

Abstract
Sympathetic neurons depend on NGF binding to TrkA for their survival during vertebrate development. NGF deprivation initiates a transcription-dependent apoptotic response, which is suggested to require activation of the transcription factor c-Jun. Similarly, apoptosis can also be induced by selective activation of the p75 neurotrophin receptor. The transcriptional dependency of p75-mediated cell death has not been determined; however, c-Jun NH2-terminal kinase has been implicated as an essential component. Because the c-jun–null mutation is early embryonic lethal, thereby hindering a genetic analysis, we used the Cre-lox system to conditionally delete this gene. Sympathetic neurons isolated from postnatal day 1 c-jun–floxed mice were infected with an adenovirus expressing Cre recombinase or GFP and analyzed for their dependence on NGF for survival. Cre immunopositive neurons survived NGF withdrawal, whereas those expressing GFP or those uninfected underwent apoptosis within 48 h, as determined by DAPI staining. In contrast, brain-derived neurotrophic factor (BDNF) binding to p75 resulted in an equivalent level of apoptosis in neurons expressing Cre, GFP, and uninfected cells. Nevertheless, cycloheximide treatment prevented BDNF-mediated apoptosis. These results indicate that whereas c-jun is required for apoptosis in sympathetic neurons on NGF withdrawal, an alternate signaling pathway must be induced on p75 activation.
Keywords: neurotrophin; apoptosis; Jun; Trk; nerve growth factor
Introduction
During normal mammalian development, approximately half of the neurons generated undergo programmed cell death before adulthood (Oppenheim, 1991). Disruption of this naturally occurring neuronal loss during development can result in embryonic lethality and abnormal apoptosis, and is associated with pathological conditions such as Parkinson's and Alzheimer's diseases as well as ischemic injury after stroke (Yuan and Yankner, 2000). During the formation of the mammalian nervous system, neurons send out processes to their target tissues where they compete for limiting amounts of trophic factors. Among these target-derived trophic factors are the neurotrophins, including NGF, brain-derived neurotrophic factor (BDNF),* neurotrophin-3 (NT3), and NT4 (Huang and Reichardt, 2001), which act through binding to two types of receptors, the Trks, a family of tyrosine kinase receptors (Barbacid, 1994), and p75, a member of the TNF receptor family (Locksley et al., 2001). The neurotrophins promote neuronal survival through binding selectively to the Trks, NGF binds to TrkA, BDNF and NT4 to TrkB, and NT3 to TrkC (Barbacid, 1994). In contrast, all of the neurotrophins bind with similar affinity to the p75 receptor (Rodriguez-Tebar et al., 1991), which can function to promote survival, or to induce cell death (Coulson et al., 1999).
One of the neuronal populations best characterized for its dependence on neurotrophins is the superior cervical ganglia (SCG). This group of sympathetic neurons expresses the TrkA and p75 receptors, and is dependent on NGF for survival and differentiation during development. Some of the developmental apoptosis of sympathetic neurons has also been suggested to be the result of BDNF binding to p75 (Bamji et al., 1998). These apoptotic responses can also be recapitulated in cultured SCG neurons by NGF withdrawal (Martin et al., 1988) or by the addition of BDNF to neurons maintained in depolarizing levels of potassium (Bamji et al., 1998).
The molecular mechanisms by which NGF removal induces apoptosis have been shown to involve cytochrome c translocation from the mitochondria to the cytosol, which is dependent on the proapoptotic BCL-2 family members, BAX (Deckwerth et al., 1996; Lentz et al., 1999) and BIM (Putcha et al., 2001; Whitfield et al., 2001). After cytochrome c release, a cascade of caspase activation occurs, resulting in the classical characteristics of apoptosis. However, microinjection of cytochrome c into neurons is not sufficient to induce cell death, thus NGF withdrawal activates additional pathways that induce a condition referred to as “competence to die” (Deshmukh and Johnson, 1998). This “competence” likely involves the up-regulation of proapoptotic genes because apoptosis induced by NGF withdrawal is transcription and translation dependent (Martin et al., 1988). The increase in transcription has been attributed to the stabilization of the tumor suppressor p53 (Aloyz et al., 1998; Pozniak et al., 2000) and activation of c-Jun, a component of the AP-1 transcription factor (Estus et al., 1994; Ham et al., 1995). Analysis of the c-jun−/− mice has not been possible due to early embryonic lethality (Hilberg et al., 1993; Johnson et al., 1993); however, an increase in both c-Jun mRNA (Estus et al., 1994) and the activated, phosphorylated form of the protein (Ham et al., 1995) have been observed after NGF withdrawal. In addition, microinjection of antibodies to c-Jun (Estus et al., 1994), or introduction of a cDNA encoding a mutant c-Jun, into SCG neurons (Ham et al., 1995; Whitfield et al., 2001) was demonstrated to protect them from death after NGF removal. However, these experiments should be interpreted with caution because there may be excessively high levels of ectopic protein and the mutant c-Jun may still dimerize with wild-type (wt) AP-1 members and alter transcription of multiple AP-1-responsive genes.
The mechanism by which activation of the p75 receptor induces apoptosis is much less understood. Similar to NGF withdrawal, neurotrophin binding selectively to p75 has been shown to increase phosphorylation of c-Jun (Bamji et al., 1998) and to activate the upstream kinase, c-Jun NH2-terminal kinase (JNK; Casaccia-Bonnefil et al., 1996). Furthermore, blocking the activation of JNK with a pharmacological inhibitor (Yoon et al., 1998) or a dominant-negative JNK (Harrington et al., 2002), prevented p75-mediated apoptosis of oligodendrocytes. However, the requirement for c-Jun in p75 signaling cell death has not been investigated, nor has it been determined whether this is a transcription-dependent process.
To address the role of c-jun in the apoptosis of SCG neurons, we generated a conditional c-jun–null allele by flanking it with loxP sites, and used adenovirally delivered Cre recombinase to delete the gene in SCG neurons. Our findings demonstrate that c-Jun is essential for neuronal cell death after NGF deprivation, but not by neurotrophin binding to p75. Nevertheless, p75-mediated apoptosis was dependent on macromolecular synthesis because it was blocked by cycloheximide. Therefore, we propose that there are divergent, transcriptionally dependent pathways initiated by these two inducers of apoptosis.
Results
Generation of c-jun fl/fl mice
It has been proposed that programmed cell death induced by NGF withdrawal in sympathetic neurons requires activation of the transcription factor c-Jun. To address the role of c-Jun in this process, we generated mice with the c-jun allele flanked by loxP sites, thus allowing its deletion at any developmental stage through the introduction of Cre recombinase. We constructed a mutant c-jun allele with the coding exon for c-Jun flanked by loxP sites (Fig. 1). ES cell clones were selected, and those with homologous recombination were identified (Fig. 1, B and C). Because the neo cassette was also flanked by loxP sites, this gene was removed by transient transfection with pCMV-Cre, and clones were selected that contained the floxed c-jun allele (c-jun fl; Fig. 1 D). These cells were used to generate c-jun fl/wt mice that were mated with c-jun+/− to create a c-jun fl/fl on a c-jun–null background (Fig. 1 E). The viability of the c-jun fl/fl mice suggests that the loxP sites do not disrupt the normal function of c-Jun because deletion of this gene results in embryonic lethality (Hilberg et al., 1993; Johnson et al., 1993).
Figure 1.

The generation of floxed c-jun mice. (A) Schematic diagrams of the c-jun gene, the targeted locus, and the final floxed allele with the neo cassette excised and the null allele resulting from Cre-mediated excision of c-jun. The probes used for screening the Southern blots for detecting the insertion of the neo cassette (probe A) and to assess the incorporation of the 3′ loxP and BamHI site (probe B) are indicated, as are the PCR primers used for detecting the incorporation of the 5′ loxP site. (B) wt (lane 1) or 2 lines of targeted ES cell (lanes 2 and 3) DNA was digested with XbaI-EcoRI and hybridized with probe A, which detects a fragment of 1.9 kb (circle) from wt, and 2.8 kb (arrowhead) from the targeted allele containing the neo gene. (C) The 3′ loxP site is intact, as shown by the presence of the adjacent BamHI site. DNA from wt ES cells (lane 4) or a targeted clone (lane 5) was digested with BamHI and hybridized with probe B to reveal a 4.9-kb band (arrowhead) from the wt locus or a 2.2-kb band (circle) from the targeted allele. (D) The neo gene was excised by transfection with pCMV-Cre. DNA from a targeted line (the same as lane 2) before (lane 6) and after (lanes 7 and 8) transfection with Cre was digested with XbaI-EcoRI and hybridized with probe A. The 1.9-kb band is the same in the wt and excised allele, whereas the loss of the 2.7-kb band (arrowhead) indicates removal of neo to give the final floxed c-jun allele. (E) Evidence for germline transmission of the floxed allele was obtained using primers flanking the loxP sites to amplify DNA from c-jun fl/wt, c-jun fl/fl, and wt mice.
c-jun is essential for sympathetic neuron death in response to NGF withdrawal
Sympathetic neurons from the SCG, isolated from postnatal day 1 mouse pups, depend on NGF for their survival. Upon removal of this trophic factor, the neurons undergo programmed cell death, which was evaluated 48 h later by examining nuclear morphology using DAPI staining. Apoptotic neurons are apparent by their condensed or fragmented nuclei (see Fig. 3). To evaluate the necessity of the AP-1 family member c-Jun in neuronal apoptosis, we infected SCG neurons from c-jun fl/fl mice with an adenovirus expressing Cre recombinase to delete the c-jun allele. To confirm that c-jun was deleted, the neurons from the c-jun fl/fl mice or wt were infected with the Cre-adenovirus overnight, and thereafter the NGF was withdrawn. 12 h after the withdrawal, the expression of c-Jun was evaluated, specifically in the Cre-expressing neurons, by coimmunostaining. At this time, c-Jun was up-regulated (see Fig. 5), but it was before the time when the nuclei became apoptotic. Fig. 2 A shows representative immunofluorescence images of Cre recombinase and c-Jun expression patterns of neurons isolated from c-jun fl/fl or wt mice after NGF withdrawal. Both Cre recombinase and c-Jun, which are nuclear proteins, were observed in the wt neurons exclusively localized to the nucleus. In contrast, c-Jun was not detected in any Cre-immunopositive sympathetic neurons isolated from c-jun fl/fl mice. To further verify the deletion of the c-jun gene, the genomic DNA was isolated from the neurons 24 h after infection with GFP or Cre-expressing adenovirus, and c-jun was amplified by PCR (Fig. 2 B). No c-jun signal was detected from the Cre-infected cultures, thus indicating that the ectopically expressed Cre recombinase successfully deleted the c-jun alleles.
Figure 3.
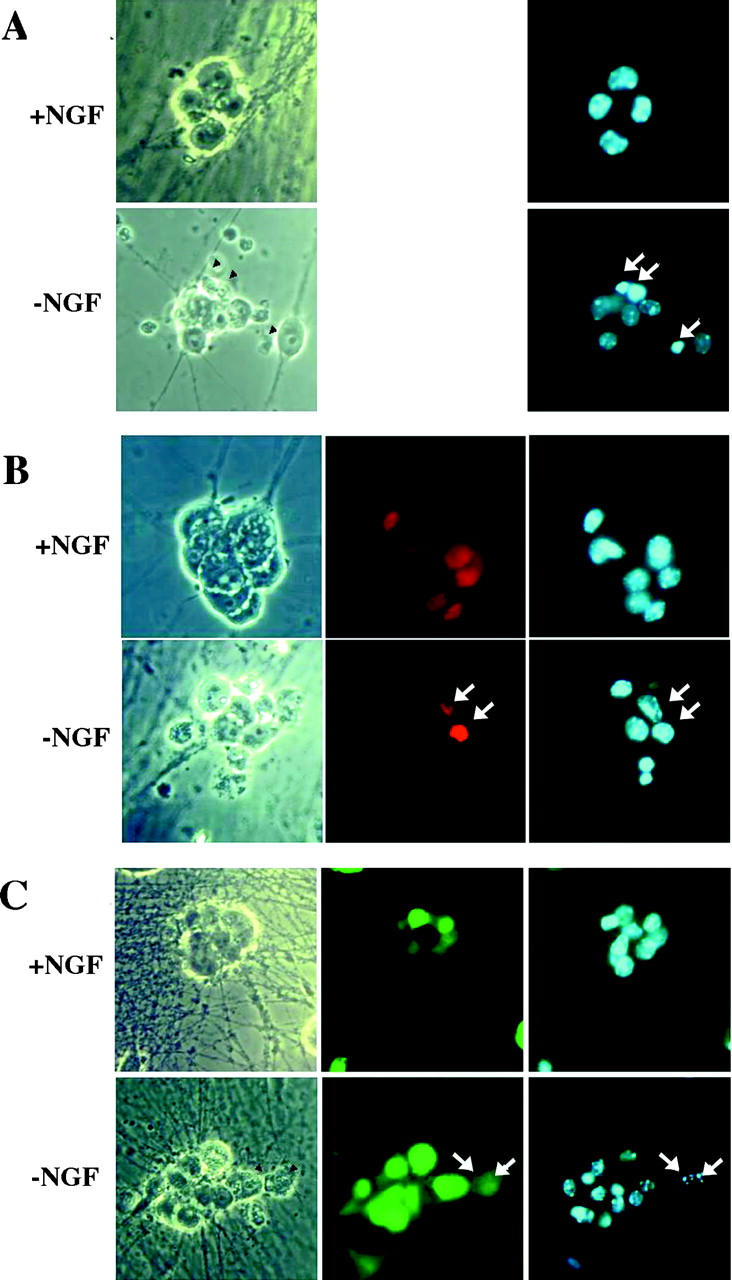

c-Jun is essential for sympathetic neuron death upon NGF withdrawal. Sympathetic neurons isolated from c-jun fl/fl mice were uninfected (A) or infected with adenovirus expressing Cre recombinase (B) or GFP (C). After an overnight incubation, the neurons were rinsed and refed with media lacking NGF and supplemented with a neutralizing anti-NGF antibody (−NGF), or with fresh NGF containing media (+NGF). After an additional 48 h, the neurons were fixed and stained with a biotinylated Cre antibody and streptavidin-Cy3 to detect Cre-positive neurons (red), or were visualized by GFP fluorescence (green). The nuclei of all neurons were costained with DAPI (blue) to evaluate apoptotic profiles. In the representative neurons depicted, the arrows indicate apoptotic, fragmented nuclei, except in Cre-infected, resistant cells. (D) Cell death expressed as percentage of apoptotic nuclei. In cells infected with the adenovirus, only GFP-expressing or Cre-immunopositive cells were considered. Shown are the mean and SEM from four independent experiments.
Figure 5.

Kinetics of c-Jun induction and apoptosis after NGF withdrawal or BDNF addition. Sympathetic neurons were cultured with NGF for 3–5 d, rinsed, and refed with media containing 20 ng/ml NGF (+NGF) or lacking NGF and containing a neutralizing anti-NGF antibody, without (−NGF) or with 12.5 mM KCl and 100 ng/ml BDNF (+BDNF), or no factor (−BDNF). After the given time, the neurons were fixed and immunostained for c-Jun and costained with DAPI to visualize the nuclei. Representative neurons stained with anti–c-Jun are depicted at various time points after NGF withdrawal (−NGF) or cultured in KCl and treated with 100 ng/ml BDNF (+BDNF) in A. The percentage of neuronal nuclei that were c-Jun immunopositive under these conditions was determined. Depicted are the mean ± SEM for three experiments, with at least 100 neurons/condition counted for each experiment (B). The percentage of neuronal nuclei that were apoptotic was determined by DAPI staining. Depicted are the means for three experiments, with at least 100 neurons/condition counted for each experiment (C). ▵, +BDNF; ▪, −NGF.
Figure 2.

Cre-mediated deletion of c-jun. Sympathetic neurons isolated from c-jun floxed (c-jun fl/fl) or wt mice were cultured for 4 d with NGF, then were infected overnight with an adenovirus carrying Cre recombinase (A). The neurons were rinsed and refed with media lacking NGF and supplemented with a neutralizing anti-NGF antibody (−NGF) and the cells were fixed 12 h later. Co-immunostaining with antibodies to Cre recombinase (green) and c-Jun (red) was performed to identify Cre-infected neurons and to detect c-Jun expression. Neurons were also stained with DAPI (blue) to detect apoptotic profiles. Note that c-Jun was not detected in Cre- immunopositive neurons from c-jun fl/fl mice. (B) Genomic DNA was isolated from sympathetic neurons, uninfected or 48 h after infection with adenovirus expressing Cre recombinase or GFP, and PCR was performed to detect the presence of c-jun and, as control, the p65 subunit of NF-κB.
The requirement for c-Jun in mediating the apoptotic response to NGF withdrawal in sympathetic neurons was demonstrated by infecting the c-jun fl/fl neurons with the Cre-adenovirus. SCG neurons isolated from c-jun fl/fl mice were uninfected, infected with Cre recombinase or, as control, with GFP-expressing adenovirus. 24 h later, the cells were rinsed to remove the NGF, maintained for an additional 48 h in medium containing NGF or NGF-free medium, and were fixed; the nuclei of the GFP-expressing or Cre-immunopositive neurons were evaluated for apoptosis. All neurons maintained in NGF contained smooth, round nuclei; however, in neurons deprived of NGF, apoptotic nuclei were observed in uninfected and GFP-expressing neurons, but not in neurons expressing Cre recombinase (Fig. 3). To quantify the extent of apoptosis, the percentage of cells with apoptotic nuclei was determined. As depicted in Fig. 3 D, 48 h after NGF removal, 59% of uninfected and 53% of GFP-expressing neurons displayed condensed or fragmented nuclei, whereas only 14% of the Cre recombinase-expressing neurons were apoptotic. The survival of the neurons was also evaluated 9 d after NGF withdrawal. Because most of the dead cells were no longer visible by this time, the number of viable, phase bright neurons containing neurites at least the length of the somal diameter was determined. Relative to cultures maintained in NGF, only 30.6% of the uninfected and 29.6% of the GFP-infected neurons were still alive; however 58.1% of the neurons infected with Cre were still alive 9 d after NGF withdrawal (at least 50 neurons were counted under each condition and experiments were done in duplicate, n = 2). Thus, deletion of the gene for the transcription factor c-Jun abrogated programmed cell death after trophic factor withdrawal in these neurons.
c-jun is not necessary for sympathetic neuron death in response to p75 activation
Neurotrophin binding selectively to the p75 receptor can activate an apoptotic program in a variety of neural cells (Barrett, 2000). In sympathetic neurons, which express only the neurotrophin receptors p75 and TrkA, p75 can be selectively activated by BDNF, and this was reported to lead to cell death (Bamji et al., 1998). To culture these neurons in the absence of NGF, the neurons were kept in mildly depolarizing media containing 12.5 mM KCl. Similar to previous findings, we observed an 87% increase in the number of apoptotic nuclei in cultured SCG neurons after a 48-h treatment with BDNF (Figs. 4–6). Moreover, this effect could be inhibited with an antibody to the extracellular domain of p75, confirming the involvement of this receptor (Fig. 4).
Figure 4.

BDNF induces apoptosis through the p75 receptor. Sympathetic neurons isolated from SCG of postnatal day 1 mice were cultured 3–5 d with 20 ng/ml NGF. The neurons were rinsed and refed with media lacking NGF and containing a neutralizing anti-NGF antibody and 12.5 mM KCl with (+BDNF) or without (−BDNF) 100 ng/ml BDNF, to activate p75. In some experiments, antibodies to the extracellular domain of p75 were included (diluted 1:500). After 48 h, the neurons were fixed and apoptotic cells were identified by their nuclear profile using DAPI staining. The percentage of apoptotic nuclei was determined by counting at least 50 neurons in six independent experiments, and the mean and SEM are shown. Only the neurons treated with BDNF and no antibody were significantly different from the others, based on an ANOVA analysis and a Tukey's multiple comparison test.
Figure 6.

Apoptosis induced by p75 activation is dependent on protein synthesis. Sympathetic neurons were cultured with NGF for 3–5 d, rinsed, and refed with media lacking NGF and containing a neutralizing anti-NGF antibody, 12.5 mM KCl, and 100 ng/ml BDNF (+BDNF), or no factor (−BDNF) in the presence (+CHX) or absence (−CHX) of 1 μg/ml cycloheximide. 2 d thereafter, the neurons were fixed and cell death was evaluated by DAPI staining. The percentage of apoptotic nuclei was determined by counting at least 50 neurons in three independent experiments. Depicted are the mean and SEM. Only the neurons treated with BDNF and no cycloheximide were significantly different from the others, based on an ANOVA analysis and a Tukey's multiple comparison test.
The mechanism by which activation of the p75 receptor induces apoptosis of sympathetic neurons is poorly understood. Similar to NGF withdrawal, neurotrophin binding selectively to p75 increases the level of phosphorylated c-Jun (Bamji et al., 1998) and the activity of the upstream kinase, c-Jun NH2-terminal kinase (JNK; Casaccia-Bonnefil et al., 1996). Furthermore, the inhibition of JNK activation prevents p75-mediated apoptosis (Yoon et al., 1998; Harrington et al., 2002). However, the requirement for c-Jun in p75 mediated cell death has not been investigated, nor has it been determined whether this is a transcription-dependent process, like trophic factor withdrawal. To characterize the induction of c-Jun after BDNF treatment, its temporal induction by p75 activation was compared with NGF withdrawal. Although the increase in c-Jun expression by BDNF slightly preceded that of NGF removal, the percentage of neurons with clearly nuclear c-Jun immunoreactivity peaked ∼18 h and was similar in magnitude following either treatment (Fig. 5, A and B). The kinetics of neuronal apoptosis were also similar after removal of NGF or addition of BDNF, peaking ∼24 h, slightly after the maximum c-Jun expression (Fig. 5 C).
To determine whether protein synthesis is required for cell death activated by p75, SCGs isolated from wt mice were incubated with cycloheximide at a concentration of 1 μg/ml at the time of NGF removal and BDNF treatment. 48 h thereafter, apoptosis was assessed by nuclear morphology after DAPI staining. In the presence of this protein translation inhibitor, BDNF-induced cell death was completely abrogated (Fig. 6). These results suggest that, like trophic factor withdrawal, apoptosis induced by p75 activation is also a transcription-dependent process.
Because c-Jun was required for the neurons to activate their cell death program after NGF removal, we wanted to determine whether this transcription factor is also an essential component for p75-induced cell death. Therefore, neurons isolated from c-jun fl/fl mice were uninfected or infected with the adenovirus expressing Cre recombinase or GFP, then maintained for 48 h in medium containing 12.5 mM KCl, with or without 100 ng/ml BDNF. The neurons were fixed and stained with an antibody against Cre recombinase to detect Cre-immunopositive neurons, and with DAPI to detect apoptotic profiles. Surprisingly, the addition of BDNF to the medium induced an equivalent amount of death in neurons that were uninfected (50.2 ± 4.9%), infected with adeno-GFP (50.5 ± 2.1%), and those expressing Cre recombinase (57.1 ± 2.7%; Fig. 7), indicating that c-Jun is not an essential component in sympathetic neuronal death induced by p75 activation.
Figure 7.

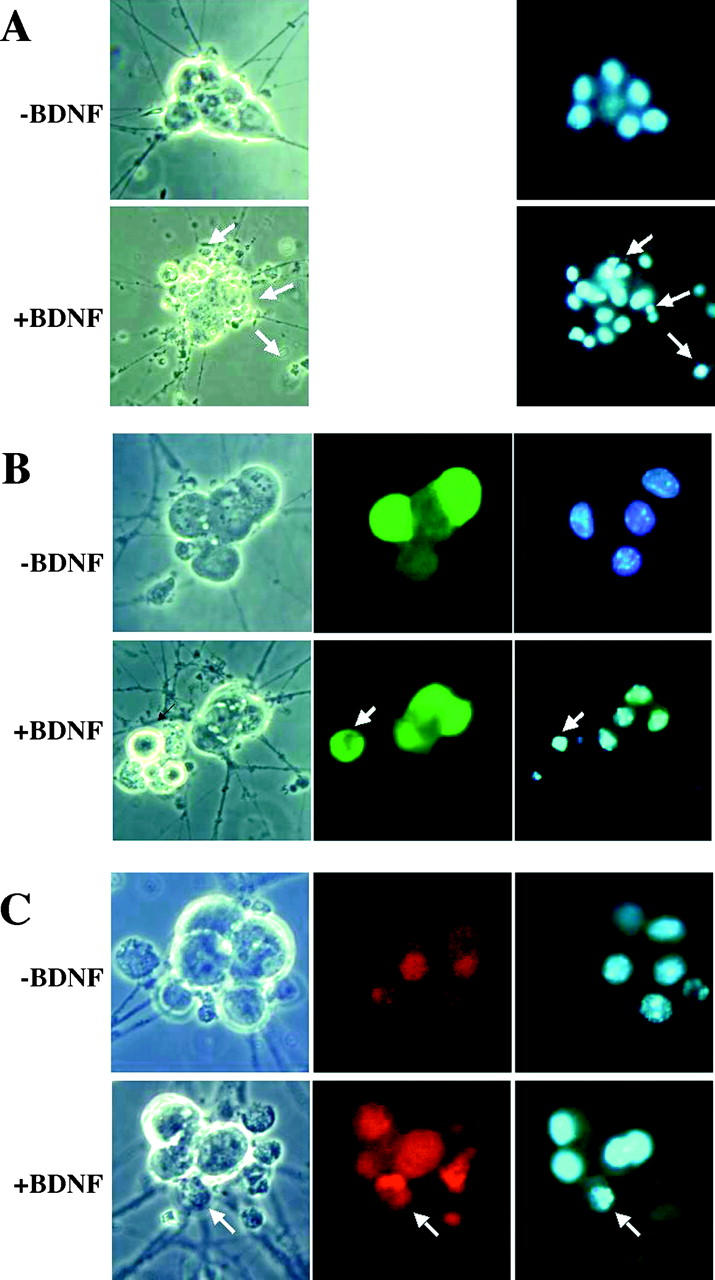
c-Jun is not required for p75-mediated apoptosis in sympathetic neurons. Sympathetic neurons isolated from c-jun fl/fl mice and cultured with NGF were uninfected (A) or infected with an adenovirus expressing GFP (B) or Cre recombinase (C). After an overnight incubation, the neurons were rinsed and refed with media lacking NGF and supplemented with a neutralizing anti-NGF antibody and 12.5 mM KCl in the absence or presence of 100 ng/ml BDNF to activate p75. After 48 h, the neurons were fixed and visualized by GFP fluorescence (green) or stained with a biotinylated anti-Cre antibody and streptavidin-Cy3 to detect Cre-positive neurons (red). The nuclei of all neurons were costained with DAPI (blue) to evaluate apoptotic profiles. In the representative neurons depicted, the arrows indicate apoptotic, fragmented nuclei. (D) Cell death is expressed as a percentage of nuclei that were apoptotic. In cultures infected with adenovirus expressing Cre or GFP, only the Cre-immunopositive or GFP-fluorescent neurons were considered. Shown are the mean and SEM from four independent experiments.
Discussion
The neuronal cell death that occurs during mammalian development has been shown to be, in part, due to competition for a limited amount of neurotrophin (Huang and Reichardt, 2001). In addition, it has also been suggested that activation of the p75 neurotrophin receptor induces cell death of inappropriately targeted or injured neurons (Bamji et al., 1998). Previous findings suggested that both of these apoptotic processes depend on activation of the JNK (Deshmukh and Johnson, 1997; Yoon et al., 1998). Moreover, we demonstrate that the kinetics and degree of c-Jun induction are similar for BDNF treatment and removal of NGF. Nevertheless, we found a differential requirement for c-Jun. The cell death after NGF removal was dependent on the presence of c-Jun; however, p75-induced apoptosis did not require this transcription factor, despite being sensitive to cycloheximide.
Sympathetic neurons have been well characterized as an in vitro model for neuronal apoptosis induced by trophic factor deprivation. Removal of NGF from the cultures activates a cell death program that is dependent on transcription and translation (Martin et al., 1988). One of the first genes shown to be induced after NGF withdrawal was the protooncogene c-jun (Estus et al., 1994). This transcription factor is an immediate early gene that up-regulates its own mRNA. It functions to drive gene expression by homo- or heterodimerizing with other members of the AP-1 family, including c-fos, Fos B, Fra 1 and 2, ATF-2, c-jun, Jun B, and Jun D. Various combinations of the AP-1 family members lead to different DNA binding specificities. Transactivation of c-jun is a two-step process requiring docking of JNK to an NH2-terminal sequence referred to as the δ-domain, followed by phosphorylation on serines 63 and 73. The ability to activate transcription depends on both the phosphorylation state of c-Jun and its dimeric partner (for review see Mechta-Grigoriou et al., 2001). Some members of the family can even antagonize transcription by potent activators such as homodimers of c-Jun (Deng and Karin, 1993) or heterodimers of c-Fos (Wisdom and Verma, 1993). Curiously, the transforming mutation of this protooncogene is a deletion of the δ-domain, although how this deletion results in unregulated growth is not clear. Hence, this immediate early gene is regulated in a complex, incompletely understood manner.
A variety of growth factors can activate the AP-1 complex and promote progression through the cell cycle, in part, through activation of cyclin D1 (Wisdom et al., 1999). In addition, many cytokines and inducers of cellular stress such as UV and γ-radiation, osmotic shock, hypoxia, and withdrawal of trophic support activate JNK, and subsequently, c-Jun. Paradoxically, this activation has been suggested to promote survival in dividing cells, such as fibroblasts, yet lead to apoptosis in post-mitotic neurons (for review see Shaulian and Karin, 2001). Inhibition of JNK activation in sympathetic neurons was shown to provide protection from NGF withdrawal (Maroney et al., 1999). Similarly, microinjection of antibodies to c-Jun (Estus et al., 1994), cDNA encoding c-Jun mutants (Ham et al., 1995), or expression of c-Jun mutants using adenovirus (Whitfield et al., 2001) attenuated apoptosis in these neurons after trophic factor deprivation, whereas overexpression of c-Jun lead to cell death (Ham et al., 1995). However, a high level expression of ectopic protein may interfere with the normal function of the neuron. For example, overexpression of mutant c-Jun could alter c-Fos dimers, which have also been implicated in regulating survival (Smeyne et al., 1993; Preston et al., 1996). Moreover, dominant-negative mutants lacking the δ-domain may well maintain the activity that leads to transformation in fibroblasts. Therefore, we chose to use a genetic approach to address the role of c-Jun in these neurons. Unfortunately, the deletion of c-jun is embryonic lethal at E14, before the sympathetic neurons undergo naturally occurring cell death (Wright et al., 1983). Therefore, we used a Cre-lox system to excise this gene. Our findings demonstrate a requirement for c-Jun in apoptosis induced by NGF removal, in agreement with previous studies.
Although neuronal apoptosis induced by trophic factor withdrawal is dependent on transcription and c-Jun, it is not clear what the relevant target genes are for this AP-1 factor. The activation of JNK in neurons has been suggested to lead to the up-regulation of two members of the proapoptotic Bcl-2 family, BAX (Miller et al., 1997) and BIM (Putcha et al., 2001; Whitfield et al., 2001). However, it is likely that c-Jun also interacts with other transcriptional elements to affect the expression of apoptotic genes. For example, the upstream kinase, JNK, has also been shown to phosphorylate p53 (Fuchs et al., 1998), which can up-regulate BAX (Miyashita and Reed, 1995). Indeed, reduced apoptosis in sympathetic ganglia was observed in p53+/− mice, and expression of the viral p53 inhibitor (E1B55K) protected these neurons in culture after NGF removal (Aloyz et al., 1998). Thus, the interplay between p53 and c-Jun in regulating neuronal survival and death genes remains to be determined. It is interesting to note that a recent study demonstrated an intimate relationship between c-Jun and p53 after UV irradiation (Shaulian et al., 2000). The induction of c-Jun after a UV response resulted in antagonism of p53 at the p21 promoter. This caused the DNA-damaged fibroblasts to reenter the cell cycle, triggering apoptosis. A similar mechanism may explain the dual dependence of neuronal apoptosis on c-Jun and p53. Both of these transcription factors may be required for the correct temporal regulation of genes key to neuronal apoptosis.
Sympathetic neurons express exclusively the neurotrophin receptors TrkA and p75, and therefore undergo apoptosis if they fail to innervate a target producing NGF (for review see Huang and Reichardt, 2001). In contrast, if these neurons come in contact with tissue producing a neurotrophin other than NGF, then p75 will be activated and apoptosis will result. Miller and colleagues have demonstrated such an effect in vivo, where mice with the BDNF gene deleted exhibit an increase in sympathetic neuron survival (Bamji et al., 1998). These authors suggest that although insufficient TrkA activation promotes cell death, neurotrophin binding selectively to p75 will result in a more rapid and efficient elimination of inappropriate connections.
There is accumulating evidence that activation of the p75 receptor induces apoptosis in a variety of neuronal (Rabizadeh et al., 1993; Barrett and Bartlett, 1994, Barrett and Georgiou, 1996; Friedman, 2000) and glial contexts (Casaccia-Bonnefil et al., 1996; Soilu-Hanninen et al., 1999), both during development (Frade et al., 1996, Frade and Barde, 1999) and after insult, such as nerve injury (Syroid et al., 2000; Harrington et al., 2002) or seizure (Roux et al., 1999). The molecular mechanisms mediating this effect, although largely undetermined, have been suggested to involve the transcription factor NF-κB and the c-Jun kinase, JNK, both of which can be activated after neurotrophin binding to p75 (for review see Barrett, 2000). Paradoxically, in a manner reminiscent of TNF receptor signaling, p75 activation of NF-κB has been shown to promote survival (Hamanoue et al., 1999; Foehr et al., 2000; Gentry et al., 2000), whereas JNK has been implicated in the apoptotic response (Casaccia-Bonnefil et al., 1996; Yoon et al., 1998). In the presence of TrkA, NGF binding to p75 will only activate the pro-survival factor NF-κB while JNK activity is inhibited (Yoon et al., 1998). Hence, when the neuron contacts the appropriate target, binding of NGF to both TrkA and p75 coordinately promotes survival. In contrast, selective activation of p75 promotes apoptosis through JNK because attenuation of this kinase activity prevented the receptor-mediated cell death (Yoon et al., 1998; Harrington et al., 2002). Thus, whether induced by p75 activation or NGF withdrawal, programmed cell death in sympathetic neurons depends on the c-Jun activating kinase, JNK. Surprisingly, however, our findings demonstrate these death signals subsequently diverge. The deletion of c-Jun protected sympathetic neurons from apoptosis caused by trophic factor withdrawal; however, the ability of p75 to kill the cells was not altered. The apoptotic signal generated from p75 was inhibited by cycloheximide, suggesting a requirement for protein translation. Miller and colleagues demonstrated that the p53 inhibitor (E1B55K) can block sympathetic neuron death caused by BDNF (Aloyz et al., 1998). Hence, it is likely that the p75 apoptotic signal is through JNK activation of p53, resulting in the up-regulation of BAX and induction of apoptosis. However, it is notable that several p75-interacting proteins have been proposed to act in the nucleus (Casademunt et al., 1999; Chittka and Chao, 1999). Whether these interactors are affected by JNK remains to be determined.
Materials and methods
Generation of c-junfl/fl mice
A 5-kb genomic fragment covering the intron-less c-Jun gene was used to construct the targeting vector (Fig. 1 A). A promoter-less neo cassette flanked by loxP sites was inserted into the EcoRI site in the 5′ UTR of the c-Jun gene; the EcoRI site only being reconstituted on the downstream side of the neo cassette. A loxP site and a BamHI site were introduced into a unique SphI site downstream of the polyadenylation signal for the c-Jun gene. Because the neo expression cassette does not contain its own promoter, integration near a functional promoter is required for expression, allowing high efficiency in selecting for homologous recombination at the c-Jun locus. After transfection into ES cells, neo-resistant clones were isolated and homologous recombinants identified by Southern blotting using probe A (Fig. 1 B) to demonstrate the insertion of neo, and probe B (Fig. 1 C) to show the presence of the loxP and adjacent BamHI site.
The insertion of the neo gene into the 5′ UTR of c-jun disrupts the expression of the gene, and therefore must be removed to allow tissue-specific activation. Hence, the targeted ES cells were transiently transfected with pCMV-Cre, and individual clones were isolated and screened by PCR to identify those with specific excision of the neo cassette. This analysis also identified some clones in which excision between the most upstream and downstream loxP sites occurred, verifying the ability of all three loxP sites to support Cre-mediated deletion. The removal of the neo gene was confirmed by Southern blotting, demonstrating that the targeted c-jun allele contains the protein coding sequence flanked by loxP sites (Fig. 1 D). The ES cells containing the floxed c-jun were then injected into blastocysts, and chimeric mice were obtained and mated. Germline transmission was confirmed by PCR using primers that flank the upstream loxP site (5′-GGAGAGTCCCTTCTCCCGCC-3′ and 5′-GCTAGCACTCACGTTGGTAGG-3′; Fig. 1 E). The mice were crossed with the c-jun+/− to eventually obtain animals homozygous for the floxed allele in place of the wt gene, thus confirming that the loxP sites introduced do not interfere with normal c-Jun function.
Neuronal cultures
SCG from wt or postnatal day 1 c-jun fl/fl pups were isolated, and the sympathetic neurons dissociated by trituration after digestion with 0.25% trypsin and 0.3% collagenase for 15 min at 37°C. The nonneuronal cells were removed by a 2-h preplating on uncoated, Falcon™ 60-mm plates (Becton Dickinson). The neurons were cultured on poly-l-ornithine and laminin-coated 4-well slides (Nalge Nunc International) at a density of 3,000–4,000 cells/well in F-14+ media (Ham's F-14 containing 5% FCS, 2 mM l-glutamine, 60 ng/ml progesterone, 16 μg/ml putrescine, 400 ng/ml l-thyroxine, 38 ng/ml sodium selenite, 340 ng/ml tri-iodothyroxine, 5 μg/ml insulin, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 μM fluorodeoxyuridine; Imperial Labs) and 20 ng/ml NGF (Regeneron Pharmaceuticals, Inc.). The neurons were maintained for 3–5 d in the presence of NGF before being used for survival assays in NGF withdrawal and in p75 activation experiments.
Survival assays
For NGF withdrawal experiments, NGF was removed by washing the cultures twice with F-14+ media lacking NGF, and once with F-14+ containing anti-NGF antibody at 1 μg/ml (Sigma-Aldrich). For the p75 activation experiments, the procedure was similar, but after the washes with media lacking NGF, neurons were switched to media containing anti-NGF antibody together with 12.5 mM KCl, to promote survival, with or without 100 ng/ml BDNF (a gift from Regeneron Pharmaceuticals, Inc.). In experiments involving the inhibition of protein synthesis, SCGs were incubated with 1 μg/ml cycloheximide (Sigma-Aldrich) at the time of NGF removal or BDNF treatment. This concentration of cycloheximide inhibited 70–80% of protein synthesis based on [35S]methionine labeling of the neurons as in Martin et al. (1988) (data not shown). Some neurons were also treated at the time of NGF withdrawal with an antiserum raised to the extracellular domain of p75 (diluted 1:500; a gift of M.V. Chao, Skirball Institute, New York University, New York, NY).
2 d after the switch to NGF-free or BDNF-containing media, or after the indicated time, the cells were fixed in 4% PFA and the number of apoptotic neurons, identified by DAPI staining, was counted in five random fields (at least 50 neurons counted/well). In the assays done with infected neurons, only Cre-immunopositive or GFP-expressing cells were considered.
Viral infections
Two different recombinant adenoviruses were used, one expressing Cre recombinase and the other expressing GFP, as a control for adenoviral infection. The GFP-adenovirus was provided by S.O. Yoon (Ohio State University, Columbus, OH) (Harrington et al., 2002) and the Cre-adenovirus was as used previously (Seagroves et al., 2001). The adenoviruses were amplified in 293 cells, the viral solution was purified on CsCl2 gradients, and viral infectivity was determined on 3T3 cells and neurons. The neurons were cultured for 3–5 d in 20 ng/ml NGF, infected with 1.6 × 109 particles/ml of recombinant adenovirus for 2 h in serum-free F-14+ media, and thereafter serum-containing F-14+ media was added. This concentration of virus was chosen in order to obtain 95% or better infection. Although there was a low level of toxicity due to the viral infection (Fig. 3 D), using 10-fold less virus did not alter the amount of cell death among infected neurons, yet the percentage of infected cells decreased to 70%. 1 d after infection (18–24 h), the virus was removed and cells were fed with fresh NGF-free or BDNF-containing F-14+ media for survival assays under conditions of NGF withdrawal or p75 activation, respectively.
Genomic PCR to detect c-jun
To confirm the deletion of c-jun, the genomic DNA was isolated from sympathetic neurons that were uninfected, or 48 h after infection with the adenovirus expressing GFP or Cre. The c-jun locus was amplified by PCR using primers flanking the loxP site (5′-AGCAACTTTCCTGACCCAGA-3′) and (5′-CGTCCCTGCTTCTGTAACAA-3′) with the following PCR conditions of: 1 cycle of 94°C for 2 min, 52°C for 30 s, and 72°C for 30 s, and then 30 cycles at 94°C for 30 s, 52°C for 30 s, and 72°C for 30 s). As a control to demonstrate the presence of genomic DNA, the NF-κB subunit p65 was amplified using primers 5′-CCTGGGGATCCAGTGTGTGAAGAAGCG-3′and 5′-AATCGGATGTGAGAGGACGACAGC-3′ (PCR conditions of the following: 94°C for 5 min, and then 40 cycles of 94°C for 1 min, 65°C for 1.5 min, and 72°C for 1 min).
Immunohistochemistry
Neurons grown as described under Neuronal cultures were rinsed in PBS, fixed in 4% PFA, and blocked with 10% goat serum in PT (PBS + 0.1% Triton X-100) followed by avidin-biotin block (Vectastain; Vector Laboratories). To detect Cre recombinase, cells were probed overnight at 4°C with a biotinylated antiserum to Cre recombinase (Covance, Inc.) diluted 1:100 in PT, followed by 20-min incubation with Cy2- or Cy3-streptavidin. To visualize c-Jun, cells were permeabilized for 2 min at 4°C in PT with 0.1% sodium citrate, blocked as for anti-Cre, and incubated with anti–c-Jun, diluted 1:500 (Cell Signaling Technology Inc.), for 1 h at RT followed by biotinylated anti–rabbit antibody and Cy3-streptavidin. Nuclei were stained with DAPI. The slides were then viewed by fluorescence microscopy (Carl Zeiss MicroImaging, Inc.).
Acknowledgments
We thank Regeneron Pharmaceuticals, Inc., for providing the neurotrophins. The authors thank other members of the Carter lab for helpful discussions, Dr. Sung Ok Yoon for critical reading of the manuscript, and Dr. Amy Sprowles for advice on the c-jun PCR.
This work was supported by a National Institutes of Health grant (NS38220) to B.D. Carter and a Juvenile Diabetes Foundation grant (3-1999-229) to S. Kanwal.
Footnotes
Abbreviations used in this paper: BDNF, brain-derived neurotrophic factor; JNK, c-Jun NH2-terminal kinase; SCG, superior cervical ganglia; wt, wild type.
References
- Aloyz, R.S., S.X. Bamji, C.D. Pozniak, J.G. Toma, J.K. Atwal, and F.D. Miller. 1998. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J. Cell Biol. 143:1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji, S.X., M. Majdan, C.D. Pozniak, D.J. Belliveau, R.K. Aloyz, C.G. Causing, and F.D. Miller. 1998. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J. Cell Biol. 140:911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbacid, M. 1994. The Trk family of neurotrophin receptors. J. Neurobiol. 25:1386–1403. [DOI] [PubMed] [Google Scholar]
- Barrett, G.L. 2000. The p75 neurotrophin receptor and neuronal apoptosis. Prog. Neurobiol. 61:205–229. [DOI] [PubMed] [Google Scholar]
- Barrett, G.L., and P.F. Bartlett. 1994. The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc. Natl. Acad. Sci. USA. 91:6501–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett, G.L., and A. Georgiou. 1996. The low-affinity nerve growth factor receptor p75NGFR mediates death of PC12 cells after nerve growth factor withdrawal. J. Neurosci. Res. 45:117–128. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil, P., B.D. Carter, R.T. Dobrowsky, and M.V. Chao. 1996. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 383:716–719. [DOI] [PubMed] [Google Scholar]
- Casademunt, E., B.D. Carter, I. Benzel, J.M. Frade, G. Dechant, and Y.A. Barde. 1999. The zinc finger protein NRIF interacts with the neurotrophin receptor p75(NTR) and participates in programmed cell death. EMBO J. 18:6050–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittka, A., and M.V. Chao. 1999. Identification of a zinc finger protein whose subcellular distribution is regulated by serum and nerve growth factor. Proc. Natl. Acad. Sci. USA. 96:10705–10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson, E.J., K. Reid, and P.F. Bartlett. 1999. Signaling of neuronal cell death by the p75NTR neurotrophin receptor. Mol. Neurobiol. 20:29–44. [DOI] [PubMed] [Google Scholar]
- Deckwerth, T.L., J.L. Elliott, C.M. Knudson, E.M. Johnson, Jr., W.D. Snider, and S.J. Korsmeyer. 1996. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 17:401–411. [DOI] [PubMed] [Google Scholar]
- Deng, T., and M. Karin. 1993. JunB differs from c-Jun in its DNA-binding and dimerization domains, and represses c-Jun by formation of inactive heterodimers. Genes Dev. 7:479–490. [DOI] [PubMed] [Google Scholar]
- Deshmukh, M., and E.M. Johnson, Jr. 1997. Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol. Pharmacol. 51:897–906. [DOI] [PubMed] [Google Scholar]
- Deshmukh, M., and E.M. Johnson, Jr. 1998. Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 21:695–705. [DOI] [PubMed] [Google Scholar]
- Estus, S., W.J. Zaks, R.S. Freeman, M. Gruda, R. Bravo, and E.M. Johnson, Jr. 1994. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 127:1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foehr, E.D., X. Lin, A. O'Mahony, R. Geleziunas, R.A. Bradshaw, and W.C. Greene. 2000. NF-kappa B signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J. Neurosci. 20:7556–7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade, J.M., and Y.A. Barde. 1999. Genetic evidence for cell death mediated by nerve growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development. 126:683–690. [DOI] [PubMed] [Google Scholar]
- Frade, J.M., A. Rodriguez-Tebar, and Y.A. Barde. 1996. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature. 383:166–168. [DOI] [PubMed] [Google Scholar]
- Friedman, W.J. 2000. Neurotrophins induce death of hippocampal neurons via the p75 receptor. J. Neurosci. 20:6340–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, S.Y., V. Adler, T. Buschmann, Z. Yin, X. Wu, S.N. Jones, and Z. Ronai. 1998. JNK targets p53 ubiquitination and degradation in nonstressed cells. Genes Dev. 12:2658–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry, J.J., P. Casaccia-Bonnefil, and B.D. Carter. 2000. Nerve growth factor activation of nuclear factor kappaB through its p75 receptor is an anti-apoptotic signal in RN22 schwannoma cells. J. Biol. Chem. 275:7558–7565. [DOI] [PubMed] [Google Scholar]
- Ham, J., C. Babij, J. Whitfield, C.M. Pfarr, D. Lallemand, M. Yaniv, and L.L. Rubin. 1995. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 14:927–939. [DOI] [PubMed] [Google Scholar]
- Hamanoue, M., G. Middleton, S. Wyatt, E. Jaffray, R.T. Hay, and A.M. Davies. 1999. p75-mediated NF-kappaB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol. Cell. Neurosci. 14:28–40. [DOI] [PubMed] [Google Scholar]
- Harrington, A.W., J.Y. Kim, and S.O. Yoon. 2002. Activation of Rac GTPase by p75 in necessary for c-jun N-terminal kinase-mediated apoptosis. J. Neurosci. 22:156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilberg, F., A. Aguzzi, N. Howells, and E.F. Wagner. 1993. c-jun is essential for normal mouse development and hepatogenesis. Nature. 365:179–181. [DOI] [PubMed] [Google Scholar]
- Huang, E.J., and L.F. Reichardt. 2001. Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24:677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, R.S., B. van Lingen, V.E. Papaioannou, and B.M. Spiegelman. 1993. A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev. 7:1309–1317. [DOI] [PubMed] [Google Scholar]
- Lentz, S.I., C.M. Knudson, S.J. Korsmeyer, and W.D. Snider. 1999. Neurotrophins support the development of diverse sensory axon morphologies. J. Neurosci. 19:1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locksley, R.M., N. Killeen, and M.J. Lenardo. 2001. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 104:487–501. [DOI] [PubMed] [Google Scholar]
- Maroney, A.C., J.P. Finn, D. Bozyezko-Coyne, T.M. O'Kane, N.T. Neff, A.M. Tolkovsky, D.S. Park, C.Y. Yan, C.M. Troy, and L.A. Greene. 1999. CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J. Neurochem. 73:1901–1912. [PubMed] [Google Scholar]
- Martin, D.P., R.E. Schmidt, P.S. DiStefano, O.H. Lowry, J.G. Carter, and E.M. Johnson, Jr. 1988. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 106:829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechta-Grigoriou, F., D. Gerald, and M. Yaniv. 2001. The mammalian Jun proteins: redundancy and specificity. Oncogene. 20:2378–2389. [DOI] [PubMed] [Google Scholar]
- Miller, T.M., K.L. Moulder, C.M. Knudson, D.J. Creedon, M. Deshmukh, S.J. Korsmeyer, and E.M. Johnson, Jr. 1997. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J. Cell Biol. 139:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita, T., and J.C. Reed. 1995. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 80:293–299. [DOI] [PubMed] [Google Scholar]
- Oppenheim, R.W. 1991. Cell death during development of the nervous system. Annu. Rev. Neurosci. 14:453–501. [DOI] [PubMed] [Google Scholar]
- Pozniak, C.D., S. Radinovic, A. Yang, F. McKeon, D.R. Kaplan, and F.D. Miller. 2000. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 289:304–306. [DOI] [PubMed] [Google Scholar]
- Preston, G.A., T.T. Lyon, Y. Yin, J.E. Lang, G. Solomon, L. Annab, D.G. Srinivasan, D.A. Alcorta, and J.C. Barrett. 1996. Induction of apoptosis by c-Fos protein. Mol. Cell. Biol. 16:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha, G.V., K.L. Moulder, J.P. Golden, P. Bouillet, J.A. Adams, A. Strasser, and E.M. Johnson, Jr. 2001. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 29:615–628. [DOI] [PubMed] [Google Scholar]
- Rabizadeh, S., J. Oh, L.T. Zhong, J. Yang, C.M. Bitler, L.L. Butcher, and D.E. Bredesen. 1993. Induction of apoptosis by the low-affinity NGF receptor. Science. 261:345–348. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Tebar, A., G. Dechant, and Y.A. Barde. 1991. Neurotrophins: structural relatedness and receptor interactions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 331:255–258. [DOI] [PubMed] [Google Scholar]
- Roux, P.P., M.A. Colicos, P.A. Barker, and T.E. Kennedy. 1999. p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. J. Neurosci. 19:6887–6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seagroves, T.N., H.E. Ryan, H. Lu, B.G. Wouters, M. Knapp, P. Thibault, K. Laderoute, and R.S. Johnson. 2001. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol. Cell. Biol. 21:3436–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian, E., and M. Karin. 2001. AP-1 in cell proliferation and survival. Oncogene. 20:2390–2400. [DOI] [PubMed] [Google Scholar]
- Shaulian, E., M. Schreiber, F. Piu, M. Beeche, E. Wagner, and M. Karin. 2000. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell. 103:897–907. [DOI] [PubMed] [Google Scholar]
- Smeyne, R.J., M. Vendrell, M. Hayward, S.J. Baker, G.G. Miao, K. Schilling, L.M. Robertson, T. Curran, and J.I. Morgan. 1993. Continuous c-fos expression precedes programmed cell death in vivo. Nature. 363:166–169. [DOI] [PubMed] [Google Scholar]
- Soilu-Hanninen, M., P. Ekert, T. Bucci, D. Syroid, P.F. Bartlett, and T.J. Kilpatrick. 1999. Nerve growth factor signaling through p75 induces apoptosis in Schwann cells via a Bcl-2-independent pathway. J. Neurosci. 19:4828–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syroid, D.E., P.J. Maycox, M. Soilu-Hanninen, S. Petratos, T. Bucci, P. Burrola, S. Murray, S. Cheema, K.F. Lee, G. Lemke, and T.J. Kilpatrick. 2000. Induction of postnatal Schwann cell death by the low-affinity neurotrophin receptor in vitro and after axotomy. J. Neurosci. 20:5741–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield, J., S.J. Neame, L. Paquet, O. Bernard, and J. Ham. 2001. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 29:629–643. [DOI] [PubMed] [Google Scholar]
- Wisdom, R., and I.M. Verma. 1993. Proto-oncogene FosB: the amino terminus encodes a regulatory function required for transformation. Mol. Cell. Biol. 13:2635–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisdom, R., R.S. Johnson, and C. Moore. 1999. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 18:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, L.L., T.J. Cunningham, and A.J. Smolen. 1983. Developmental neuron death in the rat superior cervical sympathetic ganglion: cell counts and ultrastructure. J. Neurocytol. 12:727–738. [DOI] [PubMed] [Google Scholar]
- Yoon, S.O., P. Casaccia-Bonnefil, B. Carter, and M.V. Chao. 1998. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J. Neurosci. 18:3273–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, J., and B.A. Yankner. 2000. Apoptosis in the nervous system. Nature. 407:802–809. [DOI] [PubMed] [Google Scholar]