

Abstract
Caspase-independent death mechanisms have been shown to execute apoptosis in many types of neuronal injury. P53 has been identified as a key regulator of neuronal cell death after acute injury such as DNA damage, ischemia, and excitotoxicity. Here, we demonstrate that p53 can induce neuronal cell death via a caspase-mediated process activated by apoptotic activating factor-1 (Apaf1) and via a delayed onset caspase-independent mechanism. In contrast to wild-type cells, Apaf1-deficient neurons exhibit delayed DNA fragmentation and only peripheral chromatin condensation. More importantly, we demonstrate that apoptosis-inducing factor (AIF) is an important factor involved in the regulation of this caspase-independent neuronal cell death. Immunofluorescence studies demonstrate that AIF is released from the mitochondria by a mechanism distinct from that of cytochrome-c in neurons undergoing p53-mediated cell death. The Bcl-2 family regulates this release of AIF and subsequent caspase-independent cell death. In addition, we show that enforced expression of AIF can induce neuronal cell death in a Bax- and caspase-independent manner. Microinjection of neutralizing antibodies against AIF significantly decreased injury-induced neuronal cell death in Apaf1-deficient neurons, indicating its importance in caspase-independent apoptosis. Taken together, our results suggest that AIF may be an important therapeutic target for the treatment of neuronal injury.
Keywords: neurodegeneration; neurons; apoptosis; p53; Bax
Introduction
Apoptotic cell death plays an important role in brain development as well as in neuronal injury and disease. In the developing nervous system, apoptosis is required for the establishment of appropriate cell numbers and for the elimination of improperly connected neurons (Pettmann and Henderson, 1998). In the adult nervous system, the inappropriate induction of apoptotic cell death contributes to the neuropathology associated with a number of neurodegenerative diseases (Portera-Cailliau et al., 1995; Smale et al., 1995) as well as acute neurological insults (Nitatori et al., 1995; Yakovlev et al., 1997). Therefore, identifying the molecular mechanisms that regulate neuronal apoptosis is essential for the development of therapeutic strategies for the treatment of such neurological conditions.
A number of death regulatory molecules have been implicated in neuronal injury induced by ischemia, including p53, PARP, c-jun, and plasma membrane death receptor ligand systems (Eliasson et al., 1997; Endres et al., 1997; Herdegen et al., 1998; Morrison and Kinoshita, 2000; Martin-Villalba et al., 2001). Importantly, several lines of evidence suggest that p53 is a key upstream initiator of the cell death process after neuronal injury. P53 expression has been reported to be upregulated in response to excitotoxins, hypoxia, and ischemia (Xiang et al., 1996; Banasiak and Haddad, 1998; McGahan et al., 1998). Accordingly, we and others have shown that enforced expression of p53 alone is sufficient to trigger apoptosis in postmitotic neurons (Slack et al., 1996; Xiang et al., 1998; Cregan et al., 1999). In addition, it has been demonstrated that brain damage induced by ischemia or kainic acid excitotoxicity is significantly reduced in mice carrying a null mutation for the p53 gene (Crumrine et al., 1994; Morrison et al., 1996). Furthermore, cultured neurons derived from p53-deficient mice have been shown to be resistant to excitotoxins (Xiang et al., 1996, 1998), DNA damaging agents (Johnson et al., 1998; Xiang et al., 1998; Morris et al., 2001), and hypoxia (Halterman et al., 1999).
Caspases are a family of cysteine proteases that have been implicated as key effector molecules in the execution of apoptotic cell death (Cryns and Yuan, 1998). Recent studies have demonstrated the involvement of caspases in the execution of neuronal cell death both during development and after injury. Mouse embryos deficient for apoptotic activating factor-1 (Apaf1),* caspase-9, or caspase-3 display severe craniofacial malformations and dramatically enhanced neuronal cell numbers (Kuida et al., 1996, 1998; Cecconi et al., 1998). These gross developmental defects were attributed to failed apoptosis in the neuroepithelium. The importance of the caspase signaling cascade has also been demonstrated in many models of neuronal injury, including traumatic brain injury and ischemia (Hara et al., 1997; Yakovlev et al., 1997; Cheng et al., 1998).
Although caspases have been recognized as important mediators of apoptosis, there is accumulating evidence indicating the existence of caspase-independent mechanisms of neuronal cell death (Rideout and Stefanis, 2001). For example, several groups have indicated that in excitotoxic cell death, caspases are not activated and peptide-based caspase inhibitors do not invoke neuroprotection (Johnson et al., 1999; Lankiewicz et al., 2000). Similarly, in experimental models of stroke, caspase inhibition affords protection in certain neuronal populations, but not in others (Rideout and Stefanis, 2001; Zhan et al., 2001). Furthermore, in a number of neuronal cell death paradigms in which caspases are normally activated, inhibition of caspase activity delays, but does not prevent cell death from occurring (Miller et al., 1997; Stefanis et al., 1999; D'Mello et al., 2000; Keramaris et al., 2000; Selznick et al., 2000). Thus it appears that, at least in certain neuronal death paradigms, caspase inhibition simply results in the activation or recruitment of compensatory cell death processes. Although there has been extensive investigation on caspase-mediated cell death processes, much less is known about the molecular mechanisms involved in the regulation of caspase-independent cell death.
Apoptosis-inducing factor (AIF) is a putative caspase-independent effector of cell death that has recently been cloned and characterized (Susin et al., 1999). AIF is a mitochondrial intermembrane flavoprotein that has been reported to be released from the mitochondria and to translocate to the nucleus in response to specific death signals (Daugas et al., 2000). Furthermore, this apoptotic factor has been shown to cause high molecular weight DNA fragmentation and chromatin condensation in cells and isolated nuclei in a caspase-independent manner (Susin et al., 1999, 2000; Daugas et al., 2000).
In the present study, we demonstrate that p53 can induce neuronal cell death via a caspase-mediated process in the presence of Apaf1 and via a delayed onset caspase-independent mechanism in the absence of Apaf1. More importantly, we demonstrate that AIF is an important factor involved in the regulation of caspase-independent cell death induced by p53-mediated neuronal injury.
Results
P53 can induce neuronal cell death via Apaf1-dependent and -independent pathways
We have previously reported that caspase-3–deficient neurons exhibit a significant delay in p53-induced cell death (Cregan et al., 1999; Keramaris et al., 2000). Nevertheless, in the absence of caspase-3, neurons do eventually undergo apoptosis, suggesting that either additional caspases or caspase-independent effectors could also be involved. To distinguish between these possibilities, we examined p53-induced cell death in the presence or absence of Apaf1, a critical cofactor of the mitochondrial-initiated caspase activation complex. Indeed we have recently demonstrated that p53-induced neuronal cell death involves the transcriptional upregulation of Apaf1 (Fortin et al., 2001).
In the present study, we examined long-term survival in Apaf1-deficient neurons to determine whether caspase-independent cell death occurred. We treated wild-type or Apaf1-deficient neurons with the DNA damaging agent camptothecin, which has previously been shown to induce neuronal cell death through a p53-dependent mechanism (Xiang et al., 1998; Morris et al., 2001). Camptothecin treatments were administered in the presence or absence of a broad spectrum caspase inhibitor, Boc-aspartyl (OMe)-fluoromethylketone (BAF), to account for the possible involvement of Apaf1-independent pathways of caspase activation. In wild-type neurons, treatment with camptothecin resulted in a rapid loss of neuronal survival beginning at ∼12 h, and within 24 h, survival had decreased to <20% (Fig. 1 A). In contrast, Apaf1-deficient neurons remained largely viable during the first 24 h of treatment. However, after 24 h, cell survival began to decline and by 48 h, <30% of Apaf1−/− neurons remained viable. These results demonstrate that p53 induces neuronal cell death through a rapid Apaf1-dependent pathway in wild-type neurons, and through a delayed pathway in Apaf1-deficient neurons.
Figure 1.


Camptothecin induces neuronal cell death via a caspase- mediated process in the presence of Apaf1 and via a delayed onset caspase-independent pathway in the absence of Apaf1. Wild-type and Apaf1-deficient cortical neurons were treated with camptothecin (10 μM) in the presence or absence of a broad spectrum caspase inhibitor (BAF, 50 μM). (A) Neuronal survival was measured by live/dead assay (n = 4). (B) Caspase-3 activity was determined by DEVD-AFC cleavage (n = 3). (C) The proportion of apoptotic cells was determined by TUNEL staining (n = 3). (D) Nuclear morphology was assessed by Hoechst staining. Bar, 15 μm.
These two cell death pathways could be further distinguished on the basis of caspase involvement and cellular morphology. In wild-type neurons, camptothecin-induced cell death was associated with an approximate ninefold increase in caspase-3 activity at 16 h (Fig. 1 B), and this was accompanied by a significant increase in the fraction of cells exhibiting DNA fragmentation, as detected by TUNEL assay (50.2 vs. 12.3% in controls; Fig. 1 C). Consistent with a requirement for caspases in this death process, the pan-caspase inhibitor BAF significantly reduced the fraction of TUNEL-positive cells at 16 h (11.8 vs. 50.2%; Fig. 1 B), and significantly enhanced neuronal survival at 24 h (70.7 vs. 19.8%; Fig. 1 A). In contrast, the delayed cell death induced by camptothecin in Apaf1-deficient neurons was not associated with caspase-3 activation (Fig. 1 B), and the loss of neuronal survival was not prevented by BAF (Fig. 1 A). This indicated that cell death induced in Apaf1−/− neurons did not involve additional or compensatory caspase activation pathways. Interestingly, the delayed cell death observed in Apaf1−/− neurons was associated with an increase in TUNEL-positive cells, however, this only became evident at much later times (42.2 vs. 8.5% in controls at 48 h; Fig. 1 C). Furthermore, this TUNEL labeling was not accompanied by caspase-3 activation (Fig. 1 B) and was not prevented by BAF (Fig. 1 C), indicating that, unlike in wild-type neurons, this DNA fragmentation was not caspase mediated. Cell death in wild-type and Apaf1−/− neurons could be further distinguished on the basis of nuclear morphology. Neurons undergoing Apaf1-mediated cell death displayed typical apoptotic features, including dense chromatin condensation, nuclear pyknosis, and in some cases nuclear fragmentation (Fig. 1 D, arrow). Apaf1-deficient neurons, in contrast, exhibited partial nuclear pyknosis and margination but not complete chromatin condensation (Fig. 1 D, arrowhead).
Next, to determine whether p53 could directly induce caspase-independent cell death in the absence of Apaf1, we infected Apaf1+/+ and Apaf1−/− cortical neurons with a recombinant adenoviral vector carrying an expression cassette for either p53 or the control gene LacZ. Adenoviral-mediated expression of p53 resulted in a significant induction of TUNEL-positive cell death in both wild-type (61.4 vs. 16.8%) and Apaf1-deficient neurons (44.5 vs. 14.9%) relative to Ad-LacZ–infected controls (Fig. 2 A). Cell death induced by direct p53 expression was associated with a significant induction of caspase-3 activity in wild-type neurons (4.7-fold increase), but not in Apaf1-deficient neurons (Fig. 2 B). Furthermore, Ad-p53–induced TUNEL labeling was not prevented in the presence of a pan-caspase inhibitor (unpublished data). This indicated that, similar to the delayed cell death induced by camptothecin, DNA fragmentation triggered by direct p53 expression in Apaf1-deficient neurons was induced in a caspase-independent manner. Furthermore, whereas wild-type neurons undergoing cell death exhibited classical apoptotic nuclear morphology, Apaf1-deficient neurons again displayed only peripheral chromatin condensation and intermediate pyknosis (unpublished data). These results indicate that the p53 pathway can trigger a caspase-mediated cell death process in the presence of Apaf1 and a slower caspase-independent death process in the absence of Apaf1.
Figure 2.
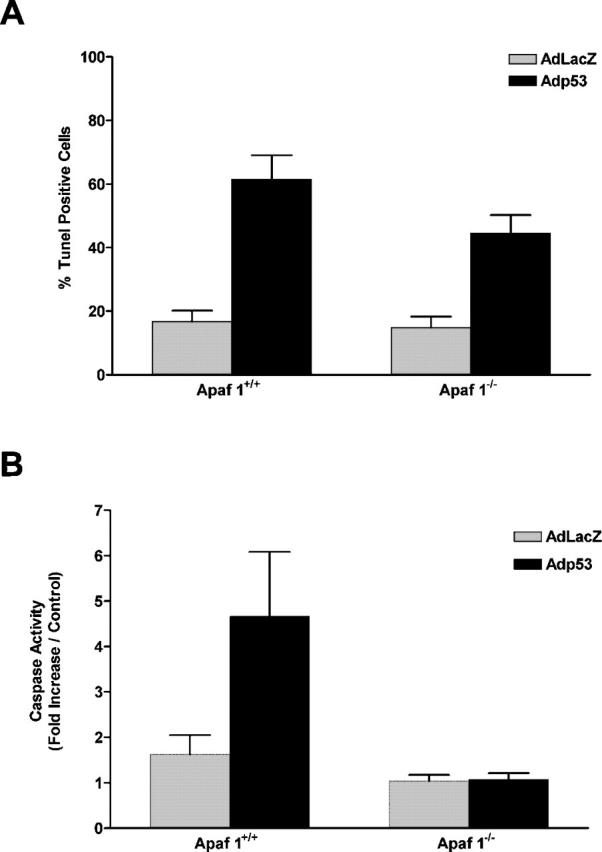
Direct expression of p53 induces neuronal cell death via a caspase-mediated process in the presence of Apaf1 and via a caspase-independent pathway in the absence of Apaf1. Wild-type and Apaf1-deficient neurons were infected with Ad-p53 or the control vector Ad-LacZ at 50 multiplicity of infection (MOI). (A) The fraction of TUNEL-positive cells was measured 72 h after infection (n = 3). (B) Caspase-3 activity was measured at 72 h by DEVD-AFC cleavage (n = 3).
P53 triggers mitochondrial release of AIF
It has previously been shown that in neurons, camptothecin triggers the release of cytochrome-c from the mitochondria (Stefanis et al., 1999). Upon release into the cytoplasm, cytochrome-c is believed to interact with Apaf1 and facilitate the formation of a caspase activating complex with caspase-9 (Li et al., 1997; Hu et al., 1999). AIF is a mitochondrial intermembrane protein that, like cytochrome-c, has been reported to be released from the mitochondria in response to specific death stimuli (Daugas et al., 2000). In contrast to cytochrome-c, however, AIF has been reported to induce apoptotic nuclear morphology independent of caspase activity (Susin et al., 2000). Therefore, in the present study, we investigated a potential role of AIF in the Apaf1/caspase-independent pathway of p53-induced neuronal cell death.
To determine whether AIF was released from the mitochondria during p53-mediated neuronal injury, we examined the cellular localization of AIF by immunofluorescence in neurons infected with Ad-p53 or Ad-LacZ. Our results demonstrate that the majority of neurons infected with the control vector displayed a punctate, cytoplasmic staining pattern that colocalized with a mitochondrial-specific marker (Fig. 3 A). In contrast, AIF staining was dramatically diminished and did not colocalize with the mitochondrial marker in Ad-p53–infected neurons exhibiting morphological features of apoptotic cell death (Fig. 3 A, arrow). Despite the significant decrease in immunostaining intensity, Western blot analysis demonstrated that the dying cells retained AIF and cytochrome-c (Fig. 3 B), suggesting that these proteins may diffuse from the cells during the fixation and permeabilization procedure. Furthermore, many Apaf1-deficient neurons infected with Ad-p53 exhibited diffuse AIF staining in the nucleus (Fig. 3 A, arrowhead). These results suggest that AIF is released from the mitochondria and can translocate to the nucleus during p53-induced neuronal cell death. It is unclear why AIF fails to translocate to the nucleus in Apaf1+/+ neurons; however, it is possible that the early activation of caspases in wild-type cells results in the inactivation of the process responsible for AIF nuclear translocation.
Figure 3.


AIF is released from the mitochondria during p53-induced neuronal cell death. (A) Apaf1+/+ and Apaf1−/− cortical neurons were infected at 50 MOI with Ad-p53 or the control vector Ad-LacZ. After 72 h, neurons were labeled with a mitochondrial-specific dye (mitotracker green FM, 0.5 μM), fixed, and immunostained for AIF. The incorporation of mitotracker green FM into mitochondria does not depend on the mitochondrial transmembrane potential. Neurons undergoing p53-mediated cell death exhibit loss of mitochondrial AIF staining (arrows) and nuclear translocation (arrowheads). Bar, 25 μm. (B) Western blot analysis of AIF and cytochrome-c levels in neurons 48 h after infection with Ad-p53 or Ad-LacZ. (C) P53−/−, Apaf1−/−, and corresponding wild-type neurons were treated with camptothecin (10 μM) and then fixed and stained for AIF after 24 h. Representative images were captured and the proportion of cells exhibiting punctate, mitochondrial AIF staining was scored (n = 3).
To ascertain whether AIF is released from the mitochondria in response to endogenous p53 activation, we examined AIF immunoreactivity after camptothecin treatment in neurons derived from p53- or Apaf1-deficient mice and corresponding wild-type littermates. Camptothecin treatment resulted in a significant decrease in the fraction of cells retaining mitochondrial AIF in wild-type (30.1 vs. 87.2% in controls) and Apaf1-deficient neuron cultures (35.2 vs. 92.2% in controls), but did not induce a significant decrease in p53-deficient cultures (88.3 vs. 91.5% in controls) (Fig. 3 C). This indicated that camptothecin triggered the release of AIF from mitochondria by a p53-dependent, but Apaf1-independent, mechanism.
We then assessed whether AIF release was associated with other mitochondrial events involved in DNA damage–induced neuronal cell death, such as mitochondrial depolarization and cytochrome-c release (Stefanis et al., 1999). Neurons treated with camptothecin were monitored for the mitochondrial release of cytochrome-c or AIF by immunofluorescence staining. In parallel cultures, mitochondrial depolarization was assessed in live cells by CMX-Ros labeling. This fluorescent dye is selectively incorporated into mitochondria with an intact transmembrane potential and therefore serves as an indicator of mitochondrial depolarization. Our results demonstrate that camptothecin induced a time-dependent decrease in the fraction of cells retaining mitochondrial transmembrane potential as well as mitochondrial cytochrome-c and AIF (Fig. 4 A). Interestingly, release of cytochrome-c appeared to precede the loss of AIF and mitochondrial membrane potential such that within 12 h of camptothecin treatment, there was already a significant decrease in the fraction of cells maintaining mitochondrial cytochrome-c staining (∼45%), but only a modest decrease in the fraction of cells exhibiting mitochondrial AIF (∼5%) and CMX-Ros (∼15%) staining (Fig. 4, A and B). These results suggest that AIF and cytochrome-c release occur by different mechanisms during p53-induced neuronal cell death.
Figure 4.


AIF and cytochrome-c are released by distinct mechanisms during camptothecin-induced neuronal cell death. (A) Apaf1-deficient neurons were treated with camptothecin (10 μM) and at the indicated times, cells were labeled with CMX-Ros to assess mitochondrial transmembrane potential, or fixed and immunostained for AIF or cytochrome-c. Images were captured and the fraction of cells retaining mitochondrial transmembrane potential or exhibiting mitochondrial cytochrome-c/AIF staining was scored (n = 4). (B) Photomicrographs of Apaf1−/− neurons treated with camptothecin for 12 h and stained for AIF, cytochrome-c, or CMX-Ros and the corresponding Hoechst or phase images. Bar, 30 μm.
Mitochondrial release of AIF and caspase-independent cell death are regulated by Bax and Bcl-2
The Bcl-2 gene family consists of proapoptotic and antiapoptotic members that are thought to regulate cell death through their opposing effects on mitochondrial-mediated death processes (Adams and Cory, 1998; Gross et al., 1999). We and others have previously shown that the proapoptotic member Bax plays a critical role in regulating p53-induced cytochrome-c release, caspase activation, and neuronal cell death (Xiang et al., 1998; Cregan et al., 1999; Keramaris et al., 2000; Morris et al., 2001). Here we investigated the role of Bax and Bcl-2 in the regulation of p53-induced mitochondrial events and caspase-independent cell death. To determine whether Bax was required for these mitochondrial death processes, Bax+/+ and Bax−/− neurons were treated with camptothecin and then examined for the loss of mitochondrial membrane potential (Fig. 5 A) and AIF release (Fig. 5 B). Camptothecin triggered mitochondrial depolarization and the release of AIF in a significant fraction of Bax+/+ neurons (∼60 and ∼50% respectively), but not in Bax-deficient neurons, suggesting that Bax mediates p53-induced mitochondrial depolarization and AIF release in neurons.
Figure 5.
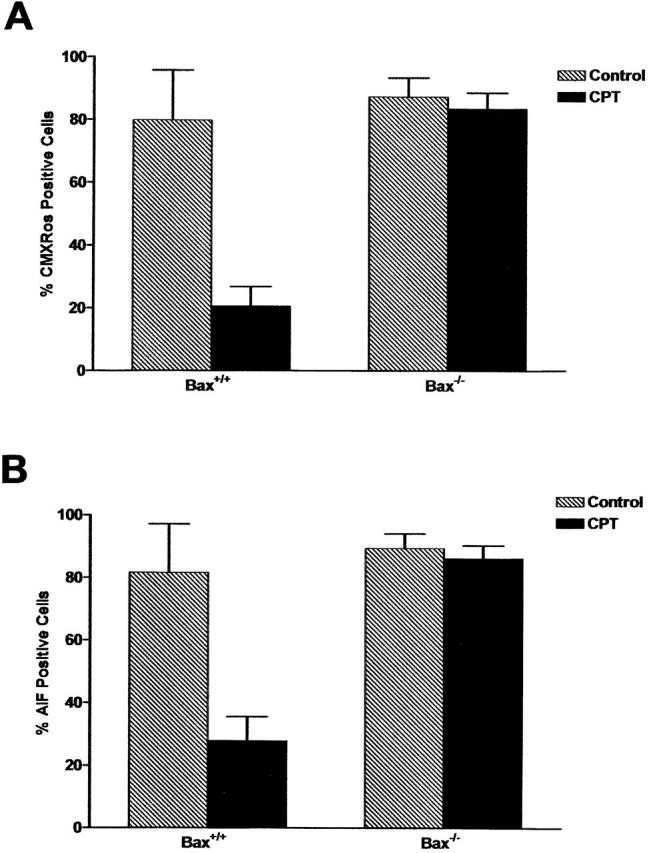
Bax mediates mitochondrial depolarization and the release of AIF during camptothecin-induced neuronal cell death. Wild-type and Bax-deficient neurons were treated with camptothecin and after 24 h, cells were either labeled with CMX-Ros to assess mitochondrial transmembrane potential or fixed and immunostained for AIF. Representative images were taken, and the proportion of cells exhibiting positive mitochondrial staining for CMX-Ros (A) or AIF (B) was scored (n = 3).
We then asked whether the antiapoptotic member, Bcl-2, could block these cell death processes. Apaf1+/+ and Apaf1−/− neurons were infected with a recombinant adenoviral vector expressing either Bcl-2 or the control gene LacZ and then treated with camptothecin. In wild-type cells, where neuronal death occurs via a caspase-mediated process, the extent of cell death at 24 h after camptothecin treatment was significantly reduced in neurons infected with Ad-Bcl-2 (75.7% survival), as compared with the control vector Ad-LacZ (20.6% survival) (Fig. 6 B). Overexpression of Bcl-2 in Apaf1−/− neurons, where neuronal cell death occurs via a slower caspase-independent mechanism, also resulted in a significant increase in neuronal survival 72 h after camptothecin treatment relative to control (71.2 vs. 17.5% in Ad-LacZ controls). Furthermore, enforced expression of Bcl-2 resulted in a marked increase in the fraction of camptothecin-treated neurons maintaining mitochondrial transmembrane potential (58.0 vs. 11.9%), and positive mitochondrial staining for cytochrome-c (68.9 vs. 8.7%) and AIF (62.3 vs. 15.8%) relative to camptothecin-treated controls (Fig. 6 C). Taken together, these results demonstrate that Bax and Bcl-2 regulate both the p53-induced release of cytochrome-c and AIF from mitochondria as well as the caspase-dependent and -independent cell death pathways.
Figure 6.
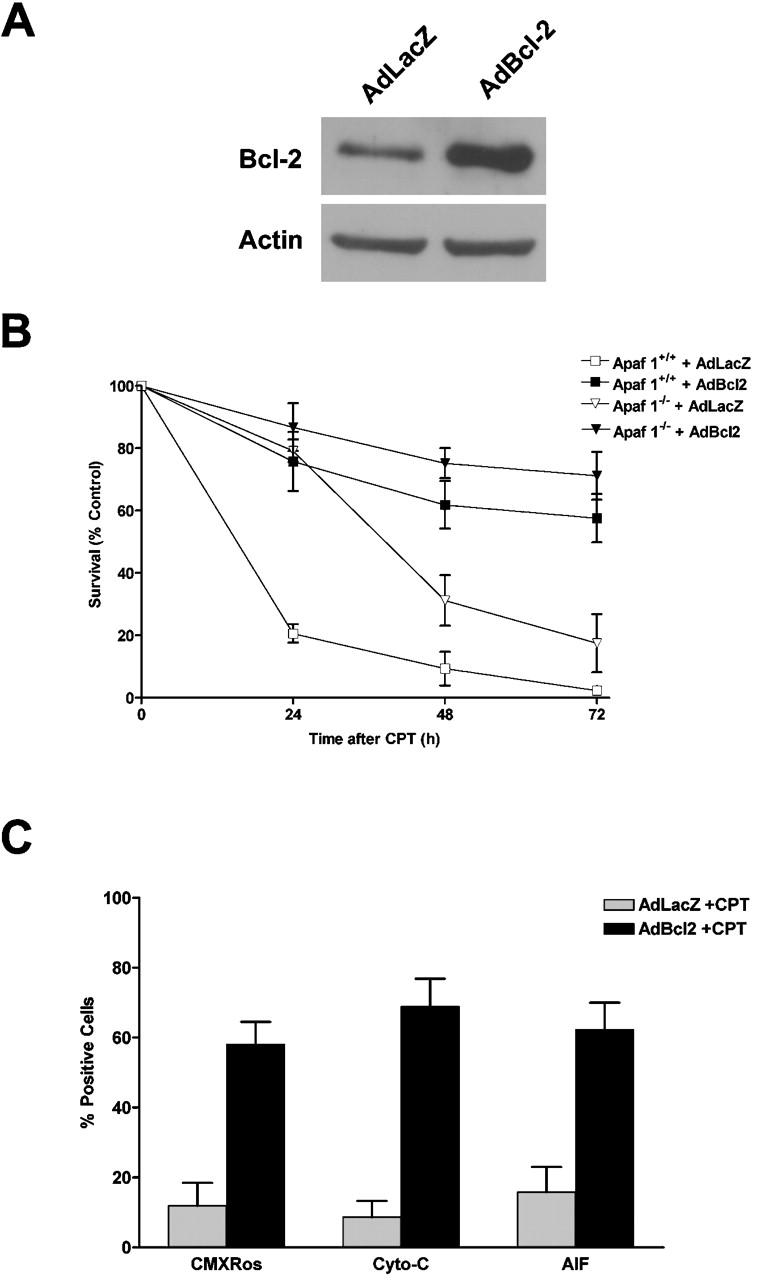
Bcl-2 inhibits camptothecin-induced AIF release and caspase-independent cell death. (A) Western blot analysis of Bcl-2 expression in cortical neurons 48 h after infection with Ad-Bcl-2 or the control vector Ad-LacZ at 50 MOI. (B) Wild-type and Apaf1-deficient cortical neurons were infected with Ad-Bcl-2 or Ad-LacZ for 48 h before treatment with camptothecin (10 μM) or vehicle control, and neuronal survival was determined at the indicated times by live/dead assay (n = 3). (C) Cortical neurons were infected with Ad-Bcl-2 or Ad-LacZ for 48 h and then treated with camptothecin or vehicle control for an additional 48 h. Cells were then labeled with CMX-Ros to assess mitochondrial transmembrane potential, or fixed and immunostained for AIF or cytochrome-c. Representative images were captured, and the fraction of cells maintaining mitochondrial membrane potential or mitochondrial cytochrome-c/AIF staining was scored (n = 3).
AIF can induce Apaf1/caspase-independent neuronal cell death
To determine whether AIF is capable of inducing caspase-independent neuronal cell death, we infected Apaf1−/− neurons with a recombinant adenoviral vector expressing AIF or LacZ as a control. AIF overexpression caused a marked reduction in neuronal survival relative to Ad-LacZ–infected controls and by 72 h, <35% of Ad-AIF–infected neurons remained viable (Fig. 7 A). Neurons undergoing AIF-induced cell death exhibited morphological features similar to that observed in p53-induced caspase-independent cell death, including intermediate nuclear pyknosis and partial chromatin condensation (Fig. 7 B, inset). In addition, Ad-AIF induced a significant increase in the fraction of TUNEL-positive neurons (46.6%) relative to Ad-LacZ–infected controls (10.1%) (Fig. 7, B and C). Interestingly, this DNA fragmentation was not associated with caspase-3 activation (unpublished data) and the fraction of TUNEL-positive cells was not significantly decreased in the presence of BAF (46.6 vs. 41.6%; Fig. 7 C). This indicated that AIF can trigger DNA fragmentation and neuronal cell death independent of caspase activation.
Figure 7.



AIF can induce neuronal cell death independent of Apaf1 and caspase activation. Apaf1-deficient neurons were infected at 75 MOI with Ad-AIF or the control vector Ad-LacZ and cultured in the presence or absence of BAF (50 μM). (A) Neuronal survival was determined at the indicated times by MTT assay (n = 4). (B) Photomicrographs of TUNEL staining and nuclear morphology in neurons 72 h after infection with Ad-LacZ or Ad-AIF. Bar, 20 μm. (C) The proportion of apoptotic cells was measured at 72 h by TUNEL staining (n = 3).
We then asked whether cell death induced by AIF was dependent on Bax. Ad-AIF induced a similar dose-dependent increase in cell death in Bax+/+ and Bax−/− neurons (Fig. 8 A). This suggests that AIF can induce cell death independently of Bax, which is consistent with the idea that AIF functions downstream of Bax in neuronal cell death. Interestingly, many (∼45%) Ad-AIF–infected cells exhibiting apoptotic nuclear morphology had maintained mitochondrial cytochrome-c staining (Fig. 8 B).
Figure 8.


AIF induces neuronal cell death independent of Bax. (A) Wild-type and Bax-deficient neurons were infected with Ad-AIF or Ad-LacZ at increasing MOI, and cell survival was determined at 72 h by MTT assay (n = 3). (B) Neurons were infected with Ad-LacZ or Ad-AIF at 75 MOI and cells were fixed and stained for cytochrome-c and counterstained with Hoechst. Bar, 15 μm.
Apaf1/caspase-independent neuronal cell death is blocked by AIF neutralizing antibodies
Finally, to determine whether AIF function is necessary for p53-mediated cell death, we microinjected Apaf1+/+ and Apaf1−/− neurons with an AIF-specific neutralizing antiserum (Susin et al., 1999) and assessed cell death after camptothecin treatment. In Apaf1+/+ neuronal cultures, the extent of camptothecin-induced cell death was not significantly different in cells microinjected with AIF antiserum, as compared with cells injected with preimmune serum (72.7 vs. 77.3%; Fig. 9 A). This indicated that AIF is not required for the caspase-dependent pathway of p53-mediated cell death. In contrast, microinjection of AIF antisera into Apaf1−/− neurons resulted in a significant decrease in camptothecin-induced cell death relative to control (34.7 vs. 53.8%; P < 0.01), suggesting that AIF plays a functional role in the caspase-independent pathway of neuronal cell death (Fig. 9 B). AIF immunostaining was performed to determine whether microinjection of AIF antisera prevented camptothecin-induced AIF translocation. As depicted in Fig. 9 C, whereas 67.3 ± 11.8% of Apaf1−/− neurons injected with preimmune serum exhibited positive nuclear staining for AIF, only 16.7 ± 4.9% of neurons injected with AIF antisera displayed distinct nuclear translocation after camptothecin treatment. These results indicated that the microinjected AIF antisera substantially diminished the ability of AIF to target the nucleus.
Figure 9.


Camptothecin-induced caspase- independent cell death and AIF translocation is inhibited by AIF neutralizing antibodies. (A) Wild-type or (B) Apaf1-deficient neurons were microinjected with either AIF antiserum or preimmune serum along with a fluorescent marker (Alexa®488-dextran) and then treated with camptothecin. After 36 h, cells were fixed, stained with Hoechst, and the fraction of Alexa®-positive cells exhibiting nuclear pyknosis was scored (n = 3). (*, P < 0.01, ANOVA). (C) AIF immunostaining in Apaf1−/− neurons microinjected with preimmune serum or AIF antisera and treated with camptothecin. Bar, 15 μm.
In summary, our results demonstrate that in addition to a caspase-mediated process activated by Apaf1, p53 can also induce cell death in Apaf1-deficient neurons via a caspase-independent mechanism. Furthermore, we have shown that AIF is an important determinant in the caspase-independent pathway and functions downstream of Bax in neuronal cell death.
Discussion
Caspases have been recognized as important mediators of apoptotic cell death after acute neuronal injury (Eldadah and Faden, 2000). However, caspase inhibition has typically resulted in limited or transient neuronal protection (Rideout and Stefanis, 2001). This lack of therapeutic efficacy has been attributed to the persistence of additional or compensatory pathways of neuronal cell death. Despite the growing recognition that caspase-independent cell death plays a significant role in neuronal injury, the molecular mechanisms regulating this cell death remain poorly defined.
P53 has been recognized as a key regulator of cell death after neuronal injury (Morrison and Kinoshita, 2000). In this paper, we have demonstrated that p53 triggers neuronal cell death via a caspase-mediated process in the presence of Apaf1, and via a caspase-independent process in the absence of Apaf1. More importantly, we provide several lines of evidence supporting a role for AIF in the regulation of caspase-independent cell death triggered by neuronal injury. We have demonstrated that (a) AIF redistributes from the mitochondria to the nucleus after p53-mediated neuronal injury, that (b) enforced expression of AIF can induce neuronal cell death in the absence of caspase activity with morphological features characteristic of caspase-independent cell death, and that (c) microinjection of neutralizing antibodies against AIF significantly decreases caspase-independent cell death induced by DNA damage. It was noted, however, that microinjected AIF antisera was moderately more effective at preventing AIF translocation (at least as detectable by immunostaining) than neuronal cell death. It is unclear whether this is due to incomplete inhibition of AIF translocation or rather to the possible involvement of other mediators of caspase-independent cell death. Indeed, the mitochondrial-derived proteins endonucleaseG and HtrA2 have recently been identified as potential caspase-independent mediators of cell death (Li et al., 2001; Suzuki et al., 2001; van Loo et al., 2001). Unfortunately, more definitive studies on the role of AIF in neuronal injury have been precluded by the lack of an AIF-null mouse model. Interestingly, AIF appears to be essential for the programmed cell death that occurs during cavitation of embryoid bodies (Joza et al., 2001). Attempts to generate chimeric mice from AIF-deficient ES cells have likely failed as a result of this defect in the early stages of embryogenesis (Joza et al., 2001). Thus, future studies of AIF function in neuronal injury in vivo will rely on the development of a conditional or tissue-specific knockout mouse model.
Two distinct stages of nuclear apoptosis have been identified during the apoptotic process (Daugas et al., 2000; Susin et al., 2000). In stage I, nuclei exhibit a wrinkled pattern of peripheral chromatin condensation, which is typically associated with high molecular weight DNA fragmentation (∼50 kb). As cell death progresses, nuclei adopt a stage II morphology, which is characterized by marked chromatin condensation and the formation of nuclear bodies. Furthermore, it has been shown that nuclear apoptosis is restricted to a stage I morphology when caspases are inhibited (Daugas et al., 2000; Susin et al., 2000). When recombinant AIF is added to isolated nuclei or injected into mouse embryo fibroblasts, nuclei adopt a stage I–type apoptotic morphology and exhibit high molecular weight DNA fragmentation, both of which occur independently of caspase activation (Susin et al., 1999, 2000; Daugas et al., 2000). In contrast, injection of active caspase-3 or caspase-activated DNase (CAD/DFF) caused a stage II–type nuclear morphology and resulted in the cleavage of DNA into oligonucleosomal fragments (Susin et al., 2000). In this paper, we have shown that in wild-type neurons, p53 initiates a caspase-mediated cell death process that manifests in a stage II–type nuclear apoptotic morphology. In contrast, p53-mediated cell death processes in Apaf1-deficient neurons resulted in a stage I–like apoptotic nuclear morphology, consistent with a caspase-independent apoptotic mechanism. Interestingly, wild-type neurons undergoing p53-mediated cell death in the presence of a caspase inhibitor also exhibited stage I apoptotic nuclei. Furthermore, we have shown that enforced expression of AIF induces stage I apoptotic morphology and DNA fragmentation in neurons in a caspase-independent manner.
The Bcl-2 protein family consists of proapoptotic and antiapoptotic members that are thought to regulate cell death through their opposing effects on mitochondrial-mediated death processes (Adams and Cory, 1998; Gross et al., 1999). Accordingly, the proapoptotic member Bax has been reported to translocate from the cytoplasm to the mitochondria in response to certain death signals (Goping et al., 1998), where it causes permeabilization of the mitochondrial membrane and the release of apoptogenic factors like cytochrome-c (Finucane et al., 1999). Upon release into the cytosol, cytochrome-c forms a complex with dATP and Apaf1, which then recruits and activates caspase-9 and initiates the caspase cascade. We and others have shown that Bax deficiency provides long-term protection against p53-induced cell death, suggesting that Bax is required for both caspase-dependent and -independent modes of neuronal cell death (Xiang et al., 1998; Cregan et al., 1999). Similarly, we report here that Bcl-2 can block both Apaf1/caspase-dependent and -independent neuronal death pathways induced by p53. We have previously demonstrated that Bax is required for p53-induced cytochrome-c release (Keramaris et al., 2000) and caspase activation in postmitotic neurons (Cregan et al., 1999). The results presented here indicate that Bax is also required for p53-induced mitochondrial depolarization and the release of AIF. Interestingly, our studies on the relative kinetics of p53-induced mitochondrial events indicated that cytochrome-c release preceded the release of AIF, at least in the absence of caspase activation. Furthermore, similar to a previous report (Stefanis et al., 1999), our results suggest that cytochrome-c is released before the collapse of mitochondrial membrane potential. In contrast, the release of AIF appears to occur after, and possibly as a consequence of, mitochondrial depolarization during p53-mediated cell death. It has previously been reported that AIF can be released from the mitochondria simultaneously with, or even before, cytochrome-c during staurosporine-induced cell death of Rat-1 fibroblasts (Daugas et al., 2000; Loeffler et al., 2001). One may hypothesize that the sequence of these events may depend upon the nature of the processes leading to mitochondrial permeabilization. Because we have shown that Bax is required for the release of both cytochrome-c and AIF, the different kinetics of their release suggests that Bax mediates the release of these factors through either distinct mechanisms or the progressive action of a common process.
The precise mechanism by which Bax mediates mitochondrial membrane permeabilization and the release of apoptogenic factors remains highly controversial. However, two main hypothetical models have been proposed. In the first, Bax is proposed to oligomerize upon insertion into the mitochondria and to directly form pores within the outer membrane. This hypothesis is supported by the finding that Bcl-2 family proteins share some structural homology with the transmembrane domain of diptheria toxin and the colicins, and that Bcl-2 family proteins can form pores in artificial membranes (Muchmore et al., 1996; Antonsson et al., 2000; Saito et al., 2000). In the other model, Bax is proposed to interact with existing membrane channels and to modulate their conductivity. Accordingly, Bax has been reported to interact with the voltage-dependent anion channel and studies in yeast cells and isolated mitochondria have suggested that this interaction is required for Bax-mediated mitochondrial effects (Shimizu et al., 1999, 2000). On the other hand, other research groups have indicated that the inner mitochondrial membrane protein, adenine nucleotide translocator (ANT), is the critical Bax target (Marzo et al., 1998). It is possible that more than one of these models is correct and Bax can form different types of channels. In this case, it is conceivable that the different apoptogenic factors could be released through distinct channels. Alternatively, Bax could alter membrane permeability either by forming pores itself or by modulating existing channels, allowing the selective release of smaller molecules like cytochrome-c. This increase in membrane permeability could lead to swelling of the mitochondrial matrix and eventual lysis of the outer mitochondrial membrane (Vander Heiden et al., 1997), resulting in the release of larger apoptogenic molecules like AIF.
In summary, we have shown that p53 induces neuronal cell death through a caspase-mediated process in the presence of Apaf1, and through a caspase-independent process in the absence of Apaf1. Furthermore, we have shown that AIF is an important regulator of the caspase-independent cell death pathway and functions downstream of Bax. The fact that blocking AIF function with neutralizing antibodies provides significant protection against cell death suggests that AIF may represent an important therapeutic target for neuroprotection after acute injury.
Materials and methods
Mice and primary neuronal cultures
P53, Bax (Jackson ImmunoResearch Laboratories), and Apaf1 (Cecconi et al., 1998) mice were maintained on a C57BL6 background to maintain genetic uniformity. Genotyping for Bax (Cregan et al., 1999), p53, and Apaf1 (Fortin et al., 2001) was done by PCR as previously described. Cortical neurons were cultured from dissociated cortices of E14.5 mice as previously described (Fortin et al., 2001).
Recombinant adenovirus infection
Total RNA was extracted from postnatal day eight mouse cortices using Trizol reagent (Invitrogen). 1 μg of total RNA was used for first strand cDNA synthesis and targeted gene amplification using Superscript One-Step RT-PCR kit (Invitrogen). cDNA synthesis was performed at 50°C for 45 min followed by a 2-min initial denaturation step at 94°C. This was followed by 37 cycles at 94°C for 30 s, 60°C for 30 s, and 72°C for 2 min using mouse-specific AIF primers: AIF forward (CCCGGGATGTTCCGGTGTGGAGG) and AIF reverse (CCCGGGTCAATCTTCATGAATG) containing Xma1 restriction sites. The resulting product was sequenced and confirmed to be AIF. Recombinant adenoviral vectors carrying human p53, Bcl-2, AIF, or LacZ expression cassettes were constructed, purified, and titered as previously described (Cregan et al., 2000). Recombinant adenoviral vectors were added to cell suspensions immediately before plating.
Cell viability assays
Neurons were infected with adenovirus at the time of plating or treated with camptothecin with or without BAF (Enzyme System Products) after 2 d in culture. Cell survival was measured by three different methods: live/dead staining, MTT assay (Cell Proliferation Kit; Promega), or TUNEL. At the times indicated, neuronal viability was determined using the Live/Dead Cytotoxicity Kit (Molecular Probes Inc.) according to the manufacturer's instructions. Representative samples were photographed using ZEISS Axiovert 100 with a Northern Eclipse Sony Power HAD 3CCD color video camera. Survival was determined as the fraction of total cells exhibiting positive staining for calcein-AM. TUNEL labeling was used to visualize cells with fragmented DNA. At the indicated times, cells were fixed in 4% paraformaldehyde for 20 min, washed in three changes of PBS, and then labeled by TUNEL, as previously described (Cregan et al., 1999), and counterstained with Hoechst 33258 (1 μg/μl) for 5 min. The fraction of TUNEL-positive cells as a percentage of total cell number was determined. For both live/dead and TUNEL assays, a minimum of 500 cells was scored for each treatment and the data represent the mean and standard deviation from three independent experiments. In certain experiments, survival was measured by colorimetric MTT assay as previously described (Cregan et al., 1999).
Antibodies, immunofluorescence staining, and cell counts
The rabbit antiserum for AIF was produced by immunizing rabbits with a mixture of synthetic peptides (coupled to keyhole limpet hemocyanin) corresponding to amino acid residues 151–170 and 181–200. The polyclonal antibody was purified through peptide-conjugated affinity chromatography and was found to specifically recognize both forms of AIF (67 and 57 kD), but predominately recognized the 57-kD truncated form of AIF. No other immunoreactive bands were detected with the AIF antibody. The specificities of anti-AIF antiserum and purified antibody were confirmed by the absence of 57- and 67-kD bands after preadsorption with peptides corresponding to amino acid residues 151–170 and 181–200 of AIF (unpublished data). Cytochrome-c and AIF immunostaining was performed as previously described (Keramaris et al., 2000). Representative fields were photographed and images were captured using a ZEISS Axioskop-II or Axiovert 100 microscope equipped with a Northern Eclipse Sony power HAD 3CCD color video camera. The fraction of cytochrome-c– or AIF-positive cells was determined as the proportion of total cells exhibiting a punctate cytoplasmic staining pattern. A minimum of 400 cells was scored per treatment and data represent the mean and standard deviation from three independent experiments.
Mitochondrial membrane potential
Loss of mitochondrial membrane potential was monitored in unfixed cells using the membrane potential–dependent dye Mitotracker CMX-Ros (Molecular Probes Inc.). This fluorescent dye is selectively incorporated into mitochondria with an intact transmembrane potential and therefore serves as an indicator of mitochondrial depolarization. Cells were incubated with CMX-Ros at 0.25 μM for 30 min at 37°C, washed in fresh media, and images were captured as described above. The fraction of cells maintaining mitochondrial transmembrane potential was determined by counting CMX-Ros–positive cells relative to total cell number in corresponding phase images. A minimum of 400 cells was scored per treatment and data represent the mean and standard deviation from three independent experiments.
Western blot analysis
Cells were lysed in RIPA buffer for 20 min on ice and the soluble extract was recovered by centrifugation. Extracts containing 30 μg of protein were separated on a 10% acrylamide gel and transferred to a nitrocellulose membrane. Membranes were blocked for 2 h in 5% skim milk and then incubated for 1 h with monoclonal antibodies directed against Bcl-2 (BD Transduction Labs)/cytochrome-c (BD Biosciences), or polyclonal antibodies to AIF (Dawson) and actin (Santa Cruz Biotechnology, Inc.) for standardization. Membranes were washed in TPBS (25 mM Na2HPO4, 5 mM NaH2PO4, 0.9% NaCl, 0.1% Tween-20) and then incubated for 1 h with appropriate secondary antibodies. Membranes were again washed and then developed by an enhanced chemiluminescence system according to the manufacturer's instructions (PerkinElmer).
Caspase activity assay
Cells were harvested and extracted for 15 min on ice in caspase lysis buffer, and 10 μg of protein was used for caspase activity assay as previously described (Cregan et al., 1999). Caspase activity is reported as the ratio of fluorescence output in treated samples relative to corresponding untreated controls.
Microinjection and cell death quantitation
Microinjection solution containing 3 mg/ml Alexa®488-dextran (Molecular Probes Inc.) and either AIF antiserum (Susin et al., 1999) or preimmune rabbit serum diluted in PBS was injected at 150 hPa (0.5 s) into neurons in 35-mm dishes using Femtotip needles (Eppendorf Inc.). Neurons were then treated with camptothecin (or vehicle control) and after 36 h, cells were fixed in 4% paraformaldehyde, washed in PBS, and stained with Hoechst 33258 (1 μg/ml). The extent of cell death was determined as the fraction of Alexa®488–positive cells exhibiting pyknotic nuclei. Where indicated, the cells were immunostained for AIF and the fraction of microinjection-positive cells exhibiting nuclear AIF staining was determined. A minimum of 200 cells were scored per treatment and data represent the mean and standard deviation from three independent experiments.
Acknowledgments
The authors are grateful to Dr. L. Sabourin and D. McBride for their assistance in certain aspects of these studies and Dr. J. Vanderluit and K. Ferguson for critical review.
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) and the Canadian Stroke Network (CSN) to R.S. Slack, a special grant from the Ligue Contre le Cancer to G. Kroemer, and grant NS43691 to T.M. Dawson. R.S. Slack is a CIHR Scholar, S.P. Cregan is supported by a CIHR fellowship, and A. Fortin is supported by a graduate scholarship from the CSN. F. Cecconi is an Assistant Telethon Scientist (grant 38/CP).
Footnotes
Abbreviations used in this paper: AIF, apoptosis-inducing factor; Apaf1, apoptotic activating factor-1; BAF, Boc-aspartyl (OMe)-fluoromethylketone; MOI, multiplicity of infection.
References
- Adams, J.M., and S. Cory. 1998. The Bcl-2 protein family: arbiters of cell survival. Science. 281:1322–1326. [DOI] [PubMed] [Google Scholar]
- Antonsson, B., S. Montessuit, S. Lauper, R. Eskes, and J.C. Martinou. 2000. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J. 345:271–278. [PMC free article] [PubMed] [Google Scholar]
- Banasiak, K.J., and G.G. Haddad. 1998. Hypoxia-induced apoptosis: effect of hypoxic severity and role of p53 in neuronal cell death. Brain Res. 797:295–304. [DOI] [PubMed] [Google Scholar]
- Cecconi, F., G. Alvarez-Bolado, B.I. Meyer, K.A. Roth, and P. Gruss. 1998. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 94:727–737. [DOI] [PubMed] [Google Scholar]
- Cheng, Y., M. Deshmukh, A. D'Costa, J.A. Demaro, J.M. Gidday, A. Shah, Y. Sun, M.F. Jacquin, E.M. Johnson, and D.M. Holtzman. 1998. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J. Clin. Invest. 101:1992–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan, S.P., J.G. MacLaurin, C.G. Craig, G.S. Robertson, D.W. Nicholson, D.S. Park, and R.S. Slack. 1999. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J. Neurosci. 19:7860–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan, S.P., J. MacLaurin, T.F. Gendron, S.M. Callaghan, D.S. Park, R.J. Parks, F.L. Graham, P. Morley, and R.S. Slack. 2000. Helper-dependent adenovirus vectors: their use as a gene delivery system to neurons. Gene Ther. 7:1200–1209. [DOI] [PubMed] [Google Scholar]
- Crumrine, R.C., A.L. Thomas, and P.F. Morgan. 1994. Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J. Cereb. Blood Flow Metab. 14:887–891. [DOI] [PubMed] [Google Scholar]
- Cryns, V., and J. Yuan. 1998. Proteases to die for. Genes Dev. 12:1551–1570. [DOI] [PubMed] [Google Scholar]
- Daugas, E., S.A. Susin, N. Zamzami, K.F. Ferri, T. Irinopoulou, N. Larochette, M.C. Prevost, B. Leber, D. Andrews, J. Penninger, and G. Kroemer. 2000. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 14:729–739. [PubMed] [Google Scholar]
- D'Mello, S.R., C.Y. Kuan, R.A. Flavell, and P. Rakic. 2000. Caspase-3 is required for apoptosis-associated DNA fragmentation but not for cell death in neurons deprived of potassium. J. Neurosci. Res. 59:24–31. [PubMed] [Google Scholar]
- Eldadah, B.A., and A.I. Faden. 2000. Caspase pathways, neuronal apoptosis, and CNS injury. J. Neurotrauma. 17:811–829. [DOI] [PubMed] [Google Scholar]
- Eliasson, M.J., K. Sampei, A.S. Mandir, P.D. Hurn, R.J. Traystman, J. Bao, A. Pieper, Z.Q. Wang, T.M. Dawson, S.H. Snyder, and V.L. Dawson. 1997. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 3:1089–1095. [DOI] [PubMed] [Google Scholar]
- Endres, M., Z.Q. Wang, S. Namura, C. Waeber, and M.A. Moskowitz. 1997. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J. Cereb. Blood Flow Metab. 17:1143–1151. [DOI] [PubMed] [Google Scholar]
- Finucane, D.M., E. Bossy-Wetzel, N.J. Waterhouse, T.G. Cotter, and D.R. Green. 1999. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J. Biol. Chem. 274:2225–2233. [DOI] [PubMed] [Google Scholar]
- Fortin, A., S.P. Cregan, J.G. MacLaurin, N. Kushwaha, E.S. Hickman, C.S. Thompson, A. Hakim, P.R. Albert, F. Cecconi, K. Helin, et al. 2001. APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J. Cell Biol. 155:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goping, I.S., A. Gross, J.N. Lavoie, M. Nguyen, R. Jemmerson, K. Roth, S.J. Korsmeyer, and G.C. Shore. 1998. Regulated targeting of BAX to mitochondria. J. Cell Biol. 143:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, A., J.M. McDonnell, and S.J. Korsmeyer. 1999. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13:1899–1911. [DOI] [PubMed] [Google Scholar]
- Halterman, M.W., C.C. Miller, and H.J. Federoff. 1999. Hypoxia-inducible factor-1alpha mediates hypoxia-induced delayed neuronal death that involves p53. J. Neurosci. 19:6818–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara, H., R.M. Friedlander, V. Gagliardini, C. Ayata, K. Fink, Z. Huang, M. Shimizu-Sasamata, J. Yuan, and M.A. Moskowitz. 1997. Inhibition of interleukin 1β converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. USA. 94:2007–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herdegen, T., F.X. Claret, T. Kallunki, A. Martin-Villalba, C. Winter, T. Hunter, and M. Karin. 1998. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J. Neurosci. 18:5124–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y., M.A. Benedict, L. Ding, and G. Nunez. 1999. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J. 18:3586–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M.D., H. Xiang, S. London, Y. Kinoshita, M. Knudson, M. Mayberg, S.J. Korsmeyer, and R.S. Morrison. 1998. Evidence for involvement of Bax and p53, but not caspases, in radiation-induced cell death of cultured postnatal hippocampal neurons. J. Neurosci. Res. 54:721–733. [DOI] [PubMed] [Google Scholar]
- Johnson, M.D., Y. Kinoshita, H. Xiang, S. Ghatan, and R.S. Morrison. 1999. Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J. Neurosci. 19:2996–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza, N., S.A. Susin, E. Daugas, W.L. Stanford, S.K. Cho, C.Y. Li, T. Sasaki, A.J. Elia, H.Y. Cheng, L. Ravagnan, et al. 2001. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 410:549–554. [DOI] [PubMed] [Google Scholar]
- Keramaris, E., L. Stefanis, J. MacLaurin, N. Harada, K. Takaku, T. Ishikawa, M.M. Taketo, G.S. Robertson, D.W. Nicholson, R.S. Slack, and D.S. Park. 2000. Involvement of caspase 3 in apoptotic death of cortical neurons evoked by DNA damage. Mol. Cell. Neurosci. 15:368–379. [DOI] [PubMed] [Google Scholar]
- Kuida, K., T.S. Zheng, S. Na, C. Kuan, D. Yang, H. Karasuyama, P. Rakic, and R.A. Flavell. 1996. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 384:368–372. [DOI] [PubMed] [Google Scholar]
- Kuida, K., T.F. Haydar, C.Y. Kuan, Y. Gu, C. Taya, H. Karasuyama, M.S. Su, P. Rakic, and R.A. Flavell. 1998. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 94:325–337. [DOI] [PubMed] [Google Scholar]
- Lankiewicz, S., C. Marc Luetjens, N. Truc Bui, A.J. Krohn, M. Poppe, G.M. Cole, T.C. Saido, and J.H. Prehn. 2000. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J. Biol. Chem. 275:17064–17071. [DOI] [PubMed] [Google Scholar]
- Li, L.Y., X. Luo, and X. Wang. 2001. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 412:95–99. [DOI] [PubMed] [Google Scholar]
- Li, P., D. Nijhawan, I. Budihardjo, S.M. Srinivasula, M. Ahmad, E.S. Alnemri, and X. Wang. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 91:479–489. [DOI] [PubMed] [Google Scholar]
- Loeffler, M., E. Daugas, S.A. Susin, N. Zamzami, D. Metivier, A.L. Nieminen, G. Brothers, J.M. Penninger, and G. Kroemer. 2001. Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J. 15:758–767. [DOI] [PubMed] [Google Scholar]
- Martin-Villalba, A., M. Hahne, S. Kleber, J. Vogel, W. Falk, J. Schenkel, and P.H. Krammer. 2001. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 8:679–686. [DOI] [PubMed] [Google Scholar]
- Marzo, I., C. Brenner, N. Zamzami, J.M. Jurgensmeier, S.A. Susin, H.L. Vieira, M.C. Prevost, Z. Xie, S. Matsuyama, J.C. Reed, and G. Kroemer. 1998. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 281:2027–2031. [DOI] [PubMed] [Google Scholar]
- McGahan, L., A.M. Hakim, and G.S. Robertson. 1998. Hippocampal Myc and p53 expression following transient global ischemia. Brain Res. Mol. Brain Res. 56:133–145. [DOI] [PubMed] [Google Scholar]
- Miller, T.M., K.L. Moulder, C.M. Knudson, D.J. Creedon, M. Deshmukh, S.J. Korsmeyer, and E.M. Johnson, Jr. 1997. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J. Cell Biol. 139:205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, E.J., E. Keramaris, H.J. Rideout, R.S. Slack, N.J. Dyson, L. Stefanis, and D.S. Park. 2001. Cyclin-dependent kinases and P53 pathways are activated independently and mediate Bax activation in neurons after DNA damage. J. Neurosci. 21:5017–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison, R.S., and Y. Kinoshita. 2000. The role of p53 in neuronal cell death. Cell Death Differ. 7:868–879. [DOI] [PubMed] [Google Scholar]
- Morrison, R.S., H.J. Wenzel, Y. Kinoshita, C.A. Robbins, L.A. Donehower, and P.A. Schwartzkroin. 1996. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J. Neurosci. 16:1337–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchmore, S.W., M. Sattler, H. Liang, R.P. Meadows, J.E. Harlan, H.S. Yoon, D. Nettesheim, B.S. Chang, C.B. Thompson, S.L. Wong, et al. 1996. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 381:335–341. [DOI] [PubMed] [Google Scholar]
- Nitatori, T., N. Sato, S. Waguri, Y. Karasawa, H. Araki, K. Shibanai, E. Kominami, and Y. Uchiyama. 1995. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J. Neurosci. 15:1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettmann, B., and C.E. Henderson. 1998. Neuronal cell death. Neuron. 20:633–647. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau, C., J.C. Hedreen, D.L. Price, and V.E. Koliatsos. 1995. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J. Neurosci. 15:3775–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout, H.J., and L. Stefanis. 2001. Caspase inhibition: a potential therapeutic strategy in neurological diseases. Histol. Histopathol. 16:895–908. [DOI] [PubMed] [Google Scholar]
- Saito, M., S.J. Korsmeyer, and P.H. Schlesinger. 2000. BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat. Cell Biol. 2:553–555. [DOI] [PubMed] [Google Scholar]
- Selznick, L.A., T.S. Zheng, R.A. Flavell, P. Rakic, and K.A. Roth. 2000. Amyloid beta-induced neuronal death is bax-dependent but caspase-independent. J. Neuropathol. Exp. Neurol. 59:271–279. [DOI] [PubMed] [Google Scholar]
- Shimizu, S., M. Narita, and Y. Tsujimoto. 1999. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 399:483–487. [DOI] [PubMed] [Google Scholar]
- Shimizu, S., T. Ide, T. Yanagida, and Y. Tsujimoto. 2000. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 275:12321–12325. [DOI] [PubMed] [Google Scholar]
- Slack, R.S., D.J. Belliveau, M. Rosenberg, J. Atwal, H. Lochmuller, R. Aloyz, A. Haghighi, B. Lach, P. Seth, E. Cooper, and F.D. Miller. 1996. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J. Cell Biol. 135:1085–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale, G., N.R. Nichols, D.R. Brady, C.E. Finch, and W.E. Horton, Jr. 1995. Evidence for apoptotic cell death in Alzheimer's disease. Exp. Neurol. 133:225–230. [DOI] [PubMed] [Google Scholar]
- Stefanis, L., D.S. Park, W.J. Friedman, and L.A. Greene. 1999. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J. Neurosci. 19:6235–6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin, S.A., H.K. Lorenzo, N. Zamzami, I. Marzo, B.E. Snow, G.M. Brothers, J. Mangion, E. Jacotot, P. Costantini, M. Loeffler, et al. 1999. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 397:441–446. [DOI] [PubMed] [Google Scholar]
- Susin, S.A., E. Daugas, L. Ravagnan, K. Samejima, N. Zamzami, M. Loeffler, P. Costantini, K.F. Ferri, T. Irinopoulou, M.C. Prevost, et al. 2000. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 192:571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, Y., Y. Imai, H. Nakayama, K. Takahashi, K. Takio, and R. Takahashi. 2001. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell. 8:613–621. [DOI] [PubMed] [Google Scholar]
- Vander Heiden, M.G., N.S. Chandel, E.K. Williamson, P.T. Schumacker, and C.B. Thompson. 1997. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 91:627–637. [DOI] [PubMed] [Google Scholar]
- van Loo, G., P. Schotte, M. van Gurp, H. Demol, B. Hoorelbeke, K. Gevaert, I. Rodriguez, A. Ruiz-Carrillo, J. Vandekerckhove, W. Declercq, et al. 2001. Endonuclease G: a mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 8:1136–1142. [DOI] [PubMed] [Google Scholar]
- Xiang, H., D.W. Hochman, H. Saya, T. Fujiwara, P.A. Schwartzkroin, and R.S. Morrison. 1996. Evidence for p53-mediated modulation of neuronal viability. J. Neurosci. 16:6753–6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, H., Y. Kinoshita, C.M. Knudson, S.J. Korsmeyer, P.A. Schwartzkroin, and R.S. Morrison. 1998. Bax involvement in p53-mediated neuronal cell death. J. Neurosci. 18:1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev, A.G., S.M. Knoblach, L. Fan, G.B. Fox, R. Goodnight, and A.I. Faden. 1997. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J. Neurosci. 17:7415–7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, R.Z., C. Wu, H. Fujihara, K. Taga, S. Qi, M. Naito, and K. Shimoji. 2001. Both caspase-dependent and caspase-independent pathways may be involved in hippocampal CA1 neuronal death because of loss of cytochrome c from mitochondria in a rat forebrain ischemia model. J. Cereb. Blood Flow Metab. 21:529–540. [DOI] [PubMed] [Google Scholar]