

Abstract
The Rce1p protease is required for the maturation of the Ras GTPase and certain other isoprenylated proteins and is considered a chemotherapeutic target. To identify new small-molecule inhibitors of Rce1p, the authors screened the National Cancer Institute Diversity Set compound library using in vitro assays to monitor the proteolytic processing of peptides derived from Ras and the yeast a-factor mating pheromone. Of 46 inhibitors initially identified with a Ras-based assay, only 9 were effective in the pheromone-based assay. The IC50 values of these 9 compounds were in the low micromolar range for both yeast (6-35 μM) and human Rce1p (0.4-46 μM). Four compounds were somewhat Rce1p selective in that they partially inhibited the Ste24p protease and did not inhibit Ste14p isoprenylcysteine carboxyl methyltransferase, 2 enzymes also involved in the maturation of isoprenylated proteins. The remaining 5 compounds inhibited all 3 enzymes. The 2 most Rce1p-selective agents were ineffective trypsin inhibitors, further supporting the specificity of these agents for Rce1p. The 5 least specific compounds formed colloidal aggregates, a proposed common feature of promiscuous inhibitors. Interestingly, the most specific Rce1p inhibitor also formed a colloidal aggregate. In vivo studies revealed that treatment of wild-type yeast with 1 compound induced a Ras2p delocalization phenotype that mimics observed effects in rce1 ste24 null yeast. The 9 compounds identified in this study represent new tools for understanding the enzymology of postisoprenylation-modifying enzymes and provide new insight for the future development of Rce1p inhibitors.
Keywords: Ras, protease, CaaX protein, posttranslational modification, isoprenylation
Introduction
ONE COMMON POSTTRANSLATIONAL MODIFICATION occurring in eukaryotic proteins is the covalent addition of an isoprenoid lipid.1 This lipid modification can influence the membrane-partitioning properties of the lipidated protein and/or the ability of the modified protein to interact with other proteins. Isoprenylated proteins function in many cellular pathways and can have key roles in signal transduction. Commonly cited examples of isoprenylated proteins are the Ras and Ras-related GTPases, Gγ subunits, nuclear lamins, and fungal mating pheromones, among others.
The precursors of certain isoprenylated proteins are readily identified by the presence of a highly degenerate C-terminally located tetrapeptide CaaX motif (where C = cysteine, a = aliphatic amino acid, and X = one of several amino acids). Proteins with this motif (i.e., CaaX proteins) undergo an ordered series of modifications (Fig. 1).1,2 The initial event is covalent attachment of either a C15 or C20 isoprenoid lipid to the cysteine of the CaaX motif. This is followed by an endoproteolytic event that removes the last 3 residues of the motif (i.e., aaX). The Ras-converting enzyme (Rce1p) and Ste24p can independently exact this cleavage event.3,4 These CaaX proteases are generally believed to have distinct substrate preferences, but at least 1 substrate, the yeast a-factor precursor, is cleaved by both enzymes.5 The final modification involves methyl esterification of the newly exposed C-terminus by an isoprenylcysteine carboxyl methyltransferase (ICMT). All of the postisoprenylation-processing enzymes are integral membrane proteins that localize to the endoplasmic reticulum.6,7
FIG. 1.

Posttranslational modifications associated with proteins bearing a C-terminal CaaX motif. Farnesyltransferase inhibitors target the first step of this pathway. This study identifies inhibitors of the proteolytic step that can be mediated by Rce1p or Ste24p.
Agents that interfere with the posttranslational modification of CaaX proteins are considered to be promising anticancer targets because CaaX proteins are involved in cellular transformation (e.g., Ras).2 Farnesyltransferase inhibitors are the most advanced agents that have been developed for such anticancer strategies.8,9 Inhibitors of the CaaX proteases and ICMT have similar therapeutic potential and are being developed, but relatively few distinct agents have been reported.10-13 To date, Rce1p has been the primary CaaX protease target for inhibitor discovery because it is specifically required for the maturation of Ras, Rho, and other CaaX proteins. Although only 1 mammalian substrate has been characterized for Ste24p, the observation that certain disease phenotypes are associated with defects in Ste24p activity clearly illustrates a need for monitoring whether Rce1p inhibitors under development display any specificity for Ste24p.14,15
The in vitro activities of the CaaX proteases have been traditionally investigated using both indirect and direct methodologies, typically using membranes enriched with the appropriate CaaX protease as the source of activity. Indirect methods rely on the observation that CaaX proteolysis and carboxyl methylation are tightly coupled sequential events.16 By following radioactive methyl tracers or monitoring the methylation-dependent activation of biologically active peptides, CaaX proteolysis can be indirectly monitored.17,18 Direct methods typically track the liberated aaX tripeptide or the processed product using high-performance liquid chromatography (HPLC) or radioactive tracers.19,20 A direct assay that monitors cleavage of a quenched fluorogenic K-Ras4b–derived peptide has also been developed.21 This fluorescence-based assay was used in this study to screen the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) Diversity Set compound library22 for small-molecule inhibitors of yeast Rce1p. This library has been used to identify inhibitors of transcription response pathways, phosphatases, antibiotic-resistant enzymes, and viral DNA synthesis, among other targets.23-29 We report the identification of compounds that inhibit the yeast Rce1p CaaX protease and describe the specificity of these compounds in the context of human Rce1p, the yeast Ste24p and ICMT enzymes, and the unrelated proteolytic enzyme trypsin.
Experimental
Yeast strains and plasmids
The yeast strains used in this study were EG123 (MATa trp1 leu2 ura3 his4 can1), SM3614 (MATa trp1 leu2 ura3 his4 can1 ste24Δ::LEU2 rce1Δ::TRP1), and RC757 (MATα sst2-1).4,30,31 Plasmid-bearing versions of SM3614 were generated according to published methods.32 Strains were routinely grown at 30 °C using YEPD or synthetic complete dropout (SC–) medium.33 The plasmids encoding yeast Rce1p (pWS479), human Rce1p (pWS335), yeast Ste24p (pSM1282), and yeast Ste14p (pSM1317) have been reported.6,18,34 pWS270 (CEN URA3 PGAL-GFP-RAS2) was created in 2 steps. First, an EcoRI-BamHI DNA fragment containing GFP (F64L S65T)35 was isolated from pBS-GFP++ (gift of E. O'shea) and subcloned into the same sites of pRS316GU (gift of P. Hieter). Second, a PCR product containing the RAS2 open reading frame and 198 base pairs of its 3′ untranslated region was introduced at the BamHI site by PCR-directed plasmid-based recombination.36 The PCR product was amplified from YEpRAS2-4 (gift of S. Powers), such that it had 39 base pair extensions on either end that were homologous to the sequences flanking the BamHI cloning junction. In addition, the PCR product contained an XbaI site that immediately preceded the start codon and an NotI site that followed the terminal sequence of the RAS2-encoding fragment.
Substrates, compound library, and other reagents
The Rce1p fluorogenic substrate ABZ-KSKTKC(farnesyl)QLIM and the Ste24p substrate ABZ-KSKTKC(farnesyl)VIQL were purchased from AnaSpec (San Jose, CA) and are based on the K-Ras4b C-terminal sequence. ABZ is an o-aminobenzoic acid, and QL is a lysine ε-dinitrophenyl. The a-factor–based substrate YIIKGVFWD-PAC(farnesyl)VIA was purchased from California Peptide (Napa, CA). Zymolyase 100T for preparation of yeast spheroplasts was purchased from Cape Cod Inc. (East Falmouth, MA). The Diversity Set compound library was obtained through the NCI DTP. Other chemicals, reagents, and enzymes were purchased from a variety of vendors (e.g., Sigma-Aldrich, VWR, Calbiochem).
In vitro fluorescence-based CaaX proteolysis assay
An established fluorescence-based assay was used to monitor CaaX protease activity in the presence of candidate inhibitors.21,37 In brief, the assay involves the mixing of a quenched fluorogenic substrate with membranes derived from yeast (SM3614) overexpressing the appropriate CaaX protease. Membranes used as the source of activity were isolated according to our reported methods and stored at −80 °C as 1 mg/ml stocks in lysis buffer (50 mM Tris, pH 7.5, 0.2 M sorbitol, 1 mM EDTA, 0.2% NaN3) containing a protease inhibitor cocktail (chymostatin, leupeptin, pepstatin, aprotinin, and phenylmethylsulphonyl fluoride) that does not inhibit either of the CaaX proteases.34,38 The membranes were typically diluted with assay buffer (100 mM HEPES, pH 7.5, 5 mM MgCl2) to 0.5 mg/ml immediately before use. The substrate was diluted to 40 μM from a 1-mM stock prepared in 4% DMSO.
Assay assembly involved the dispensing of 50-μl aliquots of diluted membranes into the wells of a black, clear-bottom, 96-well microplate. This was followed by the addition of each compound to duplicate wells and a 15-min pretreatment incubation at 30 °C. Initial screens were performed using 200 μM, reported as the pretreatment concentration, of each compound. After the pretreatment step, the assay was initiated by adding 50 μl of diluted substrate, resulting in final concentrations of 0.25 mg/ml membrane, 20 μM substrate, and 100 μM inhibitor. The sample fluorescence was measured every 30 to 60 s over a 60-min time course at 30 °C using a BioTek Synergy™ HT microplate fluorometer equipped with a 320/420 nm excitation/emission filter set. The collected data were exported to Microsoft Excel and graphed as a function of fluorescence versus time, and the initial velocities were determined. These values were used to calculate the percentage activities relative to a DMSO-treated sample, which was included as a control in each reaction set. In parallel during the primary screen, compounds were mixed with the ABZ fluorophore alone to control for the possibility of compound-induced quenching effects that would mimic a positive hit. The Z′ factor was calculated using DMSO-treated Rce1p-containing membranes and Rcep1-deficient membranes as the positive and negative controls, respectively, from more than 100 independent measurements collected during the screen. For trypsin assays, trypsin (20 μg/ml final) was added in the presence of yeast membranes devoid of CaaX proteolytic activity (0.25 mg/ml final) to mimic the standard assay conditions. For instances in which detergent effects were tested, Tween-20 (0.007% final) was added to the reaction mixture.
IC50 determinations and kinetic analyses of inhibitor interactions were conducted using a range of inhibitor concentrations (1-1000 μM for yeast Rce1p, 1-200 μM for Ste24p, and 0.05-100 μM for human Rce1p) and substrate concentrations (1.5-100 μM final), respectively. IC50 values were determined by graphing initial velocities with the aid of Prism 4.0 (GraphPad Software, Inc.). For determining kinetic parameters, data were analyzed using Prism 4.0 and nonlinear regression methods. Curves were fitted using a 4-parameter logistic equation without constraints.
In vitro α-factor–based CaaX proteolysis and methylation assays
An assay based on the in vitro production of bioactive a-factor was used to independently confirm the inhibitory properties and specificity of hits obtained from the fluorescence-based screen.18,37 The specific steps associated with the assay were as follows. First, CaaX protease–containing membranes, isolated and diluted as described above, were pretreated with 200 μM of compound for 15 min at 30 °C. The inhibitor-treated membranes (10 μl) were dispensed into the wells of a 96-well plate suitable for use in a PCR thermocycler. The proteolytic reaction was initiated by adding an equal volume of the a-factor substrate YIIKGVFWD-PAC(farnesyl)VIA, which was diluted immediately before use to 40 μM using assay buffer (defined above) from a 1-mM stock prepared in MeOH. After a 10-min incubation at 30 °C, the mixtures were heated to 95 °C for 1 min to inactivate enzymatic activity, cooled, and supplemented with a 5X Step 2 Addition mix to initiate methylation of cleaved products. The 5X Step 2 Addition mix was prepared in lysis buffer and contained Ste14p-enriched yeast membranes (0.5 mg/ml) and S-adenosylmethionine (800 μM). The Ste14p membranes were derived from a CaaX protease–deficient background (SM3614) as previously described.18 After 60 min of incubation at 30 °C, the methylation reaction was stopped by the addition of copper sulfate (2 mM final). A 2-fold serial dilution series of each sample was prepared in YPD medium, and the dilution series was spotted onto a lawn of RC757 yeast, which undergo growth arrest in the presence of 7 nM or greater concentrations of pheromone.39 After a 16- to 20-h incubation at 30 °C, the yeast growth patterns were evaluated to determine an activity profile, which was compared with that of a DMSO-treated sample to derive percentage inhibition values.
To determine whether any observed inhibitory effect involved the inhibition of Ste14p ICMT, the standard a-factor assay described above was modified such that the compounds were used to pretreat Ste14p membranes instead of the CaaX protease membranes. The inhibitor concentration during Ste14p pretreatment was twice (400 μM final) that used to pre-treat the CaaX proteases, but the final concentration in the fully assembled reaction was unchanged.
Dynamic light-scattering measurements
The propensity for compounds to form colloidal aggregates was evaluated using a dynamic light-scattering (DLS) approach. Compounds prepared as 10-mM stocks in DMSO were diluted to 200 μM into a 3:1 mixture of assay buffer and lysis buffer to mimic the reaction conditions of the fluorescence assay. The samples were centrifuged at 500g for 25 min at room temperature, and the supernatants were subjected to DLS at 30 °C using a DynaPro99 molecular sizing instrument (Protein Solutions, Piscataway, NJ) at 30% and 10% laser power and a total acquisition time of 100 s. Data were analyzed using DYNAMICS version 6.0 software. The mean particle radii were calculated from 3 or more independent sample preparations assuming a spherical shape. The presence of precipitate, which was sometimes observable by the eye, was assessed by a comparison of DLS measurements before and after the centrifugation step. Samples were deemed to have precipitates when the standard deviation value for the estimated particle radius was approximately that of the particle radius value in the prespin samples but was only a fraction of the particle radius in the post-spin sample. For the effect of detergents on aggregate formation, Triton X-100 (0.04% final) or Tween-20 (0.007%) was added to the samples and analyzed by DLS.
In vivo analysis of Rce1p inhibitors
Two methods were used to assess the in vivo effects of Rce1p inhibitors. The first was a filter disc assay that was used to determine compound toxicity. In brief, a mid-log culture of yWS133 (EG123 transformed with pWS270) was plated as a thin lawn onto SC-ura, sterile Whatman filter discs (6 mm) were applied, and 10-μl volumes of the compounds (1 mM in DMSO) were spotted onto the discs. The plates were incubated for 24 h at 30 °C and scanned to obtain an image of growth observed.
The second in vivo assessment relied on a GFP-Ras2p reporter that is mislocalized in the absence of CaaX proteolytic activity.3 In brief, mid-log yWS133 cells were treated with 25 μM compound for 1 h, harvested, washed twice with sterile H2O, and incubated in SC-ura media containing 2% galactose, 1% glycerol, and 1% ethanol for 6 to 7 h at 30 °C to induce expression of GFP-Ras2p. More than 60% of cells had associated GFP with this protocol. The induced cells were mounted on a microscope slide, and the expression pattern of GFP-Ras2p was visualized using a Zeiss Axioskop 2 Mot Plus microscope equipped with fluorescence optics. Images were captured at 100× (plan apochromat objectives, numerical aperture 1.4) using an ORCA-AG digital camera (Hamamatsu, Japan) and IPLab Spectrum Software. For each experiment, at least 5 cell fields were taken, from which representative images were selected.
Results
Identification of Rce1p inhibitors by HTS
The effect of compounds from the NCI DTP Diversity Set library on the in vitro activity of Rce1p was monitored using an established 96-well format assay that involves cleavage of a quenched fluorogenic substrate.21,37 Under the conditions of the screen, this assay had a calculated Z′ factor of 0.55. Of the 1981 compounds that were evaluated, 46 (2.3% hit rate) were found to inhibit Rce1p to 5% activity or less (Table 1). Other less potent inhibitors were also identified but not studied further. The screen was not absolutely comprehensive of the Diversity Set library because a few compounds (approximately 1%) were excluded from analysis. These included compounds observed to have quenching effects, which yield a false positive in this assay, and those observed to strongly fluoresce near the emission wavelength of the ABZ fluorophore.
Table 1.
Summary of Rce1p Inhibitor Hits from the NCI Diversity Set Library
| Fluorescence Assay | Bioassay | ||
|---|---|---|---|
| % Activity Remaining | No. of Hits | % Activity Remaining | No. of Hits |
| 0-5 | 46 | 0-5 | 8 |
| 5-10 | 11 | 5-10 | 1 |
| 10-25 | 37 | 10-25 | 7 |
| 25-50 | 124 | 25-50 | 6 |
| 50-100 | 1748 | 50-100 | 24 |
Compounds were initially evaluated at 200 μM using the fluorescence-based in vitro assay. The strongest set of inhibitors (0%-5% activity remaining) were reassessed using an a-factor–based in vitro assay (bioassay). Compounds that strongly inhibited in the a-factor assay (≤10% activity remaining) were labeled as consistently performing inhibitors.
Confirmation of primary hits using a secondary assay
We have previously observed that the inhibition of Rce1p by tosyl-L-phenylalanyl-chloromethylketone (TPCK) is assay specific.37 In part, this may be due to the fact that Rce1p is a multi-substrate enzyme having distinct affinities for different substrates and inhibitors. Thus, the hits obtained by high-throughput screening (HTS) using the fluorescence assay were reassessed using a second CaaX proteolysis assay that generates the yeast a-factor mating pheromone in an Rce1p-dependent manner. This assay is not sensitive to the effects of compounds having quenching or autofluorescent properties. The mating pheromone itself is a bioactive peptide that can be detected using a relatively straightforward bioassay.18,34,37
Analysis of the 46 strongest primary hits identified by the fluorescence assay (0%-5% category) revealed that these compounds varied significantly in their ability to inhibit a-factor maturation. Only 20% of the primary hits (n = 9) were confirmed as potent inhibitors of a-factor production (<10% activity; Fig. 2). Others inhibited only modestly (n = 13; 10%-50% activity), and yet others inhibited poorly or not at all (n = 24; >50% activity) by comparison to a DMSO-treated control. Thus, many of the primary hits from the HTS screen displayed assay-specific effects as previously observed for TPCK. The 9 compounds that potently inhibited Rce1p in both assays were operationally defined as consistently performing inhibitors (Table 2).
FIG. 2.
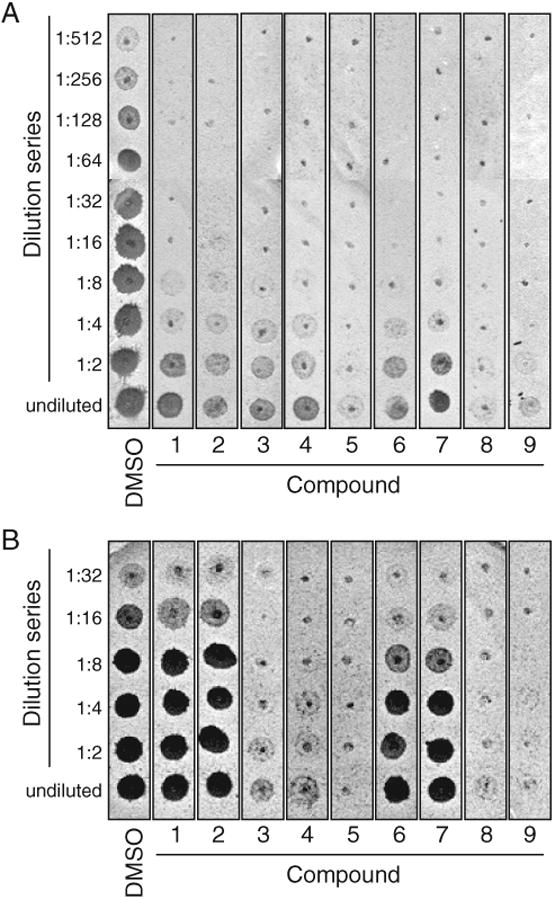
Inhibition of CaaX protease–dependent a-factor production. The inhibitory properties of the indicated compounds were evaluated using an assay that monitors the in vitro formation of the bioactive a-factor mating pheromone. Two-fold serial dilutions of each sample were spotted onto a thin lawn of RC757 (MATα sst2-1) cells that had been spread on a YEPD plate. This MATα background is supersensitive to the mating pheromone and undergoes growth arrest in its presence, as indicated by a zone of no growth (spot) in the lawn. Shown is a representative replicate where either Rce1p (A) or Ste24p (B) was used as the CaaX protease. The compounds are identified as in Figure 3.
Table 2.
Summary of Inhibitor Specificity
| Rce1p | Ste24p | Ste14p | |||||
|---|---|---|---|---|---|---|---|
| Compound | NSC No. | Fluor | Bio | Fluor | Bio | Bio | |
| DMSO | — | 100.0 ± 4.1 | 100 | 100.0 ± 0.9 | 100 | 100 | |
| 1 | 67485 | 0.2 ± 0.2 | 2 | 51.3 ± 1.7 | 100 | 100 | |
| 2 | 609974 | <0.1 | 9 | 14.3 ± 2.2 | 75 | 100 | |
| 3 | 1011 | <0.1 | 4 | <0.1 | 0 | 23 | |
| 4 | 73101 | 0.5 ± 0.5 | 3 | 1.2 ± 0.2 | 0 | 44 | |
| 5 | 270718 | 0.6 ± 0.6 | 1 | 1.1 ± 0.2 | 0 | 38 | |
| 6 | 321237 | <0.1 | 5 | 23.0 ± 3.6 | 50 | 100 | |
| 7 | 321239 | 0.3 ± 0.7 | 4 | 41.9 ± 2.9 | 37 | 100 | |
| 8 | 294526 | 0.3 ± 0.3 | 0 | 1.1 ± 0.6 | 0 | 0.5 | |
| 9 | 295642 | <0.1 | 1 | 0.7 ± 0.4 | 0 | 13 | |
For the CaaX proteases, compounds were evaluated at 200 μM (pretreatment condition) using either the fluorescence-based in vitro assay (fluor) or the a-factor–based in vitro assay (bio). For the Ste14p isoprenylcysteine carboxyl methyltransferase, compounds were evaluated at 400 μM using the a-factor assay. NSC, Cancer Chemotherapy National Service Center.
Additional material of each consistently performing Rce1p inhibitor was obtained from the NCI DTP. The inhibitory properties of the new material matched that of the initial set. The structures of the consistently performing compounds were determined by accessing the DTP structure database (Fig. 3). The compounds range in mass from approximately 300 to 800 Da. Certain conserved structural features were identified. For example, 3 of the compounds (1-3) contain a diphenylmethane substructure. Both compounds 4 and 5 contain a hexachloro-substituted bicycloheptene. Several compounds are metal chelates (compounds 6-9). Compounds 6 and 7 are nearly identical mercury-based, multi-cyclic ring structures containing purine nucleoside substructures. These differ from organomercurials previously reported as Rce1p inhibitors.20,40 Compounds 8 and 9 contain a pyridine and hydrazinecarbothioate core and are associated with distinct metals.
FIG. 3.

Chemical structures of Rce1p inhibitors. Nine compounds were identified as potent Rce1p inhibitors as judged by their ability to consistently inhibit Rce1p activity in 2 separate in vitro assays. Structures were downloaded from the Developmental Therapeutics Program structure database (http://dtp.nci.nih.gov/branches/dscb/diversity_explanation.html) and converted to ChemDraw images.
Consistently performing compounds have micromolar potency against yeast and human Rce1p
The consistent Rce1p inhibitors were evaluated in more detail to determine their in vitro potency against yeast and human Rce1p. The IC50 value of each compound was determined using the fluorescence-based assay and membranes containing the appropriate CaaX protease (Table 3, Fig. 4). For yeast Rce1p, the compounds were determined to have IC50 values at or below 35 μM, with the most potent having a value of 6 μM. The inhibitor profile for human Rce1p, which was expressed heterologously in yeast and recovered as a membrane-bound activity, paralleled that of yeast Rce1p. Generally, the most potent yeast Rce1p inhibitors (e.g., compounds 6-9) were the most potent human Rce1p inhibitors.
Table 3.
IC50 Values of Compounds for Yeast Rce1p (Sc Rce1p), Human Rce1p (Hs Rce1p), and Ste24p
| IC50 (μM) | |||
|---|---|---|---|
| Compound | Sc Rce1p | Hs Rce1p | Ste24p |
| 1 | 35 | 13 | NA |
| 2 | 30 | 46 | 17 |
| 3 | 27 | 9 | 28 |
| 4 | 32 | 12 | 8 |
| 5 | 28 | 11 | 13 |
| 6 | 8 | 4 | NA |
| 7 | 6 | 3 | NA |
| 8 | 16 | 3 | 12 |
| 9 | 8 | 0.4 | 7 |
Compounds were evaluated over a range of concentrations with the fluorescence-based in vitro assay using an appropriate combination of CaaX protease and substrate. NA, does not display dose responsiveness consistent with a sigmoidal response.
FIG. 4.

Dose-response curves. The IC50 value of each of the 9 compounds was determined using a range of concentrations and the fluorescence-based Rce1p activity assay. Data were plotted using a 4-parameter logistic equation without constraints using GraphPad Prism 4.0.
Kinetic analysis revealed that the apparent Km of inhibitor-treated Rce1p was essentially within the range observed for the untreated enzyme, except for compounds 4 and 6 (Table 4). All compounds reduced the Vmax of Rce1p below that observed for the untreated enzyme, suggesting that none of the compounds are likely to be reversible competitive inhibitors. Consistent with this observation, extensive washes of inhibitor-treated membranes with buffer did not reverse the inhibition of Rce1p (data not shown).
Table 4.
Kinetic Parameters Associated with Rce1p Inhibitors
| Compound | Km (μM) | Vmax (μmol/min) | Km/Vmax |
|---|---|---|---|
| DMSO | 6.4 ± 2.0 | 0.830 ± 0.070 | 7.71 |
| 1 | 4.3 ± 0.8 | 0.438 ± 0.022 | 9.82 |
| 2 | 4.1 ± 1.2 | 0.230 ± 0.018 | 17.83 |
| 3 | 11.8 ± 5.0 | 0.453 ± 0.060 | 26.05 |
| 4 | 2.7 ± 0.8 | 0.474 ± 0.032 | 5.70 |
| 5 | 6.2 ± 1.9 | 0.195 ± 0.024 | 31.80 |
| 6 | 3.2 ± 0.8 | 0.373 ± 0.023 | 8.58 |
| 7 | 4.4 ± 1.4 | 0.509 ± 0.042 | 8.64 |
| 8 | 8.6 ± 2.5 | 0.641 ± 0.055 | 13.42 |
| 9 | 4.1 ± 1.4 | 0.193 ± 0.017 | 21.24 |
Compounds were evaluated over a range of concentrations using the fluorescence-based assay and yeast Rce1p. Kinetic parameters were derived using nonlinear regression methods.
Certain Rce1p inhibitors inhibit the Ste24p CaaX protease
In addition to Rce1p, Ste24p functions as a CaaX protease.3,18 These 2 CaaX proteases are believed to have distinct substrates, with the a-factor precursor being the only known exception. To assess the specificity of the consistently performing set of Rce1p inhibitors, the compounds were tested for their ability to inhibit yeast Ste24p using both the fluorescence and a-factor–based assays. For the fluorescence assay, a slightly different substrate was used because quencher position appears to dictate whether the substrate is Rce1p or Ste24p specific.37 For the a-factor assay, no changes were required because the a-factor precursor is a substrate of both CaaX proteases.
Our analysis revealed a wide range of Ste24p inhibition and some assay-specific effects. As evaluated using the fluorescence-based assay, 5 of the consistently performing Rce1p inhibitors (compounds 3-5, 8, and 9) were nonselective and potently inhibited Ste24p (<5% activity remaining; Table 2). These compounds also completely inhibited Ste24p-dependent a-factor production. The remaining 4 agents (compounds 1, 2, 6, and 7) varyingly inhibited Ste24p as judged by the fluorescence-based assay (14%-50% activity remaining). Of these, several compounds appeared less potent when evaluated using the a-factor production assay. In the most extreme case (compound 1), no inhibition of Ste24p was observed.
Certain Rce1p inhibitors inhibit the yeast ICMT
Whereas the fluorescence-based assay directly monitors CaaX proteolysis, the a-factor assay involves a coupled proteolysis-methylation reaction that is used to indirectly monitor proteolysis. Therefore, it was a formal possibility that observed reductions in a-factor production seen in the presence of certain compounds could be attributed to the inhibition of the ICMT (i.e., Ste14p) and not the CaaX proteases. To investigate the inhibition of Ste14p, we repeated the a-factor assay but added compounds after the proteolysis step was complete (i.e., during step 2). This analysis revealed a range of activities (Table 2). Compounds 1, 2, 6, and 7 did not inhibit Ste14p. The remaining compounds inhibited Ste14p, albeit to different extents. Compounds 3 to 5 modestly inhibited Ste14p. The residual activity was significantly higher than observed when these compounds were added during the CaaX proteolysis step, suggesting that these compounds do indeed inhibit the CaaX proteases. Because the a-factor assay does not allow us to resolve the individual impact of CaaX protease inhibition over that of the ICMT, we assume that the observed inhibition by compounds 3 to 5 is due to synergistic inhibition of both activities. The most potent Ste14p inhibitors were compounds 8 and 9. The decrease in a-factor production observed with these compounds could thus be attributed to specific inactivation of Ste14p. Nonetheless, the further reduced a-factor production observed when these compounds were applied during step 1 and the observed inactivation of the Rce1p and Ste24p CaaX proteases in the fluorescence-based assay suggests that these compounds inhibit both CaaX protease and ICMT activities.
Probing the promiscuity of Rce1p inhibitors
High-throughput studies often yield promiscuous agents that target multiple and often unrelated enzymes. To probe this issue further, we investigated whether any of our agents could inhibit trypsin. This enzyme readily cleaves our K-Ras4b–based fluorogenic substrate and can be assayed using reaction conditions identical to those used for monitoring Rce1p and Ste24p activities. At Rce1p IC50 concentrations, only compounds 3 and 5 moderately inhibited trypsin (Table 5). At the higher concentration of 200 μM, compounds 1 and 2 had negligible effects on trypsin activity, whereas the remaining compounds inhibited to varying extents. Of the 9-compound set, compounds 1, 2, 6, and 7 had the least impact on trypsin activity, in good correlation with the modest impact of these agents on Ste24p and negligible impact on ICMT activity.
Table 5.
Effect of Compounds on Trypsin Activity
| % Activity Remaining | ||
|---|---|---|
| Compound | IC50a | 200 μM |
| DMSO | 100.0 ± 8.7 | 100.0 ± 8.5 |
| TLCK | ND | 41.5 ± 0.4 |
| 1 | 94.2 ± 2.4 | 94.5 ± 1.3 |
| 2 | 118.5 ± 2.2 | 123.6 ± 8.0 |
| 3 | 73.5 ± 3.3 | 3.0 ± 1.4 |
| 4 | 89.6 ± 3.1 | 24.9 ± 0.9 |
| 5 | 76.9 ± 2.3 | 40.8 ± 4.3 |
| 6 | 103.4 ± 1.0 | 56.5 ± 1.6 |
| 7 | 103.3 ± 1.5 | 46.8 ± 2.5 |
| 8 | 99.3 ± 2.1 | 14.4 ± 2.2 |
| 9 | 101.1 ± 3.9 | 38.3 ± 0.9 |
ND, not determined.
As reported in Table 3 for Sc Rce1p. Values are derived from 4 independent measurements.
As another approach to investigate promiscuity, we examined the potential of our agents to form colloidal aggregates. Although the underlying mechanisms of promiscuous inhibitors can vary, aggregate formation has been positively correlated with the inhibitor activity of many promiscuous compounds, including well-known drug agents.41,42 Compounds 2, 6, and 7 did not form colloidal aggregates, as judged by DLS (Table 6). These compounds had an average particle radius less than 1 nm, as expected for monomers. The remaining compounds formed aggregates having predicted radii ranging from 57 to 523 nm. Most of the compounds, including those that did not form colloidal aggregates, formed precipitates under tested conditions.
Table 6.
Dynamic Light-Scattering Observations
| Compound | Precipitate | Aggregate | Radius (nm) | Aggregate Disrupted by 0.04% TX-100 |
|---|---|---|---|---|
| DMSO | No | No | 0.4 ± 0.1 | ND |
| ANS (1 mM) | No | No | 0.5 ± 0.3 | ND |
| TX-100 (0.5%) | No | Yes | 4.7 ± 0.5 | ND |
| Congo Red (0.75 mM) | No | Yes | 14.8 ± 3.9 | No |
| 1 | Yes | Yes | 522.9 ± 126.6 | Yes |
| 2 | Yes | No | 0.4 ± 0.1 | ND |
| 3 | Yes | Yes | 227.2 ± 110.2 | Yes |
| 4 | Yes | Yes | 170.3 ± 84.9 | No |
| 5 | Yes | Yes | 165.4 ± 50.8 | Yes |
| 6 | Yes | No | 0.3 ± 0.0 | ND |
| 7 | Yes | No | 0.4 ± 0.1 | ND |
| 8 | No | Yes | 76.6 ± 5.5 | Yes |
| 9 | Yes | Yes | 57.4 ± 11.8 | No |
Compounds were evaluated at 200 μM unless otherwise noted. ANS (8-anilino-1-naphthyl-sulfonic acid) is a well-characterized nonaggregating compound.42 Triton X-100 (TX-100) and Congo Red are known aggregators. For TX-100, the results predict an average mass size of 96 kD, in good correlation with the 80-kD mass reported for a TX-100 micelle. ND, not determined.
Detergents have the propensity to neutralize the inhibitory properties of aggregation-based promiscuous inhibitors.41 Some of the aggregating compounds were indeed sensitive to 0.04% Triton X-100, a concentration previously shown to disrupt aggregates of other promiscuous inhibitors (Table 6).43 The disruption of aggregates in our case was dependent on the concentration and type of detergent. For example, compound 3 aggregates were not dissociated by Triton X-100 below 0.04%, whereas compound 5 aggregates were the only ones also readily dissociated by 0.007% Tween-20. With the exception of Tween-20, Rce1p was completely inactivated by several detergents when they were used at their critical micelle concentration (CMC) levels (e.g., Triton X-100, NP-40, and CHAPS), limiting the utility of detergents for investigating inhibitor reversibility. Despite being well tolerated by the assay, Tween-20 at its CMC level was ineffective at reactivating inhibitor-treated Rce1p for all 9 inhibitors (data not shown).
Certain Rce1p inhibitors exert in vivo effects
To assess the in vivo characteristics of the Rce1p inhibitors identified in this study, we performed 2 experiments. The first experiment was used to judge the off-target potential of Rce1p inhibitors by examining their effects on cellular viability. Using yeast as the target organism, cells were exposed to each compound using a filter disc assay. This experiment revealed that a 1-mM concentration of most compounds was toxic, as demonstrated by zones of growth inhibition surrounding the appropriate filter disc (Fig. 5A). Compound 6 produced the largest zone of growth inhibition, whereas compounds 3 and 5 produced the smallest. No growth inhibition was observed with compounds 1, 2, or 4 or with a DMSO control. Based on these results and the reported observation that Rce1p is a nonessential protein, it is likely that most of our compounds have off-targets that include essential proteins.
FIG. 5.
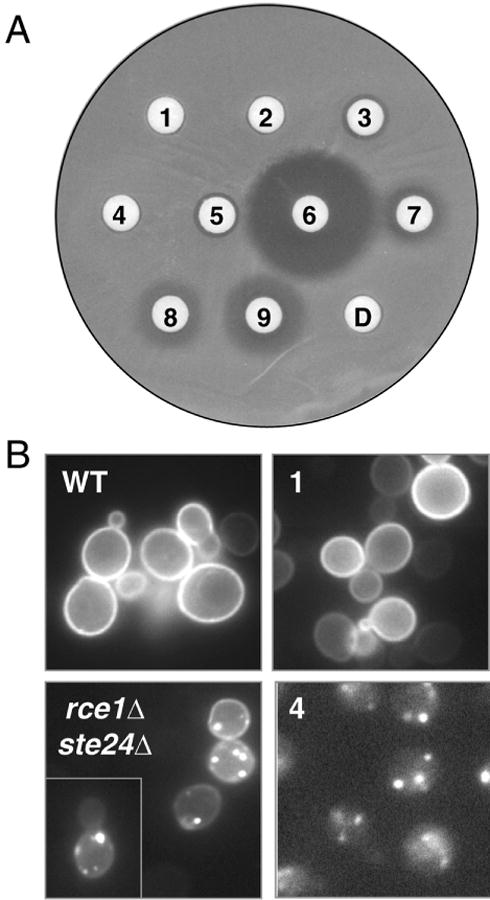
Effects of Rce1p inhibitors in vivo. (A) The 9 compounds identified in this study were spotted as 1-mM solutions onto filter discs overlaid on a yeast lawn. Zones of growth inhibition indicate compounds with toxic effects. The numbering corresponds to the compounds as identified in Figure 3, with the spot-labeled D indicating the DMSO-treated sample. (B) The subcellular localization of an induced GFP-Ras2p reporter was evaluated in CaaX protease–deficient (rce1Δ ste24Δ) and wild-type (WT) yeast. To assess the effects of compounds, WT yeast was treated with the indicated compound for 1 h prior to the induction of GFP-Ras2p expression. Because of a sparse cell distribution, 2 images were merged to form the rce1Δ ste24Δ panel.
In a second experiment to assess the in vivo characteristics of Rce1p inhibitors, we determined whether any of the apparently nontoxic compounds could disrupt GFP-Ras2p subcellular localization. This fluorescent reporter is normally localized to the plasma membrane in yeast and is delocalized to undefined endomembrane sites in the absence of CaaX proteolytic activity.3 Treatment of wild-type yeast with compounds 1 and 2 did not disrupt GFP-Ras2p localization, whereas compound 4 forced a strong GFP-Ras2p delocalization phenotype seemingly identical to that observed in the absence of the CaaX proteases (Fig. 5B, and data not shown). Based on our observations, the most parsimonious interpretation is that compounds 1 and 2 are not readily cell permeable, whereas compound 4 is cell permeable and interferes with the action of the yeast CaaX proteases.
Discussion
This study establishes proof of principle for the high-throughput in vitro identification of small-molecule inhibitors of the CaaX proteases. We expect the core structural scaffolds identified among these compounds to be useful as leads for efforts aimed at developing a highly potent and specific Rce1p inhibitor. Our future studies will address this possibility and will focus on more specific investigations into the mode of action of these inhibitors. We also expect that the assays and methods developed in this study will prove useful for the screening of larger compound libraries, which may ultimately yield even better structural scaffolds. In addition, our study strongly suggests that pharmacological inactivation of the CaaX proteases is possible, as demonstrated using our yeast system. Because delocalization of Ras and other isoprenylated proteins has been observed in rce1–/– mammalian cells44,45 and human Rce1p is equally sensitive to the yeast Rce1p inhibitors identified in this study, it seems plausible that our newly identified agents will exert similar pharmacological effects in mammalian systems.
An important aspect of this study is that it addresses the specificity of our newly identified Rce1p inhibitors in the context of other enzymes that are involved in CaaX protein maturation (i.e., Ste24p and Ste14p). The impact of previously reported Rce1p inhibitors on Ste24p and Ste14p function has generally not been addressed. Clearly, our observations establish that Rce1p inhibitors have the potential to interfere with other enzymes in the CaaX protein maturation pathway. This issue is well worth considering for future Rce1p drug design strategies because loss of Ste24p function is associated with disease phenotypes, including a progeroid syndrome.15 The specificity of compounds for multiple enzymes in the CaaX protein modification pathway is not entirely unexpected because agents that inhibit both the human farnesyltransferase and human Rce1p have been reported.11
Of the set of Rce1p-selective compounds, only compound 1 was prone to colloidal aggregation. Although several aggregators (e.g., clotrimazole, nicardipine, glyburide, and mefanamic acid) are known to inhibit membrane proteins (i.e., the sterol demethylase, Ca2+ channel, K+ channel, and cyclooxygenase, respectively), our results establish for the first time that an aggregator can inhibit membrane-bound proteolytic activity.42 Our data also support that compound 1 is relatively target specific, unlike the aforementioned aggregators that are known to be promiscuous. Although aggregation-based inhibitors are generally promiscuous, aggregators that are enzyme specific have been identified.46 Importantly, it remains to be formally resolved whether the colloidal aggregate or monomeric form of compound 1 is the inhibitory component of this agent. Because our assay is performed at a relatively high protein concentration (0.5 mg/ml at the time of compound treatment), and high protein milieus are known to neutralize aggregation-based inhibitors, the monomeric form of compound 1 could in fact be responsible for the inhibition that we observe with this compound.43 Unfortunately, the aggregation state of compound 1 could not be assessed in the presence of Rce1p-containing membranes because samples of particulate membrane material (0.25 mg/ml) overloaded the detector of our DLS instrument at its lowest sensitivity setting.
Some of the compounds identified in this study have been previously documented to inhibit enzymatic activities and to exert physiological effects not readily linked to CaaX proteolysis. In some instances, they have even been pursued as chemotherapeutic agents.29,47-49 For example, compound 4 has been identified as an inhibitor of DNA topoisomerase activity.50 Ultimately, it will be interesting to determine whether these compounds mediate part or all of their reported physiological effects through inhibition of postisoprenylation-modifying enzymes. For the present, this study has yielded compounds that represent new and useful tools for the in vitro and in vivo characterization of Rce1p and possibly other enzymes required for the production of isoprenylated proteins.
Acknowledgments
We thank the Developmental Therapeutics Program (National Institutes of Health) for providing the Diversity Set compound library; Dr. J. Prestegard, Dr. G. Wylie and Dr. S. Bansal (University of Georgia) for access to and training on the DynaPro99 Molecular Sizing instrument; Dr. M. Terns (University of Georgia) for use of the fluorescence microscope; and Dr. T. M. Dore (University of Georgia), Dr. B. Shoichet (UCSF), and members of the Schmidt laboratory for critical discussions and technical assistance. This work was supported by R01 and R03 grants from the National Institutes of Health (to W.K.S.).
References
- 1.Young SG, Ambroziak P, Kim E, Clarke S. Postisoprenylation protein processing: CXXX (CaaX) endoproteases and isoprenylcysteine carboxyl methyltransferase. In: Tamanoi F, Sigman DS, editors. The Enzymes. New York: Academic Press; 2001. [Google Scholar]
- 2.Winter-Vann AM, Casey PJ. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat Rev Cancer. 2005;5:405–412. doi: 10.1038/nrc1612. [DOI] [PubMed] [Google Scholar]
- 3.Boyartchuk VL, Ashby MN, Rine J. Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science. 1997;275:1796–1800. doi: 10.1126/science.275.5307.1796. [DOI] [PubMed] [Google Scholar]
- 4.Tam A, Nouvet F, Fujimura-Kamada K, Slunt H, Sisodia SS, Michaelis S. Dual roles for Ste24p in yeast a-factor maturation: NH2-terminal proteolysis and COOH-terminal CAAX processing. J Cell Biol. 1998;142:635–649. doi: 10.1083/jcb.142.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trueblood CE, Boyartchuk VL, Picologlou EA, Rozema D, Poulter CD, Rine J. The CaaX proteases, Afc1p and Rce1p, have overlapping but distinct substrate specificities. Mol Cell Biol. 2000;20:4381–4392. doi: 10.1128/mcb.20.12.4381-4392.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romano JD, Schmidt WK, Michaelis S. The Saccharomyces cerevisiae prenylcysteine carboxyl methltransferase Ste14p is in the endoplasmic reticulum membrane. Mol Biol Cell. 1998;9:2231–2247. doi: 10.1091/mbc.9.8.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt WK, Tam A, Fujimura-Kamada K, Michaelis S. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc Natl Acad Sci U S A. 1998;95:11175–11180. doi: 10.1073/pnas.95.19.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox A, Der C. Farnesyltransferase inhibitors: promises and realities. Curr Opin Pharmacol. 2002;2:388–393. doi: 10.1016/s1471-4892(02)00181-9. [DOI] [PubMed] [Google Scholar]
- 9.Zhu K, Hamilton AD, Sebti SM. Farnesyltransferase inhibitors as anticancer agents: current status. Curr Opin Investig Drugs. 2003;4:1428–1435. [PubMed] [Google Scholar]
- 10.Dolence EK, Dolence JM, Poulter CD. Solid-phase synthesis of a farnesylated CaaX peptide library: inhibitors of the Ras CaaX endoprotease. J Comb Chem. 2000;2:522–536. doi: 10.1021/cc000026m. [DOI] [PubMed] [Google Scholar]
- 11.Schlitzer M, Winter-Vann A, Casey PJ. Non-peptidic, non-prenylic inhibitors of the prenyl protein-specific protease Rce1. Bioorg Med Chem Lett. 2001;11:425–427. doi: 10.1016/s0960-894x(00)00685-5. [DOI] [PubMed] [Google Scholar]
- 12.Anderson JL, Henriksen BS, Gibbs RA, Hrycyna CA. The isoprenoid substrate specificity of isoprenylcysteine carboxylmethyltransferase: development of novel inhibitors. J Biol Chem. 2005;280:29454–29461. doi: 10.1074/jbc.M504982200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winter-Vann AM, Baron RA, Wong W, de la Cruz J, York JD, Gooden DM, et al. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc Natl Acad Sci U S A. 2005;102:4336–4341. doi: 10.1073/pnas.0408107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- 15.Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 2005;46:2531–2558. doi: 10.1194/jlr.R500011-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Farh L, Mitchell D, Deschenes R. Farnesylation and proteolysis are sequential, but distinct steps in the CaaX box modifcation pathway. Arch Biochem Biophys. 1995;318:113–121. doi: 10.1006/abbi.1995.1211. [DOI] [PubMed] [Google Scholar]
- 17.Otto JC, Kim E, Young SG, Casey PJ. Cloning and characterization of a mammalian prenyl protein-specific protease. J Biol Chem. 1999;274:8379–8382. doi: 10.1074/jbc.274.13.8379. [DOI] [PubMed] [Google Scholar]
- 18.Tam A, Schmidt WK, Michaelis S. The multispanning membrane protein Ste24p catalyzes CAAX proteolysis and NH2-terminal processing of the yeast a-factor precursor. J Biol Chem. 2001;276:46798–46806. doi: 10.1074/jbc.M106150200. [DOI] [PubMed] [Google Scholar]
- 19.Ashby M, King D, Rine J. Endoproteolytic processing of a farnesylated peptide in vitro. Proc Natl Acad Sci U S A. 1992;89:4613–4617. doi: 10.1073/pnas.89.10.4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolence JM, Steward LE, Dolence EK, Wong DH, Poulter CD. Studies with recombinant Saccharomyces cerevisiae CaaX prenyl protease Rce1p. Biochemistry. 2000;39:4096–4104. doi: 10.1021/bi9923611. [DOI] [PubMed] [Google Scholar]
- 21.Hollander I, Frommer E, Mallon R. Human ras-converting enzyme (hRCE1) endoproteolytic activity on K-ras-derived peptides. Anal Biochem. 2000;286:129–137. doi: 10.1006/abio.2000.4795. [DOI] [PubMed] [Google Scholar]
- 22.Holbeck SL. Update on NCI in vitro drug screen utilities. Eur J Cancer. 2004;40:785–793. doi: 10.1016/j.ejca.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 23.Lazo JS, Aslan DC, Southwick EC, Cooley KA, Ducruet AP, Joo B, et al. Discovery and biological evaluation of a new family of potent inhibitors of the dual specificity protein phosphatase Cdc25. J Med Chem. 2001;44:4042–4049. doi: 10.1021/jm0102046. [DOI] [PubMed] [Google Scholar]
- 24.Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH, et al. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002;62:4316–4324. [PubMed] [Google Scholar]
- 25.Moloughney JG, D Thomas J, Toney JH. Novel IMP-1 metallo-beta-lactamase inhibitors can reverse meropenem resistance in Escherichia coli expressing IMP-1. FEMS Microbiol Lett. 2005;243:65–71. doi: 10.1016/j.femsle.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 26.Turkson J, Zhang S, Mora LB, Burns A, Sebti S, Jove R. A novel platinum compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J Biol Chem. 2005;280:32979–32988. doi: 10.1074/jbc.M502694200. [DOI] [PubMed] [Google Scholar]
- 27.Dorjsuren D, Burnette A, Gray G, Chen X, Zhu W, Roberts P, et al. Chemical library screen for novel inhibitors of Kaposi's sarcoma-associated herpesvirus processive DNA synthesis. Antiviral Res. 2006;69:9–23. doi: 10.1016/j.antiviral.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, et al. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol. 2006;70:562–570. doi: 10.1124/mol.106.025536. [DOI] [PubMed] [Google Scholar]
- 29.Marx C, Berger C, Xu F, Amend C, Scott GK, Hann B, et al. Validated high-throughput screening of drug-like small molecules for inhibitors of ErbB2 transcription. Assay Drug Dev Technol. 2006;4:273–284. doi: 10.1089/adt.2006.4.273. [DOI] [PubMed] [Google Scholar]
- 30.Siliciano P, Tatchell K. Transcription and regulatory signals at the mating type locus in yeast. Cell. 1984;37:969–978. doi: 10.1016/0092-8674(84)90431-8. [DOI] [PubMed] [Google Scholar]
- 31.Powers S, Michaelis S, Broek D, Santa Anna S, Field J, Herskowitz I, et al. RAM, a gene of yeast required for a functional modification of RAS proteins and for production of mating pheromone a-factor. Cell. 1986;47:413–422. doi: 10.1016/0092-8674(86)90598-2. [DOI] [PubMed] [Google Scholar]
- 32.Elble R. A simple and efficient procedure for transformation of yeasts. BioTechniques. 1992;13:18–20. [PubMed] [Google Scholar]
- 33.Michaelis S, Herskowitz I. The a-factor pheromone of Saccharomyces cerevisiae is essential for mating. Mol Cell Biol. 1988;8:1309–1318. doi: 10.1128/mcb.8.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plummer LJ, Hildebrandt ER, Porter SB, Rogers VA, McCracken J, Schmidt WK. Mutational analysis of the ras converting enzyme reveals a requirement for glutamate and histidine residues. J Biol Chem. 2006;281:4596–4605. doi: 10.1074/jbc.M506284200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heim R, Tsien RY. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr Biol. 1996;6:178–182. doi: 10.1016/s0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]
- 36.Oldenburg KR, Vo KT, Michaelis S, Paddon C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res. 1997;25:451–452. doi: 10.1093/nar/25.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porter S, Hildebrandt E, Breevoort S, Dore T, Schmidt W. Inhibition of the CaaX proteases Rce1p and Ste24p by peptidyl (acyloxy)methyl ketones. Biochim Biophys Acta. 2007;1773:853–862. doi: 10.1016/j.bbamcr.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt WK, Tam A, Michaelis S. Reconstitution of the Ste24p-dependent N-terminal proteolytic step in yeast a-factor biogenesis. J Biol Chem. 2000;275:6227–6233. doi: 10.1074/jbc.275.9.6227. [DOI] [PubMed] [Google Scholar]
- 39.Marcus S, Caldwell GA, Miller D, Xue CB, Naider F, Becker JM. Significance of C-terminal cysteine modifications to the biological activity of the Saccharomyces cerevisiaea-factor mating pheromone. Mol Cell Biol. 1991;11:3603–3612. doi: 10.1128/mcb.11.7.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma YT, Gilbert B, Rando R. Inhibitors of the isoprenylated protein endoprotease. Biochemistry. 1993;32:2386–2393. doi: 10.1021/bi00060a033. [DOI] [PubMed] [Google Scholar]
- 41.McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 42.Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 43.Coan KE, Shoichet BK. Stability and equilibria of promiscuous aggregates in high protein milieus. Mol Biosyst. 2007;3:208–213. doi: 10.1039/b616314a. [DOI] [PubMed] [Google Scholar]
- 44.Bergo MO, Ambroziak P, Gregory C, George A, Otto JC, Kim E, et al. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol Cell Biol. 2002;22:171–181. doi: 10.1128/MCB.22.1.171-181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michaelson D, Ali W, Chiu VK, Bergo M, Silletti J, Wright L, et al. Postprenylation CAAX processing is required for proper localization of Ras but not Rho GTPases. Mol Biol Cell. 2005;16:1606–1616. doi: 10.1091/mbc.E04-11-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reddie KG, Roberts DR, Dore TM. Inhibition of kinesin motor proteins by adociasulfate-2. J Med Chem. 2006;49:4857–4860. doi: 10.1021/jm060115z. [DOI] [PubMed] [Google Scholar]
- 47.Bulavin D, Belova G, Fornace AJ. Compounds and methods for inhibiting Wip1 phosphatase and treating cancer, and screening method. US Pat Appl Publ. 2004:15. [Google Scholar]
- 48.Perabo FG, Wirger A, Kamp S, Lindner H, Schmidt DH, Muller SC, et al. Carboxyamido-triazole (CAI), a signal transduction inhibitor, induces growth inhibition and apoptosis in bladder cancer cells by modulation of Bcl-2. Anticancer Res. 2004;24:2869–2877. [PubMed] [Google Scholar]
- 49.Guo L, Li ZS, Wang HL, Ye CY, Zhang DC. Carboxyamidotriazole inhibits proliferation of human breast cancer cells via G2/M cell cycle arrest and apoptosis. Eur J Pharmacol. 2006;538:15–22. doi: 10.1016/j.ejphar.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 50.Bond A, Reichert Z, Stivers JT. Novel and specific inhibitors of a poxvirus type I topoisomerase. Mol Pharmacol. 2006;69:547–557. doi: 10.1124/mol.105.019067. [DOI] [PubMed] [Google Scholar]