

Abstract
In the ER, the translocon complex (TC) functions in the translocation and cotranslational modification of proteins made on membrane-bound ribosomes. The oligosaccharyltransferase (OST) complex is associated with the TC, and performs the cotranslational N-glycosylation of nascent polypeptide chains. Here we use a GFP-tagged subunit of the OST complex (GFP–Dad1) that rescues the temperature-sensitive (ts) phenotype of tsBN7 cells, where Dad1 is degraded and N-glycosylation is inhibited, to study the lateral mobility of the TC by FRAP. GFP–Dad1 that is functionally incorporated into TCs diffuses extremely slow, exhibiting an effective diffusion constant (D eff) about seven times lower than that of GFP-tagged ER membrane proteins unhindered in their lateral mobility. Termination of protein synthesis significantly increases the lateral mobility of GFP–Dad1 in the ER membranes, but to a level that is still lower than that of free GFP–Dad1. This suggests that GFP–Dad1 as part of the OST remains associated with inactive TCs. Our findings that TCs assembled into membrane-bound polysomes diffuse slowly within the ER have mechanistic implications for the segregation of the ER into smooth and rough domains.
Keywords: translocon complex; oligosaccharyltransferase; lateral mobility; FRAP; endoplasmic reticulum
Introduction
The fluid mosaic membrane model proposed about 30 years ago emphasized the free mobility of membrane proteins in the plane of the membrane (Singer and Nicolson, 1972). Since then it has been found that many membrane proteins may not be completely free to diffuse in the plane of cellular membranes (Jacobson et al., 1995). Proteins may be retarded in their lateral mobility or completely immobilized through interactions with cytoskeletal elements or extracellular matrix proteins, by being incorporated into cellular junction–forming structures, or by becoming a part of large oligomeric structures. These insights were gained through biophysical methods, including FRAP (Chazotte et al., 1998; Lippincott-Schwartz et al., 2001).
The rough ER is the site where proteins made on membrane-bound polysomes are inserted into, or translocated across the limiting membrane. A complex molecular machinery effects the signal sequence-mediated targeting, cotranslational translocation and modification of nascent polypeptide chains that are synthesized in eukaryotic cells on ribosomes bound to ER membranes (Walter and Johnson, 1994). Present evidence suggests that the proteins concerned with these functions form the TC with Sec61α at its core (Görlich et al., 1992). In recent years, several translocon complex (TC)*-associated proteins have been identified (Rapoport et al., 1996; Johnson and van Waes, 1999). The oligosaccharyltransferase (OST), which is responsible for cotranslational N-glycosylation of the nascent peptide chain, is tightly associated with the TC (Görlich et al., 1992; Wang and Dobberstein, 1999; Menetret et al., 2000). It is composed of at least four transmembrane proteins: ribophorin (R)I and II, OST48, and Dad1 (Kreibich et al., 1978; Kelleher et al., 1992; Kelleher and Gilmore, 1997).
Although much has been learned about the composition of the TC and the function of its components (Rapoport et al., 1996; Johnson and van Waes, 1999), the mechanisms underlying its spatial distribution and dynamics within ER membranes are not understood. For example, it is unknown whether in living cells the TC disassembles after a nascent polypeptide chain has been synthesized or whether it remains assembled for long periods of time. It is also unclear whether active TCs with attached ribosomes translating mRNAs freely diffuse in the plane of the ER membrane or are immobilized. Answers to these questions are fundamental to understanding the basis of the organization of the ER into rough and smooth domains, which underlies the spatial control of protein synthesis within the ER.
To investigate these questions we have examined the lateral mobility of GFP–Dad1 that has been functionally incorporated into the OST of tsBN7 cells. The tsBN7 cell line was derived from BHK-21 cells (Nishimoto and Basilico, 1978; Nishimoto et al., 1978) and has a point mutation in the DAD1 gene that is responsible for its temperature-sensitive (ts)-phenotype (Nakashima et al., 1993). The ts-form of Dad1 is completely degraded within 6 h after shifting to the nonpermissive temperature (39.5°C), which results in the inhibition of N-glycosylation of membrane and secretory glycoproteins and finally in cell death (Nakashima et al., 1993; Sanjay et al., 1998). Dad1 is a 10-kD protein containing two transmembrane domains separated by five amino acids with its NH2- and COOH-termini exposed to the cytosol (Silberstein et al., 1995; Makishima et al., 1997). We have previously shown that Dad1 is essential for the functional and structural integrity of the OST (Sanjay et al., 1998). Expression of GFP–Dad1 rescued the ts-phenotype in tsBN7 cells, indicating that at the nonpermissive temperature GFP–Dad1 was quantitatively and functionally incorporated into the OST, and therefore could be used as a marker for TCs.
To investigate the lateral mobility of the TC under various functional conditions, we have performed FRAP experiments on cells expressing GFP–Dad1. These included conditions where the TCs are either active in protein translocation, or protein synthesis is terminated, or the cells are starved of dolychol pyrophosphate oligosaccharides by tunicamycin treatment. Our results suggest that TCs remain assembled in the ER membrane as discrete complexes for long periods of time. Individual TCs are relatively mobile, but when they are engaged in cotranslational translocation and assembled into polysomes, they exhibit significantly reduced mobility. The reduced mobility of active TCs supports a mechanism for the segregation of the ER into rough and smooth domains that involves dynamic association/dissociation of TCs with relatively immobile, polysomal arrays.
Results
Isolation of clones stably expressing GFP-tagged Dad1 fusion proteins
Several lines of investigation have provided morphological and biochemical evidence that the ER forms differentiated domains characterized by the presence or absence of membrane-bound ribosomes (Kreibich et al., 1978; Baumann and Walz, 2001). For certain cell types, a very abrupt transition between these two continuous ER subcompartments have been observed, and membrane-bound polysomes have been shown to form regular patterns on the surface of the rough ER (Palade, 1955; Fawcett, 1981). These findings are consistent with the hypothesis that TCs that carry membrane-bound ribosomes are restricted in their lateral mobility. Our aim was to test this hypothesis directly by performing FRAP experiments on cells expressing a GFP-tagged protein incorporated into the TC. For these experiments to be interpretable, it is crucial that the GFP-tagged protein is in fact functional. It is equally important that the GFP-tagged protein is not overexpressed, so that it is completely incorporated into the oligomeric structure of the TC. Overexpression of the GFP-tagged subunit of the TC would severely distort the results of FRAP experiments if the oligomeric assembly has a diffusion constant different from that of the unincorporated subunit. Therefore, we decided to exploit the fact that tsBN7 cells harbor a ts-mutation in the DAD1 gene encoding one of the subunits of the OST (Nakashima et al., 1993). The aim was to replace in these cells the ts-form of Dad1 with the wild-type protein fused to GFP. This system has the distinct advantage that at the non-permissive temperature the ts-mutant of Dad1 is very quickly degraded and the cells undergo apoptotic death (Nakashima et al., 1993).
Because we could not predict whether the 27-kD GFP moiety attached to either terminus of Dad1 would interfere with the biological function of this OST subunit, we made two constructs, GFP–Dad1 and Dad1–GFP, where GFP fused with the NH2 or COOH terminus of Dad1, respectively. We isolated clones from tsBN7 cells that were stably transfected with cDNAs encoding these two constructs that were expressed under the control of an ecdysone-inducible promoter. When we used antibiotic selection only, we isolated zeocin- and neomycin-resistant clones from tsBN7 cells transfected with either the GFP–Dad1 (21 clones) or the Dad1–GFP (36 clones) construct. However, when the antibiotic selection was combined with the selection at 39.5°C, only cells expressing the GFP–Dad1 fusion protein survived (28 clones). Thus, only the GFP–Dad1 fusion protein is capable of rescuing the ts-phenotype of tsBN7 cells. In contrast, attachment of GFP at the COOH terminus of Dad1–GFP resulted in the formation of a biologically inactive fusion protein.
Four isolated clones stably expressing GFP–Dad1 were analyzed on Western blots using a pAb directed against GFP (Fig. 1 a). It was found that the level of GFP–Dad1 expression varied considerably among the clones and they responded differently to the addition of Ponasterone A. Clone M3/18 had several advantages over the other tested clones. It grew well at 39.5°C and it expressed the GFP–Dad1 fusion protein at a low level (Fig. 1 a). The clone was also highly responsive to induction by Ponasterone A. Western blot analysis of the M3/18 clone with an anti-Dad1 pAb showed that the expression level of GFP–Dad1 (Fig. 1 b, asterisk) in noninduced cells was comparable to that of the endogenous Dad1 protein at the permissive temperature (Fig. 1 b, double asterisk). In BHK-21 cells, the expression level of endogenous Dad1 (Fig. 1 b, lane 1) was slightly higher than in tsBN7 cells (lane 2). The latter cells expressed Dad1 in amounts similar to Dad1 in noninduced M3/18 cells grown at 34°C (lane 3). Furthermore, the expression level of GFP–Dad1 in noninduced M3/18 cells grown at 34°C (lane 3) was comparable to that of Dad1 in tsBN7 cells (lane 2). The endogenous ts-form of Dad1 is completely degraded in M3/18 cells grown at 39.5°C, whereas the expression level of GFP–Dad1 does not change (lane 4). These results indicate that in noninduced M3/18 cells grown at the nonpermissive temperature, GFP–Dad1 can substitute the ts-form of Dad1 in the TC. Moreover, it is expressed at levels comparable to the wild-type protein.
Figure 1.

Characterization of clones stably expressing GFP–Dad1 that is functionally incorporated into the OST. (a) Isolated clones express different levels of GFP–Dad1 and respond differently to the inducer. Permanently transfected clones isolated after antibiotic and nonpermissive temperature selection were grown in the absence (−) and presence (+) of the inducer. Equal protein amounts of total cell lysates were analyzed on Western blots using an anti-GFP pAb. (b) In clone M3/18, the expression level of GFP–Dad1 is comparable to that of endogenous Dad1. Equal amounts of protein from total lysates of cells grown at the permissive or nonpermissive temperature were analyzed on Western blots using an anti-Dad1 pAb. Lane 1, BHK-21 cells grown at 34°C; lane 2, tsBN7 cells grown at 34°C; lane 3, M3/18 cells grown at 34°C and lane 4, M3/18 cells grown at 39.5°C. An asterisk marks the position of GFP–Dad1 protein and the position of endogenous Dad1 is marked by a double asterisk. (c) N-glycosylation activity of OST is repaired in M3/18 cells grown at 39.5°C. Three different cell lines transiently expressing SEAP were grown at 34 or 39.5°C and labeled with [35S]-TransLabel. The cell lysates were immunoprecipitated with anti-SEAP pAb. The precipitates were digested with EndoH (+) or left as untreated control (−). The EndoH-resistant form of SEAP is represented by upper most band (arrow 1), the EndoH-sensitive form of SEAP is a band in the middle (arrow 2), and the nonglycosylated form of SEAP is represented by lower band (arrow 3).
The GFP–Dad1 fusion protein expressed in the M3/18 clone is functionally active
The finding that the ts-phenotype is corrected in M3/18 cells suggested that the GFP–Dad1 fusion protein is functionally active. In order to demonstrate that the N-glycosylation activity is indeed repaired, BHK-21, tsBN7, and M3/18 cells grown for 24 h at the permissive (34°C) or nonpermissive temperature (39.5°C) were transfected with a plasmid encoding a secretory form of alkaline phosphatase (SEAP). This protein has two N-glycosylation sites, which are further modified in the Golgi apparatus and become resistant to endoglycosidase H (EndoH) treatment (Martinez et al., 1994). The cells were labeled with Tran[35S]-label for 1 h, and SEAP was immunoprecipitated from detergent-solubilized cells using an anti-SEAP mAb. The immunoprecipitates were then resolved by SDS-PAGE and followed by autoradiography. The EndoH resistant (Fig. 1c, 1) and EndoH sensitive (Fig. 1 c, 2) forms of SEAP were made in all cell lines grown at 34°C, indicating that the protein is properly N-glycosylated and that some of the oligosaccharides are modified in the Golgi apparatus. Predominantly the nonglycosylated form of SEAP, but also some minor bands of lower electrophoretic mobility, some of which are not EndoH sensitive, can be detected in the immunoprecipitates obtained from tsBN7 cells grown at 39.5°C (Fig. 1 c, 3). Therefore, it appears that at the nonpermissive temperature, aside from the suppression of most N-glycosylation, some ill-defined modifications affected the SEAP polypeptide in tsBN7 cells. This is in agreement with a recent report showing that in mice homozygous for the null allele of Dad1 changes in the glycosylation pattern of laminin were observed (Hong et al., 2000). The glycosylation patterns of SEAP expressed in the M3/18 clone, even at the nonpermissive temperature, were indistinguishable from those in BHK-21 cells. Thus, these results demonstrate that N-glycosylation is restored in M3/18 cells grown at 39.5°C due to the functional incorporation of GFP–Dad1 into the OST.
In M3/18 cells, GFP–Dad1 colocalizes with other markers of the rough ER
Localization of GFP–Dad1 to the ER of M3/18 cells was confirmed by immunofluorescence microscopy. Immunolabeling of Sec61β in fixed and permeabilized M3/18 cells revealed a reticular pattern typical for the ER (Fig. 2 a). In the same cell, GFP–Dad1 displayed a similar reticular pattern (Fig. 2 b), and merging of the two images (Fig. 2 c) showed that GFP–Dad1 colocalizes with Sec61β, an integral component of the TC. In contrast, GFP–Dad1 did not colocalize with βCOPI, a marker of the Golgi apparatus (Fig. 2, d–f). We confirmed the subcellular localization of GFP–Dad1 by preparing postnuclear supernatants from BHK-21 and clone M3/18 cells and separating membranes of the Golgi apparatus and the ER-Golgi intermediate compartment (ERGIC) from those of the ER using a Nycodenz gradient. The fractions collected were analyzed on Western blots. As shown in Fig. 2 g, the ERGICp53 protein, a marker for the ERGIC (Schindler et al., 1993), was found to some extent in the fraction corresponding to the loading zone (Fig. 2 g, loading zone [LZ]), but the majority of the protein was present in the next two top fractions (Fig. 2 g, T), which were previously described as ERGIC/Golgi fractions (Helenius 1994; Greenfield and High, 1999). The subunits of the OST (OST48, RI, RII, and Dad1) as well as Sec61β were found exclusively in the heaviest fractions. The heaviest gradient fraction containing the ER markers was also examined by thin section electron microscopy. The majority of vesicles were decorated with membrane-bound ribosomes (unpublished data). From these results, we concluded that in M3/18 cells, GFP–Dad1 is confined to the ER, and its subcellular distribution is indistinguishable from that of endogenous TC components.
Figure 2.
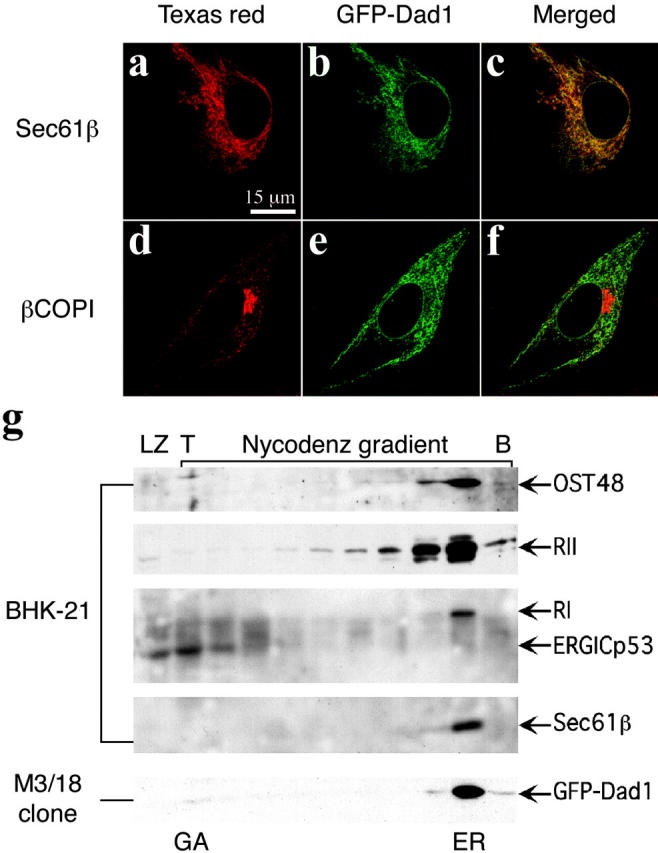
GFP–Dad1 expressed in clone M3/18 clone is located in the ER. M3/18 cell grown at 39.5°C were fixed in paraformaldehyde and permeabilized with Triton X-100. The cells were stained with primary pAbs against Sec61β (a) or βCOPI (d) and secondary antibodies were tagged with Texas red. Immunofluorescence micrographs were obtained with the LSM510 confocal microscope using the helium-neon laser. To localize GFP–Dad1 the argon-krypton laser was used (b and e). The merged images show that GFP–Dad1 is colocalized with Sec61β (c) and not with βCOPI (f). (g) GFP–Dad1 cofractionates with TC components. Postnuclear supernatants obtained from BHK-21 or M3/18 cells were fractionated on a Nycodenz gradient and equal aliquots were subjected to Western blot analysis using pAbs directed against the ER or ERGIC markers indicated next to the arrows marking the position of the respective proteins on the blots. The fractions containing Golgi and ERGIC or ER-derived membranes are indicated (GA or ER, respectively). GFP–Dad1 is only found in a fraction close to the bottom (B) of the gradient, which contains also other TC components. ERGICp53 is found exclusively in LZ and the two fractions next to the top (T) of the gradient.
GFP–Dad1 stably expressed in M3/18 cells cosediments with the OST complex
The localization of GFP–Dad1 to the ER and restoration of N-glycosylation in M3/18 cells grown at 39.5°C indicated that the fusion protein was incorporated into a biologically active OST complex. Key for the interpretation of our FRAP experiments was the demonstration that essentially all GFP–Dad1 molecules in clone M3/18 grown at the nonpermissive temperature are in fact incorporated into the OST complex. It had been previously shown by sedimentation analysis of digitonin extracts obtained from cultured cells that all four known constituents of the mammalian OST complex, namely RI and RII, OST48, as well as Dad1 cosediment as a single, enzymatically active complex (Kelleher et al., 1992; Kelleher and Gilmore, 1997). Therefore, we have used this procedure for the analysis of the M3/18 cells and the results are given in Fig. 3 (a and b). Staining of a polyacrylamide gel with Coomassie blue demonstrated that most of the total cellular protein (T) were recovered in the supernatant (S) fraction, although some distinct proteins were predominantly found in the pellet (P) (Fig. 3 a). Analysis of the gradient fractions revealed that most of the solubilized cellular proteins were found in the first four fractions, although some proteins sedimented to the bottom of the gradient and formed a pellet (GP). Western blot analysis of the gradient fractions revealed that GFP–Dad1 was mainly found in fractions 5 to 7 (Fig. 3 b). The same distribution was observed for RII, demonstrating that in M3/18 cells grown at 39°C the GFP–Dad1 fusion protein was quantitatively associated with the OST complex. The ts-form of Dad1 was completely degraded at 39.5°C and was not detected on the blot. As expected for the detergent conditions chosen, the Sec61p complex sedimented independently from the OST as indicated by the predominant localization of Sec61β in fractions 4 and 5 (Fig. 3 b).
Figure 3.

In detergent extracts from M3/18 cells grown at the nonpermissive temperature GFP–Dad1 cosediments in a glycerol gradient with the other subunits of the OST complex. M3/18 cells grown at 39.5°C were solubilized with 1.5% of digitonin in the presence of 0.5 M NaCl. The total cell lysate (T) was clarified by differential centrifugation obtaining a supernatant (S) and a pellet fraction (P). An aliquot (0.85 ml) of the supernatant fraction was layered onto a glycerol gradient (8%-30%) and after centrifugation (151,200 g for 15.5 h) the gradient was fractionated into ten 1.15-ml fractions and the LZ. Equal aliquots of the gradient fractions, the pellet formed during centrifugation (GP) as well as molecular weight markers (M) were analyzed by SDS-PAGE. The proteins on the gel were either stained with Coomassie blue (a) or transferred onto a nitrocellulose membrane and probed by Western blotting with pAbs against Dad1, RII and Sec61β (b). At the experimental conditions chosen, the OST complex (GFP–Dad1 and RII) and the Sec61 complex (Sec61β) do not cosediment. (c) In a similar experiment, tsBN7 cells transiently overexpressing GFP–Dad1 and grown at 34°C (tsBN7/pGFPDad1) were solubilized with digitonin and analyzed by Western blotting as described above. Using pAb against Dad1 to localize Dad1 and GFP–Dad1 on the glycerol gradient it is apparent, that most of the overexpressed GFP–Dad1 was not incorporated into the OST.
However, in tsBN7 cells, transiently overexpressing GFP–Dad1 and grown at the permissive temperature, the fusion protein was broadly distributed throughout the gradient and was mainly found in fractions 2 to 7 and GP (Fig. 3 c). Even the endogenous Dad1was not restricted to fractions 5 to 7 but the protein was also found in fractions 3 to 4 and GP. Considering that GFP–Dad1 is overexpressed, it is not very surprising that only a small fraction of this fusion protein is incorporated into the OST complex. Furthermore, the overexpressed GFP–Dad1 molecules compete with Dad1 for incorporation into the OST, resulting in unincorporated Dad1, slowly sedimenting in the gradient. The finding that GFP–Dad1 and to a lesser extent Dad1 were so broadly distributed throughout the gradient indicates that the nonincorporated molecular species of these membrane proteins form either aggregates or associate nonspecifically with other cellular proteins. It is unclear whether these aggregates were present in the native ER membranes or formed during solubilization.
Determination of D eff for GFP-tagged fusion proteins stably expressed in M3/18 or transiently expressed in tsBN7 cells
FRAP experiments were performed on M3/18 cells grown continuously at 39.5°C to ensure that all endogenous ts-Dad1 molecules were degraded and replaced by GFP–Dad1 (Fig. 1 b, lane 4). Under these conditions the amounts of the ts-Dad1 in M3/18 cells grown at 34°C was similar to that of GFP–Dad1 in the cells grown at the restricted temperature (Fig. 1 b, lanes 3 and 4) and essentially all GFP–Dad1 molecules were incorporated into the OST of M3/18 cells (Fig. 3 b). Therefore, for our FRAP experiments, GFP–Dad1 provides a useful reporter molecule for the OST, which is thought to be tightly associated with the TC (Görlich et al., 1992; Menetret et al., 2000). Images collected before and after photobleaching (Fig. 4 a, prebleach and 0–60 s) were used to monitor the recovery of the fluorescence into the bleached area (Fig. 4 a, arrows). The mobile fraction (M f) of GFP-tagged molecules was determined from a graphical representation of fluorescence recovery data (Fig. 4 b).
Figure 4.

Analysis of FRAP experiments by different methods. (a) tsBN7 cells transiently expressing LBR-GFP, as well as M3/18 cells kept untreated or treated with puromycin were subjected to FRAP analysis. Images were obtained before (prebleach) and after photobleaching at the indicated time points. Arrows indicate the positions of the bleached areas. The FRAP experiments were performed with wide-open pinhole to ensure that the fluorescence is collected from the entire depth of the cell. Therefore, the finest structural features of the ER (like those seen in Fig. 2, a–f) are not resolved. (b) Graphic representation of fluorescence recovery after photobleaching data. Recovery of fluorescence in the bleached box is very slow in M3/18 cells grown at 39.5°C (blue line with solid circles). If the cells are treated with puromycin (100 μM for 15 min), recovery of fluorescence in the bleached box occurs more rapidly (green line with solid squares) but still it is not as fast as that of the LBR-GFP construct expressed in tsBN7 cells (red line with solid diamonds). (c) Calculation of diffusion constants using the simulation program. Prebleach and postbleach images (like those presented in Fig. 3 a) recorded during FRAP experiments were analyzed by the simulation program described earlier (Siggia et al., 2000). The program produces a fluorescence recovery curve based on the experimental data (red solid line) and tries to fit it to a theoretical or simulated fluorescence recovery curve (blue broken line). If two curves are fitted, the simulation program calculates the diffusion constant.
In order to determine the effective diffusion constant (D eff), the collected images were analyzed by a simulation program (Siggia et al., 2000). The quantitative analysis of the FRAP experiments is summarized in Table I. At the nonpermissive temperature, GFP–Dad1 was nearly completely mobile (M f = 88%). However, the diffusion coefficient is extremely low (D eff = 0.049 μm2/s). By comparison, the lateral mobility of transiently expressed GFP-tagged lamin B receptor (LBR-GFP) (D eff = 0.331 μm2/s), which has been previously shown to diffuse freely within ER membranes (Ellenberg et al., 1997; Nehls et al., 2000), was approximately seven times higher than that of GFP–Dad1 stably expressed in M3/18 cells (Table I). By contrast, the mobility of the biologically inactive Dad1–GFP molecule transiently expressed in tsBN7 cells at 34°C (D eff = 0.315 μm2/s) was similar to that of LBR-GFP. These results suggested that GFP–Dad1 is either assembled within a large slowly moving structure or is undergoing constant association/dissociation with/from an immobile structure.
Table I. The lateral mobility of GFP-tagged fusion proteins stably expressed in M3/18 or transiently expressed in tsBN7 cells.
| Cell line | Construct | Growth temperature | Treatment | Diffusion constant (D eff), μm2/s ± SD |
Mobile fraction (Mr), % ± SD |
n |
|---|---|---|---|---|---|---|
| M3/18 | GFP–Dad1 | 39.5°C | – | 0.049 ± 0.010 | 87.9 ± 7.4 | 18 |
| Cycloheximide | 0.044 ± 0.013 | 89.6 ± 8.1 | 8 | |||
| NaCl | 0.251 ± 0.071 | 94.5 ± 5.6 | 5 | |||
| Puromycin | 0.131 ± 0.058 | 89.4 ± 5.7 | 25 | |||
| Tunicamycin | 0.048 ± 0.016 | 89.1 ± 2.5 | 10 | |||
| tsBN7 | LBR–GFP | 34°C | – | 0.331 ± 0.054 | 99.6 ± 5.2 | 4 |
| NaCl | 0.328 ± 0.032 | 97.8 ± 1.3 | 9 | |||
| Puromycin | 0.335 ± 0.029 | 96.3 ± 3.6 | 7 | |||
| 39.5°C | – | 0.331 ± 0.031 | 89.6 ± 2.9 | 8 | ||
| Dad1–GFP | 34°C | – | 0.315 ± 0.064 | 93.3 ± 5.2 | 7 | |
| 39.5°C | – | NA | NA | 15 | ||
| 34°C | – | 0.178 ± 0.037 | 89.5 ± 5.9 | 10 | ||
| GFP–Dad1 | 39.5°C | – | 0.140 ± 0.024 | 88.9 ± 1.9 | 10 |
Means ± SD denotes standard deviation of values for D eff and M f for recovery of fluorescence after photobleaching. The cells were grown overnight in glass-bottomed dishes for cell culture and pretreated as indicated with cycloheximide (80 μM for 30 min), NaCl (an extra 150 mM for 10 min), puromycin (100 μM for 15 min), or tunicamycin (1 μg/ml for 4 h) before the FRAP experiments. NA denotes that no reliable data were obtained (see Results for details).
We next investigated whether the lateral mobility of GFP–Dad1 is affected by conditions that alter the activity of the TC. Treatment of M3/18 cells with cycloheximide, an antibiotic that inhibits the elongation step of protein synthesis and therefore freezes the functional state of TCs assembled into polysomes, had no effect on the lateral mobility of GFP–Dad1 (D eff = 0.044 μm2/s and M f = 89.6%; Table I). This raised the possibility that the low diffusion coefficient of GFP–Dad1 resulted from its association with TCs in polysomal arrays.
To test this possibility, we examined the lateral diffusion of GFP–Dad1 under two conditions known to terminate protein synthesis and to disassemble polyribosomes, i.e., high salt and puromycin treatment. Increase of the NaCl concentration in the growth medium to 300 mM is known to cause a rapid inhibition of protein synthesis and results in a complete breakdown of polyribosomes (Saborio et al., 1974). This is due to an inhibition of the polypeptide initiation step of protein synthesis, which results in the read out and disassembly of polysomes. Puromycin is an antibiotic that causes the termination of nascent polypeptide chains, their release into the lumen of the ER, and the disassembly of polyribosomes (Adelman et al., 1973).
Elevating the NaCl concentration in the medium dramatically increased the lateral mobility of GFP–Dad1 in M3/18 cells (D eff = 0.251 μm2/s; M f = 94.5%; Table I). The lateral mobility of GFP–Dad1 in M3/18 cells treated with puromycin also increased significantly (D eff = 0.131 μm2/s) with no change in the mobile fraction of the protein (M f = 89.4%; Table I). In contrast, we observed no effect of the elevated NaCl concentration in the growth medium or puromycin treatment on the lateral mobility of LBR-GFP (D eff = 0.328 μm2/s and 0.335 μm2/s) or its mobile fraction (M f = 97.8% and 96.3%; Table I). This indicated that the effects of high salt and puromycin treatment on lateral diffusion were specific to GFP–Dad1. These findings are consistent with a model in which GFP–Dad1 molecules are normally associated with TCs assembled into membrane-bound polysomal arrays and that this association underlies the reduced mobility of GFP–Dad1 in the ER membranes. The fact that D eff of GFP–Dad1 in puromycin-treated cells was significantly lower than that of freely diffusing ER membrane proteins (0.131 μm2/s for GFP–Dad1 versus 0.331 μm2/s for LBR-GFP), suggests that GFP–Dad1 in these cells is associated with the TC. Our data exclude the possibilities that GFP–Dad1 is present in M3/18 cells mainly as a monomeric protein or that a large fraction of the OST complexes dissociate from inactive TCs and diffuse freely in the ER membrane.
When we compared the D eff values determined for the functional GFP–Dad1 construct expressed either stably in clone M3/18 (D eff = 0.049 μm2/s) or transiently in tsBN7 cells (D eff = 0.140 μm2/s) an almost threefold difference was observed (Table I). An explanation for this finding is that when the GFP–Dad1 construct is transiently expressed, most of the GFP–Dad1 molecules are not incorporated into the OST due to overexpression of the fusion protein (Fig. 3 c). This occurred despite our attempts to select low expressing cells with fluorescence intensity similar to that of M3/18 cells. The observed higher diffusion constants in these cells apparently resulted from over-expressed GFP–Dad1.
The lateral mobility of GFP–Dad1 is not affected in tunicamycin-treated M3/18 cells
We also examined whether there were changes in lateral mobility of GFP–Dad1 in M3/18 cells deprived of the OST substrate, dolychol pyrophosphate oligosaccharide. Treatment of cells with tunicamycin inhibits the biosynthesis of dolychol pyrophosphate oligosaccharides and N-glycosylation of proteins is thereby prevented (Leavitt et al., 1977). M3/18 cells were pretreated with tunicamycin for 4 h to starve the cells of the OST substrate but not to disturb N-glycosylation patterns of the RI and RII or other glycoproteins. FRAP analysis revealed that the lateral mobility of GFP–Dad1 in tunicamycin-treated M3/18 cells was the same (D eff = 0.048 μm2/s) as in untreated controls (D eff = 0.049 μm2/s) (Table I). This suggested that the OST remains associated with the TC even when the OST is not functioning in N-glycosylation of growing nascent peptide chains and that binding affinity of OST to TC does not depend on the OST substrate.
Discussion
In this study we have exploited FRAP to investigate the spatial and temporal regulation of the protein synthesis apparatus associated with ER membranes. This apparatus consists of at least 20 different polypeptides organized into oligomeric subcomplexes that assemble into a TC. Upon binding of a biosynthetically active ribosome, the TC functions as a molecular machine for the cotranslational translocation of proteins into the ER (Johnson and van Waes, 1999). Multiple TC-bound ribosomes translating mRNA molecules form membrane-bound polysomes. These polysomes may be further stabilized as higher order arrays that in some cells form the highly differentiated rough ER domains. This may result from interactions mediated by TC components or by not yet identified TC linker proteins. The stability of these large polysomal arrays, their mobility within ER membranes, and the kinetics of their disassembly into free TCs and/or TC components are currently unanswered questions.
To investigate the dynamics of TCs, we measured their lateral mobility under various conditions by FRAP. For a reporter of the TC, we used a GFP-tagged Dad1, a subunit of the OST, which is tightly associated with the TC. Stable expression of GFP–Dad1 in tsBN7 cell line defective in Dad1 rescued the cells at the nonpermissive temperature from inhibition of N-glycosylation and cell death. These data indicate that GFP–Dad1 is functionally incorporated into the OST, and thus could report on the behavior of TCs. The use of tsBN7 cell line harboring a mutation in the gene encoding Dad1 was essential for verifying the functional and stoichiometric incorporation of GFP–Dad1 into the TC. Comparable experiments with other TC markers, such as the core TC component, Sec61α, were not possible since no other cell lines expressing ts mutants of TC components are available. Recent studies showed that Sec61α-GFP overexpressed in COS1 cells did not crosslink to nascent polypeptide chains (Greenfield and High, 1999), indicating that it was not incorporated into TCs actively involved in protein cotranslational translocation. Our biochemical experiments have quite conclusively shown that in M3/18 cells grown at the nonpermissive temperature essentially all of the GFP–Dad1 molecules are incorporated into the OST complex. Whether all OST complexes are tightly associated with the translocon at all times is more difficult to ascertain, although biochemical and structural studies clearly support this notion (Kreibich et al., 1978; Görlich et al., 1992; Wang and Dobberstein, 1999; Menetret et al., 2000; Potter and Nicchitta, 2000).
In FRAP experiments using the M3/18 clone isolated from tsBN7 cells stably transfected with GFP–Dad1 construct, we found that GFP–Dad1 diffuses very slowly across the ER. The observed D eff for GFP–Dad1 was seven times lower than that observed for either LBR-GFP, a transmembrane protein that freely diffuses in the ER, or for Dad1–GFP, a nonfunctional fusion protein that localizes to the ER. This suggested that very little free GFP–Dad1 (which should have a D eff comparable to that of Dad1–GFP) was present in cells engaged in active protein synthesis. To determine whether the slow diffusion of GFP–Dad1 reflected its incorporation into biosynthetically active TCs, we treated cells with elevated concentration of NaCl (which inhibits initiation of translation) or with puromycin (which terminates nascent polypeptide chains). Both treatments caused a dramatic increase in the lateral diffusion of GFP–Dad1 with no effect on D eff of LBR–GFP (Table I). These data suggest that biosynthetically active TCs containing GFP–Dad1 show significantly reduced mobility compared to inactive TCs. The values of D eff for GFP–Dad1 in NaCl- or puromycin-treated cells were still two to three times lower than D eff for LBR–GFP (a fusion protein with a single transmembrane domain [TMD] that freely diffuses in the ER membrane [see Construction and expression of GFP chimeras in Materials and methods]) or nonfunctional Dad1–GFP. This indicates that GFP–Dad1 remains associated with a large complex. The data also suggest that the TC itself does not disassemble after termination of protein synthesis, in which newly synthesized polypeptides are released from ribosomes.
Assuming that the changed mobility of GFP–Dad1 in puromycin and NaCl-treated cells reflects the lateral mobility of individual TCs containing GFP–Dad1, then the measured D eff should reflect that of a complex the size of a TC. Previous estimates of the size of the TC come from models constructed from the known constituents (Rapoport et al., 1996; Johnson, and van Waes, 1999) and from structural information obtained from isolated TCs (Menetret et al., 2000). According to this information, an inactive TC contains ∼60 TMDs. The radius of the transmembrane segment usually dominates the D eff value for a membrane protein, because the membranes have a much higher viscosity than cytoplasm. Moreover, D eff of a membrane protein is proportional to ln[c/a], where a is the hydrodynamic radius of the bundle of the TMDs and c is a constant (Saffman and Delbruck, 1975; Vaz et al., 1982). In order to obtain an approximate size of the TMD bundle forming the structure with which GFP–Dad1 is associated, we compared D eff of GFP–Dad1 (in M3/18 cells treated with puromycin) with D eff for LBR–GFP. By using the simplified equation afast/aslow = eDslow/eDfast (Marguet et al., 1999), an approximate radius of 6.25 nm was calculated, corresponding to a bundle of ∼60 TMD, which is the predicted size of the TC (Johnson and van Waes, 1999). Therefore, this result is consistent with GFP–Dad1 remaining associated with the TC when protein synthesis is terminated.
Given the above results, what might account for the decreased mobility of active TCs containing GFP–Dad1? Active TCs are tethered together by mRNA to form a polysome, which take on a spiral or curved appearance, when examined by electron microscopy (Palade, 1955; Christensen and Bourne, 1999). The large and extended size of these assembled arrays would likely be a major factor reducing the mobility of active TCs observed in our FRAP experiments. This is because the polysome array enormously increases the effective radius of the diffusing species within the bilayer. An additional factor that could contribute to the reduced mobility of active TCs is the presence of nascent polypeptide chains exposed to the lumen of the ER. These chains undergo dynamic folding reactions with ER chaperones such as BiP, calnexin, and protein disulfide isomerase. Because the ER lumen has been shown to be more viscous than the cytoplasm (Dayel et al., 1999; Nehls et al., 2000), the presence of such nascent unfolded polypeptide complexes might impede the free movement of TCs across the ER.
The slow lateral mobility of active TCs may additionally be caused by interactions of membrane-bound ribosomes with cytosolic components, including cytoskeletal elements. Even if they are not directly attached to ER membranes, they may provide fence-like barriers for the diffusion of membrane-bound polysomes.
The steady state incorporation of most TCs into polysomes could provide the basis for the differentiation of the ER into rough and smooth domains. A variety of cell types exhibit well developed rough and smooth ER, which has been proposed to provide a more efficient environment for protein synthesis on membrane-bound polysomes (Rajasekaran et al., 1993). This is most apparent in hepatocytes where a sharp transition between rough and smooth ER domains is observed (Fawcett, 1981). Our data suggesting that polysome arrays diffuse extremely slowly, while individual TCs may diffuse relatively fast throughout the ER provides an explanation for how this transition might arise and be maintained. In this view, TCs would undergo constant release from and association with polysome arrays in a process coupled to termination and initiation of protein synthesis.
Materials and methods
Cell lines, antibodies, and reagents
BHK-21 and tsBN7 cells were obtained from Dr. C. Basilico (New York University Medical Center, New York, NY) and have been described earlier (Nishimoto and Basilico, 1978; Nishimoto et al., 1978). The cloned cDNA of SEAP and anti-SEAP mAb were gifts from Dr. M. Rindler (New York University Medical Center). The pAb directed against RI and RII are those previously described (Yu, et al., 1990). The antibody against ERGICp53 was obtained from Dr. H.P. Hauri (University of Basel, Basel, Switzerland). The pAb directed against GFP was purchased from CLONTECH Laboratories, Inc. The affinity-purified rabbit antibody directed against βCOPI was provided by Dr. C. DeLemos-Chiarandini (New York University Medical Center). Rabbit antiserum against Sec61β was obtained from Dr. R. Zimmermann (Universität des Saarlandes, Saarbrücken, Germany). The anti-Dad1 pAb was raised against the peptide QINPQNKADFQGISPEC, corresponding to amino acids 76 to 91 of hamster Dad1. Endonucleases, rapid ligation kit, EndoH and other supplies for molecular biology were purchased from Roche, unless otherwise stated. SeeBlue prestained standards (Novex) were used as protein molecular weight markers. Cycloheximide, tunicamycin and puromycin were obtained from Sigma-Aldrich.
Construction and expression of GFP chimeras
The cloning of the LBR–GFP, which is a fusion protein of GFP with the first 238 amino acids of human LBR was previously described (Ellenberg et al., 1997). This chimera has a single TMD with the GFP tag attached at the COOH terminus, which is facing the ER lumen. Its lateral mobility is similar to that of other ER-retained membrane proteins (Nehls et al., 2000). The cloning of human Dad1 cDNA was previously described (Fu, et al., 1997). The oligonucleotide 5′-CCGGTACCATGT CGGCGTCGGTAGTGTC-3′ was used as a sense primer to amplify the cDNA for both constructs. Oligonucleotides 5′-GGGGATCCCAGAGCGTCTAGGATGTGGT-3′ and 5′-GGGGATCCTTACAGAGCGTCTAGGATGTGGT-3′ were used as anti-sense primers to amplify the Dad1 cDNA to generate the Dad1–GFP and GFP–Dad1 constructs, respectively. The PCR products were cloned into the pEGFP-C1 or pEGFP-N3 plasmids (Clontech), using Kpn I and Bam HI cloning sites. The recombinant plasmids were named pDad1–GFP and pGFP–Dad1, respectively. LipofectAMINE PLUS reagent (GIBCO BRL) was used to transfect tsBN7 cells according to manufacturer's protocol.
Isolation of stably transfected clones
To produce stably transfected clones, the ecdysone-inducible mammalian expression system (Invitrogen) was utilized. The stably transfected cells were selected by taking advantage of the zeocin and neomycin selection markers of the plasmids. The Dad1–GFP and GFP–Dad1 constructs were sub-cloned from the corresponding recombinant plasmids into the pIND expression vector using Kpn I and Not I cloning sites. The resulting recombinant plasmids were named pINDad1-GFP and pINGFP-Dad1, respectively. To transfect 6 × 105 tsBN7 cells plated on 35-mm dishes, 1 μg of recombinant plasmid DNA was digested with Bgl I endonuclease and mixed with 2.5 μg of pVgRxR plasmid digested with Bst EI endonuclease, followed by incubation with LipofectAMINE PLUS reagent according to the protocol given by the manufacturer. The day after transfection, cells were trypsinised and plated on two 150-mm dishes containing DME (GIBCO BRL) supplemented with selective antibiotics, neomycin (1 mg/ml) and zeocin (100 μg/ml). One of the dishes was incubated at 34°C and another at 39.5°C. To maintain the effective antibiotic concentration, the medium was changed every day until clones became visible. Clones were individually transferred from 150-mm dishes to 96-well plates using cloning cylinders (Scienceware).
SDS-PAGE and Western blot
SDS-PAGE in 10–20% gradient gel and immunoblot were performed as previously described (Nikonov et al., 1992). The BM chemiluminescence Western blot kit (Roche) was used to develop the blots according to manufacturer's protocol.
Immunoprecipitation and EndoH digestion
About 3 × 106 transfected cells grown on 60-mm dishes at the appropriate temperature were incubated for 1 h in Met- and Cys-free DME, supplemented with Trans[35S]-label (400 μCi/dish) (INC Biomedicals). The 35S-labeled cells were solubilized in 400 μl of lysate buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% Triton X-100, 1 mM PMSF, 2 μg/ml leupeptin and 20 μg/ml pepstatin). Insoluble material was removed by centrifugation of the lysate at 10,000 g for 30 min. The supernatant (100 μl) was diluted with 900 μl of lysate buffer and incubated for 1.5 h with protein A-Sepharose beads, precoated with preimmune serum. The beads were recovered by brief centrifugation at 200 g. The cleared supernatant was then incubated with specific antibody for 1.5 h, followed by addition of protein A-Sepharose beads. After incubation for another 1.5 h, the beads were washed once with lysate buffer, twice with washing buffer 2 (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 0.1% NP-40, 0.05% Triton X-100), and twice with washing buffer 3 (50 mM Tris-HCl, pH 7.5, 0.1% NP-40, 0.05% Triton X-100). The beads were resuspended in EndoH buffer (50 mM sodium citrate buffer, pH 5.5, 0.02% SDS, 0.5 mM PMSF), and each sample was split into two tubes. One part of the sample was digested with EndoH, whereas the other one was left untreated. The samples were subjected to SDS-PAGE, followed by autoradiography.
Immunofluorescence microscopy
M3/18 cells were seeded and grown on glass cover slips overnight at 39.5°C. The cells were fixed in 2% paraformaldehyde in PBS for 20 min at room temperature and permeabilized in 0.1% of Triton X-100 in PBS for 5 min. Nonspecific binding sites of antibodies were blocked with 5% skimmed milk in PBS for 30 min at room temperature. The permeabilized cells were incubated with primary antibodies for 1 h, rinsed three times with PBS, incubated with anti–rabbit IgG goat antibodies conjugated to Texas red and studied under confocal microscope as described in the figure legend. The composite figures were prepared using Adobe Photoshop 6.0 (Adobe).
Subcellular fractionation on Nycodenz gradient
Subcellular fractionation was performed on Nycodenz (Accurate Chemical & Scientific Corp.) gradients as previously described (Helenius, 1994). In contrast to the original procedure, the homogenization buffer and Nycodenz gradient were supplemented with potassium acetate (50 mM) and magnesium acetate (6 mM), as we noticed that the lack of these salts in the gradient leads to stripping rough microsomes of ribosomes during ultracentrifugation (unpublished data). 50 μl of each 1-ml fraction collected was mixed with 2× SDS-PAGE sample buffer and loaded directly on the 10–20% gradient gel and subjected to SDS-PAGE omitting the TCA precipitation step described earlier (Greenfield and High, 1999).
Sedimentation analysis of OST complexes from digitonin-solubilized cell extracts
Aside from some minor changes, the sedimentation behavior of OST components was analyzed on glycerol gradients as previously described (Kelleher et al., 1992; Kelleher and Gilmore, 1997). Cells grown on 150-mm cell culture dishes (80% confluence) were washed three times with ice-cold washing buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM MgCl2, protease inhibitor cocktail (PIC) containing 0.1 μg/ml of each pepstatin A, chymostatin, leupeptin, antipain, and 1 μg/ml of aprotinin). Using rubber policemen, cells were scraped into 1 ml of homogenization buffer (20 mM Tris-HCl, pH 7.4, 500 mM NaCl, 1.5% digitonin, 1 mM MnCl2, 1 mM MgCl2, 1 mM DTT, PIC and 1mM PMSF) and passed 15 times through a ball-bearing homogenizer (clearance 16 μm). After incubation on ice for 30 min, the homogenates (T) were centrifuged for 15 min at 42,200 rpm, 4°C, using the 75 TI rotor (Beckman). Pellets (P) were resuspended in 1 ml washing buffer and saved for analysis. 850 μl of the supernatant fractions (S) were layered onto continuous glycerol gradients (8–30%) containing 20 mM Tris-HCl, pH 7.4, 50 mM NaCl, 0.125% digitonin, 25 μg/ml egg yolk L-α-lecithin, 1 mM MnCl2, 1 mM MgCl2, 1 mM DTT, PIC and 1 mM PMSF. Stock solutions of digitonin (Calbiochem, 300410) and egg yolk L-α-lecithin (Calbiochem, 524617) were prepared exactly as previously described (Kelleher et al., 1992). The gradients were centrifuged for 15.5 h at 35,000 rpm, 4°C, using a SW41 rotor (Beckman). After collecting the first 850 μl of the gradient, corresponding to the LZ, the rest of the gradient was fractionated into 10 1.15-ml fractions. Possible pellets formed during centrifugation (GP) were resuspended in 1.15 ml of washing buffer. 5 μl of total cell lysate (T), of the pellet formed during 42,200 rpm centrifugation (P) and of the supernatant (S) as well as 30 μl of each glycerol gradient fraction, including GP, were analyzed by SDS-PAGE, followed by Western blot analysis.
FRAP
FRAP experiments were performed on a LSM510 confocal microscope (Zeiss) using the 488-nm line of a 40-mW Argon laser for excitation of GFP and a 40× water-corrected, 1.2 NA objective. The microscope was operated using software (release 2.5) supplied with the instrument and supplemented by a Physiology macro (Zeiss). The digital zoom was set at 4, so that the whole cell was within an image; the detector gain value was set at 775; the amplification gain value was set to 1, and the background cut off value at 0. We have used a wide-open pinhole, corresponding to an optical focus of ∼14 μm to collect as much light as possible and to ensure that ER membranes were bleached across the entire depth of the cells. For quantitative diffusion measurements a 2-μm wide strip extending across the entire width of the cell was photobleached for four iterations (0.07 s bleach time in total) at 64% of laser power and 100% transmission. The recovery of fluorescence intensity in the bleached area was recorded at the same laser power but the transmission level was reduced to 0.4% and scan speed was set to 10. Recordings were made every 3 (clone M3/18) or 0.7 s in the case of transiently transfected cells and were continued until the fluorescence recovery reached an asymptote. To calculate values of D eff, we used a simulation code previously described (Siggia et al., 2000). This computer simulation is based on a continuum description for diffusion in a simple model for an inhomogeneous but isotropic media. In analyzing a typical FRAP experiment, the locally averaged fluorescent intensities of the diffusible GFP chimera before and during photobleach recovery was used as input. Diffusive relaxation was then simulated from the initial bleach condition to obtain the D eff. In order to obtain correctly calculated diffusion constants, it is essential that the entire bleached cell is part of the analyzed image. KaleidaGraph 3.5 (Synergy Software) was used to plot fluorescence recovery curves after photobleaching.
Acknowledgments
The gift of tsBN7 and BHK-21 cells from Dr. C. Basilico, and antibodies from several investigators as well as the advice obtained from Dr. M. Edidin (Johns Hopkins University) and Dr. R. Gilmore (University of Massachusetts) are greatly appreciated. The contributions of Dr. D. D. Sabatini (New York University School of Medicine) to the conceptual framework of this project are gratefully acknowledged.
This work was supported by a grant from the American Cancer Society (RPG-92-009-06-CB). A.V. Nikonov was supported by a National Institutes of Health training grant in Dermatology (5T32 ARO 7190), and E. Snapp is a Pharmacology Research Training Fellow.
Footnotes
Abbreviations used in this paper: D eff, effective diffusion constant; EndoH, endoglycosidase H; ERGIC, ER–Golgi intermediate compartment; LBR, lamin B receptor; LZ, loading zone; M f, mobile fraction; OST, oligosaccharyltransferase; PIC, protease inhibitor cocktail, R, ribophorin; SEAP, secreted form of bovine alkaline phosphatase; TC, translocon complex; TMD, transmembrane domain; ts, temperature sensitive.
References
- Adelman, M.R., G. Blobel, and D.D. Sabatini. 1973. An improved cell fractionation procedure for the preparation of rat liver membrane-bound ribosomes. J. Cell Biol. 56:191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann, O., and B. Walz. 2001. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 205:149–214. [DOI] [PubMed] [Google Scholar]
- Chazotte, B., J. Ellenberg, and J. Lippincott-Schwartz. 1998. Fluorescence photobleaching techniques. Cells: A Laboratory Manual. D.L. Specter, R. Goldman, and L. Leinwand, editors. Cold Spring Harbor Press, Cold Spring Harbor, NY. 79.1–79.23.
- Christensen, A.K., and C.M. Bourne. 1999. Shape of large bound polysomes in cultured fibroblasts and thyroid epithelial cells. Anat. Rec. 255:116–129. [DOI] [PubMed] [Google Scholar]
- Dayel, M.J., E.F. Hom, and A.S. Verkman. 1999. Diffusion of green fluorescent protein in the aqueous-phase lumen of endoplasmic reticulum. Biophys. J. 76:2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenberg, J., E.D. Siggia, J.E. Moreira, C.L. Smith, J.F. Presley, H.J. Worman, and J. Lippincott-Schwartz. 1997. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 138:1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett, D.W. 1981. The Cell. W.B. Saunders Company, Philadelphia, London. 827 pp.
- Fu, J., M. Ren, and G. Kreibich. 1997. Interactions among subunits of the oligosaccharyltransferase complex. J. Biol. Chem. 272:29687–29692. [DOI] [PubMed] [Google Scholar]
- Görlich, D., S. Prehn, E. Hartmann, K.U. Kalies, and T.A. Rapoport. 1992. A mammalian homolog of SEC61p and SECYp is associated with ribosomes and nascent polypeptides during translocation. Cell. 71:489–503. [DOI] [PubMed] [Google Scholar]
- Greenfield, J.J., and S. High. 1999. The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J. Cell Sci. 112:1477–1486. [DOI] [PubMed] [Google Scholar]
- Helenius, A. 1994. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. J. Cell Sci. 5:253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, N.A., M. Flannery, S.N. Hsieh, D. Cado, R. Pedersen, and A. Winoto. 2000. Mice lacking Dad1, the defender against apoptotic death-1, express abnormal N-linked glycoproteins and undergo increased embryonic apoptosis. Dev. Biol. 220:76–84. [DOI] [PubMed] [Google Scholar]
- Jacobson, K., E.D. Sheets, and R. Simson. 1995. Revisiting the fluid mosaic model of membranes. Science. 268:1441–1442. [DOI] [PubMed] [Google Scholar]
- Johnson, A.E., and M.A. van Waes. 1999. The translocon: a dynamic gateway at the ER membrane. Annu. Rev. Cell Dev. Biol. 15:799–842. [DOI] [PubMed] [Google Scholar]
- Kelleher, D.J., and R. Gilmore. 1997. DAD1, the defender against apoptotic cell death, is a subunit of the mammalian oligosaccharyltransferase. Proc. Natl. Acad. Sci. USA. 94:4994–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher, D.J., G. Kreibich, and R. Gilmore. 1992. Oligosaccharyltransferase activity is associated with a protein complex composed of ribophorins I and II and a 48 kd protein. Cell. 69:55–65. [DOI] [PubMed] [Google Scholar]
- Kreibich, G., B.L. Ulrich, and D.D. Sabatini. 1978. Proteins of rough microsomal membranes related to ribosome binding. I. Identification of ribophorins I and II, membrane proteins characteristics of rough microsomes. J. Cell Biol. 77:464–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt, R., S. Schlesinger, and S. Kornfeld. 1977. Tunicamycin inhibits glycosylation and multiplication of Sindbis and vesicular stomatitis viruses. J. Virol. 21:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., E. Snapp, and A. Kenworthy. 2001. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2:444–456. [DOI] [PubMed] [Google Scholar]
- Makishima, T., T. Nakashima, K. Nagata-Kuno, K. Fukushima, H. Iida, M. Sakaguchi, Y. Ikehara, S. Komiyama, and T. Nishimoto. 1997. The highly conserved DAD1 protein involved in apoptosis is required for N-linked glycosylation. Genes Cells. 2:129–141. [DOI] [PubMed] [Google Scholar]
- Marguet, D., E.T. Spiliotis, T. Pentcheva, M. Lebowitz, J. Schneck, and M. Edidin. 1999. Lateral diffusion of GFP-tagged H2Ld molecules and of GFP-TAP1 reports on the assembly and retention of these molecules in the endoplasmic reticulum. Immunity. 11:231–240. [DOI] [PubMed] [Google Scholar]
- Martinez, O., A. Schmidt, J. Salamero, B. Hoflack, M. Roa, and B. Goud. 1994. The small GTP-binding protein rab6 functions in intra-Golgi transport. J. Cell Biol. 127:1575–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menetret, J., A. Neuhof, D.G. Morgan, K. Plath, M. Radermacher, T.A. Rapoport, and C.W. Akey. 2000. The structure of ribosome-channel complexes engaged in protein translocation. Mol. Cell. 6:1219–1232. [DOI] [PubMed] [Google Scholar]
- Nakashima, T., T. Sekiguchi, A. Kuraoka, K. Fukushima, Y. Shibata, S. Komiyama, and T. Nishimoto. 1993. Molecular cloning of a human cDNA encoding a novel protein, DAD1, whose defect causes apoptotic cell death in hamster BHK21 cells. Mol. Cell. Biol. 13:6367–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehls, S., E.L. Snapp, N.B. Cole, K.J. Zaal, A.K. Kenworthy, T.H. Roberts, J. Ellenberg, J.F. Presley, E. Siggia, and J. Lippincott-Schwartz. 2000. Dynamics and retention of misfolded proteins in native ER membranes. Nat. Cell Biol. 2:288–295. [DOI] [PubMed] [Google Scholar]
- Nikonov, A.V., N.A. Barlev, and S.N. Borkhsenius. 1992. Molecular biological differences between strains of Mycoplasma gallisepticum. Tsitologiia. 34:107–112. [PubMed] [Google Scholar]
- Nishimoto, T., and C. Basilico. 1978. Analysis of a method for selecting temperature-sensitive mutants of BHK cells. Somatic Cell Genet. 4:323–340. [DOI] [PubMed] [Google Scholar]
- Nishimoto, T., E. Eilen, and C. Basilico. 1978. Premature of chromosome condensation in a ts DNA- mutant of BHK cells. Cell. 15:475–483. [DOI] [PubMed] [Google Scholar]
- Palade, G.E. 1955. A small particulate component of the cytoplasm. J. Biophys. Biochem. Cytol. 1:59–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter, M.D., and C.V. Nicchitta. 2000. Ribosome-independent regulation of translocon composition and Sec61alpha conformation. J. Biol. Chem. 275:2037–2045. [DOI] [PubMed] [Google Scholar]
- Rajasekaran, A.K., T. Morimoto, D.K. Hanzel, E. Rodriguez-Boulan, and G. Kreibich. 1993. Structural reorganization of the rough endoplasmic reticulum without size expansion accounts for dexamethasone-induced secretory activity in AR42J cells. J. Cell Sci. 105:333–345. [DOI] [PubMed] [Google Scholar]
- Rapoport, T.A., B. Jungnickel, and U. Kutay. 1996. Protein transport across the eukaryotic endoplasmic reticulum and bacterial inner membranes. Annu. Rev. Biochem. 65:271–303. [DOI] [PubMed] [Google Scholar]
- Saborio, J.L., S.S. Pong, and G. Koch. 1974. Selective and reversible inhibition of initiation of protein synthesis in mammalian cells. J. Mol. Biol. 85:195–211. [DOI] [PubMed] [Google Scholar]
- Saffman, P.G., and M. Delbruck. 1975. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. USA. 72:3111–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjay, A., J. Fu, and G. Kreibich. 1998. DAD1 is required for the function and the structural integrity of the oligosaccharyltransferase complex. J. Biol. Chem. 273:26094–26099. [DOI] [PubMed] [Google Scholar]
- Schindler, R., C. Itin, M. Zerial, F. Lottspeich, and H.P. Hauri. 1993. ERGIC-53, a membrane protein of the ER-Golgi intermediate compartment, carries an ER retention motif. Eur. J. Cell Biol. 61:1–9. [PubMed] [Google Scholar]
- Siggia, E.D., J. Lippincott-Schwartz, and S. Bekiranov. 2000. Diffusion in inhomogeneous media: theory and simulations applied to whole cell photobleach recovery. Biophys. J. 79:1761–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein, S., P.G. Collins, D.J. Kelleher, and R. Gilmore. 1995. The essential OST2 gene encodes the 16-kD subunit of the yeast oligosaccharyltransferase, a highly conserved protein expressed in diverse eukaryotic organisms. J. Cell Biol. 131:371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, S.J., and G.L. Nicolson. 1972. The fluid mosaic model of the structure of cell membranes. Science. 175:720–731. [DOI] [PubMed] [Google Scholar]
- Vaz, W.L., M. Criado, V.M. Madeira, G. Schoellmann, and T.M. Jovin. 1982. Size dependence of the translational diffusion of large integral membrane proteins in liquid-crystalline phase lipid bilayers. A study using fluorescence recovery after photobleaching. Biochemistry. 21:5608–5612. [DOI] [PubMed] [Google Scholar]
- Walter, P., and A.E. Johnson. 1994. Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane. Annu. Rev. Cell Biol. 10:87–119. [DOI] [PubMed] [Google Scholar]
- Wang, L., and B. Dobberstein. 1999. Oligomeric complexes involved in translocation of proteins across the membrane of the endoplasmic reticulum. FEBS Lett. 457:316–322. [DOI] [PubMed] [Google Scholar]
- Yu, Y.H., D.D. Sabatini, and G. Kreibich. 1990. Antiribophorin antibodies inhibit the targeting to the ER membrane of ribosomes containing nascent secretory polypeptides. J. Cell Biol. 111:1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]