

Abstract
13F6-1-2 is a murine monoclonal antibody that recognizes the heavily glycosylated mucin-like domain of the Ebola virus virion-attached glycoprotein (GP) and protects animals against lethal viral challenge. Here we present the crystal structure, at 2.0 Å, of 13F6-1-2 in complex with its Ebola virus GP peptide epitope. The GP peptide binds in an extended conformation, anchored primarily by interactions to the heavy chain. Two GP residues, Gln P406 and Arg P409, make extensive side chain hydrogen bond and electrostatic interactions to the antibody and are likely critical for recognition and affinity. The 13F6-1-2 antibody utilizes a rare Vλx light chain. The three light chain complementarity determining regions (CDRs) do not adopt canonical conformations and represent new classes of structures distinct from Vκ and other Vλ light chains. In addition, although Vλx had been thought to confer specificity, all light chain contacts are mediated through germline-encoded residues. This structure of an antibody that protects against the Ebola virus now provides a framework for humanization and development of a post-exposure immunotherapeutic.
Keywords: Ebola virus, Vλx light chain, glycoprotein, neutralizing antibody, new canonical structures of immunoglobulins, hypervariable loops, complementarity determining region, Fab-peptide complex
Introduction
The Ebola virus causes a severe hemorrhagic fever usually involving uncontrolled viral replication, multiple organ failure and death six-nine days after onset of symptoms 1. The 13 known outbreaks between 1976 and 2004 have resulted in 50-90% fatality 2 with the highest lethality associated with the Zaire subtype of the virus 3; 4.
An interesting feature of the Ebola virus genome is its ability to encode separate glycoproteins, which share 295 amino acids of N-terminal sequence, but have unique C-termini, which lead to different patterns of disulfide bonding and different structures. The primary gene product is a small, dimeric glycoprotein called sGP (364 amino acids), which is shed abundantly from infected cells. The secondary gene product is a larger glycoprotein called GP (676 amino acids) 5, which is proteolytically cleaved into two subunits (GP1 and GP2) connected by a disulfide linkage. Three GP1-GP2 units form a trimeric, transmembrane envelope spike which is the sole protein expressed on the surface of Ebola virus, and is involved in receptor binding, tropism, and viral entry 6-9.
It will be desirable to have well-characterized, effective immunotherapeutics against the Ebola virus available in case of exposure, outbreak, or release of the virus. Several monoclonal antibodies (mAbs) have demonstrated protection against the Ebola virus in rodent models 10-12; one mAb failed to protect in a macaque model 13. It is as yet unclear whether a different mAb, or a cocktail of mAbs might offer better protection. One challenge to the development of such immunotherapeutics is that it appears difficult to elicit an effective antibody response in natural infection: many survivors have little to no IgG 1. A second challenge is that of these antibodies that are elicited, whether by natural infection or by vaccination, the majority appear to preferentially react with sGP or cross-react between sGP and GP 10; 12; 14; 15. These antibodies may be absorbed by the much more abundant shed sGP and thus, it may be difficult to achieve concentrations required for viral neutralization.
A murine IgG2a termed 13F6-1-2 has been identified which is specific for viral surface GP, does not cross-react to shed sGP, and is protective against challenge with the highly pathogenic Zaire subtype of the Ebola virus 12. All mice (10/10) survive challenge with 300 times the lethal dose of Ebola virus when 100 μg of 13F6-1-2 is administered one day after challenge 12. The epitope has previously been mapped to nine residues (EQHHRRTDN, amino acids 405-413) within the Ebola Zaire GP mucin-like domain 12. These residues are contained within a heavily glycosylated mucin-like region unique to GP 12, which has been linked to GP-induced cytotoxicity 16.
Interestingly, 13F6-1-2 contains a rare Vλx light chain which is observed in only 0.5% of antibody sequences and is barely detectable in normal mouse serum 17-19. Vλx, first described in 1987 17-19, is conserved among mice, humans, and other mammals 20, and displays at least three features unique from Vλ1 and Vλ2. The amino acid sequence is only ~30-33% homologous to other known Vλ and/or Vκ light chains. The last codon of the Vλx gene is a TAA stop codon which must be disrupted by Jλ2 to create a functional codon (Figure 1). As a result of this V-J junction, four extra residues extend the complementarity determining region (CDR) L3. Previous studies suggest that this unusual light chain confers antigen specificity to the antibodies that contain it 21.
Figure 1. Nucleotide and translated amino acid sequences for variable regions.
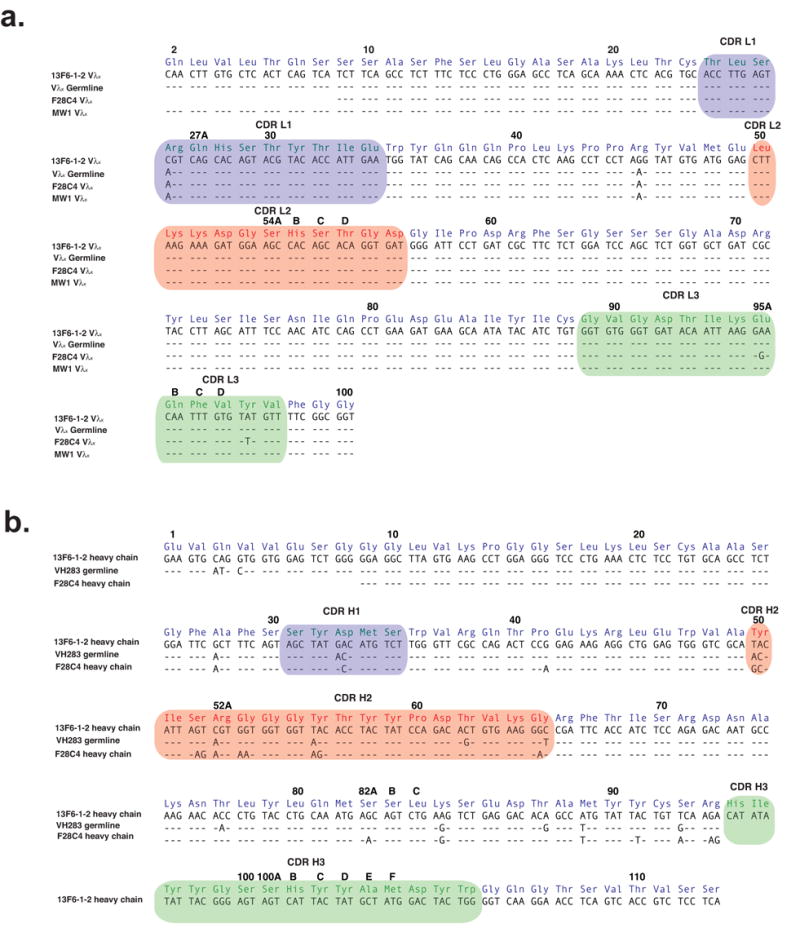
(a) The light chain variable region of 13F6-1-2 compared with the BALB/c Vλx germline (GenBank accession number M34597), and the Vλx-light chains F28C4 (GenBank accession number X76752) and MW1 (GenBank accession number M34598). 13F6-1-2 contains two mutations from the Vλx germline: a Ser to Arg mutation at residue 27 in CDR L1 and a Lys to Arg mutation in framework residue 45. (b) The heavy chain variable region of 13F6-1-2 compared with the germline VH283 and F28C4. The amino acids are labeled according to the Kabat numbering scheme and hypervariable loops are highlighted in colour.
Characterization of the mode of recognition of GP by the protective 13F6-1-2 monoclonal antibody (mAb), and successful humanization of its sequence are necessary steps toward development of this antibody as an immunotherapeutic against the Ebola virus. Here we present the crystal structure of a complex between the Fab fragment of 13F6-1-2 and its GP epitope at 2.0 Å resolution. This work provides the first available structure of an antibody directed against the Ebola virus and also illustrates unusual structural features of a Vλx-containing antibody that shed light on its immunological relevance.
Results and Discussion
13F6-1-2 contains a rare Vλx light chain
Sequencing of 13F6-1-2 revealed the usage of a rare λx light chain gene. The nucleotide sequence of the 13F6-1-2 Vλx light chain is 99.3% homologous to the previously reported BALB/c Vλx germline (GenBank accession number M34597 or IGLV3 in IMGT nomenclature; Figure 1a). Interestingly, there is only a single somatic mutation contained within all three CDRs. Indeed, Vλx-bearing light chains in general have very little somatic mutation. Only two other Vλx-bearing antibodies have been well-described. One is termed F28C4 (GenBank accession number X76752), contains just two somatic mutations in the CDR L3 region (Figure 1a) and is directed against myelin basic protein 22. The second is termed MW1 (GenBank accession number M34598), contains no somatic mutations and is directed against a poly-Gln sequence relevant for Huntington’s disease 23.
The 13F6-1-2 heavy chain belongs to the 7183 family, clone 19 (Figure 1b), and shares 88% sequence identity with the VH283 germline. Of the twelve mismatches, five reside in the CDRs (Figure 1b).
13F6-1-2 Fab structure
The crystallographic asymmetric unit contains one Fab 13F6-1-2:GP peptide complex. The polypeptides of the light and heavy chains are well defined by electron density except for five side chains (Glu H1, Gln L2, Arg L27, Gln L27A, and Asp L60), which have been truncated to alanine. Analysis of conformations by Ramachandran plot shows >99% of residues residing in favorable regions and no non-glycyl residues in disallowed regions. Note that in most Fab structures, residue 51 of the light chain exists in a γ turn in a rare region of the Ramachandran plot 24. In the structure presented here, however, CDR L2 is non-canonical and residue 51 adopts a different, favoured conformation.
The Vλx light chain and heavy chain of 13F6-1-2 form the expected overall V-type, nine-stranded β-sandwich immunoglobulin fold (Figure 2) and the light chain is, not surprisingly, similar to a recently determined Vλx-bearing variable domain (MW1) 23. To our knowledge, this is the only other Vλx antibody structure for which a structure is available. While the overall Vλx fold of these structures is similar to the V-type immunoglobulin fold, there are a few unusual differences. In MW1 and 13F6-1-2, a one residue deletion (typically, deletion of a proline or threonine) exists in the A-A′ strand which results in a smaller kink and disruption of typical A-G strand hydrogen bonding (Figure 2b) 23. In Vλx, the C″ β-strand twists towards the outer D β-strand to form a pseudo-β-barrel, but the C″ β-strand of Vλx does not make any hydrogen bond interactions to strand D, as would occur in the immunoglobulin fold in T cell receptors.
Figure 2. Overall structure of 13F6-1-2.
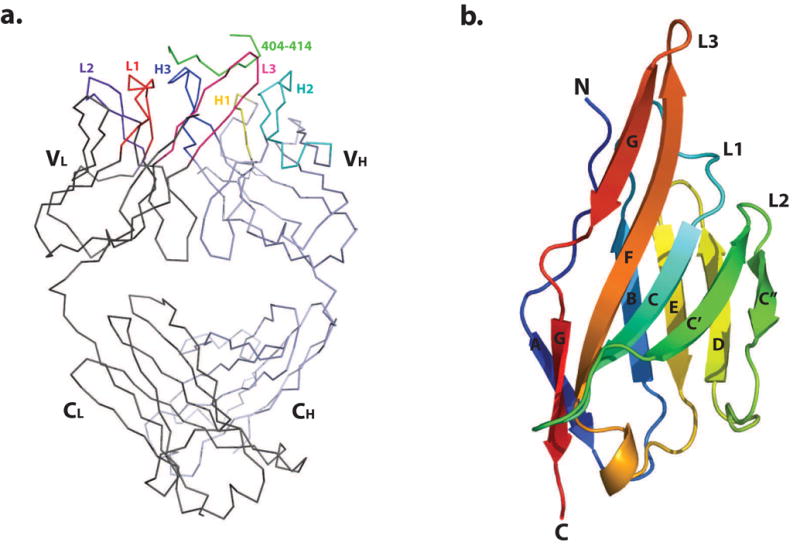
(a) Cα trace of the 13F6-1-2 Fab fragment complexed to the P404-P414 Ebola virus GP peptide. The structure is shown with bound GP peptide coloured in green, and the light and heavy chains in dark grey and light blue, respectively. CDRs L1, L2, L3, H1, H2, and H3 are coloured red, purple, pink, yellow, cyan, and blue, respectively. (b) Ribbon diagram of the 13F6-1-2 Vλx light chain variable domain. β-strands are labeled A, B, C, C′, C″, D, E, F, and G. This and subsequent figures were generated using MacPyMol 54.
The elbow angle between the variable and constant Fab domains in 13F6-1-2 is 149°. This is consistent with other Vλ light chains, which typically exhibit a bimodal distribution of elbow angles with values between 117°- 225°25. At the VL/VH interface of 13F6-1-2, ~1025 Å2 surface area is buried on VL and ~985 Å2 on VH. Interestingly, this is within the smaller range of light chain/heavy chain interfaces described for antibodies. The typical buried surface area ranges between 1068-1750 Å2 26; 27. A small VH/VL interface has been correlated with the ability to use domain rearrangements in the induced fit of the antibody to the antigen 27, although it is unclear whether an induced conformational change occurs in 13F6-1-2 upon peptide binding.
13F6-1-2 Hypervariable Loops
Complementarity determining regions (CDRs) of antibodies are typically restricted to certain conformations, depending on the length of insertion in the CDR. An analysis of 79 different hypervariable regions revealed that each can be assigned to at least one of 18 different canonical structures 24. None of the CDRs of 13F6-12 are affected by crystal contacts. The heavy chain CDRs H1 and H2 belong to canonical classes 1 and 3, respectively. CDR H3 contains 15 amino acids. Residues H92-H96 and H100e-H104 form an antiparallel β-sheet, and a type I β-turn exists between Tyr H97 and Ser H100 (i+1: φ = −60.4°, ψ = −36.5°; i+2: φ = −55°, ψ= −28.5°). By contrast, analysis of the light chain Vλx CDR loops of 13F6-1-2 and MW1 reveals a new class of hypervariable loop structures.
CDR L1
The CDR Vλ L1 region normally falls into four different canonical conformations 24; 28-30, with the region typically containing nine to eleven residues and a hydrophobic residue at center that packs into the antibody framework (Figure 3a) 24. In canonical conformation 4, residues 26 to 30 form an irregular helix. Six canonical conformations have been identified for the CDR Vκ L1 region which typically contains between six and thirteen residues 24 (Figure 3b). In all six canonical conformations, residues 26 to 29 adopt an extended conformation and residues 29 to 32 adopt a short link or hairpin loop. Almost all human germline and expressed Vκ light chains have canonical structures 2, 3, 4, or 6 31. The Vλx CDR L1 is twelve residues long and does not contain the standard helix or any other defined secondary structure element (Figure 3c). Interestingly, there is also no hydrophobic residue that packs into the framework in Vλx, as the side chain of the equivalent residue (Ser29) points away from the framework.
Figure 3. Analysis of light chain CDRs.
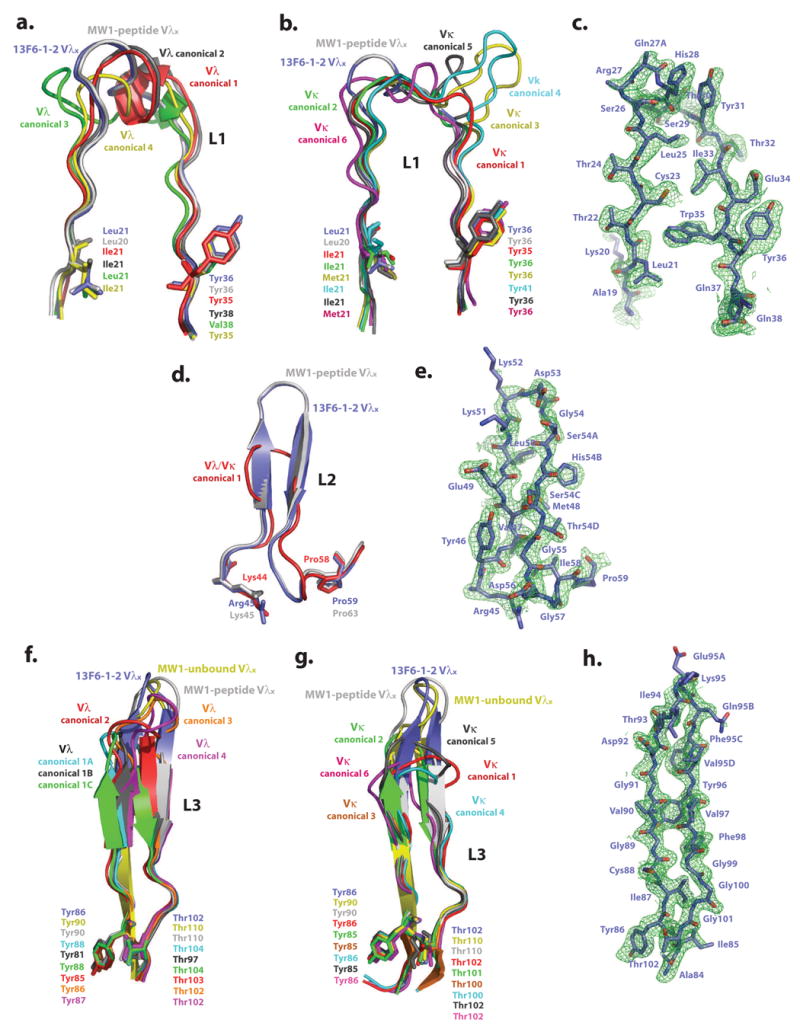
Comparison of Fab 13F6-1-2 and (a) Vλ L1, (b) Vκ L1, (d) Vλ/Vκ L2, (f) Vλ L3, and (g) Vκ L3 CDR canonical conformations. The initial difference σA-weighted Fo-Fc electron density map (green), contoured at 2σ superimposed onto the refined (c) L1, (e) L2, and (h) L3 13F6-1-2 CDR structures. Example structures of CDR canonical conformations are taken from the following antibody structures (P DB accession codes): CDR L1 Vλ canonical conformation 1: 2FB4, 2: 7FAB, 3: 1MFA, 4: 8FAB, MW1-peptide: 2OTU. CDR L1 Vκ canonical conformation 1: 2FBJ, 2: 1VFA, 3: 1HIL, 4: 1FLR, 5: 1GGI, 6: 1FIG, MW1-peptide: 2OTU. CDR L2 Vλ/κ canonical conformation 1: 2FB4, MW1-peptide: 2OTU. CDR L3 Vλ canonical conformation 1A: 1GIG, 1B: 7FAB, 1C: 1MFA, 2: 2FB4, 3: 1Q1J, 4:2B0S, MW1-peptide: 2OTU, MW1-unbound: 2GSG. CDR L3 Vκ canonical conformation 1: 1HIL, 2: 2FBJ, 3: 1BQL, 4: 1DFB, 5: 1BAF, 6: 1EAP, MW1-peptide: 2OTU, MW1-unbound: 2GSG. CDR L1 and L2 of MW1 in the unbound state (PDB code: 2GSG) are not shown in figures 3a, b, and d for figure clarity, as these CDRs are similar in conformation to those observed in the MW1 peptide-bound structure.
CDR L2
The CDR L2 for standard Vλ and Vκ light chains always contains seven residues and occurs in only one known conformation, with a three-residue γ-hairpin loop linking the C′ and C″ β-strands (Figure 3d) 24. In order to maintain the γ-hairpin loop, the residue at position 51 (usually a Val but also sometimes a Ser) typically lies in a rare conformation in the Ramachandran plot, with torsion angles of φ = +67°, ψ = -40°24. However, in Vλx-bearing antibodies, four additional residues are inserted into the CDR L2 loop, for a total of eleven residues to form a type I β-turn (Figure 3e). As a consequence, this insertion allows the formation of a longer β-sheet between β-strands C″ and C′ and allows the inner C″ β-strand to twist towards the outer β-strand D. The Vλx CDR L2 does not have a valine at position 51, but rather a lysine residue that resides in a favourable region of ϕ-ψ space and forms a hydrogen bond between its carbonyl oxygen and the amide nitrogen of Gly54.
CDR L3
The typical CDR L3 of a Vλ antibody adopts one of two main conformations (Figure 3f) 24. Canonical conformation 1 is a six-residue hairpin, with two residues at the top of the loop forming the tight turn. The high resolution structures available for canonical conformation 1 show three minor forms (A, B, and C), where the orientation of the peptide bond differs between residues 93 and 94. A hydrogen bond exists between the main chain oxygen of residue 95 and the amide nitrogen of residue 92. Canonical conformation 2 consists of eight residues in the loop, with four residues forming the turn. More recently, two additional conformations (named as canonical conformations 3 and 4 in this manuscript) have been identified for L3 in the structures of the λ-light chain containing neutralizing anti-HIV-1 antibodies 447-52D and 2219 32; 33, which have nine and ten residue L3 loops, respectively. For Vκ antibodies, CDR L3 usually belongs to one of six canonical conformations (Figure 3g). Canonical conformations 1, 3, 4, 5, and 6 do not form a β-sheet as typically seen in Vλ CDR L3, but instead form a loop (Figure 3g), with conformation 1 being the most commonly observed 24. Canonical conformation 2 forms a regular hairpin and residue 94 adopts a cis-proline conformation. By contrast, Vλx light chains have a ten residue CDR L3, which allows the loop to form a longer β-sheet. In 13F6-1-2, a type IV β-turn is found at the top of the loop (Figure 3h).
A new class of CDR canonical conformations
It has been proposed that the Vλx light chain of antibodies do not undergo significant affinity maturation via somatic mutation 20. Indeed, although very few Vλx light chains have been identified and sequenced, those that have been described do not vary significantly in sequence from germline. For example, mAb 13F6-1-2 contains only one somatic mutation (which lies in CDR L1), MW1 contains zero mutations, and mAb F28C4 contains only two somatic mutations (which lie in CDR L3). In addition, the lengths of the hypervariable loops in the Vλx light chains are conserved. The Vλx germline, mAb 13F6-1-2, MW1, and F28C4 all have CDRs L1, L2, and L3 of 12, 11, and 13 residues in length, respectively; no other antibody has been described with light chain CDRs longer than those of Vλx-bearing light chains. It appears that the hypervariable loop conformations observed in the 13F6-1-2 and MW1 Vλx structures may represent new L1, L2, and L3 canonical classes for Vλx light chains.
Analysis of the CDRs of 13F6-1-2 and bound and unbound MW1 clearly reveals that the L1 and L2 regions of these Vλx antibodies are quite similar to each other, fairly rigid, and not subject to antigen-induced conformational changes (Figure 3). Similarly, although new non-canonical conformations have been described for CDR L332; 33, fewer deviations have been noted for CDR L1 and CDR L2 of other Vλ and Vκ antibodies. In fact for CDR L2, all non-Vλx antibody structures determined to date adopt only one conformation 24, regardless of the type of light chain. Although CDRs L1 and L2 appear rigid, L3 is more flexible and the conformations of CDR L3 differ among 13F6-1-2 and the peptide-bound and unbound structures of MW1 (Figure 3f and 3g). This is rather interesting as the 13F6-1-2 and MW1 CDR L3s are identical in sequence. Hence, a single CDR sequence can adopt multiple conformations.
13F6-1-2 Fab-peptide interactions
The eleven-residue peptide used for co-crystallization (VEQHHRRTDND) corresponds to a region of the mucin-like domain of the Ebola Zaire GP. Residues P404-P414 (based on the numbering in Ebola Zaire GP) are unambiguously located in strong electron density (Figure 4a). The average B value for the peptide residues is 37 Å2, suggesting that these residues are well-defined and ordered. Surface plasmon resonance experiments were performed on the intact Ebola virus and show that IgG 13F6-1-2 has kon= 7.00 × 104 M-1s-1 and koff= 2.87 × 10-3 s-1, with a calculated affinity (Kd) of 41 nM (Figure 5a). The synthetic VEQHHRRTDND peptide used in co-crystallization competes for binding with IgG 13F6-1-2 and the full-length GP (50% inhibitory concentration (IC50) of ~3.5 μM) (Figure 5c). The competition is not as strong as expected, suggesting that additional contacts to the native GP protein may be made by 13F6-1-2, or that the conformation of the linear epitope may be stabilized in the context of the intact GP molecule.
Figure 4. Antigen binding site.
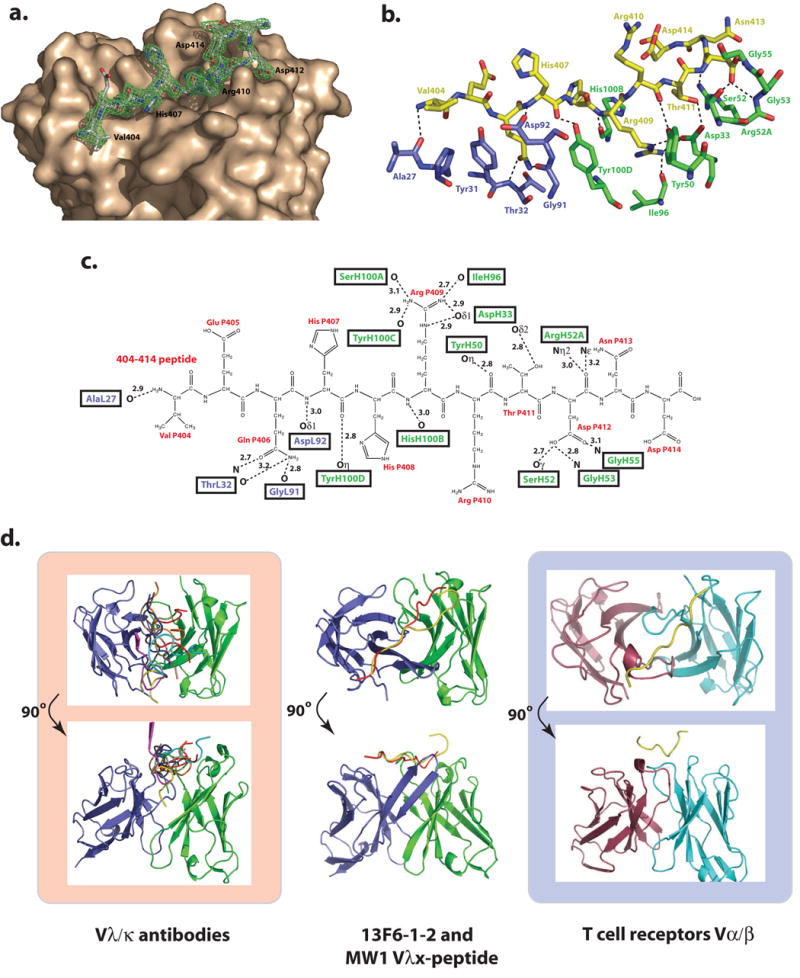
(a) Electron density for the P404-P414 GP peptide bound to 13F6-1-2. The initial difference σA-weighted Fo-Fc electron density map (green), contoured at 3σ superimposed onto the refined P404-P414 peptide and the molecular surface of 13F6-1-2. (b) Stick representation of the antigen binding site in which peptide residues are colored yellow, light chain residues are blue, and heavy chain residues are green. (c) 2-D schematic of the interactions between the peptide and Fab 13F6-1-2. Peptide resides are illustrated in black and labeled in red. (d) Comparison of general peptide binding orientations in Vλ/Vκ antibodies and Vα/β T cell receptors that bind peptides in a diagonal orientation. For all figures, the light and heavy chain and the TCR Vα and Vβ chains are coloured purple, green, red, and cyan, respectively. The light and heavy chains of all antibodies or α/β chains were superimposed. However for clarity, only the peptide is shown (PDB code 1CU4, red; 1TJG, brown; 1F58, blue; 1ACY, green; 1NAK, yellow; 1SM3, magenta; 1GGI, cyan; 1CFN, orange; and 1CE1, black). In the middle panel, the poly-Gln peptide bound to MW1 (PDB code: 2OTU), and GP peptide bound to 13F6-1-2 are shown in red and yellow, respectively.
Figure 5. Binding analysis of 13F6-1-2 to the GP peptide epitope.
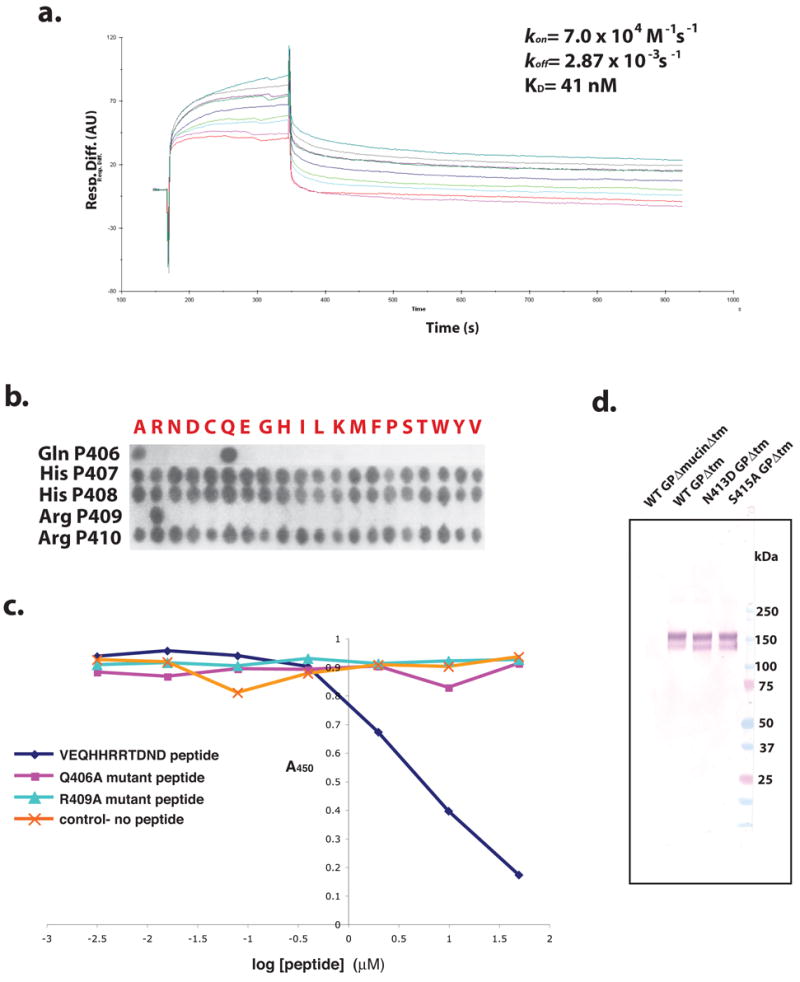
(a) 13F6-1-2 kinetic surface plasmon binding response to Ebola virus (Zaire-1995 strain). Five concentrations were injected at 30 μL/min and the kon and koff rates were calculated based on 1:1 Langmuir binding. (b) SPOTS membrane binding analysis of the 13F6-1-2 epitope. Peptides containing complete 20 amino acid replacement mutations to 13F6-1-2 peptide epitope residues Gln P406, His P407, His P408, Arg P409, and Arg P410 were synthesized onto a cellulose membrane. The membrane was incubated with IgG 13F6-1-2 and bound 13F6-1-2 was detected with an anti-mouse β-galactosidase-conjugated secondary antibody. (c) Competition ELISA of 13F6-1-2 and mutant epitope peptides. Microplates were coated with GP (transmembrane deletion) and incubated with IgG 13F6-1-2 and either the peptide epitope mimic or a mutant (Gln406Ala or Arg409Ala) epitope peptide. Bound 13F6-1-2 was detected by incubating with horse anti-mouse horseradish peroxidase secondary antibody. (d) Non-denaturing Western blot analysis of Asn413Asp and Ser415Ala GP mutants. WT GPΔmucinΔtm (wild-type GP with deletions of the mucin and transmembrane domains), WT GPΔtm, Asn413Asp GPΔtm, and Ser415Ala GPΔtm were probed for binding with IgG 13F6-1-2 antibody. WT GPΔmucinΔtm and WT GPΔtm were used as the negative and positive controls, respectively.
Antibody-peptide interactions
Nine of the eleven peptide residues (residues P404-P412) are in contact with the antibody paratope. Asn P413 and Asp P414 turn away from the antibody and form two crystal contact hydrogen bonds to symmetry-related Fab residues Asn L164 and Leu H170, respectively. The epitope mapping results (data not shown) agree with our structural model and confirm that the epitope residues 413 and 414 are not important for binding 13F6-1-2.
In total, five hydrogen bonds are formed between the Vλx-light chain and the GP peptide (Figure 4b and c). The amino group of Val P404 and the amide nitrogen of His P407 make main chain hydrogen bonds to the carbonyl oxygen of Ala L27 and the Oδ1 of Asp L92, respectively. The side chain of Gln P406 is anchored by three Vλx main chain hydrogen bonds to the main chain nitrogen and oxygen of Thr L32 and the carbonyl oxygen of Gly L91.
Fourteen hydrogen bonds are formed between the heavy chain and the GP epitope peptide (Figure 4b and c). The carbonyl oxygen of His P407 makes a hydrogen bond to the Tyr H100D Oη, and the amide nitrogen of Arg P409 forms a 3.0 Å hydrogen bond interaction to the carbonyl oxygen of His H100B. The guanidinyl group of Arg P409 is strongly anchored by five hydrogen bonds to the main chain oxygens of Ile H96, Ser H100A and Tyr H100C and the side chain Oδ1 of Asp33. The interaction of Asp H33 and the positively charged guanidinyl group of Arg P409 is likely a stabilizing salt bridge. Arg P410 only makes a main chain carbonyl hydrogen bond to the Oη of Tyr H50. The Thr P411 Oγ makes a tight hydrogen bond (2.7 Å) to the Asp H33 Oδ1. Arg H52A Nη2 and Nε make weaker hydrogen bonds (2.9 Å and 3.2 Å, respectively) to the main chain oxygen of Asp P412. The side chain of Asp P412 is stabilized by three strong hydrogen bonds to Ser H52 Oγ, and the main chain amide nitrogen atoms from Gly H53 and Gly H55.
Peptide binding mode
Peptide residues VEQHHRRTD (residues P404-P412) adopt an extended linear conformation which lies diagonally across the light and heavy chains in a groove which is ~28 Å long and 10 Å wide (Figure 4a). In anti-peptide antibody structures, the peptide typically binds at the interface between the light and heavy chains (Figure 4d). However, the peptide binding mode seen in 13F6-1-2 is rather unusual in that the peptide epitope lies diagonally across the light and heavy chains rather than at the light chain-heavy chain interface (Figure 4d). This orientation is reminiscent of some T cell receptors (TCR), in which the bound peptide binds diagonally across both the Vα and Vβ chains 34 (Figure 4d). To date, there are no other non-Vλx antibody complexes known to exhibit such an extended diagonal binding nature. In addition, peptides typically form somewhat more contacts to the heavy chain than to the light chain. 13F6-1-2 interactions are skewed more towards the heavy chain, which forms 73% of the buried surface area. By contrast, in Vλx MW1, the VH and VL domain interactions are shared about equally (Table 1).
Table 1.
Comparison of Peptide-Antibody Complexes
| PDB | Resolution (Å) | Peptide length/residues bound | Buried SA VH/VL- peptide (Å2) | Buried SA VL- peptide (Å2) | Buried SA VH- peptide (Å2) | Buried SA L1- peptide (Å2) | Buried SA L2- peptide (Å2) | Buried SA L3- peptide (Å2) | Buried SA H1- peptide (Å2) | Buried SA H2- peptide (Å2) | Buried SA H3- peptide (Å2) | Surface complementarity index (Sc) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2QHR 13F6-1-2 Vλx | 2.0 | 11/9 | 613 | 648 | 1176 | 348 | 0 | 377 | 133 | 533 | 706 | 0.78 |
| 2OTU MW1-peptide Vλx | 1.68 | 12/11 | 1687 | 885 | 855 | 553 | 0 | 358 | 467 | 90 | 430 | 0.76 |
| 1CU4 | 2.9 | 10/9 | 1428 | 596 | 919 | 345 | 55 | 259 | 427 | 342 | 264 | 0.75 |
| 1TJG | 2.0 | 7/6 | 1004 | 464 | 673 | 0 | 0 | 469 | 128 | 300 | 392 | 0.78 |
| 1F58 | 2.0 | 10/10 | 1408 | 725 | 852 | 438 | 0 | 364 | 185 | 205 | 511 | 0.79 |
| 1ACY | 3.0 | 10/7 | 1037 | 498 | 633 | 251 | 0 | 333 | 141 | 222 | 394 | 0.81 |
| 1NAK | 2.57 | 10/9 | 1074 | 553 | 623 | 225 | 171 | 230 | 267 | 0 | 410 | 0.78 |
| 1SM3 | 1.95 | 9/9 | 1031 | 444 | 757 | 160 | 106 | 235 | 150 | 95 | 64 | 0.75 |
| 1GGI | 2.8 | 9/7 | 1094 | 560 | 739 | 126 | 116 | 292 | 170 | 59 | 121 | 0.79 |
| 1CFN | 2.65 | 10/10 | 1007 | 501 | 602 | 166 | 103 | 251 | 336 | 57 | 228 | 0.56 |
| 1CE1 | 1.9 | 7/7 | 873 | 491 | 573 | 98 | 38 | 414 | 164 | 168 | 323 | 0.80 |
| Average | 2.3 | 9.5/8.5 | 1205 | 579 | 764 | 246 | 54 | 326 | 233 | 188 | 349 | 0.76 |
In 13F6-1-2, the GP peptide makes significant contacts to CDRs L1, L3, H1, H2, and H3, and ~1600 Å2 of antibody surface area is buried by peptide binding (Table 1). No contacts are formed to CDR L2 due to the diagonal orientation of the peptide. In the light chain, the peptide-binding groove is shallow and open, while in the heavy chain, the groove is more complementary to the peptide shape and contains a deep pocket to accommodate the side chain of Arg P409.
Insight into the mucin-like domain
The mucin-like domain of Ebola virus GP is heavily glycosylated with both N- and O-linked oligosaccharides. Residues encompassing the 13F6-1-2 epitope contain two potential glycosylation sites: a possible O-linked site at Thr P411 and a possible N-linked site at Asn P413. However, the NetOGlyc/NetNGlyc servers (http://www.cbs.dtu.dk/services) 35 do not predict a high glycosylation potential for either of these sites. In the crystal structure, the Thr P411 side chain is directed into a complementary-shaped site on the antibody paratope, and the restricted cavity of the antibody would prevent an O-glycosylated Thr P411 from properly binding 13F6-1-2. Asn P413 lies just outside of the antigen binding site and its side chain is pointed out into the solvent channel, so 13F6-1-2 could theoretically accommodate an oligosaccharide at this position. However, GP containing either of two point mutations which remove the N-X-S glycosylation motif (Asn413Asp and Ser415Ala) retains 13F6-1-2 binding (Figure 5d), suggesting that these residues or a glycan at this site are not involved in 13F6-1-2 binding. The crystal structure of 13F6-1-2 in complex with this extended, nonglycosylated region of the mucin-like domain suggests that there may be other similarly non-glycosylated stretches of extended structure on the surface of Ebola GP accessible to neutralizing antibodies.
Role of the Vλx-light and heavy chains in antigen binding
Most antibodies utilize residues in both the heavy and light chains for antigen binding and recognition. Our structural and functional data show that the Vλx-bearing 13F6-1-2 likely exploits both the heavy and light chains for recognition, but primarily the heavy chain for tight binding.
Only four peptide side chains interact with antibody residues: Gln P406 contacts the light chain, while Arg P409, Thr P411, and Asp P412 contact the heavy chain. Of these, Gln P406 and Arg P409 form the most extensive interactions to antibody (Figure 4b and c), and SPOTS membrane analysis and competition ELISAs of peptide mutants reveal that these two residues are important for binding. A complete amino acid replacement series mutagenesis of the 13F6-1-2 epitope by SPOTS analysis shows that positions 407, 408, and 410 can tolerate substitutions to any amino acid, but Arg P409 needs to be strictly conserved. Position 406 can only tolerate an alanine substitution (Figure 5b) by SPOTS binding; although a peptide containing a Gln406Ala mutation cannot compete with wild-type GP for 13F6-1-2 binding (Figure 5c).
Gln P406, the third residue in the epitope, makes three contacts to the light chain. Arg P409, the sixth residue in the epitope, makes five contacts to the heavy chain (Figure 4c). Interestingly, in another Vλx-bearing antibody, F28C4, NMR experiments have determined that the 3rd and 6th positions of the epitope (Gln 3 and Pro 6) are also important for recognition 22. Similarly, in the MW1-peptide complex, Gln 4 is found in the 3rd position of the epitope and makes substantial contact to the antibody. Hence, all Vλx-bearing antibodies studied require a glutamine residue at the 3rd position of the epitope for recognition. This is not surprising as all known Vλx–bearing antibodies contain very few somatic mutations and primarily utilize germline-encoded residues to mediate antigen binding (Figure 1), and binding of this glutamine in particular.
While the light chain plays a role in recognition, it is not thought to play a major role in tight binding. In 13F6-1-2, the heavy chain forms 73% of the buried surface area (Table 1). The heavy chain also forms one electrostatic interaction and thirteen hydrogen bonds to binding, while the light chain forms only five hydrogen bonds. The electrostatic salt bridge between heavy chain Asp H33 and Arg P409 likely dictates tight binding, as typical hydrogen bonds only provide 0.5-1.5 kcal/mol of binding energy 36. A simple calculation using Coulomb’s Law, by approximating a positive and negative point charge moving from infinity or an unbound state to 2.9 Å shows that this salt bridge may contribute a binding energy up to 28.6 kcal/mol (dielectric constant=4). This binding energy is more than sufficient to account for the observed 41 nanomolar affinity of 13F6-1-2 for GP.
Ebola virus vaccine development and prophylactic treatment
Although development of a vaccine against the Ebola virus is a high priority, prophylactic vaccination against a rare pathogen may not be economically feasible for much of the population. Development of immunotherapeutics would provide an immediate, valuable treatment option in event of viral exposure or release. However, there are several challenges to the development of efficacious antibody-based therapies for Ebola virus GP. Many survivors of Ebola virus infection display no or low titers of neutralizing antibody 37-40, perhaps because the disease course outcompetes the speed with which the immune system can generate virus-specific IgG. In addition, the virion-attached GP is heavily glycosylated and much of its surface may be masked from immune surveillance. Plus, those antibodies that are in survivor sera appear to preferentially recognize the secreted glycoprotein (sGP) rather than the virion-attached GP 15, even though these two proteins share the first 295 amino acids. This suggests that certain antibody epitopes may be better exposed on sGP, or that sGP competes for antibody binding as it outnumbers the virion-attached GP by approximately five-fold. Development of efficacious antibodies specific for virion-attached GP would allow more efficient immunotherapeutic targeting of virions and infected cells.
The 13F6-1-2 epitope may be a good target for vaccine and immunotherapeutic design, as it is immunodominant in a murine model and provides protection against viral challenge 12. In addition, the peptide epitope belonging to residues P404-P412 is bound in an extended conformation, and does not adopt any secondary structure (i.e. β-turns, helices, β-sheets) or posttranslational modifications that would be difficult to achieve in synthetic or recombinant systems.
When murine antibodies can be successfully humanized, they can provide clinical benefit 41. Fortunately, BLAST searches of 13F6-1-2 reveal that the human Vλ subgroup VIII light chain shares significant framework sequence identity to the rare Vλx light chain (~80%), with only 13 amino acid differences (Figure 6a). In addition, the unusual lengths of the CDR L1 and L2 regions are similar between 13F6-1-2 and the human Vλ (VIII) germline. Hence, human Vλ (VIII) could be used as a template for humanization of 13F6-1-2. Our 13F6-1-2 crystal structure will provide an excellent model to understand which interactions between the framework and the CDRs must be conserved to maintain antigen-binding affinity. From the 13 framework residues that differ between human Vλ (VIII) and 13F6-1-2, only three of these residues appear to stabilize the CDRs. Tyr L36, Asp L70, and Ser L105 hydrogen bond to key residues in the L3, L1, and L2 CDRs, respectively (Figure 6b) and may need to be maintained in any humanized molecule in order to conserve the stabilizing framework-CDR interactions necessary for peptide binding. We are currently evaluating the humanization potential of 13F6-1-2 as a post-exposure therapeutic against the Ebola virus.
Figure 6. Similarity of 13F6-1-2 Vλx and human germline Vλ subtype VIII light chains.
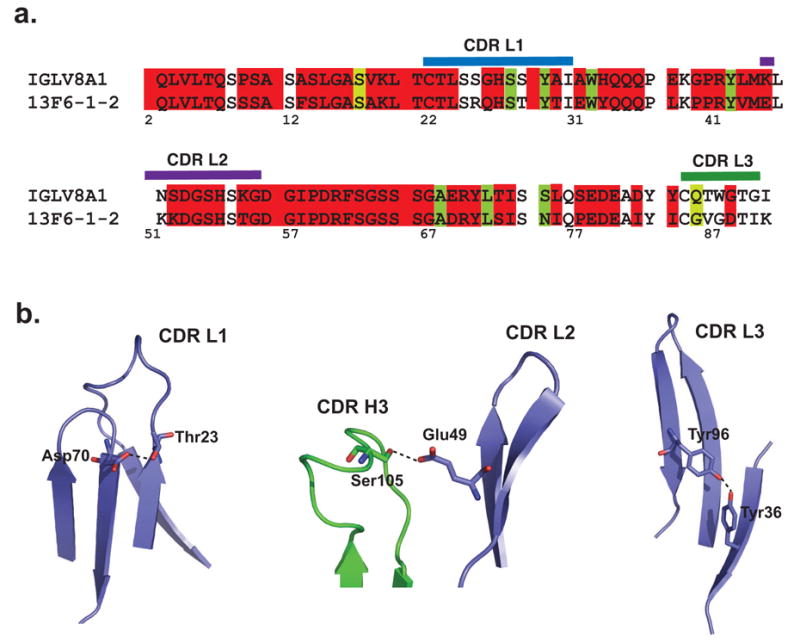
(a) Sequence alignment of the 13F6-1-2 Vλx light chain with human germline Vλ subtype VIII light chain (GenBank accession number L29806). (b) Three framework residues of 13F6-1-2 interact with the CDRs and may need to be maintained in humanization. Asp L70 hydrogen bonds to Thr L23 in CDR L1, Ser L105 hydrogen bonds to Glu L49 of CDR L2, and Tyr L36 interacts with Tyr L96 from CDR L3.
Materials and Methods
Fab 13F6-1-2 production, purification, and sequencing
BALB/c mice were vaccinated and boosted with packaged Venezuelan equine encephalitis virus replicons encoding the GP from the Mayinga isolate of Ebola virus Zaire 1976, as previously described 12. Spleen cells were harvested three days after the final injection and fused to P3X63Ag8.653 myeloma cells. Hybridoma screening was performed by ELISA and large scale production of the antibody was carried out in Integra Celline flasks in serum-free medium. MAb were purified from the supernatants on Protein G affinity columns and dialyzed in PBS. The 13F6-1-2 Fab fragments were prepared by 1% papain digestion at 37°C for one hour. Fab, Fc fragments and undigested IgG2a were separated by Protein A chromatography. Fab collected in the Protein A flow-through was further purified by size exclusion on a Superdex 200 10/300 GL column equilibrated with 10 mM Tris-HCl pH 7.5 and 150 mM NaCl. Sequencing of the variable regions was carried out as described in United States Patent #6,875,433.
Fab 13F6-1-2-peptide structure determination
Crystallization
An 11-residue peptide (VEQHHRRTDND) containing residues P404-P414 of the variable mucin-like C-terminus of GP1 was synthesized by the Center for Protein Sciences at The Scripps Research Institute. Fab 13F6-1-2 was concentrated to 15 mg/mL and pre-incubated with a five-fold molar excess of peptide (>95% purity) on ice for 30 minutes prior to crystallization by hanging drop vapour diffusion at 22°C. Crystals sufficient for data collection appeared directly from the sparse matrix screens overnight from Emerald Biosystems Wizard III condition #32 (16% (w/v) PEG 8000, 0.04 M potassium phosphate, and 20% (v/v) glycerol).
Data collection
In preparation for data collection at 100K, crystals of the Fab 13F6-1-2-GP peptide complex were gently soaked one minute in 20% (w/v) PEG 8000, 0.04 M potassium phosphate, and 25% (v/v) glycerol prior to flash cooling in liquid nitrogen. Data were measured at Beamline 8.2.2, Advanced Light Source (Lawrence Berkeley National Laboratory, Berkeley, CA) using an ADSC Q315 CCD detector. The data were indexed, integrated, and scaled with d*TREK (version 9.4) 42. Data collection statistics are presented in Table 2.
Table 2.
Data collection and refinement statistics
| Diffraction statistics | |
| Space group: | P212121 a= 68.2 Å, b=70.3 Å, c=142.1 Å, α=β=γ=90° |
| No. of observed reflections: | 188,364 |
| No. of unique reflections: | 46,873 |
| Resolution range: | 46.3-2.0 Å |
| Rsym (%)a: | 8.4 (52.3)b |
| Redundancy: | 4.0 (3.5)b |
| Completeness (%): | 99.7 (99.7) |
| Average I/σ(I): | 6.8 (1.7) |
| Refinement Statistics | |
| No. of total atoms: | 3,659 |
| No. of peptide atoms: | 98 |
| No. of waters: | 233 |
| Resolution range (Å): | 46.3-2.0 Å |
| Rcryst (%)c | 20.3 |
| Rfree (%)c | 24.5 |
| Overall B-factor (Å2) | 24.3 |
| Chain H | 20.9 |
| Chain L | 27.0 |
| Peptide | 26.0 |
| Water | 29.0 |
| r.m.s. deviation | |
| bond (Å) | 0.015 |
| angles (°) | 1.3 |
| dihedrals (°) | 11.2 |
| impropers (°) | 1.1 |
| Cross validated coordinate error | |
| Luzatti (Å) | 0.28 |
Rsym = ΣΣ ∣ I(k) - <I> ∣ / Σ I(k), where I(k) and <I> represent the diffraction intensity values of the individual measurements and the corresponding mean values. The summation is over all unique measurements.
Values given in parentheses refer to reflections in the outer resolution shell: 2.07-2.00 Å
, where Fobs and Fcalc are the observed and calculated structure factors, respectively. For Rfree the sum is extended over a subset of reflections (5%) excluded from all stages of refinement.
Structure determination and refinement
The structure of Fab 13F6-1-2-peptide complex was determined by molecular replacement using a murine IgG2a Fab fragment (PDB code: 1YEC) as the initial search model. The program CNS SOLVE (version 1.1) 43; 44 was used during all stages of structure determination and refinement. Data between 15 and 4 Å resolution were included in the cross rotation and translation functions in which a clear, unique solution was identified. Coordinates corresponding to this molecular replacement solution were subsequently subjected to a round of rigid body and torsion angle simulated annealing using all data, with no σ-cutoffs. The Rcryst and Rfree prior to the first round of manual rebuilding were 38.7% and 41.6%, respectively. The interactive computer graphics program Xfit 45 was used to rebuild the initial model to the correct Fab 13F6-1-2 sequence and alternated with rounds of torsion angle simulated annealing starting at 5000K 43; 46 using all data with no σ-cutoffs and bulk solvent correction. The progress of the refinement was monitored by reductions in Rcryst and Rfree. Difference Fourier maps calculated after changing the model to the correct sequence of Fab 13F6-1-2 revealed strong unambiguous positive electron density corresponding to the P404-P414 peptide. Water molecules were included into the model during the later rounds of refinement based on the presence of positive 3σ peaks in the σA-weighted Fo-Fc difference electron density maps and at least one hydrogen bond to a protein, peptide, or solvent atom. After the addition of water molecules and the peptide, alternating rounds of crystallographic conjugate gradient minimization refinement and model rebuilding were performed. The final model was refined in Phenix 47-49 with TLS refinement 50. Refinement statistics are reported in Table 2.
Structural superimpositions were performed using the FATCAT pairwise alignment server (http://fatcat.burnham.org/fatcat/) 51. Shape complementarity index and buried surface area were calculated using the program Sc 52 in the CCP4 suite and CNS-SOLVE 44, respectively.
Analysis of peptide and mutant peptide binding
Surface plasmon resonance binding analysis
The binding analysis was performed using a CM-5 sensor chip on a BIAcore3000 at room temperature at the USAMRIID Biosafety Level 4 laboratory (Frederick, MD). 6500 response units (RU) of sucrose gradient purified Ebola virus (Zaire-1995 strain) were immobilized using amide coupling chemistry to flow cell 1. BSA was immobilized by amide coupling in flow cell 2 and both flow cells were washed with NaOH to stabilize the surface. IgG 13F6-1-2 was diluted to 0 nM, 10 nM, 25 nM, 50 nM, 100 nM, and 500 nM. Each concentration was injected at 30 μL/min twice over flow cell 1 followed by flow cell 2 as a reference and data from flow cell 2 was subtracted out. The surface was regenerated with 10 mM glycine at pH 2.0. The final RU was monitored for any buildup or degradation of the surface. A mass transfer test was used by injecting 100 nM 13F6-1-2 at three different flow rates (5, 15, and 75 μL/min). The kon and koff values were calculated based on a 1:1 Langmuir binding curve.
SPOTS membrane peptide array
Peptides containing complete 20 amino acid replacement mutations to 13F6-1-2 epitope residues Gln P406, His P407, His P408, Arg P409, and Arg P410 (peptide sequences: QVExHHRRTDNDST, QVEQxHRRTDNDST, QVEQHxRRTDNDST, QVEQHHxRTDNDST, and QVEQHHRxTDNDST) were custom synthesized onto a cellulose membrane by Sigma-Genosys and used according to the manufacturer’s instruction. The membrane-immobilized peptides were rehydrated with 1X TBS pH 8.0 (Tris-buffered saline) and incubated at room temperature overnight with T-TBS blocking buffer (TBS, 0.05% (v/v) Tween-20, 5% (w/v) sucrose, and 10% (v/v) Genosys concentrated blocking buffer). The membranes were washed three times with T-TBS before incubation with 20 mL of IgG 13F6-1-2 at a concentration of 5 μg/mL for three hours at room temperature. Unbound antibody was removed by three washes of T-TBS and bound 13F6-1-2 was detected using an anti-mouse β-galactosidase-conjugated secondary antibody. After two hours incubation at room temperature, the membranes were washed twice with T-TBS and twice with PBS. The bound antibody was visualized using a signal development solution (potassium ferricyanide, 5-bromo-4-chloro-3-indoyl-β-D-galactopyranoside, magnesium chloride in PBS).
Competition ELISA
Peptides containing alanine point mutations were synthesized by AnaSpec (peptide sequences: VEAHHRRTDND and VEQHHARTDND). Corning Costar 96-well microplates were immobilized with recombinant, soluble full-length Ebola virus GPΔtm (lacking the transmembrane domain) (0.05 mL, 10 μg/mL) overnight at 4°C. Microplates were blocked with 0.1 mL of 3% (w/v) BSA for one hour at 22°C. 25 μL each of IgG 13F6-1-2 antibody (1.5 μg/mL final concentration in PBS with 1% (w/v) BSA) and peptide epitope mimic or mutant peptide epitope (0 to 50 μM final concentration in PBS with 1% (w/v) bovine serum albumin (BSA) were added to the microplates and incubated for two hours at 22°C. After the plates were washed with PBS with 0.05% (w/v) Tween-20, bound IgG 13F6-1-2 was detected by incubation with horse anti-mouse horseradish peroxidase-conjugated secondary antibody (0.05 mL, 1:2500 dilution) for one hour at 22°C. Each well was then washed ten times with PBS containing 0.05% (w/v) Tween-20. The product was developed using the Pierce TMB substrate kit according to the manufacturer’s protocol. Colour development was read immediately in a Molecular Devices VersaMax microplate reader at 450 nm.
Analysis of the Asn413 glycan sequon
Asn413Asp and Ser415Ala GP mutations were generated using the QuikChange II site-directed mutagenesis kit (Stratagene), according to manufacturer’s protocol. Wild-type (WT) GPΔtm, WT GPΔmucinΔtm, Asn413Asp GPΔtm, and Ser415Ala GPΔtm DNA were transiently transfected into HEK293T cells using FuGene6 in 6-well culture plates. Supernatants were harvested 4 days post-transfection. 12 μL of supernatant were loaded in each lane and analyzed by non-denaturing Western blot using mouse 13F6-1-2 monoclonal primary antibody (2 μg/mL), followed by goat anti-mouse IgG peroxidase-conjugated secondary antibody (1:1000 dilution). The signal was visualized using Sigma FAST BCIP/NBT.
Conclusions
In summary, we have determined the sequence and the structure of the anti-Ebola virus Fab 13F6-1-2 in complex with its GP epitope to 2.0 Å resolution. 13F6-1-2 utilizes a rare Vλx light chain, which displays several unusual structural features. In comparison with one other available structure of a Vλx-bearing light chain, we propose a separate class of canonical hypervariable loops for Vλx antibodies, distinct in structure from those of other Vλ and Vκ light chains. The Ebola GP peptide epitope is bound in a diagonal orientation, in which the first four peptide residues are bound by the light chain and the last five are anchored by the heavy chain. Light chain contacts to the peptide are mediated only through germline-encoded residues, yet are critical for antigen recognition. The crystal structure of the 13F6-1-2-GP peptide complex thus adds to our knowledge of antibody-antigen recognition and provides a template for development of vaccines against the Ebola virus and for humanization of the mAb as a post-exposure therapeutic.
Acknowledgments
The views of the authors are not necessarily endorsed by the U.S. Army, U.S. Department of Defense, or The Scripps Research Institute. The authors thank Dr. Corey Ralston and staff at ALS BL8.2.2 for beamline support, Brian Kearney for BIAcore/SPR binding data, and Dr. Robyn Stanfield for critical reading and comments. The authors would also like to acknowledge the insightful comments of the reviewers. J.E.L. is supported by a research fellowship from the Canadian Institutes for Health Research (CIHR). E.O.S. is supported by a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund and grants AI067927 and AI053423 from the National Institutes of Health. MKH and AK are funded by operating grants from the Defense Threat Reduction Agency #1749000 and USAMRIID #FX00604RDB. This is manuscript #18991 from The Scripps Research Institute.
Abbreviations
- CDR
complementarity determining region
- mAb
monoclonal antibody
- GP
glycoprotein
- VH
variable heavy chain
- VL
variable light chain
- TCR
T cell receptors
- H-bond
hydrogen bond
- TBS
Tris buffered saline
- MBS
membrane blocking solution
Footnotes
Nucleotide and protein accession codes- Nucleotide and protein sequences for IgG2a 13F6-1-2 light and heavy chains were published in U.S. Patent #6,875,433 and are accessible through the U.S. Patent and Trademark Office website at http://www.uspto.gov
Protein data bank accession code- The coordinates and structure factors for the Fab 13F6-1-2-peptide complex have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank 53 (http://www.rcsb.org/pdb) with PDB accession code 2QHR.
Competing interests- The authors have declared that no competing interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peters CJ, Sanchez A, Rollin PE, Ksiazek TG, Murphy GA. Filoviridae: Marburg and Ebola viruses. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. 3. Vol. 1. Lippincott-Raven Press, Inc.; Philadelphia, PA: 1996. pp. 1161–1176. [Google Scholar]
- 2.World Health Organization. Ebola haemorrhagic fever 2004 [Google Scholar]
- 3.Johnson KM, Lange JV, Webb PA, Murphy FA. Isolation and characterization of a new virus causing acute hemorrhagic fever in Zaire. Lancet. 1977;1:569–571. doi: 10.1016/s0140-6736(77)92000-1. [DOI] [PubMed] [Google Scholar]
- 4.Khan AS, Tshioko FK, Heymann DL, Le Guenno B, Nabeth P, Kerstiens B, Fleerackers Y, Kilmarx PH, Rodier GR, Nkuku O, Rollin PE, Sanchez A, Zaki SR, Swanepoel R, Tomori O, Nichol ST, Peters CJ, Muyembe-Tamfum JJ, Ksiazek TG. The reemergence of Ebola hemorrhagic fever, Democratic Republic of the Congo, 1995. Commission de Lutte contre les Epidemies a Kikwit. J Infect Dis. 1999;179(Suppl 1):S76–86. doi: 10.1086/514306. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez A, Yang ZY, Xu L, Nabel GJ, Crews T, Peters CJ. Biochemical analysis of the secreted and virion glycoproteins of Ebola virus. J Virol. 1998;72:6442–7. doi: 10.1128/jvi.72.8.6442-6447.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feldmann H, Klenk HD, Sanchez A. Molecular biology and evolution of filoviruses. Arch Virol Suppl. 1993;7:81–100. doi: 10.1007/978-3-7091-9300-6_8. [DOI] [PubMed] [Google Scholar]
- 7.Takada A, Robison C, Goto H, Sanchez A, Murti KG, Whitt MA, Kawaoka Y. A system for functional analysis of Ebola virus glycoprotein. Proc Natl Acad Sci U S A. 1997;94:14764–9. doi: 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Volchkov VE, Volchkova VA, Slenczka W, Klenk HD, Feldmann H. Release of viral glycoproteins during Ebola virus infection. Virology. 1998;245:110–9. doi: 10.1006/viro.1998.9143. [DOI] [PubMed] [Google Scholar]
- 9.Wool-Lewis RJ, Bates P. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J Virol. 1998;72:3155–60. doi: 10.1128/jvi.72.4.3155-3160.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maruyama T, Rodriguez LL, Jahrling PB, Sanchez A, Khan AS, Nichol ST, Peters CJ, Parren PW, Burton DR. Ebola virus can be effectively neutralized by antibody produced in natural human infection. J Virol. 1999;73:6024–30. doi: 10.1128/jvi.73.7.6024-6030.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takada A, Watanabe S, Ito H, Okazaki K, Kida H, Kawaoka Y. Downregulation of beta1 integrins by Ebola virus glycoprotein: implication for virus entry. Virology. 2000;278:20–6. doi: 10.1006/viro.2000.0601. [DOI] [PubMed] [Google Scholar]
- 12.Wilson JA, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn AL, Hart MK. Epitopes involved in antibody-mediated protection from Ebola virus. Science. 2000;287:1664–6. doi: 10.1126/science.287.5458.1664. [DOI] [PubMed] [Google Scholar]
- 13.Oswald WB, Geisbert TW, Davis KJ, Geisbert JB, Sullivan NJ, Jahrling PB, Parren PW, Burton DR. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLoS Pathog. 2007;3:e9. doi: 10.1371/journal.ppat.0030009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Druar C, Saini SS, Cossitt MA, Yu F, Qiu X, Geisbert TW, Jones S, Jahrling PB, Stewart DI, Wiersma EJ. Analysis of the expressed heavy chain variable-region genes of Macaca fascicularis and isolation of monoclonal antibodies specific for the Ebola virus soluble glycoprotein. Immunogenet. 2005:1–9. doi: 10.1007/s00251-005-0047-4. [DOI] [PubMed] [Google Scholar]
- 15.Maruyama T, Parren PW, Sanchez A, Rensink I, Rodriguez LL, Khan AS, Peters CJ, Burton DR. Recombinant human monoclonal antibodies to Ebola virus. J Infect Dis. 1999;179(Suppl 1):S235–9. doi: 10.1086/514280. [DOI] [PubMed] [Google Scholar]
- 16.Yang ZY, Duckers HJ, Sullivan NJ, Sanchez A, Nabel EG, Nabel GJ. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat Med. 2000;6:886–9. doi: 10.1038/78645. [DOI] [PubMed] [Google Scholar]
- 17.Dildrop R, Gause A, Muller W, Rajewsky K. A new V gene expressed in lambda-2 light chains of the mouse. Eur J Immunol. 1987;17:731–4. doi: 10.1002/eji.1830170525. [DOI] [PubMed] [Google Scholar]
- 18.Sanchez P, Cazenave PA. A new variable region in mouse immunoglobulin lambda light chains. J Exp Med. 1987;166:265–70. doi: 10.1084/jem.166.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez P, Marche PN, Le Guern C, Cazenave PA. Structure of a third murine immunoglobulin lambda light chain variable region that is expressed in laboratory mice. Proc Natl Acad Sci U S A. 1987;84:9185–8. doi: 10.1073/pnas.84.24.9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanchez P, Marche PN, Rueff-Juy D, Cazenave PA. Mouse V lambda x gene sequence generates no junctional diversity and is conserved in mammalian species. J Immunol. 1990;144:2816–20. [PubMed] [Google Scholar]
- 21.Galin FS, Maier CC, Zhou SR, Whitaker JN, Blalock JE. Murine V lambda x and V lambda x-containing antibodies bind human myelin basic protein. J Clin Invest. 1996;97:486–92. doi: 10.1172/JCI118439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maier CC, Galin FS, Jarpe MA, Jackson P, Krishna NR, Gautam AM, Zhou SR, Whitaker JN, Blalock JE. A V lambda x-bearing monoclonal antibody with similar specificity and sequence to encephalitogenic T cell receptors. J Immunol. 1994;153:1132–40. [PubMed] [Google Scholar]
- 23.Li P, Huey-Tubman KE, Gao T, Li X, West AP, Jr, Bennett MJ, Bjorkman PJ. The structure of a polyQ-anti-polyQ complex reveals binding according to a linear lattice model. Nat Struct Mol Biol. 2007;14:381–7. doi: 10.1038/nsmb1234. [DOI] [PubMed] [Google Scholar]
- 24.Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol. 1997;273:927–48. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]
- 25.Stanfield RL, Zemla A, Wilson IA, Rupp B. Antibody elbow angles are influenced by their light chain class. J Mol Biol. 2006;357:1566–74. doi: 10.1016/j.jmb.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 26.Huang M, Syed R, Stura EA, Stone MJ, Stefanko RS, Ruf W, Edgington TS, Wilson IA. The mechanism of an inhibitory antibody on TF-initiated blood coagulation revealed by the crystal structures of human tissue factor, Fab 5G9 and TF.G9 complex. J Mol Biol. 1998;275:873–94. doi: 10.1006/jmbi.1997.1512. [DOI] [PubMed] [Google Scholar]
- 27.Stanfield RL, Takimoto-Kamimura M, Rini JM, Profy AT, Wilson IA. Major antigen-induced domain rearrangements in an antibody. Structure. 1993;1:83–93. doi: 10.1016/0969-2126(93)90024-b. [DOI] [PubMed] [Google Scholar]
- 28.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196:901–17. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 29.Martin AC, Thornton JM. Structural families in loops of homologous proteins: automatic classification, modelling and application to antibodies. J Mol Biol. 1996;263:800–15. doi: 10.1006/jmbi.1996.0617. [DOI] [PubMed] [Google Scholar]
- 30.Wu S, Cygler M. Conformation of complementarity determining region L1 loop in murine IgG lambda light chain extends the repertoire of canonical forms. J Mol Biol. 1993;229:597–601. doi: 10.1006/jmbi.1993.1065. [DOI] [PubMed] [Google Scholar]
- 31.Tomlinson IM, Cox JPL, Gherardi E, Lesk AM, Chothia C. The structural repertoire of the human V kappa domain. EMBO J. 1995;14:4628–38. doi: 10.1002/j.1460-2075.1995.tb00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanfield RL, Gorny MK, Williams C, Zolla-Pazner S, Wilson IA. Structural rationale for the broad neutralization of HIV-1 by human monoclonal antibody 447-52D. Structure. 2004;12:193–204. doi: 10.1016/j.str.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Stanfield RL, Gorny MK, Zolla-Pazner S, Wilson IA. Crystal structures of human immunodeficiency virus type 1 (HIV-1) neutralizing antibody 2219 in complex with three different V3 peptides reveal a new binding mode for HIV-1 cross-reactivity. J Virol. 2006;80:6093–105. doi: 10.1128/JVI.00205-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–66. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 35.Gupta R, Jung E, Brunak S. Prediction of N-linked glycosylation sites in human proteins. in preparation. [Google Scholar]
- 36.Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, Blow DM, Brick P, Carter P, Waye MM, Winter G. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314:235–8. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 37.Centers for Disease Control and Prevention. Outbreak of Ebola Viral Hemorrhagic Fever - Zaire. Morbid Mortal Wkly Rep. 1995;44:381–382. [PubMed] [Google Scholar]
- 38.Centers for Disease Control and Prevention. Update: outbreak of Ebola Viral Hemorrhagic Fever. Morbid Mortal Wkly Rep. 1995;44:399. [PubMed] [Google Scholar]
- 39.Ksiazek TG, Rollin PE, Williams AJ, Bressler DS, Martin ML, Swanepoel R, Burt FJ, Leman PA, Khan AS, Rowe AK, Mukunu R, Sanchez A, Peters CJ. Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J Infect Dis. 1999;179(Suppl 1):S177–87. doi: 10.1086/514321. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez A, Khan AS, Zaki SR, Nabel GJ, Ksiazek TG, Peters CJ. Filoviridae: Marburg and Ebola viruses. In: Knipe DM, Howley PM, editors. Fields Virology. Lippincott, Williams, and Wilkins; Philadelphia: 2001. pp. 1279–1304. [Google Scholar]
- 41.Tsurushita N, Hinton PR, Kumar S. Design of humanized antibodies: From anti-Tac to Zenapax. Methods. 2005;36:69–83. doi: 10.1016/j.ymeth.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 42.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr. 1999;D55:1718–25. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 43.Adams PD, Pannu NS, Read RJ, Brünger AT. Cross-validated maximum likelihood enhances crystallographic simulated annealing refinement. Proc Natl Acad Sci U S A. 1997;94:5018–23. doi: 10.1073/pnas.94.10.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 45.McRee DM. Practical Protein Crystallography. Academic Press; San Diego: 1993. [Google Scholar]
- 46.Brunger AT, Krukowski A, Erickson JW. Slow-cooling protocols for crystallographic refinement by simulated annealing. Acta Crystallogr. 1990;A47:195–204. doi: 10.1107/s0108767390002355. [DOI] [PubMed] [Google Scholar]
- 47.Adams PD, Gopal K, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Pai RK, Read RJ, Romo TD, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC. Recent developments in the PHENIX software for automated crystallographic structure determination. J Sync Rad. 2004;11:53–5. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]
- 48.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. 2002;D58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 49.Afonine PD, Grosse-Kunstleve RW, Adams PD. The Phenix refinement framework. CCP4 Newsletter. 2005 July; Contribution 8. [Google Scholar]
- 50.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. 2001;D57:122–33. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 51.Ye Y, Godzik A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics. 2003;19(Suppl 2):ii246–55. doi: 10.1093/bioinformatics/btg1086. [DOI] [PubMed] [Google Scholar]
- 52.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–50. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 53.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DeLano WL. The PyMol Molecular Graphics System 2002 [Google Scholar]