

Abstract
The function of adhesion receptors in both cell adhesion and migration depends critically on interactions with the cytoskeleton. During cell adhesion, cytoskeletal interactions stabilize receptors to strengthen adhesive contacts. In contrast, during cell migration, adhesion proteins are believed to interact with dynamic components of the cytoskeleton, permitting the transmission of traction forces through the receptor to the extracellular environment. The L1 cell adhesion molecule (L1CAM), a member of the Ig superfamily, plays a crucial role in both the migration of neuronal growth cones and the static adhesion between neighboring axons. To understand the basis of L1CAM function in adhesion and migration, we quantified directly the diffusion characteristics of L1CAM on the upper surface of ND-7 neuroblastoma hybrid cells as an indication of receptor–cytoskeleton interactions. We find that cell surface L1CAM engages in diffusion, retrograde movement, and stationary behavior, consistent with interactions between L1CAM and two populations of cytoskeleton proteins. We provide evidence that the cytoskeletal adaptor protein ankyrin mediates stationary behavior while inhibiting the actin-dependent retrograde movement of L1CAM. Moreover, inhibitors of L1CAM–ankyrin interactions promote L1CAM-mediated axon growth. Together, these results suggest that ankyrin binding plays a crucial role in the anti-coordinate regulation of L1CAM-mediated adhesion and migration.
Keywords: cell migration; single particle tracking; traction force; cell adhesion; axon growth
Introduction
The establishment and maintenance of neuronal connections are essential features of nervous system function. The activity of adhesion proteins on the cell surface is essential to both of these processes. During development, the translocation of the neuronal growth cone depends on adhesion proteins that mediate the recognition of molecules in the extracellular environment (Tessier-Lavigne and Goodman, 1996). In the adult, many of the same adhesion proteins help maintain axon fascicles and synaptic contacts. To function in adhesion and migration, receptors need to regulate their distribution and movement in the membrane. Although receptors involved in adhesion maintain static connections between adjacent cells, receptors that mediate cell migration must be more dynamic, transmitting traction forces from the cell to its environment (Harris et al., 1980). Receptor-mediated traction-force generation depends on connections between adhesion receptors and the cytoskeleton (Felsenfeld et al., 1996; Choquet et al., 1997). Therefore, understanding the regulation of the cytoplasmic interactions of adhesion receptors and the resulting changes in receptor movement on the cell surface is central to characterizing their function in adhesion and migration.
A variety of adhesion receptor families have been shown to serve as receptors for permissive, substrate-bound molecules that promote axon outgrowth, including integrins, immunoglobulin superfamily cell adhesion molecules (IgCAMs), and cadherins (Kamiguchi and Yoshihara, 2001). In light of the demonstrated role of IgCAMs in the guided growth of neuronal processes during development (Kamiguchi and Lemmon, 2000; Rutishauser, 2000), understanding the biophysical properties of these of proteins may provide crucial insight into the mechanism underlying growth cone translocation.
In the vertebrate central nervous system, L1 cell adhesion molecule (L1CAM), a neuronal IgCAM, plays an essential role in the guidance of descending cortico-spinal tract neurons (Dahme et al., 1997; Cohen et al., 1998). L1CAM is the founding member of a subfamily of neuronal IgCAMs that include both vertebrate and invertebrate members (Hortsch, 2000). L1CAM mutations in humans lead to a variety of developmental defects, including corpus callosum hyperplasia, mental retardation, adducted thumbs, spastic paraplegia, and hydrocephalus (CRASH syndrome), suggesting that L1CAM plays a crucial role in the development of the central nervous system (Fransen et al., 1995). Moreover, the capacity of substrate-bound L1CAM ligands to promote neurite extension in vitro through homophilic binding (Lemmon et al., 1989; Kuhn et al., 1991; Felsenfeld et al., 1994) raises the possibility that L1CAM on the growth cone may mediate traction-force generation in a mechanism similar to that observed for integrins in other cell types.
The regulation of receptor distribution, movement, and function in adhesion and migration depends on the connection between these glycoproteins and the cytoskeleton. L1CAM and L1 family members interact with four known cytosolic binding partners through two discrete sites in the cytoplasmic tail, including ankyrin, components of the clathrin AP-2 complex, ezrin, radixin, moesin (ERM) proteins, and doublecortin (Zhang et al., 1998; Dickson et al., 2002; Kizhatil et al., 2002; Schaefer et al., 2002). The binding of L1 family members to members of the ankyrin family of cytoskeletal adaptor proteins is perhaps the best characterized of these interactions (Davis and Bennett, 1994; Garver et al., 1997; Hortsch et al., 1998). The L1 family member neurofascin binds to ankyrin through a motif that is highly conserved among L1 family members near the carboxy terminus of the cytoplasmic tail (Garver et al., 1997). The ankyrin-binding site, mapped based on the interaction between neurofascin and ankyrin G, is comprised of a core 12-aa motif that is essential for ankyrin binding, including a carboxy-terminal tyrosine (QFNEDGSFIGQY; identical in neurofascin and L1 from rat; Miura et al., 1991; Zhang et al., 1998). In neurofascin, ankyrin binds to this motif in its dephosphorylated state (Garver et al., 1997). Mutations at this site in human L1CAM lead to a similar disruption in ankyrin binding (Needham et al., 2001). However, the Drosophila L1 homologue neuroglian, although requiring the FIGQY motif for ankyrin recruitment, appears to be regulated primarily through oligomerization of the extracellular domain (Dubreuil et al., 1996). At a functional level, the binding of ankyrin to L1 family members like neurofascin plays a critical role in cell adhesion (Tuvia et al., 1997).
The work presented here is directed at understanding the regulation of L1CAM function as reflected in changes in its diffusion kinetics. Quantifying directly the movement of receptors on the upper surface of the cell provides an accurate reflection of receptor function on the lower surface, where cells exert traction forces during migration (Galbraith and Sheetz, 1999). Therefore, the detailed analysis of L1CAM kinetics in the plane of the membrane may provide crucial insight into the mechanism underlying L1CAM function in both axon growth during development and static adhesion between mature axons. Previous work has revealed that L1 family members display two discrete diffusion rates on the cell surface, consistent with protein that is either bound or unbound from the cytoskeleton (Pollerberg et al., 1990; Garver et al., 1997). However, as this work relies on photobleaching of populations of receptors, it provides no information about the directed movement of protein in the lower diffusion state. Here, we describe evidence for three distinct classes of L1CAM movement on the cell surface, including diffusing, nondiffusing with directed movement (retrograde), and nondiffusing without directed movement (stationary). Although the stationary state of L1CAM depends on ankyrin binding to the L1CAM tail, retrograde movement occurs under conditions that inhibit ankyrin binding and depends on interactions between the L1CAM cytoplasmic tail and dynamic actin in the cytosol. Ankyrin binding inhibits L1CAM retrograde movement, suggesting that ankyrin may play a crucial role in effecting the switch between the stationary and directed behavior of L1CAM on the cell surface. More significantly, peptides that inhibit ankyrin binding stimulate L1CAM-mediated neuronal extension, suggesting that the regulation of L1CAM-mediated traction-force generation is essential to the migration of neuronal growth cones on L1CAM ligands in vivo.
Results
To begin to characterize the regulation of L1CAM–cytoskeleton interactions, we examined the lateral mobility of cell surface L1CAM in cultured cell lines. Full-length rat L1CAM including the neuron-specific RSLE exon was expressed in ND-7 cells (rat DRG/neuroblastoma hybrid; Dunn et al., 1991) to provide a controlled background on which to characterize L1CAM function. These adherent cells express both endogenous L1CAM and ankyrin B (Fig. 1, A and E). Cells were transfected transiently with a wild-type rat L1CAM cDNA construct encoding an amino-terminal myc epitope to permit the detection of the transgene product in the context of endogenous L1CAM. The distribution of the epitope-tagged protein was similar to that of endogenous L1CAM, suggesting that mycL1CAM was appropriately transported and distributed on the cell surface (Fig. 1, A and B). 1-μm latex beads coated with an anti-myc antibody (9E10; Evan et al., 1985) were placed and held with an optical gradient laser trap on the cell surface between 0.5 and 1 μm from the leading edge for 2 s. To identify cells expressing the L1CAM transgene, cells were transfected with a bicistronic vector encoding both mycL1CAM and EGFP (CLONTECH Laboratories, Inc.). mycL1CAM expression, detected by indirect immunofluorescence, was well correlated with EGFP expression (unpublished data). Bead binding to the cell surface varied with antibody concentration and fell off dramatically between 0.037 and 0.0073 mg/ml beads (Fig. 2, white bars). Additionally, binding of beads coated with a high concentration of 9E10 (0.58 mg/ml beads) to cells transfected with L1CAM lacking the myc epitope was 0–20% (for each individual experiment), suggesting that bead binding is selective for myc-tagged L1CAM on the cell surface. Bound beads were subject to a second pulse from the laser trap (see Materials and methods) to test the resistance of L1CAM–bead complex to lateral displacement, a strong indicator of cytoskeletal attachment (Choquet et al., 1997; Felsenfeld et al., 1999). At the highest concentration of antibody (0.37 mg Ab/ml beads), the majority of beads were resistant to lateral displacement (Fig. 2, black bars “rigid”). The percentage of beads that were bound but not resistant to displacement (Fig. 2, gray bars, “loose”) was inversely proportional to antibody concentration. These results suggest that extracellular oligomerization of L1CAM by antibody regulates the association between L1CAM and the actin cytoskeleton, consistent with results from analyses of other L1 family members (Dubreuil et al., 1996).
Figure 1.
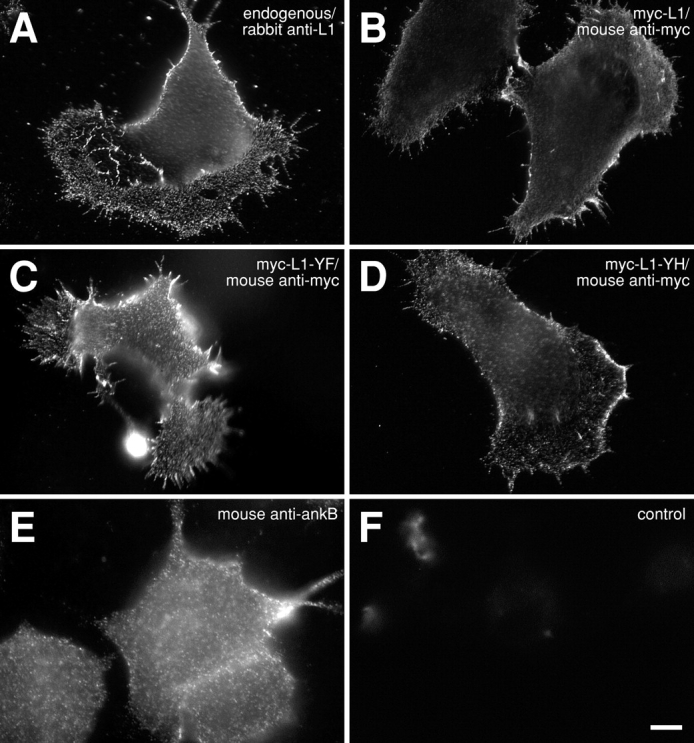
Wild-type or mutant rat L1CAM is expressed on the surface of ND7 cell lines. ND7 cells expressing cDNA constructs encoding either wild-type (B) or mutant L1CAM (C and D), tagged with a myc epitope. Wild-type myc-tagged L1CAM (B) detected by indirect immunofluorescence using an anti-myc antibody (9E10) appears on the cell surface with a distribution that is similar to that of endogenous L1CAM expressed by the cell line (A; rabbit polyclonal against L1CAM). Mutant forms of L1CAM encoding single aa substitutions at tyrosine 1229 including Y1229F (C) and Y1229H (D) are similarly indistinguishable in distribution from endogenous protein. ND-7 cells also express ankyrin B as detected by immunofluorescence (E) and immunoblot (not depicted). (F) Control image of cells expressing wild-type L1CAM stained with secondary antibody alone. Bar, 10 μm.
Figure 2.

L1CAM–cytoskeleton interactions depend on L1CAM cross-linking. 1-μm latex microspheres coated with anti-myc antibodies recognize cell surface L1CAM tagged with a myc epitope. Bead binding to the cell surface after placement with a laser trap (white bars indicate no binding) depended on antibody concentration. Once bound, beads were tested for resistance to lateral movement with the laser trap (see Materials and methods) as an indication of cytoskeletal attachment. The percentage of trials that are attached and rigid on the cell surface (black bars) or attached and but subject to displacement (gray bars) varied directly with antibody concentration.
L1CAM engages in three distinct classes of movement on the cell surface
To analyze directly the behavior of L1CAM on the upper surface of the cell, we recorded the movement of beads bound by antibody to cell surface L1CAM. Beads coated with 9E10 (0.58 mg/ml beads) bound to the cell surface and underwent rapid diffusion (Fig. 3, A, D, and G), retrograde movement (Fig. 3, B, E, and H), or remained stationary (Fig. 3, C, F, and I). For diffusing beads, the trajectory lacked any detectable directed movement with respect to the leading edge (Fig. 3 D). Similarly, mean square displacement for diffusing beads (Fig. 3 G) was linear with respect to time, consistent with diffusion in the absence of directed movement. The average rate of diffusion (0.082 μm2s−1; n = 13) suggests that the random movement of L1CAM in the bilayer occurs largely in the absence of cytoskeletal or other interactions that would immobilize the protein(s) bound to the bead. In contrast, beads bound to retrograde-moving L1CAM (Fig. 3 B) showed little or no diffusion, moving away from the leading edge of the cell with a uniform velocity and direction. This directed movement occurred largely in the absence of movement parallel to the leading edge (Fig. 3 E). For all experiments, cells were selected at a wide variety of expression levels (based on coexpression of EGFP) to reduce the possibility of biases that might arise from overexpression. Nonetheless, we cannot preclude the possibility that L1CAM transgene overexpression may affect bead binding and lateral mobility. As with integrins (Felsenfeld et al., 1996), retrograde movement of L1CAM was often proceeded by a brief (<10 s), diffusive latency period (unpublished data). The velocity of retrograde-moving beads on cells expressing wild-type L1CAM ranged from 0.6 to 2.2 μm min−1, similar to the velocity of other cell surface adhesion proteins (Felsenfeld et al., 1996; Lambert et al., 2002). Therefore, the velocity and direction of bead movement is consistent with an interaction between L1CAM and treadmilling actin in the cytosol of the lamella.
Figure 3.

Wild-type L1CAM engages in three distinct classes of kinetic behavior on the cell surface. 1-μm latex microspheres coated with anti-myc antibodies recognize cell surface L1CAM tagged with a myc epitope. Bound beads underwent diffusion (A, D, and G), retrograde movement (B, E, and H), or stationary behavior (C, F, and I). All data sets were rotated to orient the cell with its leading edge facing left. (A–C) Plots of X vs. Y coordinates of representative data sets (in μm) are consistent with either diffusive (diffusion coefficient D = 0.11 μm2s−1; v = 0 μm min−1), slow directed movement (D = 1.12 × 10−4 μm2s−1; v = 0.924 μm min−1) or stationary behavior (D = 4.18 × 10−4 μm2s−1; v = 0 μm s−1; origin indicated by blue arrow). Stationary behavior can be observed as discrete clusters in the bead trajectory (C, black circle). Plots of X and Y coordinates vs. time indicated that the diffusing bead (D) shows no detectable directed movement in the axis of cell migration as movement perpendicular (⊥, fine trace) or parallel (ll, thick trace) to the leading edge are similarly chaotic. In contrast, beads undergoing retrograde movement on the cell surface show slow, uniform movement away from the leading edge with little or no displacement parallel to the leading edge (E). For stationary beads, in the case of the bead shown here (F), diffusive movement decreased (indicated by bars; colors correspond to circles in C after a period of free diffusion [∼10 s]). Mean squared displacement (MSD) for the diffusing bead was linear with respect to time (G), confirming that the behavior is diffusive in the absence of directed movement. The black trace reflects the measured MSD. The red line reflects the best fit of this curve by linear regression. For the retrograde-moving bead (H), the MSD plot has a quadratic shape, reflecting directed movement with substantially reduced diffusion. For stationary beads (I), MSD for the period indicated by the gray bar in F shows no evidence of directed movement.
In addition to diffusion and retrograde movement, cell surface L1CAM displayed a distinct behavior characterized by a low rate of diffusion in the absence of detectable directed movement (Fig. 3, C, F, and I). The stationary behavior was transient and appeared to punctuate periods of diffusion (Fig. 3 C, black circle). Periods of stationary behavior appeared as plateaus with reduced noise in plots of displacement vs. time (Fig. 3 F, black line), consistent with a decreased rate of diffusion. Although it is difficult to analyze the velocity from the mean square displacement for short data segments, analysis of data sets with a high ratio of stationary behavior to diffusion (Fig. 3 C, data in gray circle; Fig. 3 F, data over gray bar) revealed a reduced diffusion coefficient compared with freely diffusing particles (Fig. 3, G and I) with little or no directed component. In cells expressing wild-type L1CAM, 9E10 beads underwent retrograde transport on the cell surface in 67.9% of trials, whereas 28.6% displayed stationary behavior (Fig. 4 A). Together, these results demonstrate that L1CAM is capable of three distinct classes of behavior on the cell surface, including diffusion, retrograde movement, and stationary behavior in the absence of directed movement. These distinct behaviors are consistent with interactions between L1CAM and both dynamic and static components of the cytoskeleton.
Figure 4.
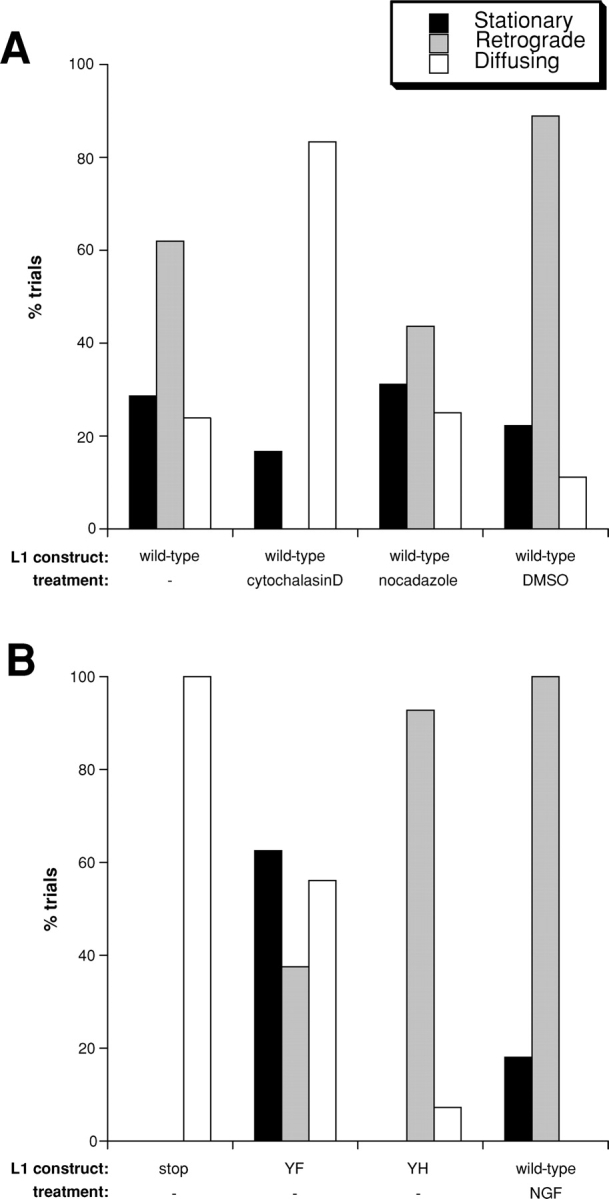
L1CAM–cytoskeleton interactions mediate retrograde and stationary behaviors. Bar graphs showing the percentage of trials engaging in stationary behavior, retrograde movement, or diffusion in cells expressing either wild-type or mutant forms of L1CAM. (A) Wild-type L1CAM treated with actin and microtubule inhibitors. In ND-7 cells expressing wild-type L1CAM, 28.6% of trials were stationary, 61.9% retrograde, and 23.8% diffused (n = 21). Cytochalasin D treatment (2 μM) eliminated retrograde movement (stationary 16.7%, retrograde 0%, diffusing 83.3%; n = 12). In contrast, nocadazole treatment (1 μM) inhibited retrograde movement slightly while leaving stationary behavior and diffusion unaffected (stationary 31.25%, retrograde 43.75%, diffusing 25%; n = 16). Treatment with DMSO alone at the same concentration used to dilute cytochalasin D and nocadazole caused a slight increase in retrograde movement (stationary 22.2%, retrograde 88.8%; diffusing 11.1%; n = 9). (B) L1CAM cytoplasmic tail mutants. In cells expressing an L1 mutant with an introduced stop codon designed to truncate the protein, leaving 4 aa of the predicted cytoplasmic tail (stop), the receptor diffused exclusively. In cells expressing the YF mutant of L1CAM (YF), 62.5% of trials were stationary and 37.5% were retrograde moving (56.2% diffused; n = 16). In cells expressing a histidine in place of Y1229 (FIGQY-H; YH, a mutation that inhibits ankyrin binding), 0% were stationary, 92.9% were retrograde-moving and 7.1% diffusing (n = 14). Finally, in cells expressing wild-type L1CAM treated with NGF to stimulate phosphorylation of Y1229, inhibiting ankyrin binding, 18.2% of trials displayed some form of stationary behavior, whereas 100% underwent retrograde transport on the cell surface (0% diffusing; n = 11).
L1CAM retrograde movement depends on the L1CAM cytoplasmic tail and dynamic pools of actin
To begin to examine the mechanism underlying the directed and stationary behavior of L1CAM on the cell surface, we observed the movement of L1CAM in the presence of various cytoskeletal inhibitors. Cytochalasin D, at a concentration that completely suppresses F-actin in the periphery of ND-7 cells (2 μM; unpublished data), abolishes the retrograde movement of L1CAM (Fig. 4 A; n = 12, P < .01). In contrast, stationary behavior was still observed in a small percentage of trials, suggesting that this behavior is either actin independent or mediated by nondynamic pools of actin that are less sensitive to cytochalasin D treatment. In contrast, both retrograde movement and stationary behavior were observed in the presence of 1 μM nocadazole (a concentration that blocks microtubule polymerization in ND-7 cells; unpublished data), although the frequency of retrograde movement was diminished as compared with untreated control cells (n = 16, P < .22). Treatment with DMSO alone at the same concentration used in the dilution of cytochalasin D and nocadazole resulted in a slight increase in retrograde movement (perhaps due to changes in membrane fluidity). These results suggest that retrograde movement of cell surface L1CAM is actin-mediated, although microtubules may also contribute indirectly to this process.
To determine whether the low diffusive states of L1CAM on the cell surface are mediated directly by the L1CAM cytoplasmic tail, we generated a truncation mutant of L1CAM that interrupts the cytoplasmic tail with a stop mutation 4 aa after the predicted transmembrane domain. Beads bound to truncated L1CAM on the cell surface diffused in 100% of trials (Fig. 4 B; n = 17, P < .01), suggesting that both retrograde movement and stationary behavior depend on interactions between the L1CAM cytoplasmic tail and the cytoskeleton.
Mutations that affect ankyrin binding modulate L1CAM movement in the plane of the membrane
To examine directly the role of L1CAM–cytoskeleton interactions in L1CAM movement on the upper surface, we introduced a series of point mutations into the region of the L1CAM tail that has been implicated in ankyrin binding. Mutant constructs were generated encoding single aa substitutions for tyrosine1229 to either phenylalanine, a mutation that induces constitutive ankyrin binding in other vertebrate L1 family members (L1-YF; Garver et al., 1997; Tuvia et al., 1997), or to histidine to inhibit ankyrin binding (L1-YH; a naturally occurring MASA mutation in humans; Garver et al., 1997; Tuvia et al., 1997; Needham et al., 2001). Each of these constructs was expressed in ND-7 cells and displayed cell surface distribution comparable to that seen for wild-type L1CAM (Fig. 1, B–D). In culture, 9E10 beads placed on the upper surface of the cell with a laser trap bound with a frequency similar to that seen in cells expressing wild-type mycL1CAM. Like wild-type L1CAM, L1-YF displayed a combination of diffusive, retrograde, and stationary behaviors. However, the ratio of these behaviors was different from that of the wild-type receptor, showing an increase in stationary behavior (62.5%) with a commensurate decrease in retrograde movement (37.5%; Fig. 4 B; n = 16, P < .01). In contrast, beads bound to the L1-YH mutant showed a large increase in the percentage of trials undergoing retrograde movement (92.9%) and a complete loss of stationary behavior (Fig. 4 B; n = 14, P < .011). These results suggest that L1CAM stationary behavior is mediated by ankyrin binding, whereas the retrograde movement of L1CAM on the cell surface is ankyrin independent.
Growth factor treatment inhibits ankyrin recruitment and L1CAM stationary behavior at the cell surface
To test further this hypothesis, we examined the behavior of wild-type L1CAM (including the myc-epitope tag) after growth factor treatment. It has been reported previously that tyrosine phosphorylation of L1 family members at the FIGQY motif is modulated by activation of a variety of membrane-linked tyrosine kinase receptors, including receptors for NGF, FGF, EGF (Garver et al., 1997), and by the Eph kinase Cek5 (Zisch et al., 1997). L1CAM-transfected 293 cells recruited ankyrin to the membrane in an L1CAM-dependent manner (Fig. 5, A, C, and E). Treatment of these cells with EGF (50 ng/ml; 1 h) inhibited ankyrin membrane localization (Fig. 5, B, D, and F), similar to the behavior of other L1 family members (Zhang et al., 1998) and consistent with a phosphorylation-dependent inhibition of L1CAM–ankyrin binding. Measurement of ankyrin immunolocalization along a line drawn across the junction of L1CAM-positive cells (Fig. 5 H; Oancea et al., 1998) demonstrates a quantifiable and significant change in ankyrin–membrane association (Fig. 5 G; P < .01). Similar results were obtained using ND-7 cells treated with NGF (unpublished data), suggesting that these cells, derived from primary sensory neurons, have maintained their sensitivity to NGF.
Figure 5.
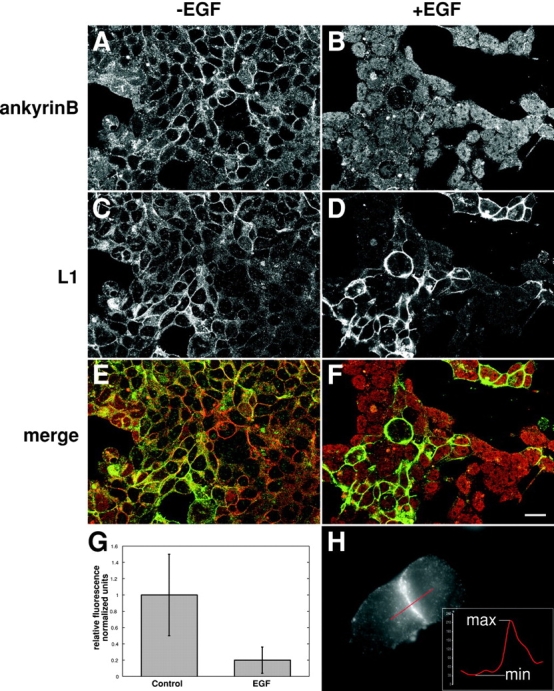
Growth factor treatment inhibits ankyrin B binding to L1CAM. 293 cells transfected with a cDNA encoding full-length rat L1CAM in the absence (A, C, and E) or presence (B, D, and F) of EGF. Ankyrin (A and B) and L1CAM (C and D) were detected by indirect immunofluorescence in double-labeled confocal sections through cell aggregates to permit the visualization of L1CAM and ankyrin B at the cell membrane. (E) Combined micrographs indicating ankyrin staining (red) and L1CAM staining (green) reveal clear codistribution of signal in the absence of EGF. In contrast, in the presence of EGF (F), L1CAM staining remains at the membrane, but ankyrin B staining is largely absent, appearing in a more uniform distribution throughout the cytosol. (G) Direct quantification shows a significant reduction of ankyrin B colocalization with L1CAM at the membrane in the presence of EGF (mean ± SD; P < 0.01). (H) The method for quantifying ankyrin B localization to the membrane uses densitometry of a line scan (red) across a cell–cell junction where L1CAM is expressed (ankyrin B staining is shown here). The resulting intensity profile is used to determine a minimum signal for comparison to the value of the ankyrin B signal at the point where the maximum signal in the L1CAM channel occurs. These values are combined to give an index value using the equation index = (max−min)/min. Index values were averaged and normalized with respect to the control values. Error bars ± SD. Bar, 10 μm.
Treatment of ND-7 cells expressing myc-tagged wild-type L1CAM with NGF caused a shift in the ratio of stationary to retrograde-moving beads similar to that seen in cells expressing L1-YH (18.2% stationary, 100% retrograde; Fig. 4 B; n =11, P < .02; for some trials, beads exhibited both forms of behavior). Together, these results suggest that L1CAM–ankyrin interactions mediate the stationary behavior of cell surface L1CAM. Moreover, the increase in the percentage of beads undergoing retrograde movement in conditions that perturb ankyrin binding raises the possibility that ankyrin may negatively modulate L1CAM-mediated traction-force generation.
Peptides derived from the L1CAM tail inhibit ankyrin binding and stationary behavior by L1CAM on the cell surface
To examine independently the role of ankyrin binding in the directed movement of L1, we designed peptides directed at inhibiting L1–ankyrin interactions in live cells. The inhibitory peptide is a fusion between the ankyrin-binding region of the L1CAM tail and the membrane-permeable penetratin domain of antennapedia (Derossi et al., 1998). The inhibitory region of this peptide was derived from the 12-aa conserved ankyrin-binding domain of the L1CAM tail (Zhang et al., 1998) including a Y to F substitution (QFNEDGSFIGQF; AP-YF). Peptide activity was compared with that of a peptide in which the inhibitory sequence was reversed (AP-Scramble).
To test the function of the AP-YF in situ, we examined its capacity to inhibit L1CAM-mediated recruitment of ankyrin to the cell membrane. In the presence of peptide AP-YF, ankyrinB was almost entirely absent from sites of cell–cell contact (Fig. 6 A). In contrast, in the absence of peptide or in the presence of the control, scrambled peptide, ankyrinB appeared at the cell membrane where L1CAM was expressed (Fig. 6 A; unpublished data). Quantification of ankyrin colocalization with L1CAM at the membrane revealed a significant reduction in the junctional distribution of ankyrin in AP-YF–treated cells (Fig. 6 A; P < 0.01). These results suggest that the AP-YF peptide is an effective inhibitor of L1CAM–ankyrin interactions in live cells.
Figure 6.

FIGQF peptides inhibit L1CAM–ankyrin interactions, stationary behavior of cell surface L1CAM, and increase the velocity of L1CAM retrograde movement. Peptides derived from the sequence of the ankyrin-binding domain of L1CAM conjugated to the membrane-permeant domain of antennapedia (AP-YF; see text) inhibit L1CAM–ankyrin interactions in membrane recruitment assays (A; P < 0.01). Scrambled peptides (AP-Scramble) where the ankyrin-binding domain is reversed have no detectable effects on L1CAM–ankyrin interactions. (B) Quantification of bead movement shows that the AP-YF peptides inhibit the stationary behavior of beads bound to cell surface L1CAM as compared with treatment with either AP-Scramble peptide or control (untreated) conditions. (C) AP-YF peptides also increase the mean velocity of retrograde movement by approximately twofold as compared with either AP-Scramble (P < 0.01) or control conditions (P < 0.01). A similar increase in the velocity of bead movement is observed using L1CAM mutants that inhibit ankyrin binding (FIGQH; Y1229-H substitution; P < 0.01). Error bars ± SD.
As was the case with the L1-YH mutation, the AP-YF inhibitory peptide reduced the percentage of beads showing stationary behavior on the cell surface with an accompanying increase in the percentage of trials undergoing retrograde movement (Fig. 6 B; n = 18, P < .05). Cells treated with the control, AP-Scramble peptide behaved in a manner similar to untreated cells, although there may be a slight (though not significant) increase in retrograde movement (n = 9; P > .05). These results confirm that experimental treatments that interfere with L1CAM–ankyrin binding inhibit selectively the low diffusion stationary state observed in wild-type L1CAM on the cell surface. This observation strongly suggests that ankyrin mediates L1CAM interactions with stationary components of the cytoskeleton. Moreover, the increase in the percentage of trials undergoing retrograde movement on the cell surface after inhibition of L1CAM–ankyrin interactions raises the possibility that ankyrin binding may also inhibit the directed movement of L1CAM on the cell surface.
To address this question, we quantified the velocity of bead movement in trials undergoing translocation on the cell surface in the presence of AP-YF or control peptides. Cells cultured in the presence of inhibitory peptide showed, on average, a twofold increase in the velocity of L1CAM-directed movement on the cell surface as compared with cells treated with control peptide (Fig. 6 C; P < 0.01) or untreated cells (P < 0.01). The movement of L1CAM in the presence of control peptide was largely unaffected. Similarly, analysis of mutant L1-YH, which is also deficient in ankyrin binding, displays a significant increase in the rate of directed protein movement on the cell surface as compared with untreated cells expressing wild-type L1CAM (P < 0.01). The change in mean velocity does not merely reflect the decrease in the percentage of stationary beads, as beads with a mean velocity of 0 were not included in the calculated average velocity. Together, these results implicate L1CAM–ankyrin interactions in the regulation of L1CAM-directed movement on the cell surface.
Inhibitors of ankyrin binding stimulate L1CAM-mediated neurite outgrowth
The changes in bead kinetics on the upper surface of the cell raise the possibility that the role of ankyrin binding in vivo may be to differentially regulate the adhesion and migration of growing neurons. To address this question directly, we cultured mouse cerebellar granular neurons in the presence of either inhibitory AP-YF or control peptides (Fig. 7). These neurons use cell surface L1CAM as the primary receptor for substrate-bound L1CAM ligands (Dahme et al., 1997), permitting us to test directly L1CAM function in neurite extension. Neurons grown on NgCAM, a chick homologue of L1CAM, extend 21 μm (± 2) after 24 h in culture in the presence of control peptides. In contrast, neurons cultured in the presence of AP-YF extend 55% above control levels (32 ± 2 μm; P < 0.01). Axon extension on laminin, which promotes outgrowth through interactions with cell surface integrins (Felsenfeld et al., 1994), was not significantly affected by peptide treatment (P > 0.05). These results suggest that L1CAM-dependent neuronal growth is modulated by changes in L1CAM–ankyrin interactions. Additionally, these results support the idea that traction-force generation in the neuronal growth cone plays a role in neurite extension.
Figure 7.
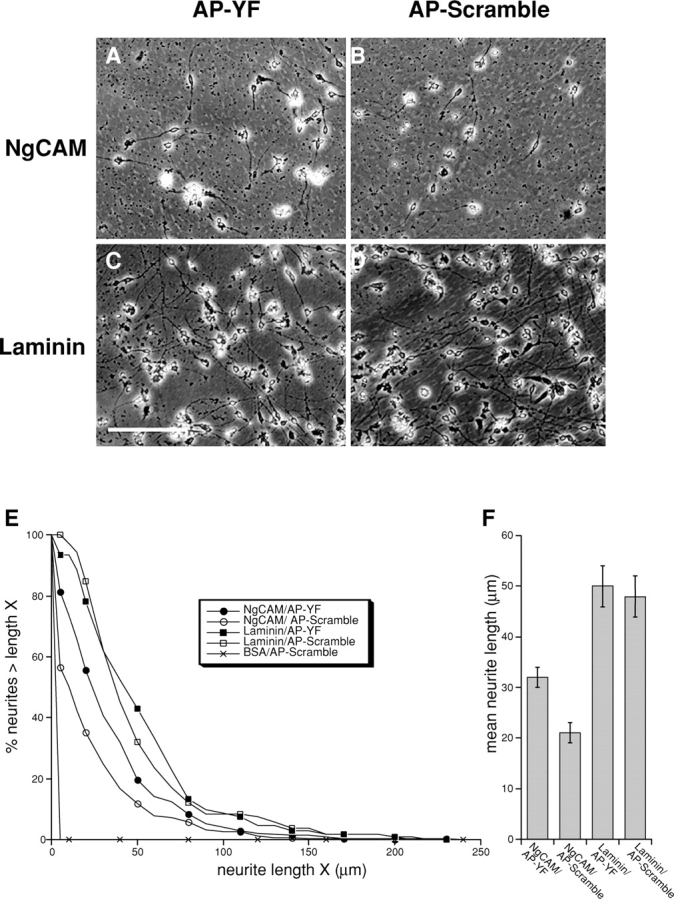
Peptide inhibitors of L1CAM–ankyrin interactions selectively stimulate L1CAM-mediated neuronal growth. Cerebellar cells prepared from P4 mouse (1.25 × 105 cells) were plated on chick NgCAM (A and B) or on mouse laminin (C and D). Cultures were fixed after 24 h and images were collected. Cultures were treated with either AP-YF (A and C) or AP-Scramble (B and D) peptides. Measured cell numbers are 105 (laminin + AP-YF), 106 (laminin + AP-Scramble), 322 (NgCAM + AP-YF), and 273 (NgCAM + AP-Scramble). Bar, 125 μm. (E) The percentage of neurons (y-axis, %) with neurites greater than length X (x-axis, μm). Note that treatment of neurons on NgCAM substrates with AP-YF peptide (closed circle) shifted the profile plot to the right compared with control treatment with AP-Scramble peptide (open circle), whereas peptide treatment did not affect profile plot of laminin substrate (squares). Control (BSA) substrate did not promote neurite extension (x). (F) Bar graph showing mean neurite length (μm; error bars represent SEM) for neurons grown under each condition (indicated at bottom).
Discussion
Here, we have presented evidence for multiple, discrete modes of kinetic behavior for L1CAM in the lipid bilayer, consistent with the roles of L1CAM in adhesion and migration. L1CAM function, as detected by changes in the diffusion kinetics of L1CAM in the bilayer, depends on at least two distinct interactions between L1CAM and the cytoskeleton, a conclusion supported by the following observations. First, stationary behavior is exquisitely sensitive to mutations and treatments that inhibit ankyrin binding, suggesting that ankyrin is a primary mediator of this activity. Second, L1CAM retrograde movement is completely inhibited by cytochalasin D or L1CAM cytoplasmic tail truncations, indicating that dynamic actin at the front of the cell drives L1CAM force generation. Third, treatments that inhibit ankyrin binding activate retrograde movement. Finally, ankyrin binding appears to play a role in regulating axon extension, raising the possibility that an increase in receptor retrograde movement on the cell surface is directly linked to cell motility.
Both retrograde and stationary behaviors depend on the interactions of the L1CAM cytoplasmic tail. L1CAM truncation mutants (Stop) diffuse freely on the cell surface, demonstrating that the L1CAM cytoplasmic domain is crucial for these phenomena, and suggesting that cis interactions with other receptors in the bilayer contribute little to the restricted movement of L1CAM in our assay system. The effects of cytochalasin D treatment suggest that dynamic actin in the cytosol mediates L1CAM retrograde movement. In combination with the velocity and direction of retrograde movement, these results strongly suggest that L1CAM, like other cell surface glycoproteins, associates with treadmilling actin in the lamella of the cell (Fig. 8 B, I; Felsenfeld et al., 1996; Suter et al., 1998; Lambert et al., 2002). However, the observed decrease in retrograde movement after nocadazole treatment raises the possibility that microtubules may also play some part in this process. Conditions that inhibit ankyrin binding, including L1-YH mutants, consistently stimulate retrograde movement, suggesting that ankyrin is not the primary adaptor in this process. In contrast, stationary behavior is independent of dynamic actin and microtubules, suggesting that a distinct cytoskeletal pool, perhaps the membrane skeleton, serves as a transient attachment site for L1CAM in the cytosol (Fig. 8 B, III). Mutations and treatments that inhibit or block ankyrin binding including L1-YH, growth factors, and the AP-YF peptide all inhibit stationary behavior of L1CAM, indicating that ankyrin binding to the FIGQY motif in L1CAM plays a primary role in this process.
Figure 8.

Model describing the regulation of L1CAM dynamics and function by ankyrin binding. (A) Schematic diagram illustrating the leading edge (lamella) of a cell in profile (gray) spread on a two-dimensional surface (black line with crosshatching). Cell is oriented toward the right-hand edge of the page. Treadmilling actin in the lamella of the cell (indicated by yellow chevrons and arrow) provides the force for moving cell surface glycoproteins like L1CAM (light blue) in a retrograde direction on the cell surface. The movement of L1CAM in the plane of the membrane is monitored by beads bound to L1CAM on the upper surface of the cell through a selective antibody (green). (B) Expanded view of area indicated by box in A. Ankyrin-binding domain of L1CAM (light blue) is indicated by oval in cytoplasmic tail. L1CAM–ankyrin interactions mediate the static behavior of L1CAM on the upper surface of the cell. This may be accomplished through ankyrin (red) binding to the membrane/spectrin cytoskeleton. Additionally, L1CAM interacts with dynamic components of the cytoskeleton, presumably treadmilling actin (yellow). These interactions offer a potential explanation for the distinct classes of L1CAM movement that we have observed. The indirect interaction between L1CAM and treadmilling actin in the absence of ankyrin binding results in the retrograde movement of cell surface L1CAM (i). In contrast, ankyrin binding in the absence of interactions with treadmilling actin gives rise to L1CAM that remains stationary on the cell surface (iii). Finally, both interactions together (ii) results in a regulated traction force generation where ankyrin binding modulates the ankyrin-independent generation of traction forces through cell surface L1CAM (smaller black arrow).
Analyses of L1CAM and L1 family members provide a list of potential candidates for adaptor proteins mediating retrograde movement. The ankyrin binding site in neurofascin, when phosphorylated, serves as a target for the protein doublecortin (Kizhatil et al., 2002). Directed movement can be detected in L1-YF mutants that are likely to be deficient in doublecortin binding (Kizhatil et al., 2002). Therefore, it is unlikely that doublecortin contributes to the retrograde movement of L1CAM. A distinct phosphorylation site in the L1CAM cytoplasmic tail (YRSLE) upstream of the ankyrin site binds both the μ2 chain of the AP-2 clathrin complex (Schaefer et al., 2002) and ERM proteins (Dickson et al., 2002). Although binding to AP-2 plays a critical role in the endocytosis and recycling of L1CAM at the back of the growth cone (Kamiguchi and Yoshihara, 2001), inhibition of the clathrin complex protein α-adaptin has no significant effect on L1CAM interactions with force-generating components of the cytoskeleton, as detected by the movement of L1CAM-bound beads on the cell surface (Kamiguchi and Yoshihara, 2001). However, in light of their known interaction with actin (Bretscher et al., 2002), ERM proteins remain potential candidates as adaptors between L1CAM and treadmilling actin.
The anti-coordinate regulation of retrograde and stationary behavior by ankyrin binding has important implications for the function of L1CAM in live cells. Although it is reasonable to assume that ankyrin binding modulates the activity of an independent cytoskeletal adaptor protein responsible for the interaction between L1CAM and treadmilling actin, the mechanism underlying this regulation has not been determined. First, ankyrin could serve as a competitive inhibitor for a distinct adaptor protein that binds at the same site on L1CAM. Although it is unlikely that doublecortin mediates L1CAM retrograde movement, we cannot preclude the existence of other proteins that bind at or near the FIGQY motif in the L1CAM tail. Second, ankyrin binding could induce a change in the conformation of the L1CAM tail that inhibits interactions with other adaptor proteins at a distance. Finally, the L1CAM tail may serve as a mechanical integrator of traction forces and as the restraining force provided by ankyrin binding (Fig. 8 B, II). According to this model, ankyrin binding would retard the movement of L1CAM being dragged backward through the membrane through an indirect interaction with treadmilling actin.
Previous work focusing on traction-force generation alone has shed little light on how cells regulate the transition between adhesion and migration (Felsenfeld et al., 1996; Choquet et al., 1997), a common occurrence in vivo. The experiments described here raise the possibility that the modulation of adhesion receptor–cytoskeleton interactions plays an essential role in the regulation of cell migration and adhesion by effecting changes in the capacity of these proteins to transfer traction forces to the extracellular environment. Moreover, the observation of a single receptor with three discrete kinetic behaviors on the cell surface is, to the best of our knowledge, entirely novel. The versatility of L1CAM in this respect may reflect a critical feature of the biology of L1CAM; the repertoire of L1 family members is relatively limited compared with other families of adhesion receptors (e.g., integrins, cadherins). Therefore, modulation of L1CAM-mediated neuronal growth may require post-translational regulation rather than a change in receptor expression. This type of direct modulation would also provide a means for rapidly promoting or arresting cell movement, which may be required during axon guidance. The capacity of a single cytosolic interaction to modulate the function of a receptor between static adhesion and traction-force generation raises the possibility that ankyrin binding may serve as a master switch for L1 function in these two classes of cell behavior, an idea that is strongly supported by the nerve growth–promoting properties of the inhibitory peptide. This form of direct modulation has important implications for the regulation of the shift from nerve growth to static adhesion during neural development.
Materials and methods
cDNA constructs
cDNA encoding full-length rat L1CAM including the neuron-specific RSLE exon were a gift of A. Furley (University of Sheffield, Sheffield, UK). The cDNA was modified by PCR to include a 10-aa myc epitope (EQKLISEEDL) 4 aa after the predicted amino terminus of the mature protein (IPDEQKLISEEDLYKGHH; inserted aa's indicated in bold case). The insertion is upstream of mini-exon 2, which has been shown to play a role in L1CAM binding to neural ligands (De Angelis et al., 2001). Mutations in the cytoplasmic tail were introduced by PCR using the QuikChange® protocol (Stratagene). Substitutions at tyrosine 1229 were effected by replacing codon TAC with either TTC (Y to F) or CAC (Y to H). All constructs were expressed using the bicistronic vector pIRES2-EGFP (CLONTECH Laboratories, Inc.).
L1CAM-GFP was generated by linking GFP2 (PerkinElmer) in frame to the carboxy terminus of the full-length wild-type L1CAM construct by PCR.
Ankyrin cell membrane recruitment assay
Constructs encoding full-length wild-type L1CAM including the RSLE mini-exon and either by an amino-terminal myc-epitope tag or by a carboxy-terminal GFP tag (see cDNA constructs above) were introduced by LipofectAMINE™ plus (Invitrogen) transfection into human kidney 293 cells. Cells were used 24–48 h after transfection or as stable pooled lines of L1CAM-expressing cells. L1CAM expression was detected by indirect immunofluorescence using an antibody against either L1CAM (rabbit anti-L1CAM; gift of Carl Lagenaur, University of Pittsburgh, Pittsburgh, PA), by 9E10 (mouse anti-myc; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), or by GFP distribution. In all cases, the results were indistinguishable. Ankyrin B was detected by indirect immunofluorescence using a mouse mAb (BD Biosciences). Confocal micrographs (Olympus) were collected at a plane intersecting cell–cell junctions. Control images collected by exciting fluorophores with the inappropriate laser line revealed no detectable crosstalk between channels.
Images were analyzed using NIH ImageJ (National Institutes of Health (NIH), Bethesda, MD) under Macintosh OSX. Densitometry was performed using a 5 pixel-wide line scan normal to the interface between two L1CAM-positive cells. Signal maximum for ankyrin staining at junction between cells was determined at the position of the maximal L1CAM staining to ensure that we were quantifying membrane rather that juxtamembrane staining. Minima were determined from the regions of the line overlapping the cytoplasm of either of the two cells. Membrane localization index was determined using the equation index = max/(max−min) (Oancea et al., 1998).
Immunofluorescence
For immunolocalization, cells were fixed for 10 min using 1% PFA in 60 mM Pipes, 25 mM Hepes, 10 mM EGTA, and 2 mM MgCl2 (PHEM; Schliwa and van Blerkom, 1981). Staining was performed as described previously (Felsenfeld et al., 1999). Micrographs were collected on a microscope (Axiovert 100TV; Carl Zeiss MicroImaging, Inc.) using a 100× plan neofluor objective (NA 1.4). Antigens were detected by indirect immunofluorescence using primary antibodies described in figure legends and secondary antibodies (either donkey anti–rabbit or donkey anti–mouse) conjugated to indocarbocyanin Cy3 (Jackson ImmunoResearch Laboratories). Micrographs were collected using a cooled CCD camera (CoolSNAP HQ™; Roper Scientific) under the control of ISee imaging software (ISee Imaging Systems). Images were subsequently processed in Photoshop® (Adobe) to maximize contrast, and were subject to an unsharp mask.
Antennapedia peptides
Inhibitory peptides were generated as a fusion between the 16-aa penetratin domain of antennapedia at the amino end and the ankyrin-binding domain of L1CAM with the carboxy-terminal tyrosine modified to phenylalanine (inhibitory peptide sequence RQIKIWFQNRRMKWKKQFNEDGSFIGQF). Control peptides were identical with a reversed ankyrin-binding domain (control peptide sequence RQIKIWFQNRRMKWKKFQGIFSGDENFQ). Both peptides included an amino-terminal biotin. Peptides were synthesized by FastMoc chemistry (Tufts University Core Facility, Boston, MA) and purified by HPLC yielding >97% purity as determined by mass spectrometry. Peptides were dissolved in HBSS at 1 mg/ml and diluted into cell culture medium at a final concentration of 1.4 μg/ml.
Bead preparation
Beads were prepared as described previously (Choquet et al., 1997; Felsenfeld et al., 1999). 1-μm carboxylated latex microspheres (Polyscience) were covalently coupled to ovalbumin (fraction VII; Sigma-Aldrich) using a carbodiimide linkage to neutralize the bead surface. Ovalbumin-coated beads were derivatized with Sulpho-NHS-LC-biotin (Pierce Chemical Co.). Beads at this stage were used fresh or stored for up to 2 wk at 4°C. Biotinylated beads were subsequently incubated with an excess of neutravidin (Molecular Probes, Inc.) overnight at 4°C. Beads were washed extensively, and a 15-μl aliquot (based on starting concentration) was incubated with biotinylated 9E10 antibody for 1 h (at RT) or overnight (at 4°C). Unreacted sites were blocked with biotin-BSA (BSA-biotinamidocaproyl; Sigma-Aldrich). Beads were sonicated for 5 s in a 0°C bath sonicator before experiments.
Cell culture and transfection
Neuroblastoma/DRG hybrid cells (ND-7) were transfected with constructs encoding either wild-type or mutant forms of L1CAM expressed in a bicistronic vector encoding EGFP after an internal ribosomal entry site (pIRES2-EGFP; CLONTECH Laboratories, Inc.). ND-7 cells were plated in L15 buffered for CO2 (supplemented with 1:1:2 glucose/glutamine/Pen-Strep and with dimethlytetrahydropterine, glutathione, and ascorbic acid; Cell and Molecular Technologies) containing 10% bovine calf serum (Hyclone) on coverslips 24 h before transfection. For video microscopy, medium was replaced before moving cells to the microscope with phenol red–free, serum-free L15 (air buffered) with 20 mM Hepes, 0.1% BSA, and 0.5% ovalbumin. Coverslips were silanized and laminin-coated for video microscopy as described previously (Felsenfeld et al., 1996). Alternatively, cells were plated on coverslips coated with poly-d-lysine and laminin sealed to the bottom of 35-mm culture dishes (MatTek Corporation). Transfections were performed using LipofectAMINE™ plus (Invitrogen), and cells were used 24–36 h later either live for video microscopy or fixed for immunohistochemistry.
Video microscopy, laser tweezers, and data analysis
Video microscopy was performed largely as described previously (Felsenfeld et al., 1999). All experiments were performed on a microscope (Axiovert 100 TV; Carl Zeiss MicroImaging, Inc.). Cells on laminin-coated coverslips were cultured in sealed chambers permitting illumination with a high resolution, oil immersion condenser. Images were collected and laser trap formed through a 100× plan neofluor NA 1.4 objective. The laser trap consisted of a Titanium Sapphire laser (model 890; Coherent) pumped by a 5-W Neodymium Vanadate laser (Coherent; Verdi) and tuned to 800 nm. Laser power at the output of the Ti:Sapp was 40 mW, and beads were placed and held on the cell surface for <3 s to further reduce the possibility of heating artifacts. Video images were collected using a Newvicon camera (VE-1000N; Dage MTI), and were subsequently digitized onto an Intel processor-based computer running the ISee software (ISee Imaging) for quantification. Diffusion analysis was performed using a custom spreadsheet in Excel (Microsoft). Individual traces were scored blind for classes of behavior. Statistical analysis of percentages of trials was performed using chi-squared analysis or a Fisher's exact probability test (Fig. 6 B). For cytoskeleton attachment assays (Fig. 2), beads were retested with a second pulse from the laser trap applied 0.5 μm from the bead center. Lateral movement of beads <0.2 μm was scored as rigidly attached.
Neurite outgrowth assays
For neurite outgrowth assays, 50 μg/ml purified chick NgCAM or 100 μg/ml mouse laminin (Invitrogen) was spotted on a 35-mm plastic culture dish at RT for 1 h. After washing with PBS, the plastic surface was blocked with 1% BSA/PBS at RT. Cerebellar cells were prepared from postnatal day 4 (P4) mouse by trypsinization followed by trituration. Cells were resuspended in BME/B27 with Pen/Strep (GIBCO BRL) at 5 × 105 cells/ml, and 250 μl was plated on the dishes. Cultures were incubated at 37°C for 24 h in 5% CO2. Antennapedia peptides dissolved in HBSS (GIBCO BRL) were added to the cultures at final concentration of 30 μg/ml when cells were plated. Cultures were fixed with 4% PFA, and images were captured as described earlier in Materials and methods. Neurite outgrowth measurements were performed by NIH image software and processed with Microsoft Excel. P values were determined using t test analysis.
Acknowledgments
The authors would like to thank Vann Bennett, Deanna Benson, Andrew Furley, Catherine Galbraith, and John Whittard for comments on the manuscript, and Vann Bennett for many helpful conversations during the preparation of this work. The generation of L1CAM mutant constructs was carried out in the laboratory of Vann Bennett (Department of Cell Biology, Duke University, Durham, NC), and early work on this project was carried out with support from Michael P. Sheetz. The monoclonal mouse anti-ankyrin B, developed by V. Bennett and coworkers, was obtained from BD Biosciences. The monoclonal antibody 9E10, developed by J.M. Bishop and coworkers, was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biological Sciences (Iowa City, IA). The authors also wish to acknowledge the technical support of Mihaela Gazdoiu and the help of Barney Yoo synthesizing the early versions of the inhibitory peptides. Inhibitory peptides used in this manuscript were synthesized by the Tufts University Core Facility.
This work was supported by NIH grant GM63192-01 and by a grant from the Speaker's Fund of the New York Academy of Medicine. D.P. Felsenfeld is the Dale F. and Betty Ann Frey Scholar of the Damon Runyon Cancer Research Foundation.
A.E. Bradley's present address is Department of Environmental Health, University of Washington, Seattle, WA 98195.
J.A. Kuo's present address is Medical College of Georgia, Augusta, GA 30912.
Abbreviations used in this paper: ERM, ezrin, radixin, moesin; IgCAM, immunoglobulin superfamily cell adhesion molecule; L1CAM, L1 cell adhesion molecule.
References
- Bretscher, A., K. Edwards, and R.G. Fehon. 2002. ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3:586–599. [DOI] [PubMed] [Google Scholar]
- Choquet, D., D.P. Felsenfeld, and M.P. Sheetz. 1997. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 88:39–48. [DOI] [PubMed] [Google Scholar]
- Cohen, N.R., J.S. Taylor, L.B. Scott, R.W. Guillery, P. Soriano, and A.J. Furley. 1998. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr. Biol. 8:26–33. [DOI] [PubMed] [Google Scholar]
- Dahme, M., U. Bartsch, R. Martini, B. Anliker, M. Schachner, and N. Mantei. 1997. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat. Genet. 17:346–349. [DOI] [PubMed] [Google Scholar]
- Davis, J.Q., and V. Bennett. 1994. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J. Biol. Chem. 269:27163–27166. [PubMed] [Google Scholar]
- De Angelis, E., T. Brummendorf, L. Cheng, V. Lemmon, and S. Kenwrick. 2001. Alternative use of a mini exon of the L1 gene affects L1 binding to neural ligands. J. Biol. Chem. 276:32738–32742. [DOI] [PubMed] [Google Scholar]
- Derossi, D., G. Chassaing, and A. Prochiantz. 1998. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 8:84–87. [PubMed] [Google Scholar]
- Dickson, T.C., C.D. Mintz, D.L. Benson, and S.R. Salton. 2002. Functional binding interaction identified between the axonal CAM L1 and members of the ERM family. J. Cell Biol. 157:1105–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil, R.R., G. MacVicar, S. Dissanayake, C. Liu, D. Homer, and M. Hortsch. 1996. Neuroglian-mediated cell adhesion induces assembly of the membrane skeleton at cell contact sites. J. Cell Biol. 133:647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, P.M., P.R. Coote, J.N. Wood, G.M. Burgess, and H.P. Rang. 1991. Bradykinin evoked depolarization of a novel neuroblastoma x DRG neurone hybrid cell line (ND7/23). Brain Res. 545:80–86. [DOI] [PubMed] [Google Scholar]
- Evan, G.I., G.K. Lewis, G. Ramsay, and J.M. Bishop. 1985. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 5:3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenfeld, D.P., M.A. Hynes, K.M. Skoler, A.J. Furley, and T.M. Jessell. 1994. TAG-1 can mediate homophilic binding, but neurite outgrowth on TAG-1 requires an L1-like molecule and beta 1 integrins. Neuron. 12:675–690. [DOI] [PubMed] [Google Scholar]
- Felsenfeld, D.P., D. Choquet, and M.P. Sheetz. 1996. Ligand binding regulates the directed movement of beta1 integrins on fibroblasts. Nature. 383:438–440. [DOI] [PubMed] [Google Scholar]
- Felsenfeld, D.P., P.L. Schwartzberg, A. Venegas, R. Tse, and M.P. Sheetz. 1999. Selective regulation of integrin–cytoskeleton interactions by the tyrosine kinase Src. Nat. Cell Biol. 1:200–206. [DOI] [PubMed] [Google Scholar]
- Fransen, E., V. Lemmon, G. Van Camp, L. Vits, P. Coucke, and P.J. Willems. 1995. CRASH syndrome: clinical spectrum of corpus callosum hypoplasia, retardation, adducted thumbs, spastic paraparesis and hydrocephalus due to mutations in one single gene, L1. Eur. J. Hum. Genet. 3:273–284. [DOI] [PubMed] [Google Scholar]
- Galbraith, C.G., and M.P. Sheetz. 1999. Keratocytes pull with similar forces on their dorsal and ventral surfaces. J. Cell Biol. 147:1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garver, T.D., Q. Ren, S. Tuvia, and V. Bennett. 1997. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J. Cell Biol. 137:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, A.K., P. Wild, and D. Stopak. 1980. Silicone rubber substrata: a new wrinkle in the study of cell locomotion. Science. 208:177–179. [DOI] [PubMed] [Google Scholar]
- Hortsch, M. 2000. Structural and functional evolution of the L1 family: are four adhesion molecules better than one? Mol. Cell. Neurosci. 15:1–10. [DOI] [PubMed] [Google Scholar]
- Hortsch, M., K.S. O'Shea, G. Zhao, F. Kim, Y. Vallejo, and R.R. Dubreuil. 1998. A conserved role for L1 as a transmembrane link between neuronal adhesion and membrane cytoskeleton assembly. Cell Adhes. Commun. 5:61–73. [DOI] [PubMed] [Google Scholar]
- Kamiguchi, H., and V. Lemmon. 2000. IgCAMs: bidirectional signals underlying neurite growth. Curr. Opin. Cell Biol. 12:598–605. [DOI] [PubMed] [Google Scholar]
- Kamiguchi, H., and F. Yoshihara. 2001. The role of endocytic L1 trafficking in polarized adhesion and migration of nerve growth cones. J. Neurosci. 21:9194–9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizhatil, K., Y.X. Wu, A. Sen, and V. Bennett. 2002. A new activity of doublecortin in recognition of the phospho-FIGQY tyrosine in the cytoplasmic domain of neurofascin. J. Neurosci. 22:7948–7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, T.B., E.T. Stoeckli, M.A. Condrau, F.G. Rathjen, and P. Sonderegger. 1991. Neurite outgrowth on immobilized axonin-1 is mediated by a heterophilic interaction with L1(G4). J. Cell Biol. 115:1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, M., D. Choquet, and R.M. Mege. 2002. Dynamics of ligand-induced, Rac1-dependent anchoring of cadherins to the actin cytoskeleton. J. Cell Biol. 157:469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon, V., K.L. Farr, and C. Lagenaur. 1989. L1-mediated axon outgrowth occurs via a homophilic binding mechanism. Neuron. 2:1597–1603. [DOI] [PubMed] [Google Scholar]
- Miura, M., M. Kobayashi, H. Asou, and K. Uyemura. 1991. Molecular cloning of cDNA encoding the rat neural cell adhesion molecule L1. Two L1 isoforms in the cytoplasmic region are produced by differential splicing. FEBS Lett. 289:91–95. [DOI] [PubMed] [Google Scholar]
- Needham, L.K., K. Thelen, and P.F. Maness. 2001. Cytoplasmic domain mutations of the L1 cell adhesion molecule reduce L1-ankyrin interactions. J. Neurosci. 21:1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oancea, E., M.N. Teruel, A.F. Quest, and T. Meyer. 1998. Green fluorescent protein (GFP)-tagged cysteine-rich domains from protein kinase C as fluorescent indicators for diacylglycerol signaling in living cells. J. Cell Biol. 140:485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollerberg, G.E., J. Davoust, and M. Schachner. 1990. Lateral mobility of the cell adhesion molecule L1 within the surface membrane of morphologically undifferentiated and differentiated neuroblastoma cells. Eur. J. Neurosci. 2:712–717. [DOI] [PubMed] [Google Scholar]
- Rutishauser, U. 2000. Defining a role and mechanism for IgCAM function in vertebrate axon guidance. J. Cell Biol. 149:757–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, A.W., Y. Kamei, H. Kamiguchi, E.V. Wong, I. Rapoport, T. Kirchhausen, C.M. Beach, G. Landreth, S.K. Lemmon, and V. Lemmon. 2002. L1 endocytosis is controlled by a phosphorylation-dephosphorylation cycle stimulated by outside-in signaling by L1. J. Cell Biol. 157:1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliwa, M., and J. van Blerkom. 1981. Structural interaction of cytoskeletal components. J. Cell Biol. 90:222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter, D.M., L.D. Errante, V. Belotserkovsky, and P. Forscher. 1998. The Ig superfamily cell adhesion molecule, apCAM, mediates growth cone steering by substrate-cytoskeletal coupling. J. Cell Biol. 141:227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier-Lavigne, M., and C.S. Goodman. 1996. The molecular biology of axon guidance. Science. 274:1123–1133. [DOI] [PubMed] [Google Scholar]
- Tuvia, S., T.D. Garver, and V. Bennett. 1997. The phosphorylation state of the FIGQY tyrosine of neurofascin determines ankyrin-binding activity and patterns of cell segregation. Proc. Natl. Acad. Sci. USA. 94:12957–12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X., J.Q. Davis, S. Carpenter, and V. Bennett. 1998. Structural requirements for association of neurofascin with ankyrin. J. Biol. Chem. 273:30785–30794. [DOI] [PubMed] [Google Scholar]
- Zisch, A.H., W.B. Stallcup, L.D. Chong, K. Dahlin-Huppe, J. Voshol, M. Schachner, and E.B. Pasquale. 1997. Tyrosine phosphorylation of L1 family adhesion molecules: implication of the Eph kinase Cek5. J. Neurosci. Res. 47:655–665. [DOI] [PubMed] [Google Scholar]