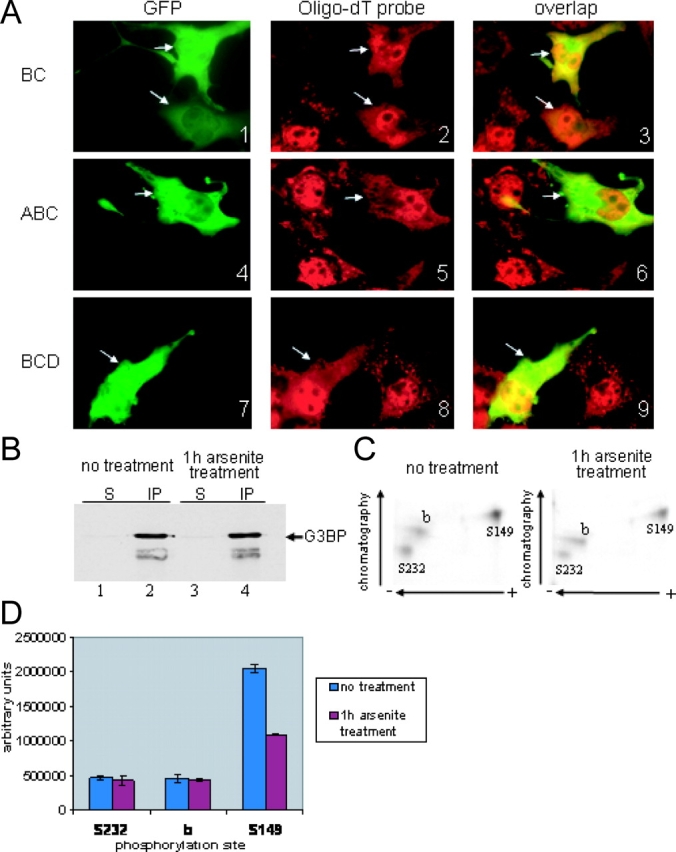
G3BP BC domain inhibits arsenite-induced assembly of SGs. (A) Cos cells were transiently transfected with GFP-BC, GFP-ABC, or GFP-BCD, treated with arsenite before fixation, and double stained with fluorescent oligo-dT probe (red). GFP, green. Arrows indicate transfected cells expressing GFP fusion. (B) Equal amounts of whole-cell extracts prepared from metabolically labeled CCL39 cells were immunoprecipitated with anti-G3BP antibody. Proteins were revealed by immunoblot analysis using the same antibody. S, supernatant; IP, immunoprecipitation. (C) Phosphotryptic peptide mapping of immunopurified 32P-labeled G3BP, shown in A, from untreated (left) and arsenite-treated (right) CCL39 cells. The identification of phosphorylation sites Ser 149 and Ser 232 was previously reported (Tourrière et al., 2001). (D) The intensity of each spot from untreated (blue) and arsenite-treated (red) was quantitated by densitometry scanning of the chromatography plates using ImageQuant™ software version 5.2. Error bars resulting from two independently performed experiments are shown.
Domain C corresponds to the RasGAP-interacting domain, and domain B contains the conserved Ser 149 that is phosphorylated in a RasGAP-dependent manner. Thus, we asked whether phosphorylation of Ser 149 was affected in cells treated with arsenite. For this purpose, proteins were metabolically labeled with [32P]orthophosphate, immunopurified with anti-G3BP antibody, and subjected to phosphotryptic mapping. To achieve efficient metabolic labeling of G3BP, CCL39 cells were used instead of Cos cells. Consistent with previous work (Gallouzi et al., 1998; Tourrière et al., 2001), three labeled phosphopeptides characterized the tryptic pattern of immunopurified G3BP (Fig. 3 C). Two of them correspond to phosphorylation at Ser 149 and Ser 232 (Tourrière et al., 2001). Although the same amount of G3BP was immunopurified from arsenite-treated and untreated cells (Fig. 3 B), phosphorylation at Ser 149 was twofold less in treated cells compared with untreated cells, whereas phosphorylation of Ser 232 was unchanged (Fig. 3 D). These results clearly establish that arsenite treatment induces specific dephosphorylation of G3BP at Ser 149.
Wild-type G3BP (but not a phosphomimetic S149E mutant) dominantly assembles SGs
The dominant-negative effect of G3BP BC domain on SG formation, and the dephosphorylation of Ser 149 after arsenite treatment prompted us to ask whether phosphorylation of Ser 149 affected the recruitment of G3BP to SGs. The GFP was fused in frame to the amino terminus of several phosphorylation mutants of G3BP, and the proteins were transiently expressed in Cos cells. In all cases, arsenite stress resulted in the accumulation of G3BP mutants in SGs, including S149E (Fig. 2 A and Fig. 4 B, panel 10) and S149A (Fig. 2 A).
Figure 4.
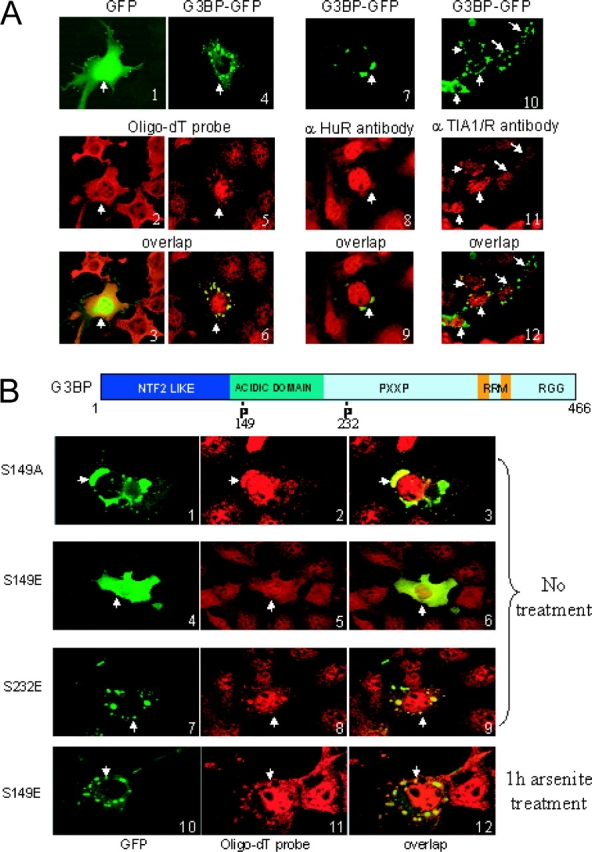
G3BP assembles SGs. (A) Untreated Cos cells transfected with GFP or G3BP-GFP (green) were counterstained with either fluorescent oligo-dT (2 and 5, red) or anti-HuR antibody (8, red). Untreated HeLa cells transfected with G3BP-GFP (10, green) were counterstained with anti-TIA1/R antibody (11, red). (B) GFP-S149E does not assemble SGs. Top, schematic representation of G3BP with the positions of the major phosphorylation sites. Bottom, localization of G3BP phosphorylation mutants in Cos cells. GFP fluorescence (green) of GFP-S149A, GFP-S149E, and GFP-S232 was acquired 20 h after transfection on cells fixed and counterstained with fluorescent oligo-dT to reveal SGs (red). Cells in 10–12 were transfected with GFP-S149E, and were treated with arsenite before fixation. Arrows indicate transfected cells expressing GFP fusions.
During the course of these experiments, we noticed that cells transfected with G3BP-GFP concentrated the protein in aggregates resembling SGs, even in absence of any arsenite treatment (Fig. 4 A, panel 4). Furthermore, G3BP aggregates were enriched in poly(A)+ RNA (Fig. 4 A, 5 and 6) and also contained HuR (Fig. 4 A, 8 and 9), and thus likely corresponded to bona fide SGs. Induction of SG assembly was not due to the transfection itself because GFP alone (Fig. 4 A, 1–3) induced formation of SGs to much lower levels (93% of the transfected cells for G3BP versus 28% for GFP, Fig. 2 A). Furthermore, this effect is specific for G3BP because truncation mutants, or RRMs of unrelated proteins, were unable to assemble SGs, although G3BP domain D had a weak effect (Fig. 2 A). However, not all G3BP-induced SGs colocalized with TIA1/R in HeLa cells, implying that the assembly of a subset of these SGs did not require TIA1/R (Fig. 4 A, 10–12). Remarkably, although all other phosphorylation mutants induced SGs as efficiently as wild-type G3BP (Fig. 2 A and Fig. 4 B, 1–3 and 7–9), the phosphomimetic S149E mutant did not induce SGs at all (Fig. 2 A and Fig. 4 B, 4–6).
Together, the data indicate that overexpression of G3BP can dominantly induce SG assembly, and that this requires dephosphorylation of Ser 149. This data fully support the proposal that G3BP is a regulated effector of SG assembly.
Phosphorylation of G3BP at Ser 149 prevents its oligomerization
Ser 149 is 20 amino acids COOH-terminal to the NTF2-like domain, which plays a key role in mediating protein–protein interactions (see previous paragraph). Thus, we tested the capacity of G3BP to form multimers and determined the effect of phosphorylation on multimerization. Extracts prepared from cells expressing GFP-G3BP or its phosphorylation mutants were subjected to immunoprecipitation with anti-GFP antibody, and G3BP was revealed by Western blot using monoclonal anti-G3BP antibody. Fig. 5 A clearly established that endogeneous G3BP was efficiently coimmunoprecipitated with GFP-G3BP (lane 4), GFP-S149A (lane 6), GFP-S332A (lane 10), and GFP-S232E (lane 12), but not GFP-S149E (lane 8). The lack of association could be due to either preferential binding of S149E with different partners, or to phosphorylation-mediated changes of the protein conformation incompatible with G3BP self-association. To distinguish between these possibilities, recombinant G3BP S149A and S149E phosphorylation mutants were purified from baculovirus-infected insect cells, and subjected to glutaraldehyde cross-linking. Untreated and cross-linked proteins were analyzed by Western blot (Fig. 5 B). Untreated samples revealed only monomeric S149A and S149E proteins (lanes 1 and 7, respectively). In contrast, glutaraldehyde cross-linked samples of S149A contained species of molecular masses corresponding to monomers, dimers, trimers, and tetramers (lanes 2–6), which accumulated with incubation time. However, with the S149E mutant, no homo-oligomers were detected (lanes 8–11). In this case, disappearance of the monomers with time may be due to the inactivation of the epitope as a result of glutaraldehyde treatment. The propensity of the S149A (but not S149E) mutant to form homo-oligomers provides direct evidences that phosphorylation at Ser 149 induces a conformational change that prevents G3BP self-association.
G3BP as a regulated effector of SG assembly
Our data suggest that G3BP is an effector molecule promoting SG assembly, similar to the role previously proposed for TIA-1 (Kedersha et al., 1999). First, endogenous G3BP is very efficiently recruited to SGs after stress. Second, overexpression of G3BP (but not of other RNA-binding proteins) can efficiently trigger assembly of SGs. Third, G3BP BC domain, which contains an acidic region and the RasGAP binding sequences, can dominantly inhibit SG formation.
Phosphorylation of G3BP on Ser 149 appears to be critical for G3BP to assemble SG because a G3BP phosphomimetic mutant of Ser 149 can no longer trigger SGs assembly. This phosphomimetic mutant also fails to undergo multimerization in vivo and in vitro, suggesting that one function of G3BP during SGs assembly is to multimerize and thus cross-link mRNA molecules, similar to the mechanism proposed for TIA-1 (Kedersha et al., 1999). Importantly, we show that endogenous G3BP becomes partially dephosphorylated on Ser 149 during formation of SGs, consistent with the idea that this event participates in the regulation of SG assembly in vivo.
It is remarkable that Ser 149 is precisely the residue that is phosphorylated in a RasGAP-dependent manner, and that it becomes dephosphorylated after activation of Ras (Gallouzi et al., 1998; Tourrière et al., 2001). This raises the possibility that Ras may participate in the control of SG assembly. Indeed, oxidative stress has been shown to activate Ras and several receptor tyrosine kinases like EGF (Rao, 1996), and could thus lead to G3BP dephosphorylation at Ser 149, thereby promoting SG assembly. Consistent with this possibility, we found that SGs assemble more rapidly in fibroblasts transformed with Ha-Ras compared with untransformed fibroblasts (Fig. 1 C). With other types of stress that do not activate Ras, SGs may be assembled by other possibly redundant effectors such as TIA1/R. However, in this case, G3BP monomers containing phosphorylated Ser 149 could still be recruited to SGs through their RRM domain, mimicking the recruitment of the S149E mutant after arsenite treatment. Given that the RRM of G3BP alone induced only weak SG formation (Fig. 2 A), both oligomerization and the RRM domain of G3BP appear be required for efficient recruitment.
In conclusion, this work provides the first evidence that ribonucleases are recruited to SGs. With the addition of G3BP, SGs contain a variety of factors involved at all major levels of mRNA regulation; translation (TIA1/R; Kedersha et al., 1999), stability (HuR; Kedersha et al., 2002), and degradation (G3BP; this work). This fully supports the idea that SGs are the sites where mRNA fates are determined during stress (Kedersha et al., 1999, 2002).
Materials and methods
Plasmids
All DNA manipulations were performed according to standard techniques. G3BP-GFP and its phosphorylation mutants, as well as the RSF1-RRM and dmSF2-RRM GFP fusions, have been described previously (Tourrière et al., 2001; Allemand et al., 2002).
Cell culture, immunoprecipitations, and phosphopeptide analysis
Cos-7 cells were grown in DME containing 10% FCS, and were transfected by calcium phosphate coprecipitation as described previously (Samarsky et al., 1998). HeLa cells were grown in RPMI 1640 (GIBCO BRL) supplemented with 10% FCS. Chinese hamster lung fibroblasts (CCL39) and CCL39 derivative Ha-Ras were provided by J. Pouysségur (Université de Nice, Nice, France) and were maintained in DME supplemented with 10% FCS and antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin) at 37°C in 5% CO2/95% air (Seuwen et al., 1988). For arsenite-induced stress, cells were exposed to 0.5 mM sodium arsenite for 1 h, except as otherwise stated. Total extracts were prepared according to Gallouzi et al. (1998), and G3BP immunoprecipitations were performed with anti-G3BP antibodies (1F1; Gallouzi et al., 1998), or with anti-GFP antibodies (Roche).
Proteins were analyzed by Western blotting using dilutions of primary antibodies as follows: 1/3,000 for anti-G3BP (200 ng/ml), 1/3,000 for anti-HuR (3A2), 1/1000 for anti-Ran (Santa Cruz Biotechnology, Inc.), 1/1,000 for anti-eIf2α (Santa Cruz Biotechnology, Inc.), 1/200 for anti-Rac, and 1/200 for anti-RhoA. Anti-HuR antibody was a gift from Drs. I. Gallouzi and J. Steitz (Yale University, New Haven, CT). Anti-Rac and anti-RhoA antibodies were provided by P. Fort (Centre de Recherches de Biochimie Macromoléculaire, Montpellier, France). 32P-labeling and phosphopeptide mapping were performed according to Gallouzi et al. (1998).
Immunofluorescence and in situ hybridization
Transfected Cos or HeLa cells were grown on coverslips and treated with 0.5 mM sodium arsenite 48 h after transfection. Cells were washed twice with PBS, fixed for 15 min at RT with 4% PFA, and permeabilized overnight in 70% ethanol/PBS. In situ hybridization was done with fluorescent oligo-dT probe synthesized and labeled with Cy3 as described previously (Samarsky et al., 1998). Immunofluorescence was done as described previously (1;2) with the following dilutions of primary antibody: 1/100 for anti-G3BP (1F1), and 1/300 for anti-HuR (3A2). For anti-TIA1/R (3E6) antibody, immunofluorescence was performed as described previously (Kedersha et al., 1999). Anti-TIA1/R antibody was a gift from Drs. N. Kedersha and P. Anderson (Brigham and Women's Hospital Hospital, Boston, MA).
Purification of S149A and S149E recombinant proteins and chemical cross-linking
Full-length cDNAs corresponding to G3BP phosphorylation mutants S149E and S149A were cloned in pBlueBacHis2 B vector (Invitrogen), and recombinant proteins were produced and purified from baculovirus-infected SF21 cells according to Gallouzi et al. (1998). For cross-linking, proteins were dialyzed against 50 mM triethanolamine, pH 7.9, mixed at 37°C with 0.05% of glutaraldehyde in the same buffer, and the reactions were stopped with 4× Laemmli loading buffer.
Acknowledgments
We thank J. Derancourt for phosphopeptide analyses, E. Prieto for her help with the analysis of GFP mutants, and R. Hipskind for his advice and critical reading of the manuscript. A special thanks to I. Gallouzi and J. Steitz for providing anti-HuR antibodies. We are also grateful to N. Kedersha and P. Anderson for providing anti-TIA1/R antibody.
The work was supported by grants from l'Association pour la Recherche sur la Cancer (ARC) and the CNRS. H. Tourrière was supported by a graduate fellowship from the Ministère de L'Éducation Nationale, de la Recherche et de la Technologie and benefited from graduate training fellowships from the ARC and Foundation par le Recherche Médicale.
H. Tourrière and K. Chebli contributed equally to this paper.
Footnotes
Abbreviations used in this paper: RRM, RNA recognition motif; Ser 149, serine 149; SG, stress granule.
References
- Allemand, E., R. Gattoni, J. Stevenin, H.M. Bourbon, J. Caceres, J. Soret, and J. Tazi. 2001. Distinctive features of Drosophila SF2/ASF splicinf factor RS domain: implication for specific phosphorylation, shuttling, and splicing activation. Mol. Cell. Biol. 21:1345–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allemand, E., S. Dokudovskaya, R. Bordonne, and J. Tazi. 2002. A conserved Drosophila transportin-serine/arginine-rich (SR) protein permits nuclear import of Drosophila SR protein splicing factors and their antagonist repressor splicing factor 1. Mol. Biol. Cell. 13:2436–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock, T.L., W.D. Clarkson, H.M. Kent, and M. Stewart. 1996. The 1.6 angstroms resolution crystal structure of nuclear transport factor 2 (NTF2). J. Mol. Biol. 260:422–431. [DOI] [PubMed] [Google Scholar]
- Gallouzi, I.E., F. Parker, K. Chebli, F. Maurier, E. Labourier, I. Barlat, J.P. Capony, B. Tocque, and J. Tazi. 1998. A novel phosphorylation-dependent RNase activity of GAP-SH3 binding protein: a potential link between signal transduction and RNA stability. Mol. Cell. Biol. 18:3956–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha, N.L., M. Gupta, W. Li, I. Miller, and P. Anderson. 1999. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 147:1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha, N., M.R. Cho, W. Li, P.W. Yacono, S. Chen, N. Gilks, D.E. Golan, and P. Anderson. 2000. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 151:1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha, N., S. Chen, N. Gilks, W. Li, I.J. Miller, J. Stahl, and P. Anderson. 2002. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol. Biol. Cell. 13:195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, H.M., W.D. Clarkson, T.L. Bullock, and M. Stewart. 1996. Crystallization and preliminary X-ray diffraction analysis of nuclear transport factor 2. J. Struct. Biol. 116:326–329. [DOI] [PubMed] [Google Scholar]
- Morimoto, R.I. 1998. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 12:3788–3796. [DOI] [PubMed] [Google Scholar]
- Nover, L., K.D. Scharf, and D. Neumann. 1983. Formation of cytoplasmic heat shock granules in tomato cell cultures and leaves. Mol. Cell. Biol. 3:1648–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nover, L., K.D. Scharf, and D. Neumann. 1989. Cytoplasmic heat shock granules are formed from precursor particles and are associated with a specific set of mRNAs. Mol. Cell. Biol. 9:1298–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, F., F. Maurier, I. Delumeau, M. Duchesne, D. Faucher, L. Debussche, A. Dugue, F. Schweighoffer, and B. Tocque. 1996. A Ras-GTPase-activating protein SH3-domain-binding protein. Mol. Cell. Biol. 16:2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazman, C., C.A. Mayes, M. Fanto, S.R. Haynes, and M. Mlodzik. 2000. Rasputin, the Drosophila homologue of the RasGAP SH3 binding protein, functions in ras- and Rho-mediated signaling. Development. 127:1715–1725. [DOI] [PubMed] [Google Scholar]
- Petersen, R., and S. Lindquist. 1988. The Drosophila hsp70 message is rapidly degraded at normal temperatures and stabilized by heat shock. Gene. 72:161–168. [DOI] [PubMed] [Google Scholar]
- Rao, G. 1996. Hydrogen peroxide induces complex formation of SHC-Grb2-SOS with receptor tyrosine kinase and activates Ras and extracellular signal-regulated protein kinases group of mitogen-activated protein kinases. Oncogene. 13:713–719. [PubMed] [Google Scholar]
- Samarsky, DA., M.J. Fournier, R.H. Singer, and E. Bertrand. 1998. The snoRNA box C/D motif directs nucleolar targeting and also couples snoRNA synthesis and localization. EMBO J. 17:3747–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seuwen, K., A. Lagarde, and J. Pouyssegur. 1988. Deregulation of hamster fibroblast proliferation by mutated ras oncogenes is not mediated by constitutive activation of phosphoinositide-specific phospholipase C. EMBO J. 7:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodorakis, N.G., and R.I. Morimoto. 1987. Posttranscriptional regulation of hsp70 expression in human cells: effects of heat shock, inhibition of protein synthesis, and adenovirus infection on translation and mRNA stability. Mol. Cell. Biol. 7:4357–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, Q., M. Streuli, H. Saito, S.F. Schlossman, and P. Anderson. 1991. A polyadenylate binding protein localized to the granules of cytolytic lymphocytes induces DNA fragmentation in target cells. Cell. 67:629–639. [DOI] [PubMed] [Google Scholar]
- Tourrière, H., I.E. Gallouzi, K. Chebli, J.P. Capony, J. Mouaikel, P. van der Geer, and J. Tazi. 2001. RasGAP-associated endoribonuclease G3BP: selective RNA degradation and phosphorylation-dependent localization. Mol. Cell. Biol. 21:7747–7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourrière, H., K. Chebli, and J. Tazi. 2002. mRNA degradation machines in eukaryotic cells. Biochimie. 84:821–837. [DOI] [PubMed] [Google Scholar]
- van der Houven van Oordt, W., M.T. Diaz-Meco, J. Lozano, A.R. Krainer, J. Moscat, and J.F. Caceres. 2000. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J. Cell Biol. 149:307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]