

Abstract
By tethering intermediate filaments (IFs) to sites of intercellular adhesion, desmosomes facilitate formation of a supercellular scaffold that imparts mechanical strength to a tissue. However, the role IF–membrane attachments play in strengthening adhesion has not been directly examined. To address this question, we generated Tet-On A431 cells inducibly expressing a desmoplakin (DP) mutant lacking the rod and IF-binding domains (DPNTP). DPNTP localized to the plasma membrane and led to dissociation of IFs from the junctional plaque, without altering total or cell surface distribution of adherens junction or desmosomal proteins. However, a specific decrease in the detergent-insoluble pool of desmoglein suggested a reduced association with the IF cytoskeleton. DPNTP-expressing cell aggregates in suspension or substrate-released cell sheets readily dissociated when subjected to mechanical stress whereas controls remained largely intact. Dissociation occurred without lactate dehydrogenase release, suggesting that loss of tissue integrity was due to reduced adhesion rather than increased cytolysis. JD-1 cells from a patient with a DP COOH-terminal truncation were also more weakly adherent compared with normal keratinocytes. When used in combination with DPNTP, latrunculin A, which disassembles actin filaments and disrupts adherens junctions, led to dissociation up to an order of magnitude greater than either treatment alone. These data provide direct in vitro evidence that IF–membrane attachments regulate adhesive strength and suggest furthermore that actin- and IF-based junctions act synergistically to strengthen adhesion.
Keywords: desmosome; desmoplakin; intermediate filaments; cadherins; adherens junction
Introduction
Cytoskeletal–cell membrane attachments are critical for establishing the proper relationships between cells during tissue morphogenesis and for maintaining the integrity of tissues during differentiation. In vertebrates, intermediate filaments (IFs)* provide the tensile strength required for tissue integrity (Coulombe and Omary, 2002). In complex epithelia and other tissues such as heart that experience mechanical stress, the association of IFs with intercellular adhesive junctions known as desmosomes generates a stress-resistant scaffolding that integrates all of the cells within the tissue (Green and Gaudry, 2000; McMillan and Shimizu, 2001). Perturbation of either the IF cytoskeleton or its desmosome anchor compromises tissue integrity (Fuchs and Cleveland, 1998; McMillan and Shimizu, 2001); however, the respective contributions and molecular mechanisms by which these integrated components serve their mechanical roles have not been fully elucidated. Specifically, the question of whether IFs strengthen desmosomal cadherin-mediated adhesion, much in the way actin has been proposed to strengthen classical cadherin-mediated adhesion, has not been directly addressed.
Diseases targeting IFs as well as proteins that tether IFs to desmosomes demonstrate the importance of the desmosome–IF scaffolding. On one hand, point mutations that interfere with the assembly of IFs into networks lead to epidermolysis bullosa simplex, which is characterized by cell fragility and cytolysis in response to mild trauma (Epstein, 1992; Fuchs and Coulombe, 1992; McLean and Lane, 1995). On the other hand, patients with mutations in desmosomal plaque components involved in IF tethering also exhibit skin fragility, accompanied by retraction of IFs from the cell surfaces and perinuclear accumulation (McMillan and Shimizu, 2001). Histological observations also show widened intercellular spaces, raising the possibility that such mutations might cause an adhesive defect, but making such a conclusion is complicated by possible reductions in desmosome number (McMillan and Shimizu, 2001).
Desmosomes are highly organized structures composed of members from three families, the cadherins, armadillo proteins, and plakins (for reviews see Schmidt et al., 1994; Garrod et al., 1996; Burdett, 1998; Kowalczyk et al., 1999a; Green and Gaudry, 2000). The desmosomal cadherins, subdivided into the desmocollins and desmogleins, form the adhesive core of the desmosome (Koch and Franke, 1994; Angst et al., 2001). The cadherins interact, in turn, with members of the armadillo family, including plakoglobin and, in certain cases, the plakophilins (Gelderloos et al., 1997; Hatzfeld, 1999). The latter comprises a subclass of armadillo proteins related to the classic cadherin-associated protein p120ctn (Hatzfeld, 1999). Armadillo family members bind to desmoplakin (DP) and possibly other members of the plakin family of cytolinkers, which also includes plectin, envoplakin, and periplakin (Ruhrberg and Watt, 1997; Fuchs and Yang, 1999; Leung et al., 2001). DP is an obligate desmosomal component that closes the link by mediating interactions with the IF cytoskeleton (Green and Bornslaeger, 1999). The submembranous desmosomal plaque is further bolstered by interactions of a lateral nature among armadillo family members from different subclasses, between DP and the cadherins and possibly between IFs and armadillo family members (Green and Gaudry, 2000).
DP plays an essential role in anchoring IFs to the membrane through its COOH-terminal plakin repeat domain (Stappenbeck and Green, 1992; Kouklis et al., 1994). DP's role is underscored by the existence of a dominantly inherited DP haploinsufficiency (Armstrong et al., 1999) as well as recessively inherited truncations within the COOH-terminal IF-binding domain of DP, which lead to striate palmoplantar keratoderma and skin, hair, and cardiac defects, respectively (Norgett et al., 2000). Genetically engineered mice have provided additional insight into the fundamental importance of DP. DP-null mice are unable to proceed past day 6.5 of embryonic development due to defects of the embryonic endoderm and failure of the egg cylinder to elongate (Gallicano et al., 1998). Chimeric morulae expressing DP in extraembryonic tissue survived somewhat longer, until E10.5–E12.5, exhibiting defects in heart, neuroepithelium, and epidermis and finally dying as a result of defects in the microvascular system (Gallicano et al., 2001). Most recently, conditional knockouts in which DP is specifically ablated in the epidermis provided compelling data that this molecule plays a key role in epidermal sheet formation (Vasioukhin et al., 2001). However, keratinocytes derived from conditional null animals have a greatly reduced number of desmosomal junctions, precluding a determination of whether severing the IF–desmosome connection, while still leaving the remaining plaque intact, affects cell integrity versus adhesive strength.
DP-null keratinocytes also displayed abnormalities in microfilament organization, suggesting that the proper maturation of the cortical actin cytoskeleton is dependent on the presence of desmosomes (Vasioukhin et al., 2001). Other studies have suggested that the reverse is also true; that is, desmosome assembly is dependent on the prior initiation of adhesion through the classic cadherin system (Gumbiner et al., 1988; Lewis et al., 1997), which triggers assembly of actin and reorganization of the cortical actin cytoskeleton (Vasioukhin et al., 2000; Vasioukhin and Fuchs, 2001; Kovacs et al., 2002). In addition, studies have reported that loss or mutation of desmosomal plaque constituents can result in the intermingling of adherens junction and desmosomal components (Bornslaeger et al., 1996; Ruiz et al., 1996). The observed interdependence of these junction–cytoskeleton systems raises the question of their relative contributions to adhesive strength.
To address these questions and to specifically test the role of the IF connection in cellular integrity and adhesion, we developed Tet-On A431 cell lines expressing a dominant-negative DP polypeptide (DPNTP) that uncouples IFs from the desmosome while leaving junctional plaques intact (Bornslaeger et al., 1996). Cell aggregates in hanging drop culture as well as epithelial cell sheets generated from cells expressing this polypeptide both exhibited increased dissociation without evidence of cytolysis when subjected to mild mechanical stress. In addition, time-lapse imaging analysis revealed that dissociation of DPNTP-expressing cells occurred more rapidly and with less cell distension compared with control cells. This adhesive defect occurred without alteration of the cell surface expression of desmosomal or classic cadherins or effect on E-cadherin–dependent adhesion in capillary shear flow assays. However, the adhesive defect was accompanied by an approximately twofold decrease in desmoglein 2 (Dsg 2) present in the detergent-insoluble pool, suggesting that desmosomal cadherins are specifically affected by loss of cytoskeletal attachment. Latrunculin A (LtnA), which interferes with the cortical actin cytoskeleton and associated adherens junctions, decreased adhesive strength as well, but importantly, DPNTP expression together with LtnA treatment resulted in an almost complete dissociation of the cell sheet. Keratinocytes derived from patients with a COOH-terminal truncation of DP also exhibit an adhesive defect, supporting the physiological relevance of these findings. Together these data provide the first direct in vitro evidence for a specific role of IF attachment in cell–cell adhesive strength.
Results
DPNTP expression leads to uncoupling of IFs from cell junctions without altering cell surface expression of desmosomal or classic cadherins
Previous studies demonstrated that ectopic expression of a DP deletion mutant, DPNTP, which retains plakoglobin and plakophilin binding sites but lacks the central rod and COOH-terminal IF-binding domains (Bornslaeger et al., 1996) (Fig. 1 A), results in uncoupling of IFs from the plasma membrane while leaving the IF network intact. To further study the consequences of severing the IF–desmosome connection without potential complications arising from clonal variability, tetracycline-inducible cell lines were generated (Tet-On; CLONTECH Laboratories, Inc.). Single stable (SS) A431 clones expressing the tetracycline-dependent transactivator were first generated and then selected for optimum morphology coupled with the ability to robustly induce expression of a luciferase reporter construct in a tightly regulated fashion. A431/SS clones were stably transfected with an expression cassette driven by a tetracycline response element containing a coding sequence for a COOH-terminally FLAG-tagged DPNTP. Double stable clones were obtained by puromycin selection and induction of DPNTP expression was scored by Western blot analysis. Of the stable clones identified, two clones, A431/C2 and G4, were characterized in detail. These clones behaved similarly in all assays used in this study but occasionally differed in the magnitude of response (e.g., see Fig. 5 B). The observed differences might be explained by the fact that the G4 clone tended to exhibit a low level of leaky expression of DPNTP and, in addition, exhibited a lower level of E-cadherin expression compared with the C2 line. Data for the C2 line are shown throughout, and where instructive, we have included and discussed representative data for the G4 line.
Figure 1.

Characterization of inducible A431 cell lines expressing DPNTP. (A) Comparison of DPNTP and human JD-1 deletion mutant DP to wild-type DP. DPNTP encodes the NH2-terminal 584 amino acids of DP containing binding sites for both plakoglobin and the plakophilins. The JD-1 mutation removes the C subdomain and includes 18 amino acids not found in wild-type DP. (B) Dose response of DPNTP expression. A431/C2 cells were induced for 48 h with increasing concentrations of Dox and then analyzed by Western blotting using antibodies directed against the NH2 terminus of DP. (C) Western blot analysis of junctional proteins in Dox-treated and untreated A431/C2 cells. Whole cell lysates were analyzed using antibodies directed against Dsg 2, E-cadherin, α-catenin, β-catenin, plakoglobin, and DP/DPNTP. Immunoblotting for keratin 18 was used as a loading control in both B and C. Note that the levels of desmosomal and classic cadherins and catenins are comparable in DPNTP-expressing and control cells.
Figure 5.
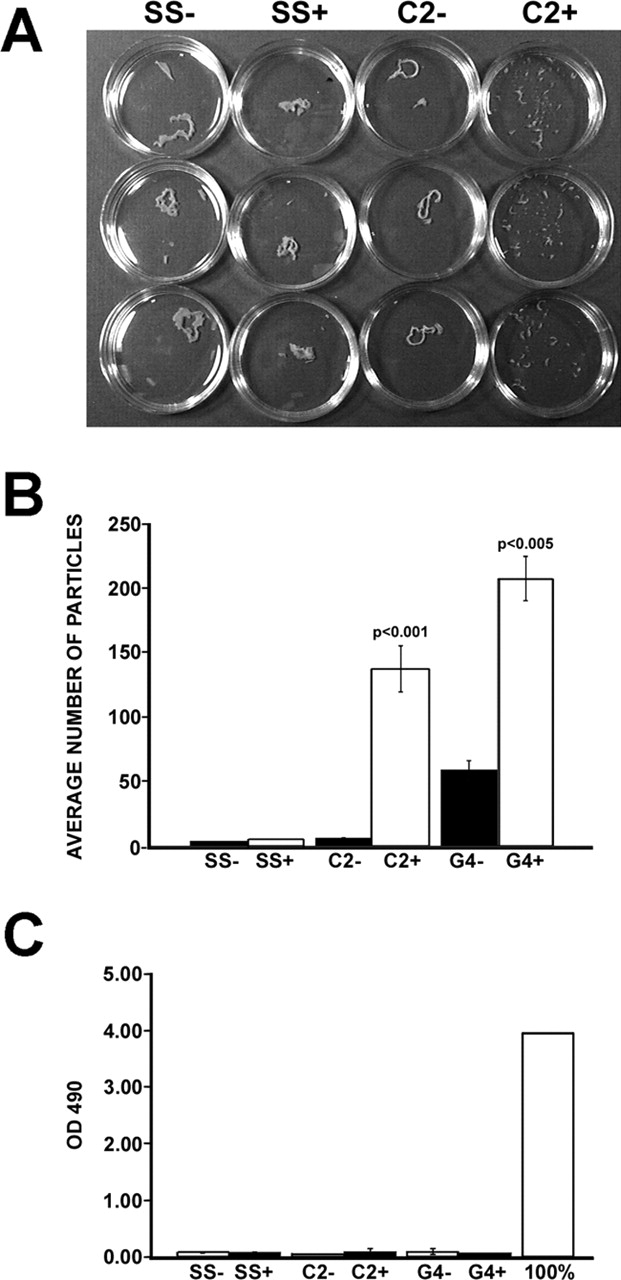
DPNTP weakens adhesion in epithelial sheets. (A) Dox-treated and untreated monolayers, plated in triplicate, were separated from culture dishes via incubation with dispase. Monolayers were transferred to 15-ml conical tubes containing 5 ml of DPBS. After 50 inversions, the degree of fragmentation of the monolayer was observed. (B) The dissociation assay was quantified by counting the number of total particles. Dox-treated A431/C2 and G4 monolayers showed increased dissociation relative to uninduced and A431/SS monolayers (t test, P < 0.001 and P < 0.005, respectively). Each bar represents the SEM from three independent experiments in which each condition was tested in triplicate. (C) LDH release was not increased despite increased fragmentation of monolayers expressing DPNTP. Values are compared with controls obtained from a mechanically lysed cell population.
Northern and Western blot analysis showed that in the absence of doxycycline (Dox), a tetracycline analogue, DPNTP mRNA and protein were undetectable in the A431/C2 cell line (unpublished data). Incubation of cells for 24–48 h with 2 μg/ml of Dox led to robust expression of DPNTP mRNA, and protein could be detected as early as 12 h after induction (Gaudry et al., 2001). Maximum levels of DPNTP expression were observed at Dox concentrations between 2 and 6 μg/ml (Fig. 1 B) and reached levels comparable to endogenous DP expression. Levels of endogenous DP decreased up to twofold upon expression of DPNTP, consistent with our previous observations (Fig. 1 B) (Bornslaeger et al., 1996).
To examine whether DPNTP affected the expression of other junctional proteins, immunoblot analysis was performed using antibodies against the desmosomal molecules DP, Dsg 2, desmocollin 2, and plakoglobin and the adherens junction proteins E-cadherin, β-catenin, and α-catenin (Fig. 1 C; unpublished data). No detectable changes in expression of any of the desmosomal components, besides DP, or of the adherens junction components were observed. Similar data were obtained for the G4 line (unpublished data). In addition, no discernible difference in total classic or desmosomal cadherin turnover was observed over a period of 2 d in 10 μg/ml cycloheximide to block protein synthesis (unpublished data).
In the absence of Dox, endogenous DP was localized at cell borders in a punctate pattern typical of parental A431 cells (Fig. 2). Using an antibody directed against the FLAG epitope on DPNTP, cell border staining colocalizing with endogenous desmosomes was rarely detected in the C2 line and it was only occasionally detected in the leaky G4 line. Staining for IFs revealed an intact network extending out to and anchoring at desmosomes at sites of cell–cell contact. After Dox treatment, DPNTP was localized at cell borders in a punctate pattern, whereas endogenous DP largely disappeared from cell borders. IF bundles were dramatically uncoupled from membrane sites where DPNTP was localized, although a few plasma membrane sites still retained some endogenous DP with associated IF bundles.
Figure 2.

DPNTP localizes to intercellular contacts in a punctate pattern and uncouples IFs from the cell surface. A431/C2 cells were induced for 24 h in 2 μg/ml Dox and either immunostained using antibodies directed against endogenous DP (DP2.15) or the FLAG epitope (poly-FLAG) on the COOH terminus of DPNTP (A and B), or antibodies directed against DP/DPNTP (NW161) and keratins (KS-B17.2) (C and D). Dox induction led to the accumulation of DPNTP in a junctional distribution at cell–cell borders accompanied by loss of endogenous DP and detachment of K8/18-containing IF bundles.
Our results described above indicated that total expression levels of desmosomal and classic cadherins did not change in DPNTP-expressing cells. However, because previous observations showed that expression of DPNTP can cause intermixing of E-cadherin and Dsg 2 (Bornslaeger et al., 1996), we sought to ensure that cadherins were properly delivered to the plasma membrane and present in a normal distribution. We compared the steady-state expression level and distribution of desmosomal and classic cadherins on the cell surface in uninduced and induced cells by labeling cell surface proteins with a membrane-impermeable biotin reagent. Levels of biotinylated Dsg 2 and E-cadherin from induced and uninduced A431/SS and C2 cells were compared after precipitation with streptavidin-conjugated agarose beads. Cell surface levels of both cadherins remained the same after DPNTP expression (Fig. 3 A). In addition, the amounts of cadherin-associated β-catenin and plakoglobin were unchanged.
Figure 3.
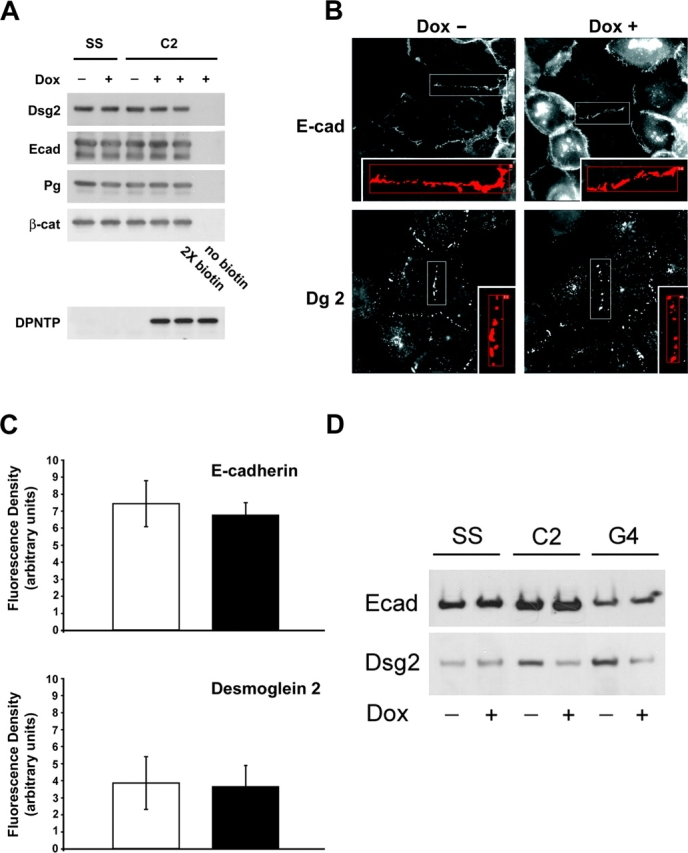
Cell surface distribution of desmosomal and classic cadherins is unaltered but Dsg 2 is reduced in the detergent-insoluble fraction of DPNTP-expressing cells. (A) Dox-induced and uninduced A431/SS and A431/C2 were biotinylated using a cell-impermeable biotin reagent. After precipitation using streptavidin beads, precipitates were separated by SDS-PAGE and immunoblotted using antibodies against E-cadherin (795), Dsg 2 (6D8), plakoglobin (1407), and β-catenin (C2206). To verify complete biotinylation, twice the concentration of biotin reagent was used in a separate experiment. Induction of DPNTP was confirmed by immunoblot analysis of whole cell lysates using an anti-FLAG antibody. No difference was seen in the amount of either E-cadherin or Dsg 2 or their associated proteins at the cell surface. (B) Immunofluorescence analysis was performed in Dox-induced and uninduced A431/C2 cells using antibodies against E-cadherin (HECD-1) and desmoglein (Dg3.10). (C) Fluorescence density was determined by dividing the fluorescent signal at cell borders (area in square pixels, see boxed examples) by the length of the border (pixels). The distribution and intensity for these cadherins was not detectably altered by DPNTP expression. (D) The detergent-insoluble fractions from Dox-treated and untreated cells were analyzed by SDS-PAGE and immunoblotted using antibodies against E-cadherin (795) and Dsg 2 (6D8). Note the decrease in the Triton-insoluble pool for Dsg 2, but not E-cadherin.
The fluorescence intensity distribution of E-cadherin and Dsg 2 at regions of cell–cell contact was determined by immunofluorescence and morphometric analysis (Fig. 3, B and C). Fluorescence intensity was quantified and normalized for the length of the region of cell contact. The distributions (Fig. 3 B) and fluorescence intensities (Fig. 3 C) of E-cadherin and Dsg 2 in Dox-treated and untreated cells were both comparable. Similar data were obtained for desmocollin 2 (unpublished data).
A decrease in the Triton-insoluble fraction of desmoglein in DPNTP-expressing cells is accompanied by reductions in intercellular adhesion and cell sheet integrity
As described above, DPNTP did not detectably alter the cell surface expression of classic or desmosomal cadherins. However, as IFs are uncoupled from these adhesive structures, we predicted that the loss of IF attachment might alter the amount of Dsg 2 in the IF-associated Triton-insoluble pool. To test this possibility, untreated or treated A431/SS, C2, and G4 lines were lysed in 1% Triton X-100 buffer and the insoluble pellet was analyzed by SDS-PAGE (Fig. 3 D). Both the A431/C2 and G4 lines exhibited an approximately twofold decrease in the Triton-insoluble fraction, whereas no change was detected in the solubility of E-cadherin.
We next sought to test whether this desmosomal cadherin-specific alteration in solubility translated into a functional difference in intercellular adhesion and epithelial sheet integrity. We first employed an adaptation of a previously described hanging drop aggregation assay using EDTA-dissociated single cells to generate clusters of cells not adherent to the substrate (Thoreson et al., 2000). Cells allowed to aggregate for at least 6 h in a hanging drop on the underside of a culture dish were subjected to trituration through a 200-μl Gilson pipette tip in an attempt to disrupt intercellular adhesion. The degree of dissociation was quantified by counting the particles that dissociated from the original cluster. Cells induced with Dox to express DPNTP were more susceptible to dissociation than untreated cells (Fig. 4 A) and exhibited on average an approximately fourfold increase in particle number when compared with the untreated cells after trituration (Fig. 4 B). Dox-treated and untreated A431/SS cells did not show any differences in the degree of dissociation, which was comparable to untreated C2 cells.
Figure 4.

DPNTP weakens adhesion in cell clusters formed in hanging drop culture. (A) 1 × 103 cells that had previously been Dox induced for at least 24 h or left untreated were seeded into hanging drop cultures without (top left) or with Dox-containing (top right) media and allowed to aggregate for 6 h. After trituration by passing the cell cluster 30 times through a 200-μl pipette tip, the degree of dissociation of the cell cluster was visualized by microscopy (A) and quantified by manual counting in a dissecting microscope (B). Note that cell clusters were imaged while suspended in a hanging drop and that dark patterns at the edge of field are caused by refraction of light. Due to cropping of the images, some of the dissociated small particles at the periphery in the bottom right panel of A are not visible but are still reflected in the graph in B. Error bars represent the standard deviation from an experiment in which each condition was tested in triplicate.
Because cell clusters grown in suspension were not well suited for analysis by immunofluorescence microscopy to monitor desmosome status, we sought to use an assay in which we could both verify formation of intercellular junctions and test adhesive strength. Confluent monolayers of Dox-induced and uninduced A431/SS and C2 cells were harvested from tissue culture dishes by incubation with dispase as previously described (Calautti et al., 1998). Monolayers were then transferred to 15-ml conical tubes containing 5 ml of Dulbecco's PBS (containing 0.9 mM calcium chloride and 0.5 mM magnesium chloride [DPBS]). After inverting the tubes 50 times on a rocker, monolayer fragments were counted (Fig. 5 A). DPNTP-expressing monolayers dissociated into numerous smaller fragments, whereas both Dox-treated and untreated control cell lines and uninduced A431 monolayers exhibited only minimal dissociation. Quantification of the number of fragments showed a 23-fold (t test, P < 0.001) and fourfold (P < 0.005) increase in fragmentation by the DPNTP-expressing A431/C2 and G4 monolayers, respectively (Fig. 5 B).
DPNTP-dependent dissociation of cell sheets is not accompanied by lactate dehydrogenase (LDH) release
To address whether the observed increase in fragmentation in DPNTP-expressing monolayers was due to increased cellular fragility and cytolysis, we assessed cellular damage by measuring the release of lactate dehydrogenase (LDH) from monolayers after application of mechanical stress. LDH release measured from DPNTP-expressing monolayers after mechanical disruption was not statistically altered compared with that released from the uninduced and control cell lines, which had minimal fragmentation (Fig. 5 C). Thus, despite a dramatically increased degree of monolayer dissociation, the DPNTP-expressing cell lines did not exhibit a corresponding increase in LDH release.
Time-lapse microscopy of living cells reveals that DPNTP-expressing cells dissociate more rapidly and with less cell distortion under stress than control cells
To examine at higher resolution the behavior of IF-uncoupled cells undergoing stress within epithelial cell sheets, time-lapse imaging of living cells was performed. Tet-On cell lines expressing DPNTP COOH-terminally tagged with EGFP were generated to confirm that DPNTP was being expressed and incorporated into cell junctions specifically in the cells under observation. These cell lines behave similarly in all respects to the DPNTP–FLAG-expressing cells. One cell line, termed A431/I3, which homogeneously expressed DPNTP–EGFP and exhibited the best morphological characteristics for imaging, was selected for this analysis. Data obtained with Dox-induced I3 cells were compared with either uninduced cells or control cell lines treated with Dox to rule out Dox-dependent effects.
The I3 cell line inducibly expressing DPNTP–EGFP was grown to confluence in the presence or absence of Dox, wounded, and then mounted in a living cell viewing chamber as described in the Materials and methods. To initiate stress-induced tearing of the cell sheet, the wounded monolayers were released from the center of the coverslip by flowing dispase through the chamber. The edges of the cell sheet remained tethered by the circumferential gasket associated with the living cell chamber; thus, cells at the edge of the wound began to stretch and tear due to contraction of the sheet as it lifted from the coverslip. Time-lapse images were captured at 4- (Fig. 6 A) or 8-s (Fig. 6 B) intervals and are shown here as selected individual still images and as four supplemental video clips (Videos 1–4, available online at http://www.jcb.org/cgi/content/full/jcb.200206098/DC1). Aspect ratios, which describe the geometry or “squareness” of a cell, were determined by dividing the diameter along the axis of stretch by the diameter orthogonal to the axis of stretch.
Figure 6.


DPNTP expression leads to rapid dissociation of cells within an epithelial sheet with less cell distortion than control cells. (A) The A431 I3 DPNTP–EGFP line treated with Dox (top panels) or without (bottom panels) was imaged during stress-induced tearing. Expression of DPNTP was confirmed by visualizing EGFP fluorescence (top, second panel from right). Individual still images from the time-lapse record have been enlarged here to include the boxed regions and are shown with a time stamp. The entire time-lapse record for each movie can be viewed at http://www.jcb.org/cgi/content/full/jcb.200206098/DC1 as Video 1 (top panels) and Video 2 (bottom panels). (B) A Dox-induced DPNTP–EGFP line (top panels) was imaged during stress-induced tearing and compared with control cells treated with Dox (bottom panels) to rule out Dox- dependent effects. Individual still images from the time-lapse record are shown here and the complete movies can be viewed as Video 3 (top panels) and Video 4 (bottom panels). Expression of DPNTP was confirmed by visualizing GFP fluorescence (unpublished data). Quantitative information for the cells marked with the asterisk is shown in C, left. Note that in the examples in both A and B, cell sheets expressing DPNTP tear more rapidly, and cells become less distended before cell–cell dissociation occurs, reflecting that less stress is required for dissociation to occur. This can be appreciated best by watching the video clips. (C) Cell aspect ratios for the cells in B were determined as described in the Materials and methods and plotted versus time. Arrows represent the time points shown in B. Note that the DPNTP–EGFP cells dissociated more quickly with less distension along the axis of stretch. Population data summarizing the average time to breakage are provided in C, right. Error bars represent the SEM for a population of 10–15 cells.
Although the precise degree of stretch could not be controlled using this method, we observed a dramatic difference between DPNTP-expressing cells and control cells, both in the length of time it took for tearing to occur and the degree of cell distortion observed at the time of dissociation (Fig. 6, A–C). DPNTP-expressing cells dissociated much more rapidly than control cells (Fig. 6, C and D) and did so without undergoing as much distension along the axis of stretch (Fig. 6 C).
DPNTP expression does not alter E-cadherin function
As shown above, total and cell surface expression of E-cadherin were not altered by expression of DPNTP. To verify that DPNTP did not interfere with E-cadherin function, a capillary tube flow assay was employed (Yap et al., 1997; Gottardi et al., 2001). Uninduced and induced A431/C2 and G4 cells were first allowed to adhere to the inner surface of capillary tubes precoated with the extracellular domain of E-cadherin and then subjected to shear flow through the capillaries after either 10 or 45 min (Fig. 7), the latter time frame having previously been shown to allow for a strengthening phase of adhesion (Yap et al., 1997). The percentage of cells still adhering to the capillary tube after being subjected to various flow rates was recorded. CHO cells, which do not express E-cadherin, were used as a negative control, whereas transfected CHO cells expressing E-cadherin were used as a positive control. Although previous studies suggested that the β-catenin binding site is not necessary for adhesion or strengthening, when assays were performed in the presence of LtnA, E-cadherin–dependent adhesion was abrogated, supporting the importance of the actin cytoskeleton in this assay (Fig. 7). Both uninduced and induced A431/C2 and G4 cells exhibited adhesion comparable to the E-cadherin–expressing CHO cells at both time points (unpublished data). These results suggest that DPNTP expression does not impair the early phase of E-cadherin–dependent adhesion or the strengthening phase, both of which are also dependent on an intact actin cytoskeleton.
Figure 7.
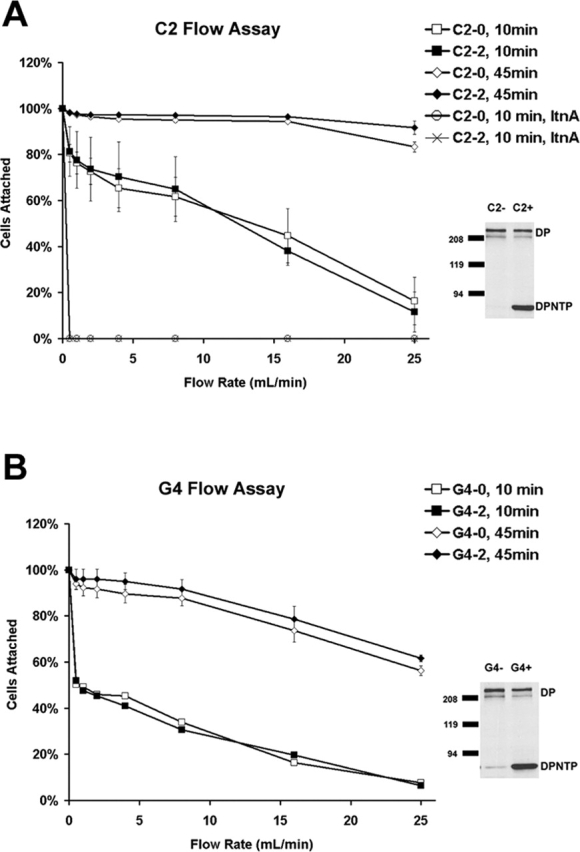
DPNTP expression does not alter the adhesive function of E-cadherin. Capillary flow assays were performed on induced (2 μg/ml Dox) and uninduced (0 μg/ml Dox) A431/C2 and G4 cells using glass capillary tubes coated with recombinant E-cadherin extracellular domain as described in the Materials and methods. Adhesion of both treated and untreated cells was similar under various flow rates. In addition, no difference was seen in the strengthening of adhesion after cells were allowed to adhere to capillary tubes for longer periods of time (45 min). Note that pretreatment with 2 μM latrunculin A abrogated E-cadherin–dependent adhesion. Note that the G4 line exhibited a somewhat greater loss of binding in response to flow than the C2 line in both the Dox-treated cultures and controls. It is possible this could be explained in part by the lower expression of E-cadherin in this line.
Contribution of microfilaments to adhesive strength in uninduced and induced cell lines
To further verify that DPNTP expression did not interfere with the cortical actin cytoskeleton–dependent strengthening of adhesion, Dox-treated and untreated A431 monolayers were incubated with LtnA before application of mechanical strain. LtnA disrupts the actin cytoskeleton by sequestering G-actin, which subsequently causes depolymerization of F-actin and disrupts adherens junctions (Cai et al., 2000). If the observed DPNTP-dependent loss of adhesive strength was solely due to interfering with adherens junction function, one would not expect a dramatic increase in stress-induced dissociation when DPNTP is expressed in the presence of LtnA.
As expected, Dox-treated A431/C2 monolayers exhibited significant dissociation (Fig. 8 A). LtnA alone also resulted in the dissociation of cell sheets. When the A431/C2 line was treated with 2 μM LtnA after Dox treatment to induce DPNTP expression, the monolayer dissociated into >500 particles. Measurement of LDH release indicated that there was no statistical difference among the samples and, in all cases, the amount of LDH released after mechanical strain was <5% of the total amount of LDH expressed.
Figure 8.
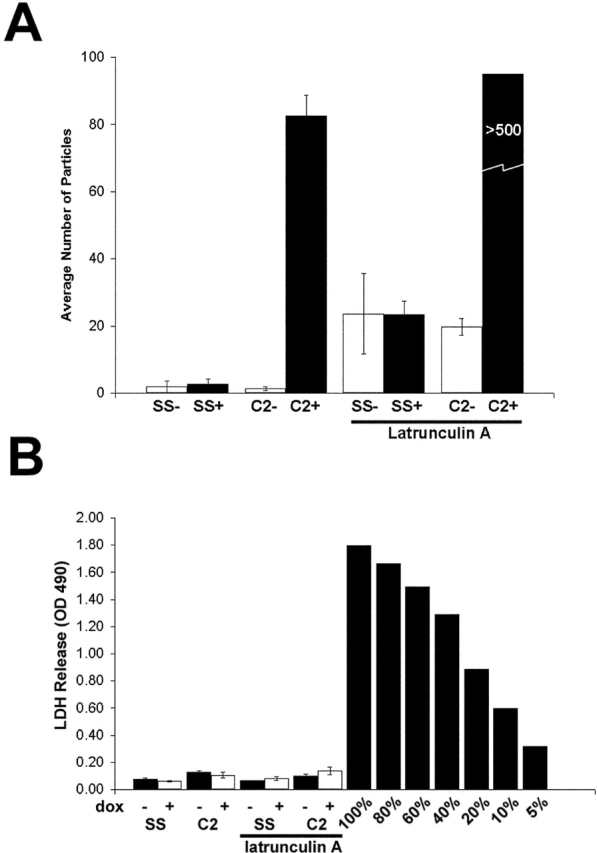
LtnA and DPNTP act synergistically to weaken adhesion. (A) Dox-treated and untreated A431/SS and A431/C2 monolayers were treated with dispase and subjected to mechanical stress as described above for Fig. 4, with or without a 30-min treatment with 2 μM LtnA. (B) Again, LDH release was not significantly increased despite the dramatic fragmentation of LtnA-treated monolayers expressing DPNTP. Error bars represent the SEM from an experiment in which each condition was tested in triplicate.
Keratinocytes derived from patients with a recessively inherited COOH-terminal truncation of DP exhibit adhesive defects in vitro
To further examine the physiological relevance of COOH-terminal DP truncations, we employed keratinocytes that had been isolated from patients with a COOH-terminal truncation of DP due to deletion of the G at position 7622 within the coding sequence of DP (position 7901 of Genbank/EMBL/DDBJ accession no. M77830) (Norgett et al., 2000). Histological analysis of patient epidermis revealed widening of intercellular spaces, suggesting the possibility that this truncated form of DP might lead to an adhesive defect. This mutation deletes the C-subdomain containing the plakin repeats and part of the upstream linker region and introduces 18 new amino acids downstream of the deletion (Fig. 1 A). Based on earlier work, we would predict that this deletion would compromise interactions between keratin IFs and the plaque (Stappenbeck et al., 1993; Meng et al., 1997). However, the remaining upstream plakin repeat domain and linking motifs may be sufficient to allow a partial interaction with IFs, whereas the recruitment of plakophilins and/or plakoglobin via the DP NH2 terminus may provide additional stabilization of IFs (Smith and Fuchs, 1998; Kowalczyk et al., 1999b; Hofmann et al., 2000).
Consistent with this, immunofluorescence analysis of unstressed keratinocytes growing in vitro did not reveal a dramatic uncoupling of IF tonofibrils from the desmosomal plaque, even though the presence of the truncated DP was confirmed by immunoblot analysis (unpublished data). In addition, no difference in the fluorescence intensity of plaque or transmembrane cadherins was detected (Fig. 9 A) in these cells compared with normal human epidermal keratinocytes (NHEK). However, when subjected to mechanical stress, dispase-released monolayers generated from these keratinocytes exhibited dissociation into particles greater than that seen with NHEK cells (Fig. 9 B). In contrast to A431 cell monolayers, both the control NHEK cells and JD-1 cells exhibited release of LDH, indicating that some cytolysis was occurring (Fig. 9 C). However, there was no difference in the degree of cytolysis between these cultures, suggesting that the greater particle number was due to an additional adhesive defect existing in the JD-1, and not the control, cell lines.
Figure 9.

JD-1 keratinocytes derived from patients harboring a COOH-terminal truncation of DP exhibit an adhesive defect. (A) Fluorescence density measurements performed on JD-1 and NHEK cell lines using antibodies directed against α-catenin (C2081) and DP (DP2.15) were found to be comparable. Confluent JD-1 and NHEK monolayers were analyzed using the dissociation assay. Particle numbers were quantified (B) and LDH release was measured (C). Although both NHEK and JD-1 keratinocytes exhibited more cell lysis compared with the DPNTP-expressing A431 cells, JD-1 cells fragmented into a larger number of particles, supporting the existence of an adhesive defect in the JD-1 cells. Error bars represent the SEM from an experiment in which each condition was tested in triplicate.
Discussion
Role of the cytoskeleton in adhesion: uncoupling IFs from desmosomes decreases intercellular adhesive strength
Several lines of evidence support a role for the actin cytoskeleton in strengthening adhesion. The catenin complex of intercellular adherens junctions, which mediates the link with actin, is required for E-cadherin–dependent adhesion in vitro and in vivo (Ozawa et al., 1990; Vasioukhin et al., 2000), and the actin-capping drug cytochalasin D prevents cadherin-mediated adhesion (Angres et al., 1996; Imamura et al., 1999). These data are consistent with our observation that LtnA abrogates adhesion in the capillary flow assay. Each of the three major catenins, α-catenin (Watabe et al., 1994), β-catenin (Chen et al., 1997), and p120ctn (Thoreson et al., 2000; Yap et al., 1998), has been implicated in playing important roles in cell–cell adhesion, although the regulatory role of p120ctn has been somewhat controversial (Ohkubo and Ozawa, 1999). In addition, determining the respective contributions of lateral cadherin clustering and physical linkage to the cytoskeleton has been challenging. It has been proposed that linkage to the actin cytoskeleton through α- and β-catenin may strengthen initial p120ctn -dependent clustering by subsequent recruitment or nucleation of actin filament attachments and that continued tension between contacting cells requires associated myosin activity (Adams and Nelson, 1998).
Less attention has been focused on the role of IF–plasma membrane associations in regulating adhesive strength. Previous studies demonstrated that desmosomal cadherin tails play a critical role in regulating IF–desmosomal connections and demonstrated that a desmoglein 1–connexin chimera can act in a dominant-negative fashion to uncouple IFs from cell–cell contacts (Troyanovsky et al., 1993, 1994). In that case, however, a disappearance of endogenous desmosomes was observed. The strategy used here, which employed a truncated DP molecule, allowed us to test the role of the IF–membrane connection in epithelial sheet integrity and adhesive strength in vitro by severing the IF connection while retaining clustered desmosomal cadherins associated with their armadillo family members. Furthermore, as E-cadherin function appeared to be normal in this system, it allowed us to test the relative contributions of IFs and actin cytoskeleton linkages to epithelial sheet integrity. A431 cells induced to express dominant-negative DPNTP exhibited a decreased ability to withstand trituration after aggregation in cell suspension. In addition, DPNTP-expressing confluent cell sheets demonstrated a decreased ability to withstand turbulent mechanical strain as compared with uninduced monolayers, dissociating into many fragments without an increase in cell lysis. In other experiments not shown here, even greater cell sheet dissociation was achieved by using a trituration protocol; however, data from these experiments were difficult to interpret, as trituration often led to cell lysis. Here we show that gentle inversion of the epithelial sheets is sufficient to cause cell detachment without resulting in lysis. Furthermore, higher resolution time-lapse analysis of strained cell sheets revealed that dissociation of DPNTP-expressing cells is more rapid and occurs with less cell distension than control cells. Together these data suggest that although the presence of an intact IF cytoskeleton plays a role in maintaining cellular integrity, connection of this network to the plasma membrane via desmosomes provides strong intercellular adhesion.
Contributions of the desmosomal plaque and IF attachment to adhesive strength
Drawing a parallel to adherens junctions, one might envision that desmosomal strengthening of adhesion would involve at least two major steps: (1) lateral clustering and proper orientation of desmosomal cadherins through proper assembly of the plaque and (2) physical anchoring of the cadherin complex via IFs. In the case of adherens junctions, it has been suggested that lateral cadherin clustering of the extracellular domain, whether through artificial means or via p120ctn, can increase adhesive strength (Brieher et al., 1996; Yap et al., 1997, 1998; Thoreson et al., 2000). We showed previously that the p120ctn-related protein plakophilin 1 cooperates with plakoglobin to cluster desmosomal cadherins in the presence of DP (Bornslaeger et al., 2001). Furthermore, the NH2-terminal 584 amino acids of DP, which comprise DPNTP, are sufficient for such clustering in L cell fibroblasts (Kowalczyk et al., 1997). It seems likely that this clustering event constitutes a necessary step in desmosomal cadherin–dependent adhesive strengthening. Consistent with this idea is the observation that the DP NH2 terminus appeared to restore sealing of epithelial sheets and possibly even some measure of IF attachment in DP-null keratinocytes (Hatsell and Cowin, 2001; Vasioukhin et al., 2001). However, it is clear from the present work that even though DPNTP is capable of clustering desmosomal cadherins and forming junctional plaques in A431 cells, adhesive strength is severely compromised in epithelial sheets derived from these cells.
As DPNTP-dependent clustering is not sufficient to support adhesion comparable to wild-type DP, this ability must require the remaining DP domains, which include the rod and the COOH-terminal IF-binding domains. Previous work suggests that the DP rod contributes to the formation of oligomeric coassemblies with IF polypeptides that build the innermost portion of the plaque (Stappenbeck and Green, 1992; Kouklis et al., 1994). Thus, it could be envisioned that loss of these domains may compromise the ability of the plaque to withstand mechanical stress. However, the JD-1 cell experiments suggest that even small deletions of the DP COOH terminus that interfere with IF anchorage are enough to compromise adhesive strength when stress is applied to cells in vitro (Fig. 9 A) and in vivo (Norgett et al., 2000).
IF–membrane attachments may enhance adhesive strength in several ways. As has been suggested for actin, IFs may stabilize desmosomal cadherin clusters formed by the armadillo–DP NH2-terminal complex, and thus, the proper disposition of desmosomal cadherin extracellular domains. Another possibility is that IFs anchor desmosomal cadherins within the plane of the membrane and that uncoupling of this connection results in molecular extraction of desmosomal cadherins out of the plane of the membrane. Similar molecular extraction has been reported previously for agglutinin receptors from red blood cells (Evans et al., 1991), L-selectin from neutrophils (Shao and Hochmuth, 1999), and integrins from fibroblasts moving across a substratum (Palecek et al., 1996). Another possibility is that the entire plaque might be extracted, as might be suggested by the plaque-associated membrane blebs seen in electron micrographs of the epidermis of DP-null animals (Vasioukhin et al., 2001). Whether such blebs seen in vivo would be enough to lead to cytolysis is not clear, but if this sort of extraction occurred in A431 cells, membrane resealing would have to be rapid so that LDH release would not occur.
The actin and IF cytoskeletons contribute synergistically to provide strong intercellular adhesion through classic and desmosomal cadherins
DPNTP was previously shown in stable A431 clones to lead to the codistribution of certain adherens junction and desmosome components (Bornslaeger et al., 1996), raising the possibility that DPNTP might interfere with adhesive strength solely by interfering with E-cadherin function. However, as shown here, the fluorescence patterns for E-cadherin and Dsg 2 did not detectably differ in uninduced and induced cells (Fig. 3, B and C), and the dramatic codistribution between desmosomal and adherens junction components that was previously reported in those clonal lines was not observed here. The basis for this difference is not currently known, although it may be related to DPNTP expression levels, clonal variability, or both.
Nevertheless, we sought to ensure that the observed adhesive defect was not through effects on E-cadherin. The experiments outlined here show that, in addition to the fact that neither cell surface expression nor distribution was altered, the fraction of E-cadherin present in the detergent-insoluble pool remained constant. This was in contrast to Dsg 2, which was reduced in the Triton-insoluble pool, likely due to the loss of attachment to the IF cytoskeleton. The data presented here also support the idea that E-cadherin is still functional in DPNTP-expressing cells. The ability of DPNTP-expressing cells adhering to capillary tubes coated with the E-cadherin extracellular domain to resist shear stress was no different than that of uninduced cells at early or later time points, even though adhesion was totally abrogated by LtnA. In addition, DPNTP-expressing epithelial sheets treated with LtnA exhibited a dramatically increased dissociation, which would be predicted only if IF attachments have an additional strengthening effect beyond that contributed by the cortical actin–adherens junction complex. Thus, the decreased structural integrity of DPNTP-expressing monolayers is not simply due to interference with E-cadherin function.
In fact, as the combined effect of LtnA treatment and DPNTP expression is much greater than the sum of the effects of the separate treatments, our data suggest that the actin and IF cytoskeletons function synergistically to provide strong intercellular adhesion and maintain structural integrity of cell sheets. These data are consistent with earlier work demonstrating that the microfilament and IF cytoskeletons are physically associated at the ultrastructural level in cells. Indeed, our earlier work in primary mouse keratinocytes demonstrated an intimate relationship between the cortical actin circumferential ring associated with the perpendicular bundles emanating from adherens junctions and keratin IF bundles that looped in and out of the cortical actin (Green et al., 1987). More recent work suggested that F-actin serves as a template for cytokeratin organization in vitro (Weber and Bement, 2002), and such interactions might in fact be stabilized by other members of the plakin family of cytolinkers, which may cross-link these systems laterally (Fuchs and Yang, 1999).
Observations from DP-null keratinocytes support the idea that the DP–IF network plays an important role in the maturation of the keratinocyte microfilament cytoskeleton (Vasioukhin et al., 2001). Cells cultured from these animals had severely reduced desmosome numbers and, in addition, failed to undergo the normal maturation of the cortical actin cytoskeleton, which might in turn contribute to the observed failure to form epithelial sheets in vitro and skin fragility in vivo. In the current work, the actin microfilament system did not appear to be significantly altered in DPNTP-expressing cells or JD-1 keratinocytes (unpublished data), possibly due to restoration of certain DP NH2-terminal–dependent functions (Hatsell and Cowin, 2001). Together these previous observations and the data presented here suggest two related but independent points. First, DP, possibly through its NH2-terminal head domain, plays an important role in the development and maturation of the actin cytoskeleton. Second, actin and IF cytoskeleton connections to cadherins function to synergistically strengthen adhesion, possibly through anchoring cadherins in the plane of the membrane, thus maintaining epithelial cell sheet integrity.
DP truncations in human disease
Epithelial sheets formed from JD-1 keratinocytes derived from patients harboring a short COOH-terminal truncation of the IF-binding domain also show an adhesive defect. This is supported by data showing an increase in cell sheet dissociation compared with normal keratinocytes, without any additional increase in LDH release. That some LDH release occurs in this case is not surprising based on the fact that these cells have a much milder defect than the genetically modified A431 cells, and thus some cells may break apart during the increased number of inversions required to release particles in the assay. The JD-1 data suggest that perturbation of the cytoplasmic plaque and its IF connection leads to the weakening of cell adhesion that underlies the cell separation observed in patients harboring this truncated protein (Norgett et al., 2000) and that the cellular consequences are distinct from the largely cytolytic defects caused by mutant IF proteins.
This idea is consistent with observations seen in patients with epidermolytic palmoplantar keratoderma (EPPK) or non-EPPK (NEPPK), a noncytolytic form of EPPK. Some EPPK patients possess mutations in keratin K9 that cause skin blistering on the palms of the hand and soles of the feet with characteristic cytolysis in the suprabasal layers (Kobayashi et al., 1996). On the other hand, in one NEPPK study, a mutation was discovered in keratin K1 at a position in the head domain that, in keratin K5, is believed to interact with DP (Kimonis et al., 1994; Kouklis et al., 1994). These patients do not appear to undergo the same sort of skin blistering exhibited by the EPPK patients. Similar phenotypic traits were observed in patients with mutations in desmosomal molecules, such as plakophilin 1 (McGrath et al., 1997). These observations are consistent with a hypothesis that the presence of an IF cytoskeleton is responsible for maintaining the structural integrity of the cell, whereas connection of this network to the plasma membrane via DP may be responsible for maintaining or modulating intercellular adhesion.
Together the results presented in this paper demonstrate the first direct physical evidence to support a role for the IF–desmosome connection, independent of desmosomal cadherin clustering, in regulating the adhesive strength of desmosomes. Furthermore, the data suggest that the IF–desmosome connection works synergistically with the actin–adherens junction complex in providing adhesive strength to epithelia.
Materials and methods
Generation of cDNA constructs
p700, a CMV promotor–based construct expressing DPNTP with a COOH-terminal FLAG tag (DPNTP–FLAG), was previously described (Kowalczyk et al., 1999b). DPNTP–FLAG was inserted into a shuttle vector by subcloning the EcoRI–HindIII fragment of p700 into pGEM7Zf (p766) (Promega). To generate the construct with DPNTP–FLAG under the control of the tetracycline-responsive element, p766 was digested with EcoRI and BamHI, and the resulting fragment was ligated into the same sites in the MCS of pTRE (CLONTECH Laboratories, Inc.), yielding p801.
To create DPNTP–EGFP under the control of the tetracycline response element, PCR was used to amplify a 1.755-kb DPNTP-containing fragment from pTRE-DPNTP–FLAG (p801). This fragment was cloned into the EcoRI/KpnI sites of pEGFP-N1 (CLONTECH Laboratories, Inc.) to create pEGFP-N1 DPNTP. Next, a 2.5-kb DNA fragment containing DPNTP plus EGFP was removed from pEGFP-N1 DPNTP by sequential NotI/NheI digestion. The resulting fragment was subcloned into the NotI/NheI sites of the pTRE2 response plasmid (CLONTECH Laboratories, Inc.), producing vector p992. Constructs were DNA sequenced to verify correct structure.
Cell culture and generation of inducible lines
Parental A431 epithelial cells were cultured as previously described (Bornslaeger et al., 1996). These cells were transfected with pTet-On (CLONTECH Laboratories, Inc.) using calcium phosphate precipitate and selected using 400 μg/ml G418 (Mediatech). Expression of the tetracycline-responsive transactivator in drug-resistant lines was determined by a luciferase assay (pTRE-Luc; CLONTECH Laboratories, Inc., and pRL-CMV; Dual-Luciferase Reporter Assay System; Promega). To establish tetracycline-inducible DPNTP–FLAG lines, a single stable line, 68Q71, was cotransfected with p801 and pSV2pacΔP. After selection with 400 μg/ml G418 and 1 μg/ml puromycin, resistant clones were screened for DPNTP–FLAG by treating with 2 μg/ml Dox for 24 h. The cells were then lysed in Laemmli sample buffer and analyzed via SDS-PAGE and immunoblotting. To establish a cell line inducibly expressing EGFP-tagged DPNTP, the 68Q71 cell line was transfected with vectors p992 and pSV2pacΔP, and puromycin-resistant clones were selected as described above.
JD-1 keratinocytes isolated from patients were immortalized with an HPV16 plasmid (pJ45216) that has early region genes driven by MoMLV-LTR as previously described (Storey et al., 1988). Cells were cultured in DME/Ham's F12 (3:1) supplemented with 10% FCS, 4 mM glutamine, 0.4 μg/ml hydrocortisone, 0.1 nM cholera toxin, 5 μg/ml insulin, and 10 ng/ml EGF. NHEK were isolated from neonatal foreskin as previously described (Halbert et al., 1992) and were cultured in media M154 (Cascade Biologics, Inc.).
Antibodies
Full-length DP and DPNTP–FLAG were detected using a rabbit polyclonal antibody, NW161, against the DP NH2 terminus (Bornslaeger et al., 1996) or poly-FLAG (Sigma-Aldrich) against the FLAG-tagged constructs. The following rabbit polyclonal antibodies were also used: C2206 against β-catenin (Sigma-Aldrich), C2081 against α-catenin (Sigma-Aldrich), 795 against E-cadherin (provided by R. Marsh and R. Brackenbury, University of Cincinnati, Cincinnati, OH), and NW6 against the DP COOH terminus (Angst et al., 1990). 1407 is a polyclonal chicken antibody recognizing plakoglobin (Gaudry et al., 2001). Mouse monoclonal antibodies used in this study were: DP2.15 (a gift from P. Cowin, New York University, New York, NY) against the central rod domain of DP, Dg3.10 against Dsg 2 (Schmelz et al., 1986), HECD-1 against human E-cadherin (Shimoyama et al., 1989), 6D8 against Dsg 2, 11E4 against plakoglobin, 5H10 against β-catenin (gifts from M. Wheelock, University of Nebraska, Omaha, NE), and KSB17.2 against keratin 18 (Sigma-Aldrich).
Preparation of whole cell lysates and Triton-insoluble pool
To obtain whole cell lysates, cells were homogenized in Laemmli sample buffer, resolved on 7.5% SDS-PAGE gels, transferred to nitrocellulose, and immunoblotted using the following antibodies: 6D8 (1:1,000), 795 (1:1,000), C2081 (1:2,000), 5H10 (1:100), 11E4 (1:1,000), and NW161 (1:5,000). Sample loading was normalized to keratin that was visualized using KSB17.2 (1:2,000). For analysis of the Triton X-100–insoluble pool, cells were lysed in 1% Triton X-100 buffer (1% Triton X-100, 145 mM NaCl, 10 mM Tris-HCl, pH 7.4, 5 mM EDTA, 2 mM EGTA, and 1 mM PMSF) followed by centrifugation (16000 g, 30 min). The Triton X-100–insoluble pellet was solubilized in Laemmli sample buffer, resolved by SDS-PAGE, transferred to nitrocellulose, and labeled using 795 (1:1,000) and Dg3.10 (1:100).
Immunofluorescence analysis
Cells were seeded onto poly-l-lysine–coated (0.1 mg/ml, 3 h) glass coverslips. 1 d after plating, samples were induced with Dox (2 μg/ml, 24 h). Coverslips were then rinsed in DPBS and fixed in methanol (−20°C, 2 min). Indirect immunofluorescence was performed using KSB17.2 (1:200), poly-FLAG (1:50), NW6 (1:50), DP2.15 (1:100), Dg3.10 (1:10), and HECD-1 (undiluted supernatant). Primary antibodies were detected using Alexa Fluor®488 goat anti–mouse IgG and Alexa Fluor®568 goat anti–rabbit IgG at a dilution of 1:400 (Molecular Probes). Images were captured using a Leica DMR microscope/Orca Hamamatsu digital camera system and analyzed using OpenLab software (Improvision).
Fluorescence density at cell borders was determined by measuring fluorescent signal (OpenLab) from regions of intercellular contact in induced and uninduced A431/SS, C2, and G4 cells stained using 795 (1:1,000) and Dg3.10 (1:10) or JD-1 and NHEK cells stained using DP2.15 (1:500) and C2081 (1:2,000). Fluorescence density was calculated by dividing the fluorescent signal (pixels2) by the length (pixels) of the measured cell border. Calculations were determined from 10 representative regions of intercellular contact for each cell line.
Cell surface biotinylation
Cells treated with or without 2 μg/ml Dox were grown to confluency in 60-mm dishes. Cultures were washed twice with DPBS and labeled with cell-impermeable EZ-Link™ Sulfo-NHS-LC-Biotin (4 ml at 500 μg/ml in DPBS, 4°C, 30 min) (Pierce Chemical Co.) on a rocker. Cultures were then washed twice with DPBS and incubated in 4 ml of DPBS containing 100 mM glycine (4°C, 20 min) on a rocker. Finally, cells were washed twice with DPBS and lysed with 1 ml RIPA buffer (10 mM Tris-HCl, pH 7.5, 140 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 5 mM EDTA, 2 mM EGTA, 1mM PMSF). Biotin-labeled proteins were precipitated using 40 μl of UltraLink™ Immobilized Streptavidin (Pierce Chemical Co.) in 1.5-ml Eppendorf tubes overnight at 4°C on a rotator. The streptavidin beads were washed five times using lysis buffer and then centrifuged (10,000 g, 1 min, 4°C). Precipitated proteins were eluted using Laemmli buffer, boiled, resolved by SDS-PAGE, and immunoblotted using 795 (1:1,000), C2206 (1:4,000), Dg3.10 (1:100), and 1407 (1:5,000).
Hanging drop assay
Hanging drop cultures of aggregated cells were generated from 1 × 103 cells that were either untreated or pretreated with Dox for 24 h and then dissociated in 1mM EDTA in DPBS without Ca2+ and Mg2+. Cells were allowed to aggregate over times ranging from 2 h to overnight on the underside of a culture dish as previously described (Thoreson et al., 2000). Resulting cell clusters were subjected to 30 rounds of pipetting through a 200-μl Gilson pipette, and the degree of dissociation was quantified by counting the particles after trituration.
Dispase-based dissociation assay
A431/SS, C2, and G4 cultures were seeded in triplicate onto 60-mm dishes. At ∼50% confluency, cells were treated with or without 2 μg/ml Dox. 24 h after reaching confluency, cultures were washed twice in DPBS and then incubated in 2 ml of dispase (2.4 U/ml; Roche Diagnostics GmbH) for more than 30 min. Released monolayers were carefully washed twice with DPBS and transferred to 15-ml conical tubes. Enough DPBS was added for a final volume of 5 ml. Tubes were secured to a rocker and subjected to 50 inversion cycles. Fragments were counted using a dissecting microscope (Leica MZ6). For experiments using LtnA, cell monolayers released by dispase were further incubated for 30 min in 2 ml of dispase solution containing 2 μM LtnA. Statistical analysis was performed on the average of three separate experiments in the cases of the A431/SS and C2 cell lines and one experiment using G4 cells. Statistical significance (t test) was defined as P < 0.05. For experiments with JD-1 and NHEK, application of mechanical stress included 50 inversion cycles with either five additional inversion cycles in which the sample tubes were turned rapidly end over end or shaking at 400 rpm for 1 min in a rotary shaker.
LDH release measurements
LDH measurements using the Cytotoxicity Detection Kit (Roche Diagnostics GmbH) were performed according to the manufacturer's instructions on 200 μl of cell-free supernatant collected after monolayers were subjected to the dispase-based dissociation assay.
Videomicroscopy of dispase-treated wounded cell monolayers
Cell lines inducibly expressing DPNTP–FLAG or DPNTP–EGFP were seeded onto 40-mm diameter circular glass coverslips and allowed to grow to confluence either in the presence or absence of 2–4 μg/ml of Dox. Monolayers were then wounded at the center of the coverslips using a razor blade. Coverslips were then quickly mounted into the FCS2 closed system live-cell chamber (Bioptechs). The cell chamber was then filled with DPBS containing 48 U/ml dispase. Phase contrast and/or fluorescence time-lapse record of tearing of the monolayer was obtained at 40× magnification using a Leica DMIRBE inverted microscope fitted with an Orca Hamamatsu (C4742–95) digital camera and analyzed using Openlab software. Images were captured at 4- or 8-s intervals. Aspect ratios of measured cells were determined by dividing the diameter along the axis of stretch by the diameter orthogonal to the axis of stretch. Quicktime movies of the time-lapse record were assembled using Adobe Premier software.
Shear flow assay
Glass capillary tubes coated with 1 mg/ml protein A (Amersham Biosciences) in DPBS (4–7 h, 4°C) and blocked with 0.5% casein enzymatic hydrolysate were coated with 25 μg/ml E-cadherin extracellular domain–Fc fusion protein in HBSS plus 1 mM calcium chloride (HBSS+) (overnight, 4°C). Tubes were then blocked with 5% nonfat dry milk (in HBSS+, 2 h, 4°C) followed by a wash in HBSS+. A431/C2 or G4 cells treated with 2 μg/ml Dox for 36 h were dispersed with 2 mM EDTA in PBS without calcium or magnesium (37°C, 15 min) and washed in HBSS+ containing 0.2 mg/ml RGD peptide. Cells were loaded onto tubes and allowed to incubate for 10 or 45 min. Flow of HBSS+ started at 0.5 ml/min and doubled every 30 s, ending with a final flow rate of 25 ml/min. The percentage of cells bound to substrate was determined at each flow rate by video microscopy. For experiments involving LtnA, cells were incubated in 2 μM LtnA for 30 min before loading onto coated capillary tubes.
Online supplemental material
The supplemental videos are available at http://www.jcb.org/cgi/content/full/jcb.200206098/DC1. Videos 1 and 2 correspond to the top and bottom panels of Fig. 6 A, respectively. Dox-treated or untreated DPNTP–EGFP I3 line monolayers were wounded and treated with dispase to release the sheets from the substrate and initiate contraction of the cells surrounding the wound. Tearing of the sheet and cell–cell dissociation were recorded in exposures taken every 4 s with a display rate of 15 frames per second for both Videos 1 and 2. The time stamp in the lower right hand corner of each video clip is displayed in min:sec. See Fig. 6 A for static images at selected time points. Videos 3 and 4 correspond to the top and bottom panels of Fig. 6 B, respectively. Dox-treated DPNTP–EGFP I3 line and a Dox-treated control line were wounded and treated with dispase to release the epithelial sheets as described above for Fig. 6 A. Exposures were taken every 8 s and the movie is displayed at 15 frames per second. See Fig. 6 B for static images at selected time points and Fig. 6 C for quantitative analysis of the cells indicated with asterisks.
Acknowledgments
The authors would like to thank all those who have generously contributed antibodies, plasmids, and other reagents, including P. Cowin, R. Marsh, R. Brackenbury, M. Wheelock, K. Johnson, and P. Purkis. Thanks also S. Getsios and A. Kowalczyk for helpful discussion and critical reading of the manuscript.
This work is supported by National Institutes of Health grants RO1 AR43380 and PO1 DE12328 (project No. 4) to K. Green and R01 GM52717 to B. Gumbiner.
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: Dox, doxycycline; DP, desmoplakin; Dsg 2, desmoglein 2; EPPK, epidermolytic palmoplantar keratoderma; IF, intermediate filament; LDH, lactate dehydrogenase; Ltn A, latrunculin A; NEPPK, non-EPPK; NHEK, normal human epidermal keratinocytes; SS, single stable.
References
- Adams, C.L., and W.J. Nelson. 1998. The cytomechanics of cadherin-mediated cell-cell adhesion. Curr. Opin. Cell Biol. 10:572–577. [DOI] [PubMed] [Google Scholar]
- Angres, B., A. Barth, and W.J. Nelson. 1996. Mechanism for transition from initial to stable cell–cell adhesion: kinetic analysis of E-cadherin–mediated adhesion using a quantitative adhesion assay. J. Cell Biol. 134:549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angst, B., C. Marcozzi, and A. Magee. 2001. The cadherin superfamily: diversity in form and function. J. Cell Sci. 114:629–641. [DOI] [PubMed] [Google Scholar]
- Angst, B.D., L.A. Nilles, and K.J. Green. 1990. Desmoplakin II expression is not restricted to stratified epithelia. J. Cell Sci. 97:247–257. [DOI] [PubMed] [Google Scholar]
- Armstrong, D.K.B., K.E. McKenna, P.E. Purkis, K.J. Green, R.A.J. Eady, I.M. Leigh, and A.E. Hughes. 1999. Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum. Mol. Genet. 8:143–148. [DOI] [PubMed] [Google Scholar]
- Bornslaeger, E., L. Godsel, C. Corcoran, J. Park, M. Hatzfeld, A. Kowalczyk, and K. Green. 2001. Plakophilin 1 interferes with plakoglobin binding to desmoplakin, yet together with plakoglobin promotes clustering of desmosomal plaque complexes at cell-cell borders. J. Cell Sci. 114:727–738. [DOI] [PubMed] [Google Scholar]
- Bornslaeger, E.B., C.M. Corcoran, T.S. Stappenbeck, and K.J. Green. 1996. Breaking the connection: displacement of the desmosomal plaque protein desmoplakin from cell–cell interfaces disrupts anchorage of intermediate filament bundles and alters intercellular junction assembly. J. Cell Biol. 134:985–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieher, W.M., A.S. Yap, and B.M. Gumbiner. 1996. Lateral dimerization is required for the homophilic binding activity of C-cadherin. J. Cell Biol. 135:487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdett, I.D.J. 1998. Aspects of the structure and assembly of desmosomes. Micron. 29:309–328. [DOI] [PubMed] [Google Scholar]
- Cai, S., X. Liu, A. Glasser, T. Volberg, M. Filla, B. Geiger, J.R. Polansky, and P.L. Kaufman. 2000. Effect of latrunculin-A on morphology and actin-associated adhesions of cultured human trabecular meshwork cells. Mol. Vis. 6:132–143. [PubMed] [Google Scholar]
- Calautti, E., S. Cabodi, P.L. Stein, M. Hatzfeld, N. Kedersha, and G.P. Dotto. 1998. Tyrosine phosphorylation and src family kinases control keratinocyte cell–cell adhesion. J. Cell Biol. 141:1449–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H., N.E. Paradies, M. Fedor-Chaiken, and R. Brackenbury. 1997. E-cadherin mediates adhesion and suppresses cell motility via distinct mechanisms. J. Cell Sci. 110:345–356. [DOI] [PubMed] [Google Scholar]
- Coulombe, P.A., and M.B. Omary. 2002. “Hard” and “soft” principles defining the structure, function and regulation of keratin intermediate filaments. Curr. Opin. Cell Biol. 14:110–122. [DOI] [PubMed] [Google Scholar]
- Epstein, E.H. 1992. Molecular genetics of epidermolysis bullosa. Science. 256:799–804. [DOI] [PubMed] [Google Scholar]
- Evans, E., D. Berk, and A. Leung. 1991. Detachment of agglutinin-bonded red blood cells. I. Forces to rupture molecular-point attachments. Biophys. J. 59:838–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, E., and D.W. Cleveland. 1998. A structural scaffolding of intermediate filaments in health and disease. Science. 279:514–519. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., and P.A. Coulombe. 1992. Of mice and men: genetic skin diseases of keratin. Cell. 69:899–902. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., and Y. Yang. 1999. Crossroads on cytoskeletal highways. Cell. 98:547–550. [DOI] [PubMed] [Google Scholar]
- Gallicano, G.I., P. Kouklis, C. Bauer, M. Yin, V. Vasioukhin, L. Degenstein, and E. Fuchs. 1998. Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J. Cell Biol. 143:2009–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallicano, G.I., C. Bauer, and E. Fuchs. 2001. Rescuing desmoplakin function in extra-embryonic ectoderm reveals the importance of this protein in embryonic heart, neuroepithelium, skin and vasculature. Development. 128:929–941. [DOI] [PubMed] [Google Scholar]
- Garrod, D., M. Chidgey, and A. North. 1996. Desmosomes: differentiation, development, dynamics and disease. Curr. Opin. Cell Biol. 8:670–678. [DOI] [PubMed] [Google Scholar]
- Gaudry, C.A., H.L. Palka, R.L. Dusek, A.C. Huen, M.J. Khandekar, L.G. Hudson, and K.J. Green. 2001. Tyrosine phosphorylated plakoglobin is associated with desmogleins but not desmoplakin after EGFR activation. J. Biol. Chem. 276:24871–24880. [DOI] [PubMed] [Google Scholar]
- Gelderloos, J.A., L. Witcher, P. Cowin, and M.W. Klymkowsky. 1997. Plakoglobin, the other “Arm” of vertebrates. Cytoskeletal-Membrane Interactions and Signal Transduction. P. Cowin and M. Klymkowsky, editors. Landes Bioscience, Austin. 12–30.
- Gottardi, C.J., E. Wong, and B.M. Gumbiner. 2001. E-cadherin suppresses cellular transformation by inhibiting β-catenin signaling in an adhesion-independent manner. J. Cell Biol. 153:1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, K.J., and E.A. Bornslaeger. 1999. Desmoplakin. Guidebook to the Extracellular Matrix, Anchor, and Adhesion Proteins. T. Kreis and R. Vale, editors. Oxford University Press, Oxford. 102–105.
- Green, K.J., and C.G. Gaudry. 2000. Are desmosomes more than cell-type specific tethers for intermediate filaments? Nat. Rev. Mol. Cell Biol. 1:208–216. [DOI] [PubMed] [Google Scholar]
- Green, K.J., B. Geiger, J.C.R. Jones, J.C. Talian, and R.D. Goldman. 1987. The relationship between intermediate filaments and microfilaments prior to and during the formation of desmosomes and adherens-type junctions in mouse epidermal keratinocytes. J. Cell Biol. 104:1389–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner, B., B. Stevenson, and A. Grimaldi. 1988. The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J. Cell Biol. 107:1575–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert, C., G. Demers, and D. Galloway. 1992. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J. Virol. 66:2125–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsell, S., and P. Cowin. 2001. Deconstructing desmoplakin. Nat. Cell Biol. 3:E270–E272. [DOI] [PubMed] [Google Scholar]
- Hatzfeld, M. 1999. The armadillo family of structural proteins. Int. Rev. Cytol. 186:179–224. [DOI] [PubMed] [Google Scholar]
- Hofmann, I., C. Mertens, M. Brettel, V. Nimmrich, N. Schnolzer, and H. Herrmann. 2000. Interaction of plakophilins with desmoplakin and intermediate filament proteins: an in vitro analysis. J. Cell Sci. 113:2471–2483. [DOI] [PubMed] [Google Scholar]
- Imamura, Y., M. Itoh, Y. Maeno, S. Tsukita, and A. Nagafuchi. 1999. Functional domains of α-catenin required for the strong state of cadherin-based cell adhesion. J. Cell Biol. 144:1311–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimonis, V., J.J. DiGiovanna, J.-M. Yang, S.Z. Doyle, S.J. Bale, and J.G. Compton. 1994. A mutation in the V1 end domain of keratin 1 in non-epidermolytic palmar-plantar keratoderma. J. Invest. Dermatol. 103:764–769. [DOI] [PubMed] [Google Scholar]
- Kobayashi, S., T. Tanaka, N. Matsuyoshi, and S. Imamura. 1996. Keratin 9 point mutation in the pedigree of epidermolytic hereditary palmoplantar keratoderma perturbs keratin intermediate filament network formation. FEBS Lett. 386:149–155. [DOI] [PubMed] [Google Scholar]
- Koch, P.J., and W.W. Franke. 1994. Desmosomal cadherins: another growing multigene family of adhesion molecules. Curr. Opin. Cell Biol. 6:682–687. [DOI] [PubMed] [Google Scholar]
- Kouklis, P.D., E. Hutton, and E. Fuchs. 1994. Making a connection: direct binding between keratin intermediate filaments and desmosomal proteins. J. Cell Biol. 127:1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs, E.M., M. Goodwin, R.G. Ali, A.D. Paterson, and A.S. Yap. 2002. Cadherin-directed actin assembly. E-cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr. Biol. 12:379–382. [DOI] [PubMed] [Google Scholar]
- Kowalczyk, A.P., E.A. Bornslaeger, J.E. Borgwardt, H.L. Palka, A.S. Dhaliwal, C.M. Corcoran, M.F. Denning, and K.J. Green. 1997. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin–plakoglobin complexes. J. Cell Biol. 139:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk, A.P., E.A. Bornslaeger, S.M. Norvell, H.L. Palka, and K.J. Green. 1999. a. Desmosomes: intercellular adhesive junctions specialized for attachment of intermediate filaments. Int. Rev. Cytol. 185:237–302. [DOI] [PubMed] [Google Scholar]
- Kowalczyk, A.P., M. Hatzfeld, E.A. Bornslaeger, D.S. Kopp, J.E. Borgwardt, C.M. Corcoran, A. Settler, and K.J. Green. 1999. b. The head domain of plakophilin-1 binds to desmoplakin and enhances its recruitment to desmosomes. Implications for cutaneous disease. J. Biol. Chem. 274:18145–18148. [DOI] [PubMed] [Google Scholar]
- Leung, C.L., R.K. Liem, D.A. Parry, and K.J. Green. 2001. The plakin family. J. Cell Sci. 114:3409–3410. [DOI] [PubMed] [Google Scholar]
- Lewis, J.E., J.K. Wahl III, K.M. Sass, P.J. Jensen, K.R. Johnson, and M.J. Wheelock. 1997. Cross-talk between adherens junctions and desmosomes depends on plakoglobin. J. Cell Biol. 136:919–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J.A., J.R. McMillan, C.S. Shemanko, S.K. Runswick, I.M. Leigh, E.B. Lane, D.R. Garrod, and R.A.J. Eady. 1997. Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat. Genet. 17:240–244. [DOI] [PubMed] [Google Scholar]
- McLean, W.H.I., and E.B. Lane. 1995. Intermediate filaments in disease. Curr. Opin. Cell Biol. 7:118–125. [DOI] [PubMed] [Google Scholar]
- McMillan, J.R., and H. Shimizu. 2001. Desmosomes: structure and function in normal and diseased epidermis. J. Dermatol. 28:291–298. [DOI] [PubMed] [Google Scholar]
- Meng, J.-J., E.A. Bornslaeger, K.J. Green, P.M. Steinert, and W. Ip. 1997. Two hybrid analysis reveals fundamental differences in direct interactions between desmoplakin and cell type specific intermediate filaments. J. Biol. Chem. 272:21495–21503. [DOI] [PubMed] [Google Scholar]
- Norgett, E.E., S.J. Hatsell, L. Carvajal-Huerta, J.C. Cabezas, J. Common, P.E. Purkis, N. Whittock, I.M. Leigh, H.P. Stevens, and D.P. Kelsell. 2000. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 9:2761–2766. [DOI] [PubMed] [Google Scholar]
- Ohkubo, T., and M. Ozawa. 1999. p120(ctn) binds to the membrane-proximal region of the E-cadherin cytoplasmic domain and is involved in modulation of adhesion activity. J. Biol. Chem. 274:21409–21415. [DOI] [PubMed] [Google Scholar]
- Ozawa, M., M. Ringwald, and R. Kemler. 1990. Uvomorulin-catenin complex formation is regulated by a specific domain in the cytoplasmic region of the cell adhesion molecule. Proc. Natl. Acad. Sci. USA. 87:4246–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek, S.P., C.E. Schmidt, D.A. Lauffenburger, and A.F. Horwitz. 1996. Integrin dynamics on the tail region of migrating fibroblasts. J. Cell Sci. 109:941–952. [DOI] [PubMed] [Google Scholar]
- Ruhrberg, C., and F.M. Watt. 1997. The plakin family: versatile organisers of cytoskeletal architecture. Curr. Opin. Genet. Dev. 7:392–397. [DOI] [PubMed] [Google Scholar]
- Ruiz, P., V. Brinkmann, B. Ledermann, M. Behrend, C. Grund, C. Thalhammer, F. Vogel, C. Birchmeier, U. Gunthert, W.W. Franke, and W. Birchmeier. 1996. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J. Cell Biol. 135:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelz, M., R. Duden, P. Cowin, and W.W. Franke. 1986. A constitutive transmembrane glycoprotein of Mr 165,000 (desmoglein) in epidermal and non-epidermal desmosomes. II. Immunolocalization and microinjection studies. Eur. J. Cell Biol. 42:184–199. [PubMed] [Google Scholar]
- Schmidt, A., H.W. Heid, S. Schafer, U.A. Nuber, R. Zimbelmann, and W.W. Franke. 1994. Desmosomes and cytoskeletal architecture in epithelial differentiation: cell type-specific plaque components and intermediate filament anchorage. Eur. J. Cell Biol. 65:229–245. [PubMed] [Google Scholar]
- Shao, J.Y., and R.M. Hochmuth. 1999. Mechanical anchoring strength of L-selectin, β2 integrins, and CD45 to neutrophil cytoskeleton and membrane. Biophys. J. 77:587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoyama, Y., S. Hirohashi, S. Hirano, M. Noguchi, Y. Shimosato, M. Takeichi, and O. Abe. 1989. Cadherin cell-adhesion molecules in human epithelial tissues and carcinomas. Cancer Res. 49:2128–2133. [PubMed] [Google Scholar]
- Smith, E.A., and E. Fuchs. 1998. Defining the interactions between intermediate filaments and desmosomes. J. Cell Biol. 141:1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stappenbeck, T.S., and K.J. Green. 1992. The desmoplakin carboxyl terminus coaligns with and specifically disrupts intermediate filament networks when expressed in cultured cells. J. Cell Biol. 116:1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stappenbeck, T.S., E.A. Bornslaeger, C.M. Corcoran, H.H. Luu, M.L.A. Virata, and K.J. Green. 1993. Functional analysis of desmoplakin domains: specification of the interaction with keratin versus vimentin intermediate filament networks. J. Cell Biol. 123:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey, A., D. Pim, A. Murray, K. Osborn, L. Banks, and L. Crawford. 1988. Comparison of the in vitro transforming activities of human papillomavirus types. EMBO J. 7:1815–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreson, M.A., P.Z. Anastasiadis, J.M. Daniel, R.C. Ireton, M.J. Wheelock, K.R. Johnson, D.K. Hummingbird, and A.B. Reynolds. 2000. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J. Cell Biol. 148:189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troyanovsky, S.M., L.G. Eshkind, R.B. Troyanovsky, R.E. Leube, and W.W. Franke. 1993. Contributions of cytoplasmic domains of desmosomal cadherins to desmosome assembly and intermediate filament anchorage. Cell. 72:561–574. [DOI] [PubMed] [Google Scholar]
- Troyanovsky, S.M., R.B. Troyanovsky, L.G. Eshkind, V.A. Krutovskikh, R.E. Leube, and W.W. Franke. 1994. Identification of the plakoglobin-binding domain in desmoglein and its role in plaque assembly and intermediate filament anchorage. J. Cell Biol. 127:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin, V., and E. Fuchs. 2001. Actin dynamics and cell-cell adhesion in epithelia. Curr. Opin. Cell Biol. 13:76–84. [DOI] [PubMed] [Google Scholar]
- Vasioukhin, V., C. Bauer, M. Yin, and E. Fuchs. 2000. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 100:209–219. [DOI] [PubMed] [Google Scholar]
- Vasioukhin, V., E. Bowers, C. Bauer, L. Degenstein, and E. Fuchs. 2001. Desmoplakin is essential in epidermal sheet formation. Nat. Cell Biol. 3:1076–1085. [DOI] [PubMed] [Google Scholar]
- Watabe, M., A. Nagafuchi, S. Tsukita, and M. Takeichi. 1994. Induction of polarized cell-cell association and retardation of growth by activation of the E-cadherin–catenin adhesion system in a dispersed carcinoma line. J. Cell Biol. 127:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, K.L., and W.M. Bement. 2002. F-actin serves as a template for cytokeratin organization in cell free extracts. J. Cell Sci. 115:1373–1382. [DOI] [PubMed] [Google Scholar]
- Yap, A.S., W.M. Brieher, M. Pruschy, and B.M. Gumbiner. 1997. Lateral clustering of the adhesive ectodomain: a fundamental determinant of cadherin function. Curr. Biol. 7:308–315. [DOI] [PubMed] [Google Scholar]
- Yap, A.S., C.M. Niessen, and B.M. Gumbiner. 1998. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J. Cell Biol. 141:779–787. [DOI] [PMC free article] [PubMed] [Google Scholar]