

Abstract
Centromere-associated protein-E (CENP-E) is an essential mitotic kinesin that is required for efficient, stable microtubule capture at kinetochores. It also directly binds to BubR1, a kinetochore-associated kinase implicated in the mitotic checkpoint, the major cell cycle control pathway in which unattached kinetochores prevent anaphase onset. Here, we show that single unattached kinetochores depleted of CENP-E cannot block entry into anaphase, resulting in aneuploidy in 25% of divisions in primary mouse fibroblasts in vitro and in 95% of regenerating hepatocytes in vivo. Without CENP-E, diminished levels of BubR1 are recruited to kinetochores and BubR1 kinase activity remains at basal levels. CENP-E binds to and directly stimulates the kinase activity of purified BubR1 in vitro. Thus, CENP-E is required for enhancing recruitment of its binding partner BubR1 to each unattached kinetochore and for stimulating BubR1 kinase activity, implicating it as an essential amplifier of a basal mitotic checkpoint signal.
Keywords: kinetochore; mitosis; cell cycle; LENP-E; BubR1
Introduction
Successful cellular propagation requires faithful replication and equal segregation of genetic information. Loss or gain of even a single chromosome during meiosis most often results in the production of gametes that are unable to produce viable offspring (for review see Cohen, 2002). Loss or gain of chromosomes during mitotic divisions leads to the production of cells with a DNA content greater or less than 2N, a condition known as aneuploidy, which is a hallmark of cancer cells. Maintenance of ploidy is ensured through action of the mitotic checkpoint, which prevents the transition to anaphase until all chromosomes have made productive, bipolar attachments through their kinetochores to the microtubules of the mitotic spindle.
Six genes have been identified in Saccharomyces cerevisiae as essential for the kinetochore-dependent mitotic checkpoint: MAD1, MAD2 and MAD3 (Li and Murray, 1991), BUB1 and BUB3 (Hoyt et al., 1991), and the Mono Polar Spindles gene MPS1 (Weiss and Winey, 1996). Homologues of each have been identified in higher eukaryotes, although the vertebrate Mad3 homologue, BubR1, contains a kinase domain not found in the budding yeast protein (Cahill et al., 1998; Chan et al., 1998; Taylor et al., 1998; Kaplan et al., 2001). Although initially named a “checkpoint” because of its activation in response to spindle damage in yeast, in mammals it is an essential mechanism that serves to control advance to anaphase during every mitosis. Gene inactivation of Mad2 (Dobles et al., 2000) or Bub3 (Babu et al., 2003) produces lethality in mice and rapid acquisition of aneuploidy in cell culture. Heterozygous mutations in Mad2 (Michel et al., 2001) or Bub3 (Babu et al., 2003) promote tumorigenesis in mice, presumably by enhancing the rate of chromosome missegregation.
The checkpoint signal is generated by unattached kinetochores. Even a single kinetochore is sufficient to prevent progression to anaphase, as demonstrated by classic laser irradiation and micromanipulation experiments (Rieder et al., 1994, 1995; Li and Nicklas, 1995). The signal or signals produced by these kinetochores has not been identified, but it inhibits the Cdc20-activated form of a ubiquitin ligase, the anaphase promoting complex/cyclosome (APC/C), and prevents the ubiquitination of substrates whose destruction is required for advance to anaphase (for review see Cleveland et al., 2003).
Centromere-associated protein-E (CENP-E) is a large (∼300 kD), essential, kinesin-like protein that accumulates in G2, is used throughout mitosis, and is degraded in telophase (Brown et al., 1994). Localized at kinetochores from prometaphase through anaphase A, CENP-E stabilizes microtubule capture by kinetochores (Putkey et al., 2002). This is required for complete chromosome alignment at metaphase in multiple contexts, as inhibition of CENP-E function leads to a failure of metaphase alignment of chromosomes in Xenopus laevis extracts (Wood et al., 1997), Drosophila melanogaster embryos (Yucel et al., 2000), and primary or transformed human and mouse cells grown in culture (Schaar et al., 1997; Yao et al., 2000; McEwen et al., 2001; Putkey et al., 2002). Because CENP-E also coimmunoprecipitates with the checkpoint kinase BubR1 (Chan et al., 1998; Yao et al., 2000) and extends at least 50 nm away from the outer surface of each kinetochore (Yao et al., 1997), it is appropriately positioned to serve as a sensor linking microtubule capture to underlying kinetochore-bound checkpoint components.
Efforts to determine whether absence or inhibition of CENP-E affects kinetochore-dependent mitotic checkpoint signaling have come to sharply divergent conclusions. Xenopus extracts in which all kinetochores are unattached (after nocodazole-induced microtubule depolymerization) no longer recruit Mad1 and Mad2 to their kinetochores and fail to arrest after immunodepletion of CENP-E (Abrieu et al., 2000). In contrast, HeLa cells injected with CENP-E antibodies (Schaar et al., 1997; McEwen et al., 2001) or depleted of CENP-E with antisense oligonucleotides (Yao et al., 2000) arrest in mitosis with many misaligned chromosomes, although these reports conflict about whether or not sister chromatids separate during this arrest.
To extend these earlier efforts, selective gene inactivation in primary cells in vitro and regenerating liver in vivo is now used to test the role of CENP-E in the mitotic checkpoint in normally cycling mammalian cells.
Results
Efficient Cre recombinase–mediated disruption of the single functional murine CENP-E gene in primary mouse fibroblasts
The gene encoding murine CENP-E is located on mouse chromosome 3 (Fig. 1 A), which is syntenic to the previously identified location of human CENP-E on human chromosome 4 (Testa et al., 1994). A full-length murine CENP-E cDNA was identified and sequenced (see Materials and methods). The 7,425-base cDNA encodes a protein of 287 kD (2474 amino acids; Fig. 1 A), including a kinesin-like motor domain, a central domain (predicted to be a 220 nm long coiled coil), three cdc2–cyclin B consensus phosphorylation sites, a second microtubule-binding domain, and a terminal CAAX box that may direct farnesylation (Ashar et al., 2000). Recently, we have reported that CENP-E is essential using gene targeting in mice to produce null or conditional alleles of CENP-E (Putkey et al., 2002). Recognition sequences (lox P sites) for the Cre recombinase were inserted into intronic sequences on either side of an early exon in the CENP-E gene (identified here to be exon 4) to create the conditional allele (lox P). The null allele (Δ) was created by Cre-mediated excision of exon 4, which introduces a premature stop codon at amino acid 82 and truncates all of the known functional domains of CENP-E (Putkey et al., 2002; Fig. 1 B).
Figure 1.
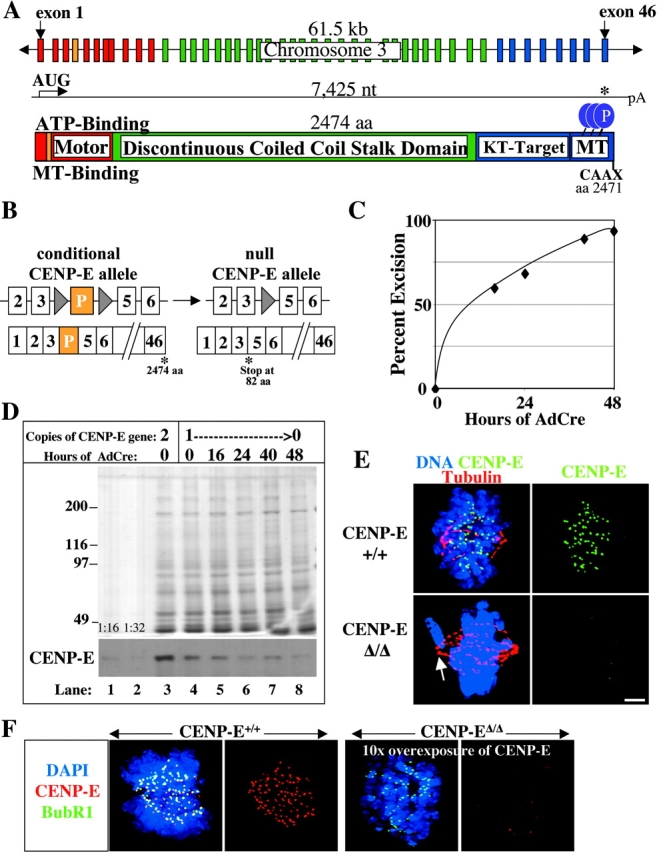
Murine CENP-E is efficiently removed after recombinase-mediated excision of the single murine CENP-E gene. (A) Schematic of the murine CENP-E gene, mRNA, and protein. The 46 exons of the CENP-E gene span 61.5 kb and encode a 7,425-nucleotide mRNA that produces a 2,474-aa protein. Apparent functional domains are labeled. KT, kinetochore; MT, microtubule; purple circles denote cdc2–cyclin B consensus phosphorylation sites. (Red) The kinesin-like motor region and associated exons; (orange) exon 4 encodes the ATP-binding consensus site of the motor domain and is selectively deleted (see B); (green) the discontinuous α-helical coiled-coil stalk region and associated exons; (blue) the carboxy-terminal globular domain and associated exons; CAAX, the terminal CAAX motif that may direct farnesylation. (B) Conversion of the conditional CENP-E allele to the null allele by Cre recombinase–mediated excision of exon 4 (exon P, shown in orange), due to lox P sites (denoted by triangles) incorporated into the adjacent introns, which introduces a premature stop codon at aa 82 (out of 2,474). (C) Real-time PCR analysis of a single experiment indicates that excision of the CENP-E gene reaches >90% by 48 h after addition of AdCre. (D) Immunoblot showing that CENP-E protein levels are diminished ∼16-fold in CENP-EΔ/Δ cells (lane 8) as compared with control cells (lane 3). Lane 1 is a 16-fold dilution of lane 3. Coomassie stain is shown as a loading control. (E) Immunofluorescence detection of CENP-E (green) in mitotic CENP-E+/+ or CENP-EΔ/Δ cells. CENP-EΔ/Δ MEFs acquire misaligned chromosomes that appear abnormally close to the spindle poles (white arrow). Tubulin, red; DNA stained with DAPI, blue. Bar, 2.5 μm. (F) Immunofluorescence detection of CENP-E (red) in mitotic CENP-E+/+ or CENP-EΔ/Δ cells. The CENP-E image in the CENP-EΔ/Δ cells was exposed 10 times longer than the CENP-E image in the CENP-E+/+ cells. CENP-E is still undetectable at kinetochores (marked by BubR1, green) in CENP-EΔ/Δ cells. DAPI, blue.
Primary mouse embryonic fibroblasts (MEFs) derived from d 14 embryos were obtained by mating CENP-E+/Δ mice with CENP-E+/loxP mice. Individual embryos were genotyped by PCR to identify MEFs that were of the wild type (CENP-E+/+) or were CENP-E–conditional/null (CENP-EloxP/Δ). The CENP-EloxP/Δ cells were converted to CENP-E–null cells (CENP-EΔ/Δ) by infection with a replication-defective adenovirus expressing the Cre recombinase (AdCre; Anton and Graham, 1995). Real-time PCR was used to determine that excision of the conditional CENP-E allele reached ∼90% (87 ± 6%, n = 4) within 48 h after addition of AdCre (Fig. 1 C). CENP-E protein levels, as measured by quantitative immunoblotting, were diminished 16-fold compared with the wild type (Fig. 1 D). At the single-cell level, CENP-E was completely undetectable by immunofluorescence in >85% of CENP-EloxP/Δ cells 48 h after addition of AdCre (Fig. 1 E), even after 10× overexposure (Fig. 1 F). In light of the quantitative loss of the CENP-E gene and protein by 48 h after addition of AdCre, subsequent experiments were undertaken at this time point in cells hereafter described as CENP-EΔ/Δ.
CENP-E–deleted fibroblasts do not sustain a mitotic checkpoint despite unattached kinetochores
In agreement with previous experiments in other systems (Schaar et al., 1997; Wood et al., 1997; Yao et al., 2000; Yucel et al., 2000; McEwen et al., 2001), a high percentage of mitotic CENP-EΔ/Δ fibroblasts have misaligned chromosomes. In two independent experiments, 69 (±4) percent of CENP-EΔ/Δ cells with the majority of their chromosomes aligned had at least one chromosome juxtaposed to a spindle pole (Fig. 1 E, arrow; Fig. 2 A, arrows), a condition we define as “pseudo metaphase.” Most of the CENP-EΔ/Δ cells in pseudo metaphase contained only one or a few polar chromosomes; 55% had only one or two and 90% had five or fewer (Fig. 2 B).
Figure 2.
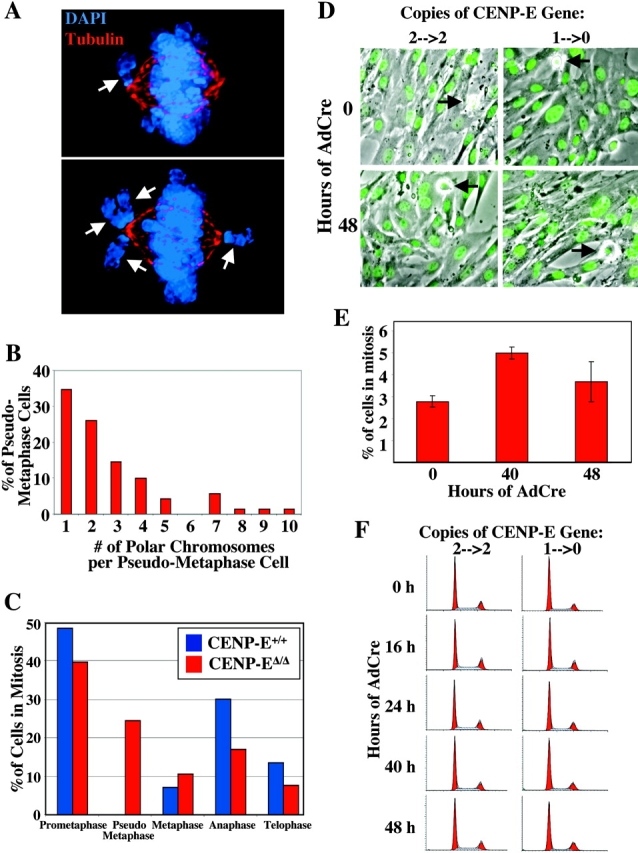
CENP-E Δ/Δ MEFs do not mount a robust cell cycle arrest despite the presence of unattached kinetochores. (A) Immunofluorescence of CENP-EΔ/Δ cells (48 h after addition of AdCre). Arrows denote polar chromosomes. DAPI, blue; tubulin, red. (B) Histogram showing the number of polar chromosomes per pseudo metaphase cell. (C) Graph showing the percentage of cells in various stages of mitosis. CENP-E+/+ cells are depicted by blue bars and CENP-EΔ/Δ cells are depicted by red bars. Pseudo metaphase cells are those in which the majority of chromosomes are aligned at the metaphase plate, but one or more chromosomes are at the poles. (D) Live cells whose DNA has been visualized by Hoechst 33258 do not exhibit marked mitotic arrest due to loss of CENP-E. Arrows denote mitotic cells. (E) Quantitation of the mitotic index of MEFs with one conditional and one null allele of CENP-E (CENP-EloxP/Δ) treated with AdCre for 0, 40, and 48 h. At least 1,000 MEFs were counted at each time point. (F) FACS® profiles of CENP-E+/+ cells (left column) and CENP-EloxP/Δ cells (right column) stained with propidium iodide at various times after addition of AdCre, indicating that loss of CENP-E does not induce robust mitotic arrest.
Previous EM performed in CENP-EΔ/Δ fibroblasts in pseudo metaphase have shown that both kinetochores on the misaligned chromosomes at the poles are unattached to spindle microtubules (Putkey et al., 2002). If loss of CENP-E left mitotic checkpoint signal generation intact, these unattached kinetochores would be expected to generate a sustained mitotic checkpoint arrest, forcing accumulation in mitosis after CENP-E gene disruption. Consistent with this, a transient preanaphase delay was indeed observed, as there was an accumulation of mitotic CENP-EΔ/Δ cells in pseudo metaphase (24% as opposed to 0% CENP-E+/+ cells) and a relative reduction in fractions of mitotic cells in anaphase and telophase (Fig. 2 C). However, despite chronic polar chromosomes, inspection of live cells revealed only a very modest increase in the mitotic index of CENP-EΔ/Δ MEFs relative to control cells treated with AdCre (5.0 ± 0.3 versus 2.4 ± 0.3%; Fig. 2 D) or to CENP-EloxP/Δ MEFs untreated with AdCre (5.0 ± 0.3 versus 2.8 ± 0.2%; Fig. 2 E). FACS® profiles taken at various times after addition of AdCre confirmed that CENP-EΔ/Δ cells with a 4N DNA content never accumulated to high levels (Fig. 2 F), as would have occurred if a sustained checkpoint signal was generated.
CENP-E is essential to prevent aneuploidy resulting from the loss or gain of one (or a few) chromosome(s)
The high frequency of polar chromosomes in CENP-EΔ/Δ MEFs and the absence of a sustained mitotic arrest raised the question of whether these chromosomes were eventually captured and aligned before the cells entered anaphase, or whether cells entered anaphase despite the presence of polar chromosomes. Examination of CENP-EΔ/Δ MEFs in anaphase revealed that 13% (12 of 90) of such cells had paired sister chromatids at one or both poles (Fig. 3, A-C, arrows; Fig. 3 A, inset). However, because the chromosome masses move to the poles during anaphase, only polar chromosomes whose arms are directed away from the spindle could be scored with certainty. Those that have their kinetochores at the pole but their arms directed into the spindle (see Fig. 5 E′, at right in panel 3) are obscured by the chromosome mass adjacent to the pole. For this reason, the observed frequency of 13% is probably an approximate twofold underestimate. Each CENP-EΔ/Δ anaphase cell with polar chromosomes had only one or two. No polar chromosomes were observed in CENP-E+/+ cells, either in ≥100 metaphase cells or in 63 anaphase figures.
Figure 3.
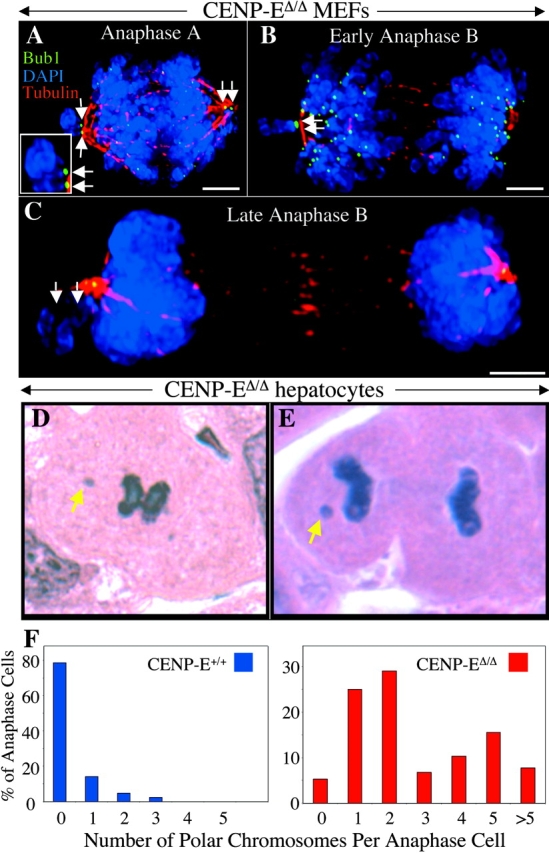
Absence of CENP-E causes cells to enter anaphase in the presence of one or a few polar chromosomes both in vitro and in vivo. (A–C) Immunofluorescence images of primary CENP-EΔ/Δ MEFs that have entered and proceeded through anaphase in the presence of one (B and C) or two (A) polar chromosomes that were never properly bioriented and aligned and will be missegregated. Bub1, green; DAPI, blue; tubulin, red. Bars, 2.5 μm. The inset in A is an enlargement of the polar chromosome at the left spindle pole showing the double dot pattern of Bub1 staining on the paired sister chromatids. (D and E) Regenerating hepatocytes that have entered (D) and proceeded through (E) anaphase in the presence of polar chromosomes (yellow arrows) in vivo after AdCre-mediated deletion of the single functional CENP-E gene in CENPElox P/Δ mice. (F) Histograms showing the number of polar chromosomes per anaphase figure in CENP-E+/+ (blue bars) and CENP-EΔ/Δ (red bars) hepatocytes in vivo. CENP-EΔ/Δ numbers were normalized to reflect the 70% excision rate.
Figure 5.

CENP-E is required for efficient kinetochore targeting of BubR1, Mad1, and Mad2. (A–E′) Kinetochore localization of CENP-E (A and A′), BubR1 (B and B′), Mad1 (C and C′), Mad2 (D and D′), and Bub1 (E and E′) in CENP-E+/+ (A–E) and CENP-EΔ/Δ (A′–E′) cells. Kinetochore proteins are shown in green. DNA, blue; tubulin, red. Bar, 2.5 μm. (B′′–E′′) Quantitation of the normalized integrated intensity of BubR1 (B′′), Mad1 (C′′), Mad2 (D′′), and Bub1 (E′′) signals at kinetochores in CENP-E+/+ (blue bars) and CENP-EΔ/Δ (red bars) prometaphase and metaphase cells. Kinetochore signals on aligned chromosomes in CENP-EΔ/Δ pseudo metaphase cells are also shown. 20 –1,750 kinetochores from 2 to 19 different cells were quantitated for each bar. *, P < 0.05; **, P < 0.001. Error bars represent standard error.
The continued localization of Bub1 (Fig. 3, A and B) or Mad1 and Mad2 checkpoint proteins (unpublished data) in a double-dot pattern confirmed that the polar chromosomes seen in anaphase were paired sister chromatids, and suggested that each kinetochore was attempting to generate a mitotic checkpoint signal. However, any inhibitor produced was insufficient to prevent anaphase onset. With the exception of the continued presence of polar chromatid pairs, subsequent mitotic steps, including early (Fig. 3 B) and late (Fig. 3 C) anaphase B, appeared to proceed normally. Thus, the polar chromosomes in CENP-EΔ/Δ cells do not cause sustained mitotic arrest and are ultimately missegregated at high frequency.
One or a few unattached kinetochores do not sustain an in vivo mitotic checkpoint in the absence of CENP-E
Recently, we have reported that administration of the Cre recombinase (using tail vein injection of AdCre) successfully deleted the one functional CENP-E gene in ∼70% of liver cells in CENP-EloxP/Δ mice (Putkey et al., 2002). Using this approach, we examined regenerating hepatocytes in CENP-EΔ/Δ livers from four different animals after damage to those livers was induced by exposure to carbon tetrachloride. Absence of CENP-E led to pseudo metaphase and aberrant anaphase figures in hepatocytes. 95% of anaphase figures exhibited polar chromosomes (Fig. 3, D–F, right), whereas very few anaphase cells from wild-type animals had such chromosomes (Fig. 3 F, left). As in the MEFs, despite their presence in the majority of the CENP-EΔ/Δ hepatocytes, there were few polar chromosomes per cell. 55% of CENP-EΔ/Δ hepatocytes had one or two polar chromosomes, and 85% had five or fewer (Fig. 3, D–F). Some of these hepatocytes also had lagging chromosomes, but most of those were distributed along spindle fibers and appeared to represent previously attached chromosomes that were released during anaphase due to the unstable microtubule capture in the absence of CENP-E. Thus, in this in vivo example, one or a few unattached kinetochores depleted of CENP-E are insufficient to prevent entry into anaphase.
CENP-EΔ/Δ cells sustain the mitotic checkpoint when all kinetochores are unattached
That murine CENP-EΔ/Δ cells lose chromosomes in vitro and in vivo raised the question of whether the mitotic checkpoint can maintain an arrest in these cells in the absence of CENP-E. To test this, CENP-E+/+ and CENP-EΔ/Δ MEFs were incubated with the microtubule-depolymerizing agent colcemid to inhibit spindle assembly. The mitotic index was then scored either by FACS® or by direct visualization of live cells with Hoechst-stained DNA. As expected, wild-type MEFs initiated and sustained checkpoint signaling efficiently, yielding a chronic checkpoint response in a majority of cycling cells after 16 h of drug-induced microtubule disassembly (Fig. 4 A). When all kinetochores were unattached, CENP-EΔ/Δ cells also showed a sustained arrest, with 60% of cycling cells accumulated with 4N DNA after 16 h of treatment (Fig. 4 A). A similar mitotic arrest in both CENP-E+/+ and CENP-EΔ/Δ cells was also observed after taxol-induced suppression of microtubule dynamics (Fig. 4 A). However, even under conditions when all kinetochores are sending a signal, the arrest in the absence of CENP-E appears to be less robust than in wild-type cells because the 4N DNA peak of CENP-EΔ/Δ cells is somewhat smaller at both 8 and 16 h after addition of either microtubule-disrupting drug (Fig. 4 A).
Figure 4.

Microtubule depolymerization causes CENP-E Δ/Δ cells to arrest in mitosis with reduced amounts of BubR1, Mad1, and Mad2 on their kinetochores. (A) FACS® profile of propidium iodide–stained CENP-E+/+ cells (left column) and CENP-EΔ/Δ cells (right column) after microtubule depolymerization with colcemid or microtubule stabilization with taxol. 75% of these primary cells are cycling. (B) Immunofluorescence of CENP-E+/+ cells (left column) and CENP-EΔ/Δ cells (right column) after a 30-min treatment with 200 ng/ml colcemid, showing that CENP-E is required for maximal kinetochore targeting of BubR1, Mad1, and Mad2 (green). DNA is shown in blue. Bar, 2.5 μm. (C) Quantitation of the normalized integrated intensities of the kinetochore signals in B. ≥800 kinetochores from ≥10 cells were quantitated for each bar. Error bars represent standard errors. **, P < 0.001.
CENP-E is required for efficient recruitment of BubR1, Mad1, and Mad2 to attached and newly unattached kinetochores
To generate a checkpoint signal, each kinetochore must recruit checkpoint proteins and modify one or more of them in such a way as to generate an inhibitor of Cdc20-APC/C. To test if CENP-E enhanced recruitment of known checkpoint components, thereby amplifying a basal CENP-E–independent kinetochore-derived signal, quantitative immunofluorescence was used to determine levels of kinetochore-bound BubR1, Mad1, and Mad2 after rapidly forcing spindle microtubule disassembly with colcemid. Compared with CENP-E+/+ cells, kinetochores in mitotic CENP-EΔ/Δ cells (Fig. 4, B and C) recruited two-to fourfold lower levels (2.1-, 3.7-, and 3.2-fold, respectively) of all of these checkpoint components. Thus, at least three essential checkpoint proteins are inefficiently recruited to kinetochores in the absence of CENP-E.
Kinetochore levels of Bub1, BubR1, Mad1, and Mad2 were also determined in the absence of spindle inhibitors. Relative to CENP-E–containing cells, CENP-EΔ/Δ prometaphase kinetochores were diminished in BubR1, Mad1, and Mad2. Metaphase kinetochores in CENP-EΔ/Δ cells were also diminished in BubR1 and Mad1, whereas Mad2 levels were largely undetectable during metaphase in both CENP-E+/+ and CENP-EΔ/Δ cells (Fig. 5, A–D′′; Table I). However, Bub1, which is recruited to kinetochores before CENP-E (Jablonski et al., 1998; unpublished data), was not diminished on prometaphase or metaphase kinetochores in the absence of CENP-E (Fig. 5 E′′; Table I).
Table I. Diminished kinetochore recruitment of BubR1 in the absence of CENP-E.
| Prometaphase | Metaphase | Pseudo meta aligned | Pseudo meta polar | |||||
|---|---|---|---|---|---|---|---|---|
| CENP-E genotype | +/+ | Δ/Δ | +/+ | Δ/Δ | +/+ | Δ/Δ | +/+ | Δ/Δ |
| BubR1 | 1.00 (0.13) | 0.52 (0.06) | 0.35 (0.05) | 0.13 (0.02) | NA | 0.03 (0.01) | NA | 0.52 (0.08) |
| Bub1 | 1.00 (0.16) | 0.95 (0.09) | 0.41 (0.04) | 0.36 (0.04) | NA | 0.33 (0.05) | NA | 2.00 (0.14) |
| Mad1 | 1.00 (0.18) | 0.53 (0.13) | 0.37 (0.08) | 0.17 (0.10) | NA | 0.03 (0.01) | NA | 1.88 (0.31) |
| Mad2 | 1.00 (0.10) | 0.48 (0.14) | NA | NA | NA | NA | NA | 2.62 (0.40) |
Values represent normalized, integrated intensities of kinetochore-associated BubR1, Bub1, Mad1, and Mad2, as detailed in Materials and methods. Standard errors are in parentheses.
Specific reduction in BubR1 recruitment to polar kinetochores in the absence of CENP-E
The increase in pseudo metaphase figures coupled with the decrease in anaphase and telophase figures in cycling CENP-EΔ/Δ cells (Fig. 2 C) suggested that the polar chromosomes in these cells transiently delayed progression into anaphase. During such a delay, aligned chromosomes that attached to the spindle with normal kinetics would be expected to completely silence checkpoint signaling. Consistent with this, BubR1 and Mad1 signals on many aligned kinetochores in CENP-EΔ/Δ pseudo metaphase cells dropped below the level of detection (Fig. 5, B′ and C′). BubR1 and Mad1 signals on these kinetochores were, on average, ≥10-fold less intense than on aligned chromosomes in CENP-E+/+ metaphase cells, and four- to sixfold less intense than on aligned chromosomes in CENP-EΔ/Δ metaphase cells (Fig. 5, B′′ and C′′; Table I).
Conversely, the unattached kinetochores on the polar chromosomes in CENP-EΔ/Δ pseudo metaphase cells would be expected not only to continue checkpoint signaling, but to recruit checkpoint proteins to levels higher than during a normal prometaphase, a phenomenon observed in many contexts during mitotic delay (Rieder and Palazzo, 1992; Thrower et al., 1996; Hoffman et al., 2001). Consistent with this, unattached kinetochores on polar chromosomes continued to recruit Mad1, Mad2, and Bub1, with levels of these proteins rising two- to sixfold over their levels on prometaphase kinetochores in CENP-EΔ/Δ cells (Fig. 6, B–F; Table I) and two- to threefold higher than on prometaphase kinetochores in CENP-E+/+ cells (Fig. 5, C–E′; Fig. 6 G). In contrast, BubR1 levels on those same polar kinetochores were unchanged relative to CENP-EΔ/Δ prometaphase levels (Fig. 6, A and F). Both polar and prometaphase kinetochores in CENP-EΔ/Δ cells recruited half the BubR1 of prometaphase kinetochores in wild type cells (Fig. 6 G). Thus, there is a specific defect in recruitment of BubR1 to CENP-E–depleted kinetochores.
Figure 6.

Kinetochores on polar chromosomes exhibit a specific defect in recruiting BubR1. (A–E) Polar kinetochores in pseudo metaphase cells (right) have higher levels of Bub1 (C), Mad1 (B and D), and Mad2 (E), but not BubR1 (A and B) than prometaphase kinetochores (left). Insets show higher magnification images of designated kinetochores. Arrows indicate polar chromosomes. DNA, blue; tubulin, red; checkpoint proteins, green, except in B, in which Mad1 is green and BubR1 is in red. (F and G) Comparison of the normalized integrated intensity of BubR1, Bub1, Mad1, and Mad2 signals at kinetochores. Prometaphase kinetochores in CENP-EΔ/Δ cells (striped bars in F) or in CENP-E+/+ cells (blue bars in G) are compared with kinetochores of polar chromosomes in CENP-EΔ/Δ pseudo metaphase cells (red bars). At least 53 kinetochores from ≥12 different cells were quantified for each bar. *, P < 0.05; **, P < 0.001. Error bars represent standard errors.
HeLa cells depleted of CENP-E also recruit reduced amounts of BubR1 to their kinetochores, but arrest because they have a large number of misaligned chromosomes
CENP-E–depleted HeLa cells are checkpoint arrested (Schaar et al., 1997; Yao et al., 2000; Harborth et al., 2001; McEwen et al., 2001) whereas, as we have shown here, primary CENP-EΔ/Δ MEFs (in the absence of spindle poisons) are not. To clarify these paradoxical findings, HeLa cells were depleted of CENP-E using RNAi. Quantitative immunofluorescence revealed that, like CENP-EΔ/Δ MEFs, colcemid-treated HeLa cells with undetectable levels of CENP-E (Fig. 7 A) recruit only half as much BubR1 to their kinetochores (Fig. 7, A and B). Thus, kinetochores in HeLa cells depleted of CENP-E send a reduced checkpoint signal, similar to kinetochores in CENP-EΔ/Δ MEFs. However, compared with the MEFs, there were many more polar chromosomes in the CENP-E–depleted HeLa cells. Although the MEFs had 1–10 polar chromosomes with one being the most common, the HeLa cells contained 1–25 misaligned chromosomes, with an average of 7.75 per cell (Fig. 7 C). Thus, unattached CENP-E–depleted kinetochores send a weaker BubR1-dependent checkpoint signal in both MEFs and HeLa cells, but the large number of unattached kinetochores in the HeLa cells is sufficient for a sustained checkpoint arrest, whereas the small number of unattached chromosomes in CENP-EΔ/Δ mouse cells in vitro or in vivo is not.
Figure 7.
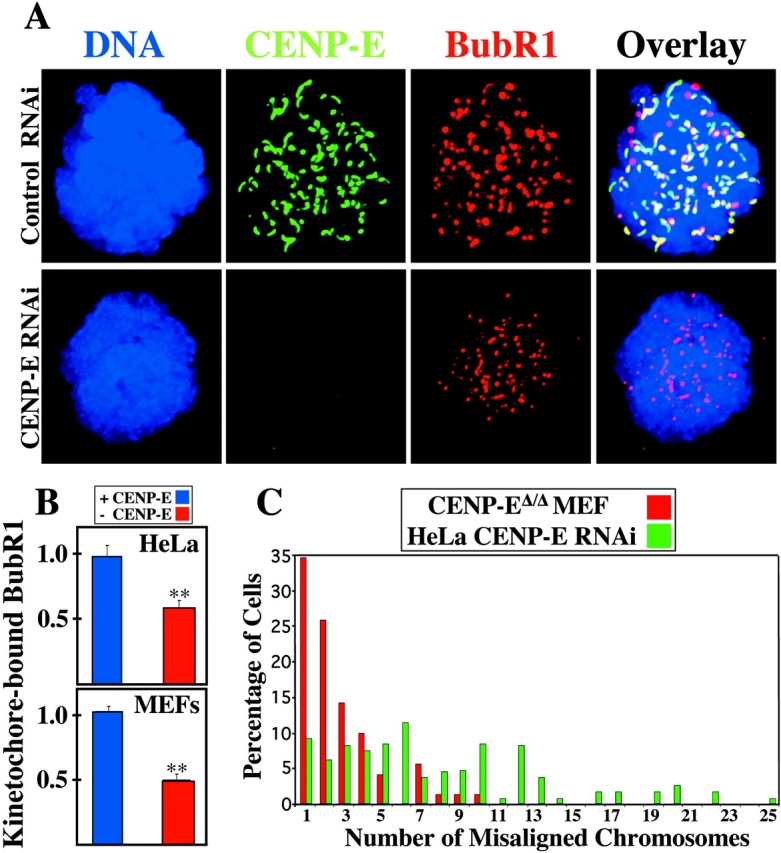
HeLa cells depleted of CENP-E recruit reduced amounts of BubR1 to their kinetochores and have many misaligned chromosomes. (A) HeLa cells depleted of CENP-E (green) by RNAi (bottom) recruit less BubR1 (red) to their kinetochores than HeLa cells treated with control RNAi (top) after a 30-min treatment with 200 ng/ml colcemid. DNA, blue. (B) Quantitation of the normalized integrated intensity of BubR1 signals at kinetochores in HeLa cells (top) and primary MEFs (bottom). Control kinetochores (blue bars) recruit more BubR1 than CENP-E–depleted kinetochores (red bars). ≥800 kinetochores from ≥10 cells were quantitated for each bar. **, P < 0.001. Error bars represent standard error. (C) Histogram showing the number of misaligned chromosomes per pseudo metaphase cell in CENP-EΔ/Δ MEFs (red bars) and in HeLa cells depleted of CENP-E by RNAi (green bars).
CENP-E stimulates mammalian BubR1 kinase activity
Mammalian CENP-E interacts with the checkpoint kinase BubR1, as has been shown by a yeast two-hybrid assay (Chan et al., 1998) and by coimmunoprecipitation from mitotic cell extracts (Chan et al., 1998; Yao et al., 2000). This interaction led us to test whether BubR1 kinase activity is affected by the loss of CENP-E. BubR1 was immunoprecipitated from CENP-E+/+ or CENP-EΔ/Δ cells and was assayed for kinase activity, using histone H1 as an in vitro substrate. This revealed that murine BubR1 kinase activity is sharply elevated in wild-type cells enriched in mitosis (Fig. 8 A, top, lane 2 vs. lane 3), as was previously demonstrated for human BubR1 (Chan et al., 1999). However, elevated BubR1 kinase activity during mitosis was not observed in the absence of CENP-E (Fig. 8 A, top panel, lane 5 vs. lane 6), although comparable levels of BubR1 were precipitated from cells with and without CENP-E (Fig. 8 A, bottom). Thus, mitotic stimulation of BubR1 kinase activity is dependent on CENP-E in primary MEFs (Fig. 8 A, compare lane 3 with lane 6).
Figure 8.
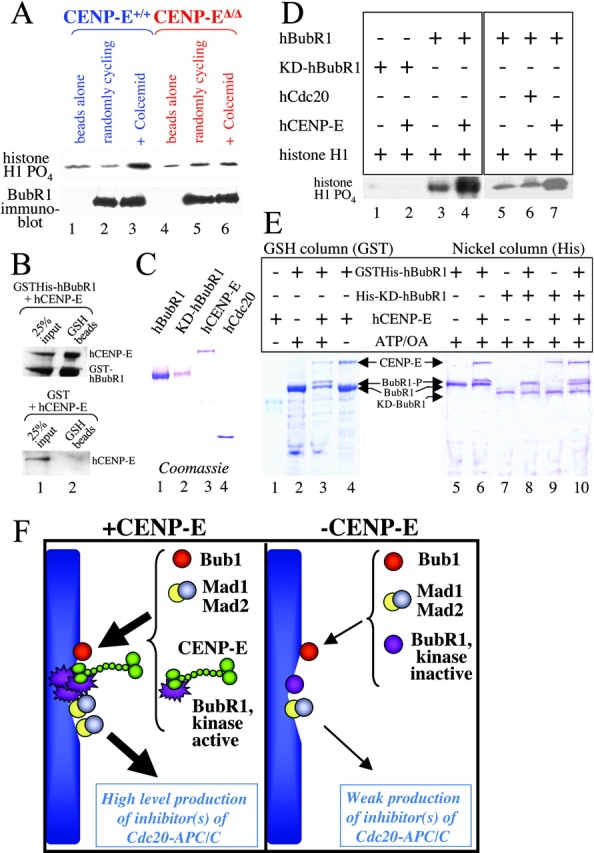
Mammalian BubR1 kinase activity is stimulated by CENP-E. (A) BubR1 was immunoprecipitated from CENP-E+/+ (lanes 1–3) and CENP-EΔ/Δ MEFs (lanes 4–6), and was assayed for kinase activity using histone H1 as a substrate (top). Equivalent levels of BubR1 were confirmed by immunoblot (bottom). Lane 1 and lane 4, beads alone control; lane 2 and lane 5, BubR1 was immunoprecipitated from randomly cycling cells; lane 3 and lane 6, BubR1 was immunoprecipitated from cells enriched in mitosis by a 16-h treatment with 50 ng/ml of the microtubule-depolymerizing agent colcemid. (B) Purified hCENP-E interacts with purified GST-hBubR1 bound to GSH beads (lane 2, top), but not to GST alone (lane 2, bottom). 25% of input was loaded in lane 1. (C) Coomassie stain of purified recombinant human BubR1, kinase-dead BubR1 (KD-BubR1), CENP-E, and Cdc20 that were expressed in insect cells using baculovirus. (D) In vitro kinase activity of purified hBubR1 by itself (lane 3 and lane 5), KD-hBubR1 by itself (lane 1), BubR1 + hCdc20 (lane 6), BubR1 + hCENP-E (lane 4 and lane 7), and BubR1 kinase-dead + hCENP-E (lane 2). Histone H1 was used as a substrate. (E) Coomassie stains of GSTHis-hBubR1, His-KD-hBubR1, and hCENP-E that were expressed singly or in combination in Hi5 cells and purified using GSH-Sepharose (left) or Ni-NTA agarose (right). Proteins in all lanes except 1 and 4 were purified in the presence of okadaic acid and 2 mM ATP. (F) Model of CENP-E as an activator of BubR1 in checkpoint signaling. In the presence of CENP-E (green, left) unattached kinetochores recruit large amounts of Bub1 (red), Mad1 (yellow), Mad2 (blue), and BubR1 (purple) whose kinase activity is stimulated by CENP-E (purple star). The unattached kinetochores assemble large quantities of Cdc20-APC/C inhibitors, which permit each kinetochore to delay anaphase onset until it has become attached. In the absence of CENP-E (right), unattached kinetochores recruit reduced amounts of Mad1 (yellow), Mad2 (blue), and BubR1 (purple) with reduced kinase activity (no star). Each kinetochore produces fewer molecules of Cdc20-APC/C inhibitors, which are no longer sufficient to meet the threshold required to prevent premature anaphase onset.
To verify that CENP-E and BubR1 do directly bind in a high affinity complex, insect cells were infected with baculoviruses expressing CENP-E and GSTHis-BubR1 or CENP-E and GST alone. Newly made proteins were [35S]-labeled with methionine and were recovered by incubation with glutathione beads. CENP-E was efficiently recovered when it was coexpressed with GSTHis-BubR1 (Fig. 8 B, top), but as expected did not bind to GST alone (Fig. 8 B, bottom).
To test if CENP-E directly stimulates BubR1 kinase activity, recombinant human GSTHis-BubR1 (110 kD; Fig. 8 C, lane 1) and human CENP-E (312 kD; Fig. 8 C, lane 3) were expressed and purified from insect cells. Kinase activity of GSTHis-BubR1 was then measured in the presence or absence of added CENP-E, again using histone H1 as a substrate. Although a basal level of kinase activity was associated with BubR1 by itself, this was stimulated about fivefold by addition of CENP-E (Fig. 8 D, compare lane 3 with lane 4; lane 5 with lane 7). CENP-E binding selectively stimulated the GSTHis-BubR1 kinase inasmuch as addition of another BubR1-binding partner, hCdc20, did not affect GSTHis-BubR1 kinase activity (Fig. 8 D, compare lane 5 and lane 6). BubR1 with a point mutation in the ATP-binding domain that was predicted to disrupt kinase activity (K795R—to be referred to as KD-BubR1), was expressed and purified in parallel (Fig. 8 C, lane 2). Only trace kinase activity was detectable for this KD-BubR1 mutant either in the presence or absence of CENP-E (Fig. 8 D, lane 1 and lane 2), indicating that the activity detected in purified wild-type BubR1 was indeed from BubR1 and not due to a contaminating kinase.
To test if BubR1 phosphorylates itself and whether CENP-E enhances this activity, GSTHis-BubR1 and His-KD-BubR1 were expressed by themselves or in the presence of CENP-E. The proteins were purified in the presence of ATP and the phosphatase inhibitor okadaic acid using GSH-Sepharose (Fig. 8 E, left) or Ni-NTA (Fig. 8 E, right). The purified BubR1s were then tested for autokinase activity. Both GSTHis-BubR1 and His-KD-BubR1 migrated as single bands when expressed alone (Fig. 8 E, lane 2 and lane 5 for GSTHis-BubR1; lane 7 for His-KD-BubR1). However, coexpression of wild-type GSTHis-BubR1 with CENP-E resulted in the production of a more slowly migrating species of BubR1 (Fig. 8 E, lane 3 and lane 6) that was dependent on the continued presence of ATP (Fig. 8 E, lane 4 vs. lane 3). This shift in migration was not observed for His-KD-BubR1, though it was still capable of interacting with CENP-E (Fig. 8 E, lane 9). Furthermore, His-KD-BubR1 was not modified to a more slowly migrating form when coexpressed with GSTHis-BubR1 (Fig. 8 E, lane 8) or GSTHis-BubR1, whose kinase activity had been stimulated by the additional coexpression of CENP-E (Fig. 8 E, lane 10). Thus, CENP-E directly stimulates intramolecular autophosphorylation of kinase competent BubR1.
Discussion
CENP-E has previously been shown to bind to kinetochores and microtubules and stabilize the interaction between them (Putkey et al., 2002), thereby contributing to silencing mitotic checkpoint signaling. Now, we have shown that CENP-E is also required in vitro and in vivo for maximal mitotic checkpoint signal generation at individual kinetochores, and is thus bifunctional in checkpoint signaling. We find that CENP-E stimulates recruitment of its binding partner BubR1 to kinetochores in HeLa cells and in MEFs. In addition, CENP-E directly stimulates the kinase (and autokinase) activity of BubR1 in vitro and in primary MEFs (Fig. 8). The simplest view is that CENP-E amplifies a basal mitotic checkpoint that is sufficient for long-term arrest when large numbers of kinetochores are unattached, but is of insufficient strength for one or a few kinetochores to produce a checkpoint signal that is able to sustain mitotic arrest.
Several lines of evidence offer strong support for our proposal that CENP-E is essential for enhancing checkpoint signaling to suppress the mitotic loss of one chromosome (or a few chromosomes) that would arise from advance to anaphase in the presence of one unattached kinetochore (or a few kinetochores). First, in each of the two systems in which direct tests have been feasible using laser irradiation or micromanipulation (Rieder et al., 1994, 1995; Li and Nicklas, 1995), a single unattached kinetochore has been found sufficient to prevent progression to anaphase. Second, wild-type primary MEFs do not generate aneuploidy at a significant rate in vitro (Babu et al., 2003), but CENP-EΔ/Δ primary MEFs missegregate one or two chromosomes in ∼25% of divisions (and enter anaphase with only one or a few kinetochores signaling). Third, weakening of checkpoint signaling by loss of one copy of the Bub3 gene produces a similar rate of chromosome loss (Babu et al., 2003) to that seen in CENP-EΔ/Δ MEFs. Fourth, CENP-E–deleted but not CENP-E+/+ hepatocytes in vivo missegregate one or two chromosomes during most mitoses. This is consistent with a checkpoint response that initially delays anaphase but cannot be sustained after attachment of most kinetochores.
With all of this in mind, we propose that, instead of acting as a simple molecular switch that toggles between on and off, the intensity of the mitotic checkpoint signal generated at individual kinetochores is dependent on CENP-E. Without CENP-E, the checkpoint signal generated by individual kinetochores is weakened such that the checkpoint cannot prevent anaphase onset if only one or a few kinetochores are unattached. This results in an effective loss/gain of one or two chromosomes in ∼25% of cell divisions in vitro and in at least ∼95% of divisions in hepatocytes in vivo (Fig. 3).
Immunodepletion of BubR1 from Xenopus extracts (Chen, 2002; Mao et al., 2003) or expression of dominant BubR1 mutants in mammalian cells (Chan et al., 1999) have implicated BubR1 as an essential checkpoint protein. As BubR1 is the primary checkpoint protein whose function is diminished in the absence of CENP-E (other than CENP-E itself), our evidence supports a CENP-E dependency in this essential BubR1 role. Because BubR1 has been shown to be essential for kinetochore localization of Mad1 and Mad2 (Chen, 2002; Mao et al., 2003), we predict that CENP-E indirectly affects kinetochore targeting and activation of Mad1 and Mad2 through its direct action on BubR1 localization and kinase activity.
BubR1 kinase activity is apparently essential for the checkpoint in mammalian cells (Chan et al., 1999) and Xenopus extracts (Mao et al., 2003; see following paragraph), but is not required for BubR1 to inhibit Cdc20-APC/C (Tang et al., 2001). Kinase activity correlates with the presence of CENP-E, whose cell cycle–dependent accumulation yields a maximum of ∼5,000 molecules during mammalian mitosis, almost all of which are kinetochore associated (Brown et al., 1994). This contrasts with BubR1, whose mitotic level has been estimated to be 50 times higher (Tang et al., 2001), and most of which is soluble. Therefore, with little correspondingly soluble CENP-E, it is most plausible that CENP-E stimulation of the BubR1 kinase is primarily at kinetochores, although the direct stimulation of BubR1 kinase by CENP-E in vitro demonstrates that such activity does not require kinetochore binding.
One report has argued that BubR1 kinase activity is unnecessary for the checkpoint because Xenopus extracts depleted of BubR1 displayed efficient checkpoint signaling after addition of kinase-inactive mutants (Chen, 2002). However, kinetochore-bound wild-type BubR1 was still present in those experiments (see Fig. 3 of Chen, 2002). A subsequent series of immunodepletion and readdition experiments has shown that only a small proportion of BubR1 needs to be kinase active to restore checkpoint activity (Mao et al., 2003). Therefore, we predict that kinase-active BubR1 at kinetochores participates in the generation of the diffusible checkpoint inhibitor(s). Such an interpretation of CENP-E–stimulated BubR1 kinase activity also offers an attractive explanation for an initial conundrum concerning BubR1 kinase activity. The homologue of BubR1 in both budding and fission yeasts (Mad3), as well the homologue in Caenorhabditis elegans, lacks a kinase domain, clearly demonstrating that BubR1 kinase activity is not essential for the checkpoint in these organisms. However, in all species for which the data are available (S. cerevisiae, Schizosaccharomyces pombe, C. elegans, Drosophila, Xenopus, Arabidopsis, mice, and human), when the BubR1 homologue contains a kinase domain, a CENP-E homologue is present as well. Thus, it seems plausible that during evolution of more complex eukaryotes in which the checkpoint became essential, BubR1 acquired a CENP-E–stimulated catalytic role to amplify signal generation per unattached kinetochore.
One surprising feature of this work is that, without CENP-E, the known Cdc20-APC/C inhibitor Mad2 is robustly recruited to one or a few polar, unattached kinetochores, but this is insufficient to prevent anaphase entry. In the presence of CENP-E, Mad2 rapidly cycles on and off unattached kinetochores (Howell et al., 2000) and has been shown to bind and inhibit Cdc20-APC/C (Li et al., 1997; Fang et al., 1998; Kallio et al., 1998; Luo et al., 2000). For these reasons, Mad2 has frequently been interpreted to be the downstream effector molecule of the mitotic checkpoint, and its presence at kinetochores has often been taken as a measure of ongoing checkpoint signal generation. However, we find that despite the presence of Mad2 at unattached kinetochores in CENP-E–deleted cells, the strength of the inhibitory signal sent by these Mad2-rich kinetochores is insufficient to prevent cell cycle advance, and cells lacking CENP-E produce aneuploid progeny at high frequency. This is similar to the situation with two other proteins, Rod and ZW10, whose inhibition yields an inactive checkpoint despite prominent Mad2 binding at kinetochores (Chan et al., 2000). On the other hand, diminution of another kinetochore component, Hec1, has been reported to yield a chronically activated checkpoint without obvious Mad2 bound to kinetochores (Martin-Lluesma et al., 2002). Though Mad2 intensity was not quantified in either of these analyses, it now seems clear that the detectable presence of Mad2 at kinetochores is not a faithful reporter for successful checkpoint activation.
Finally, our efforts here indicate that CENP-E is essential for maximal mitotic checkpoint signaling per individual kinetochore in all contexts. In the absence of CENP-E, the checkpoint response per kinetochore is diminished in Xenopus extracts (Abrieu et al., 2000), primary mouse cells (Fig. 3 through Fig. 6, and Fig. 8 A), murine hepatocytes in vivo (Fig. 3, D–F), and HeLa cells (Fig. 7, A and B). However, HeLa cells exhibit a much stronger dependence on CENP-E for capture and alignment of chromosomes, thus yielding a larger number of misaligned chromosomes (averaging >7) per cell. Though each kinetochore still generates a diminished signal, it appears that the sum of these unattached kinetochores is sufficient to sustain checkpoint arrest. Thus, in each of these systems, one unattached kinetochore (or in some cases a few unattached kinetochores) depleted of CENP-E do not recruit and activate enough BubR1 to generate a sufficiently robust checkpoint signal to prevent anaphase onset and the consequent development of aneuploidy.
Materials and methods
Library screening
The full-length murine CENP-E cDNA was cloned from an 8.5-d λgt10 mouse cDNA library using previously obtained 5′ and 3′ murine cDNA fragments as probes. Additional sequences were obtained from murine ESTs, RT-PCR products from ES cell RNA, and PCR products from assorted cDNA libraries. All regions of the cDNA were confirmed by two or more independent sequences.
MEF preparation, culture, and excision
MEFs were prepared from d 14 embryos as described previously (Putkey et al., 2002). For excision, cells were infected with an adenovirus encoding Cre recombinase (a gift from Kenneth Chien, University of California, San Diego, La Jolla, CA) at a multiplicity of infection of 10. Excision was tested by real-time PCR (Putkey et al., 2002). Reduction in CENP-E protein levels was tested by immunoblotting using the Hpx anti-CENP-E antibody (Brown et al., 1996) diluted 1:200 in 5% milk in 1× TBS plus 0.1% Tween 20. ECL blotting was made quantitative by running serial dilutions of control extract.
Immunofluorescence
Cells were washed with 37°C microtubule stabilizing buffer (MTSB; 100 mM Pipes, 1 mM EGTA, 1 mM MgSO4, and 30% glycerol) and then extracted before fixation with 0.5% Triton X-100 in MTSB at 37°C for 5 min. After extraction, cells were washed again and fixed in 4% formaldehyde (Tousimis Research Corporation) for 10 min. 0.1% Triton X-100, 2.5% FBS, and 200 mM glycine in PBS was used to block cells and dilute antibodies. The Hpx antibody to CENP-E (Brown et al., 1996) was diluted 1:200, the DM1α antibody to α-tubulin (Blose et al., 1984) 1:1,000, the 5F9 BubR1 antibody (Taylor et al., 2001) 1:200, the Mad1 antibody (Campbell et al., 2001) 1:250, Bub1 1:200, and XMad2 (Waters et al., 1998) 1:60. Secondary antibodies (Jackson ImmunoResearch Laboratories) were diluted 1:200.
Deconvolution microscopy and quantitation
Deconvolution images were collected using a DeltaVision wide-field deconvolution microscope system built on an inverted microscope (model TE200; Nikon) base. Optical sections were taken at 0.2-μm intervals and images were processed using DeltaVision SoftWoRx™ software. Comparable images were taken at the same exposure times on the same day. Figures were generated by projecting the sum of the optical sections. Kinetochore fluorescence was quantified using the integrated intensity of three-dimensional polygons surrounding each kinetochore.
FACS® analysis
Adherent and nonadherent cells were collected and washed 3× in cold PBS before being resuspended in 200 μl cold PBS and fixed with 800 μl cold 100% ethanol while being vortexed mildly. Cells were stored at 4°C, resuspended in 20 μg/ml propidium iodide and 40 μg/ml RNase, incubated in the dark for ≥30 min before being sorted (20,000 events/sample) on a cytometer (FACSort™; Becton Dickinson) by Dr. Frank Furnari using CellQuest™ software (BD Biosciences).
Immunoprecipitation and kinase assay
Adherent and nonadherent CENP-E+/+ and CENP-EΔ/Δ MEFs were grown for 16 h with or without 50 ng/ml colcemid and were washed 2× with cold PBS on ice before being lysed in 50 mM Tris 7.4, 100 mM NaCl, 1% Triton X-100, 2 mM MgCl2, 5 mM EDTA with 400 μM pefablock, 100 μg/ml leupeptin, pepstatin, and chymostatin, 10 mM sodium fluoride, and 20 mM sodium orthovanadate. Cell extracts were spun for 10 min at 4°C at full speed in a microfuge and the supernatant was incubated with Gammabind G beads (Amersham Biosciences) ± the 5F9 BubR1 antibody (see Immunofluorescence section) at 4°C for 2–3 h. Pellets were washed 3× with lysis buffer, and phosphorylation reactions were performed at 30°C for 30 min in a 40-μl reaction containing histone H1 kinase buffer (20 mM Tris-Cl, pH 7.4, 50 μM cold ATP, 10 mM MgCl2, 10 mM EGTA, and 750 μg/ml histone H1) together with 5 μCi of γ[32P]ATP per sample.
Protein expression and purification
Cdc20, hBubR1, and BubR1 kinase-dead (K795R) in pFB-NHis10-HA vectors for baculovirus expression were gifts from Dr. Peter Sorger (Massachusetts Institute of Technology, Cambridge, MA). GST-BubR1 was recloned into pFB and hCENP-E into pFB-HTb (both 6xHis vectors for baculovirus expression). Hi5 cells (∼3 × 107 cells) were infected with the appropriate virus. After 42–48 h of infection, cells were harvested and lysed in 50 mM Tris-Cl, pH 7.0, 250 mM NaCl, 2 mM MgCl2, 0.3% Triton X-100, and 5 mM β-mercaptoethanol containing PMSF and leupeptin, pepstatin, and chymostatin as protease inhibitors. The supernatant was incubated with Ni-NTA. After washing, proteins were eluted with 500 mM imidazole in the same buffer. All eluates were dialyzed against lysis buffer with 150 mM NaCl.
Phosphorylation reactions
Phosphorylation reactions contained purified hBubR1/KD-hBubR1 proteins (∼25 ng) in the presence or absence of hCENP-E (∼20 ng) or Cdc20 (∼40 ng) in a 40-μl reaction containing histone H1 kinase buffer (20 mM Tris-Cl, pH 7.4, 50 μM cold ATP, 10 mM MgCl2, 10 mM EGTA, and 750 μg/ml histone H1) together with 5 μCi of γ[32P]ATP per sample. The same buffer was added to the BubR1 immunoprecipitates from the mouse fibroblasts. The reaction was then performed at 30°C for 30 min.
GST pulldown assay
Hi5 cells were infected with His6-hCENP-E and His6-GST-BubR1 viruses singly or together in the presence of 35S-labeled methionine (50 μCi per plate). The cells were lysed in buffer containing 50 mM Tris-Cl, pH 7.0, 100 mM NaCl, and 0.1% Triton X-100 plus protease inhibitors. The supernatant in each case was divided into two aliquots. One half was incubated with Ni-NTA beads to purify total CENP-E and BubR1. The other half was incubated with glutathione beads to purify GST-BubR1 and any CENP-E bound to it. After SDS-PAGE, the gel was subjected to 35S-labeled fluorography.
Autokinase assay
Hi5 cells were infected with GST-His6-hBubR1 alone, His6-KD-hBubR1 alone, hCENP-E alone, or combinations of viruses. The proteins were purified in PBS in the presence or absence of okadaic acid and 2 mM ATP using GSH-Sepharose or Ni-NTA agarose. Proteins were subjected to SDS-PAGE followed by Coomassie staining.
RNAi
RNAi was performed in HeLa cells using siRNAs or a plasmid-based approach. The CENP-E siRNAs used were described in Harborth et al. (2001). 5 × 104 cells were seeded into 6-well plates ∼20 h before transfection of RNAs with OligofectAMINE™, as recommended by the manufacturer (Invitogen). Alternatively, the oligonucleotide 5′- GATCCCCGCAGAGAGAAGGGTGAACCTTCAAGAGAGGTTCACCCTTCTCTCTGCTTTTTGGAAA-3′ was annealed to 5′-AGCTTTTCCAAAAAGCAGAGAGAAGGGTGAACCTCTCTTGAAGGTTCACCCTTCTCTCTGCGGG-3′, phosphorylated by T4 polynucleotide kinase, and ligated to HindIII/BglII-cut pSUPER (Brummelkamp et al., 2002) to make pS-hCENP-E. HeLa cells were transfected with pS-hCENP-E and pCMV-CD20 in a ratio of 10:1. 48 h after transfection, cell pellets were incubated with mouse anti-CD20 (DakoCytomation) for 20 min on ice, washed with PBS, and incubated with goat anti–mouse microbeads (Miltenyi Biotec) for 20 min on ice, recovered on an MS column in a magnetic stand, eluted by removing the column from the magnet, and replated onto poly-l-lysine–coated coverslips. 24 h after replating, cells were fixed for immunofluorescence.
Acknowledgments
We thank Dr. Frank Furnari for assistance with the FACS® analysis; Dr. Tim Yen (Fox Chase Cancer Center, Philadelphia, PA) for the Mad1 antibody; Dr. Bonnie Howell and Dr. E.D. Salmon (University of North Carolina, Chapel Hill, NC) for the Mad2 antibody; Dr. Stephen Taylor (University of Manchester, Manchester, UK) for the gift of the BubR1 antibody; Dr. Paul Maddox for helpful comments on the manuscript; and Dr. Anjon Audhya for useful discussions. We also thank Dr. James Feramisco and Steve McMullen for use of and assistance with the deconvolution microscope.
This work has been support by a grant from the National Institutes of Health to D.W. Cleveland (R37 GM25913). B.A.A. Weaver has been supported by a predoctoral training grant from the National Cancer Institute and the Ludwig Institute for Cancer Research (T32 CA67754). Z.Q. Bonday was supported by a postdoctoral fellowship from the Tobacco Initiative. G.J. Kops was supported by a fellowship from the Dutch Cancer Society. Salary support for D.W. Cleveland was provided by the Ludwig Institute for Cancer Research.
Abbreviations used in this paper: AdCre, adenovirus expressing the Cre recombinase; APC/C, anaphase-promoting complex/cyclosome; CENP-E, centromere-associated protein-E; MEF, mouse embryonic fibroblast.
References
- Abrieu, A., J.A. Kahana, K.W. Wood, and D.W. Cleveland. 2000. CENP-E as an essential component of the mitotic checkpoint in vitro. Cell. 102:817–826. [DOI] [PubMed] [Google Scholar]
- Anton, M., and F.L. Graham. 1995. Site-specific recombination mediated by an adenovirus vector expressing the Cre recombinase protein: a molecular switch for control of gene expression. J. Virol. 69:4600–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashar, H.R., L. James, K. Gray, D. Carr, S. Black, L. Armstrong, W.R. Bishop, and P. Kirschmeier. 2000. Farnesyl transferase inhibitors block the farnesylation of CENP-E and CENP-F and alter the association of CENP-E with the microtubules. J. Biol. Chem. 275:30451–30457. [DOI] [PubMed] [Google Scholar]
- Babu, J.R., K.B. Jeganathan, D.J. Baker, X. Wu, N. Kang-Decker, and J.M. van Deursen. 2003. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J. Cell Biol. 160:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blose, S.H., D.I. Meltzer, and J.R. Feramisco. 1984. 10-nm filaments are induced to collapse in living cells microinjected with monoclonal and polyclonal antibodies against tubulin. J. Cell Biol. 98:847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, K.D., R.M. Coulson, T.J. Yen, and D.W. Cleveland. 1994. Cyclin-like accumulation and loss of the putative kinetochore motor CENP-E results from coupling continuous synthesis with specific degradation at the end of mitosis. J. Cell Biol. 125:1303–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, K.D., K.W. Wood, and D.W. Cleveland. 1996. The kinesin-like protein CENP-E is kinetochore-associated throughout poleward chromosome segregation during anaphase-A. J. Cell Sci. 109:961–969. [DOI] [PubMed] [Google Scholar]
- Brummelkamp, T.R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science. 296:550–553. [DOI] [PubMed] [Google Scholar]
- Cahill, D.P., C. Lengauer, J. Yu, G.J. Riggins, J.K. Willson, S.D. Markowitz, K.W. Kinzler, and B. Vogelstein. 1998. Mutations of mitotic checkpoint genes in human cancers. Nature. 392:300–303. [DOI] [PubMed] [Google Scholar]
- Campbell, M.S., G.K. Chan, and T.J. Yen. 2001. Mitotic checkpoint proteins HsMAD1 and HsMAD2 are associated with nuclear pore complexes in interphase. J. Cell Sci. 114:953–963. [DOI] [PubMed] [Google Scholar]
- Chan, G.K., B.T. Schaar, and T.J. Yen. 1998. Characterization of the kinetochore binding domain of CENP-E reveals interactions with the kinetochore proteins CENP-F and hBUBR1. J. Cell Biol. 143:49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, G.K., S.A. Jablonski, V. Sudakin, J.C. Hittle, and T.J. Yen. 1999. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J. Cell Biol. 146:941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, G.K., S.A. Jablonski, D.A. Starr, M.L. Goldberg, and T.J. Yen. 2000. Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat. Cell Biol. 2:944–947. [DOI] [PubMed] [Google Scholar]
- Chen, R.H. 2002. BubR1 is essential for kinetochore localization of other spindle checkpoint proteins and its phosphorylation requires Mad1. J. Cell Biol. 158:487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland, D.W., Y. Mao, and K.F. Sullivan. 2003. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 112:407–421. [DOI] [PubMed] [Google Scholar]
- Cohen, J. 2002. Sorting out chromosome errors. Science. 296:2164–2166. [DOI] [PubMed] [Google Scholar]
- Dobles, M., V. Liberal, M.L. Scott, R. Benezra, and P.K. Sorger. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 101:635–645. [DOI] [PubMed] [Google Scholar]
- Fang, G., H. Yu, and M.W. Kirschner. 1998. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12:1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harborth, J., S.M. Elbashir, K. Bechert, T. Tuschl, and K. Weber. 2001. Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci. 114:4557–4565. [DOI] [PubMed] [Google Scholar]
- Hoffman, D.B., C.G. Pearson, T.J. Yen, B.J. Howell, and E.D. Salmon. 2001. Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol. Biol. Cell. 12:1995–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell, B.J., D.B. Hoffman, G. Fang, A.W. Murray, and E.D. Salmon. 2000. Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J. Cell Biol. 150:1233–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt, M.A., L. Totis, and B.T. Roberts. 1991. S. cerevisiae genes required for cell cycle arrest in response to loss of microtubule function. Cell. 66:507–517. [DOI] [PubMed] [Google Scholar]
- Jablonski, S.A., G.K. Chan, C.A. Cooke, W.C. Earnshaw, and T.J. Yen. 1998. The hBUB1 and hBUBR1 kinases sequentially assemble onto kinetochores during prophase with hBUBR1 concentrating at the kinetochore plates in mitosis. Chromosoma. 107:386–96. [DOI] [PubMed] [Google Scholar]
- Kallio, M., J. Weinstein, J.R. Daum, D.J. Burke, and G.J. Gorbsky. 1998. Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase-promoting complex, and is involved in regulating anaphase onset and late mitotic events. J. Cell Biol. 141:1393–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, K.B., A.A. Burds, J.R. Swedlow, S.S. Bekir, P.K. Sorger, and I.S. Nathke. 2001. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat. Cell Biol. 3:429–432. [DOI] [PubMed] [Google Scholar]
- Li, R., and A.W. Murray. 1991. Feedback control of mitosis in budding yeast. Cell. 66:519–531. [DOI] [PubMed] [Google Scholar]
- Li, X., and R.B. Nicklas. 1995. Mitotic forces control a cell-cycle checkpoint. Nature. 373:630–632. [DOI] [PubMed] [Google Scholar]
- Li, Y., C. Gorbea, D. Mahaffey, M. Rechsteiner, and R. Benezra. 1997. MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc. Natl. Acad. Sci. USA. 94:12431–12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, X., G. Fang, M. Coldiron, Y. Lin, H. Yu, M.W. Kirschner, and G. Wagner. 2000. Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat. Struct. Biol. 7:224–229. [DOI] [PubMed] [Google Scholar]
- Mao, Y., A. Abrieu, and D.W. Cleveland. 2003. Activating and silencing the mitotic checkpoint through CENP-E-dependent activation/inactivation of BubR1. Cell. 114:87–98. [DOI] [PubMed] [Google Scholar]
- Martin-Lluesma, S., V.M. Stucke, and E.A. Nigg. 2002. Role of hec1 in spindle checkpoint signaling and kinetochore recruitment of mad1/mad2. Science. 297:2267–2270. [DOI] [PubMed] [Google Scholar]
- McEwen, B.F., G.K. Chan, B. Zubrowski, M.S. Savoian, M.T. Sauer, and T.J. Yen. 2001. CENP-E is essential for reliable bioriented spindle attachment, but chromosome alignment can be achieved via redundant mechanisms in mammalian cells. Mol. Biol. Cell. 12:2776–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel, L.S., V. Liberal, A. Chatterjee, R. Kirchwegger, B. Pasche, W. Gerald, M. Dobles, P.K. Sorger, V.V. Murty, and R. Benezra. 2001. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 409:355–359. [DOI] [PubMed] [Google Scholar]
- Putkey, F.R., T. Cramer, M.K. Morphew, A.D. Silk, R.S. Johnson, J.R. McIntosh, and D.W. Cleveland. 2002. Unstable kinetochore-microtubule capture and chromosomal instability following deletion of CENP-E. Dev. Cell. 3:351–365. [DOI] [PubMed] [Google Scholar]
- Rieder, C.L., and R.E. Palazzo. 1992. Colcemid and the mitotic cycle. J. Cell Sci. 102:387–392. [DOI] [PubMed] [Google Scholar]
- Rieder, C.L., A. Schultz, R. Cole, and G. Sluder. 1994. Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J. Cell Biol. 127:1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder, C.L., R.W. Cole, A. Khodjakov, and G. Sluder. 1995. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J. Cell Biol. 130:941–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaar, B.T., G.K. Chan, P. Maddox, E.D. Salmon, and T.J. Yen. 1997. CENP-E function at kinetochores is essential for chromosome alignment. J. Cell Biol. 139:1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z., R. Bharadwaj, B. Li, and H. Yu. 2001. Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev. Cell. 1:227–237. [DOI] [PubMed] [Google Scholar]
- Taylor, S.S., E. Ha, and F. McKeon. 1998. The human homologue of Bub3 is required for kinetochore localization of Bub1 and a Mad3/Bub1-related protein kinase. J. Cell Biol. 142:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, S.S., D. Hussein, Y. Wang, S. Elderkin, and C.J. Morrow. 2001. Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J. Cell Sci. 114:4385–4395. [DOI] [PubMed] [Google Scholar]
- Testa, J.R., J.Y. Zhou, D.W. Bell, and T.J. Yen. 1994. Chromosomal localization of the genes encoding the kinetochore proteins CENPE and CENPF to human chromosomes 4q24→q25 and 1q32→q41, respectively, by fluorescence in situ hybridization. Genomics. 23:691–693. [DOI] [PubMed] [Google Scholar]
- Thrower, D.A., M.A. Jordan, and L. Wilson. 1996. Modulation of CENP-E organization at kinetochores by spindle microtubule attachment. Cell Motil. Cytoskeleton. 35:121–133. [DOI] [PubMed] [Google Scholar]
- Waters, J.C., R.H. Chen, A.W. Murray, and E.D. Salmon. 1998. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell Biol. 141:1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, E., and M. Winey. 1996. The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J. Cell Biol. 132:111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, K.W., R. Sakowicz, L.S. Goldstein, and D.W. Cleveland. 1997. CENP-E is a plus end-directed kinetochore motor required for metaphase chromosome alignment. Cell. 91:357–366. [DOI] [PubMed] [Google Scholar]
- Yao, X., K.L. Anderson, and D.W. Cleveland. 1997. The microtubule-dependent motor centromere-associated protein E (CENP-E) is an integral component of kinetochore corona fibers that link centromeres to spindle microtubules. J. Cell Biol. 139:435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, X., A. Abrieu, Y. Zheng, K.F. Sullivan, and D.W. Cleveland. 2000. CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat. Cell Biol. 2:484–491. [DOI] [PubMed] [Google Scholar]
- Yucel, J.K., J.D. Marszalek, J.R. McIntosh, L.S. Goldstein, D.W. Cleveland, and A.V. Philp. 2000. CENP-meta, an essential kinetochore kinesin required for the maintenance of metaphase chromosome alignment in Drosophila. J. Cell Biol. 150:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]