

Abstract
Directed cell movement is a multi-step process requiring an initial spatial polarization that is established by asymmetric stimulation of Rho GTPases, phosphoinositides (PIs), and actin polymerization. We report that the Na-H exchanger isoform 1 (NHE1), a ubiquitously expressed plasma membrane ion exchanger, is necessary for establishing polarity in migrating fibroblasts. In fibroblasts, NHE1 is predominantly localized in lamellipodia, where it functions as a plasma membrane anchor for actin filaments by its direct binding of ezrin/radixin/moesin (ERM) proteins. Migration in a wounding assay was impaired in fibroblasts expressing NHE1 with mutations that independently disrupt ERM binding and cytoskeletal anchoring or ion transport. Disrupting either function of NHE1 impaired polarity, as indicated by loss of directionality, mislocalization of the Golgi apparatus away from the orientation of the wound edge, and inhibition of PI signaling. Both functions of NHE1 were also required for remodeling of focal adhesions. Most notably, lack of ion transport inhibited de-adhesion, resulting in trailing edges that failed to retract. These findings indicate that by regulating asymmetric signals that establish polarity and by coordinating focal adhesion remodeling at the cell front and rear, cytoskeletal anchoring by NHE1 and its localized activity in lamellipodia act cooperatively to integrate cues for directed migration.
Keywords: cell polarity; cytoskeleton; focal adhesion; ERM proteins; calpain
Introduction
In directed cell migration, acquiring spatial asymmetry is a necessary early event to establish polarized leading and trailing edges. In response to the activation of plasma membrane receptors, cell polarity is established by a leading-edge amplification of signaling by phosphoinositides (PIs;* Parent and Devreotes, 1999; Chung et al., 2001) and Rho family GTPases (Stowers et al., 1995; Etienne-Manneville and Hall, 2001), and accumulation of F-actin, which is a driving force for membrane protrusion (Borisy and Svitkina, 2000). However, how early polarity signals are preferentially amplified at the leading edge is unclear because receptors activated by migratory cues, including receptor tyrosine kinases and G protein–coupled receptors, are uniformly distributed at the plasma membrane (Xiao et al., 1997; Servant et al., 1999; Bailly et al., 2000). Therefore, a spatially localized integrator at the leading-edge membrane might act downstream of membrane receptors to asymmetrically amplify signaling and actin polymerization and generate polarity.
Properties of the ubiquitously expressed plasma membrane Na-H exchanger isoform 1 (NHE1) indicate that it could act as a spatially restricted integrator of migratory cues at the leading-edge membrane. In fibroblasts, NHE1 is predominantly localized in lamellipodia, where it anchors the actin cytoskeleton to the plasma membrane by its direct binding of ezrin/radixin/moesin (ERM) actin-binding proteins (Denker et al., 2000). In addition to its role as a cytoskeletal anchor, NHE1 is an ion transport protein, catalyzing an electroneutral exchange of extracellular Na+ for intracellular H+ and regulating intracellular pH homeostasis. Ion transport by NHE1 is stimulated by membrane receptors that respond to migratory cues, including integrins (Schwartz et al., 1991; Tominaga and Barber, 1998), receptor tyrosine kinases (Wakabayashi et al., 1992; Ma et al., 1994; Yan et al., 2001), and G protein–coupled receptors (Bertrand et al., 1994; Hooley et al., 1996; Tominaga et al., 1998). Moreover, NHE1 is necessary for dynamic reorganization of the actin-based cytoskeleton. The assembly of focal adhesions and actin filaments by the activation of integrins and Rho is impaired in fibroblasts lacking NHE1, but rescued by its stable expression (Vexler et al., 1996; Tominaga and Barber, 1998; Tominaga et al., 1998; Denker et al., 2000). Thus, through its localization and its functions as a cytoskeletal anchor and an ion exchanger, NHE1 could integrate migratory cues and spatially amplify asymmetric signaling and actin reorganization in lamellipodia.
We now show that both cytoskeletal anchoring and ion transport by NHE1 are necessary for establishing polarity and for directed cell migration. Using a fibroblast wounding assay, cells expressing NHE1 with mutations that independently impair either ERM binding and cytoskeletal anchoring or ion transport migrate slower than cells expressing wild-type (WT) NHE1. Both mutations result in loss of cell polarity, as indicated by impaired directional movement, misorientation of the Golgi apparatus away from the direction of migration, and inhibition of PI signaling. Additionally, both functions of NHE1 regulate the remodeling of cell adhesions at the cell front and rear, albeit through distinct mechanisms. Cytoskeletal anchoring by NHE1 promotes the assembly of focal complexes and ion translocation is necessary for de-adhesion at the front and rear. These findings suggest that NHE1 functions as a spatially restricted integrator of migratory cues to regulate early polarity signals and to coordinate events occurring at the leading and trailing edges of the cell.
Results
Both cytoskeletal anchoring and ion translocation by NHE1 are necessary for efficient cell migration
The independent effects of cytoskeletal anchoring and ion translocation by NHE1 on cell migration were assessed using NHE-null PS120 fibroblasts stably expressing either WT NHE1, a mutant NHE1 with impaired ERM binding and cytoskeletal anchoring that contains alanine substitutions of lysine and arginine residues in the COOH-terminal juxtamembrane domain (KR/A), or a mutant, ion translocation–defective NHE1 containing an isoleucine substitution for glutamine 266 (Fig. 1 A, E266I). Cell clones expressing similar amounts of WT and mutant NHE1 were selected (Fig. 1 B). As described previously (Denker et al., 2000), the localization of WT and ion translocation–defective NHE1-E266I is primarily restricted to membrane protrusions or lamellipodia, whereas NHE1-KR/A with impaired ERM binding and cytoskeletal anchoring is more uniformly distributed along the plasma membrane (Fig. 1 C).
Figure 1.
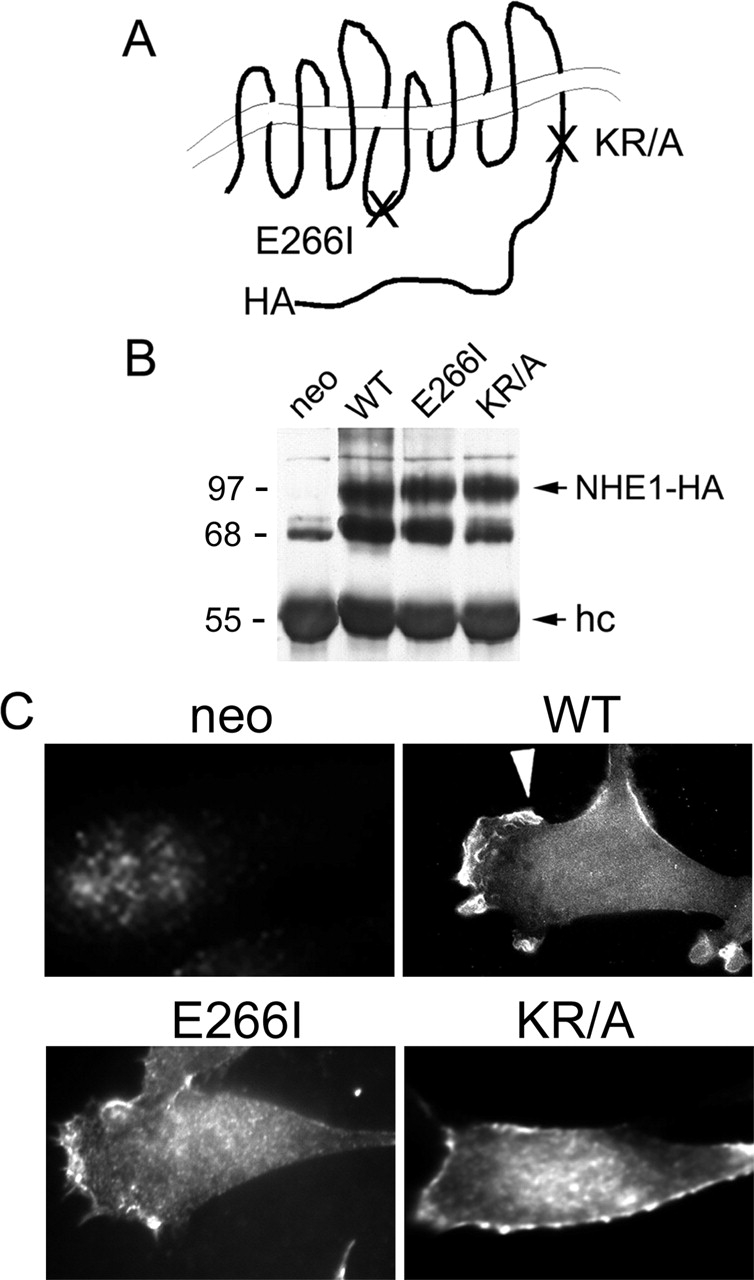
Characterization of cell lines. (A) Schematic diagram of NHE1 indicating positions of the HA-epitope tag, the KR/A mutation (amino acids 553–564 of rat sequence, alanines substituted for lysine and arginine residues) that abolishes ERM binding, and the E266I mutation that abolishes ion translocation. (B) Expression and (C) localization of NHE1 wild-type (WT; arrowhead) and mutant proteins in PS120 fibroblasts. Expression of the selection plasmid alone (neo) served as a control in initial studies. hc, IgG heavy chain.
Migratory rate was monitored in an in vitro wounding assay. Before wounding and throughout the wounding assay, cells were maintained in the presence of serum and 5% CO2, and the medium contained 25 mM NaHCO3 to ensure the function of anion exchangers. Cells expressing WT NHE1 migrated together from opposite sides of the wound edge, and at 10 h had traversed 50% of the original wound width (Fig. 2 A). By 24 h,WT cells had reestablished a monolayer. Cells expressing either the KR/A mutation, which impairs ERM binding and cytoskeletal anchoring, or the E266I mutation, which impairs ion translocation, had decreased migratory rates. At 10 h, KR/A cells had migrated only 30% of the distance of the wound, and by 24 h had migrated over 60–70% of the wound width (Fig. 2 A). Like WT cells, KR/A cells were able to reestablish a monolayer, but only after 30–32 h. E266I cells migrated significantly slower than either WT or KR/A cells. By 10 h, E266I cells lacking NHE1 activity had migrated 10–15% of the wound width, and by 24 h they had migrated over only 20–25% of the wound width (Fig. 2 A). Unlike WT and KR/A cells, E266I cells failed to reestablish a monolayer even after 48 h. Experiments were not extended beyond 48 h because abundant cell death, probably due to high confluency, began to occur within the monolayer. Thus, both functions of NHE1 (cytoskeletal anchoring through ERM binding and ion translocation activity) are required for maximally efficient migration of fibroblasts in a wounding assay.
Figure 2.
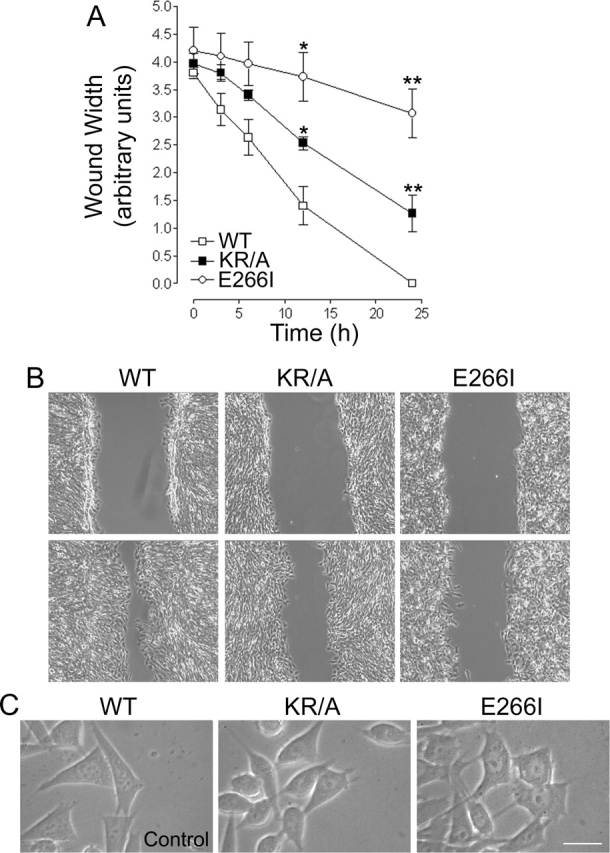
Wounding assays demonstrate that mutations in NHE1 impair cell migration. (A) The migratory rate of fibroblasts in a wounding assay was determined by measuring wound width as a function of time for cells plated on gridded coverslips. Wound closure at 24 h was 100% for WT cells, 60–70% for KR/A cells, and 20–25% for E266I cells. Data are expressed as the mean ± SEM of three experiments. Asterisks indicate a significant difference from WT cells. *, 0.05%; **, 0.01%. (B) Images from time-lapse videos acquired at 0 and 12 h. WT cells migrated together as a sheet, whereas both KR/A and E266I cells displayed asynchronous movement. Videos 1–3 were taken with a 10× phase-contrast objective. (C) High magnification images (63×) of cells at the wound edge demonstrate the fusiform shape of KR/A cells and extended tails in E266I cells. Bar, 10 μm. Videos available at http://www.jcb.org/cgi/content/full/jcb.200208050/DC1.
Time-lapse video microscopy revealed that KR/A and E266I cells had distinct morphological phenotypes. WT cells moved together at a rate of 19.34 ± 1.02 μm/h (mean ± SEM; n = 3 separate wounding assays) as a sheet to reestablish the monolayer (Fig. 2 B and Video 1). Movement was primarily perpendicular to the wound, with very few cells changing their direction to migrate away from the opposing side of the wound. Transient membrane ruffles and lamellipodia, which rapidly (<1 min) retracted with protrusive movement, were observed. The pyramidal cell shape at the migratory front (Fig. 2 C) was retained by de-adhesion and retraction of the trailing edge. The decreased migratory rate of 12.31 ± 1.08 μm/h of KR/A cells was associated with impaired cell polarity and directionality (Fig. 2 B and Video 2). After an initial phase of cohesive movement, cell–cell contacts were lost and the cells often turned parallel to the wound, rather than persisting in a direction perpendicular to the wound. Lamellipodia were not as well developed as in WT cells (Fig. 2 B and Video 2), and cell shape was more fusiform (Fig. 2 C), as described previously (Denker et al., 2000). E266I cells had a markedly decreased migratory rate of 2.88 ± 0.42 μm/h and a dramatically altered migratory phenotype (Fig. 2 B and Video 3). Large fan-shaped lamellipodia and extensive membrane ruffling persisted for 6–8 min. The persistence of lamellipodia appeared to result from sustained adhesion at the leading edge. Like KR/A cells, E266I cells had impaired polarity and directionality, and spent less time moving perpendicular toward the center of the wound and more time moving parallel to the wound edge. Although the cells were motile, net displacement was inhibited by impaired rear de-adhesion, resulting in long, trailing tails that failed to retract (Fig. 2 C).
Loss of NHE1-dependent cytoskeletal anchoring and ion translocation impairs directionality
The impaired directionality of cells expressing mutant NHE1 observed in time-lapse images was confirmed by cell tracking. Tracking of individual cells at the wound edge was used to reveal migratory distance and path (Fig. 3). WT cells traveled along an axis perpendicular to the wound; at 15 h the distance traveled by four representative cells ranged between 85 and 110 pixel units, with an average distance of 98 units. The tracked paths of WT cells showed few turns. In contrast, paths of both KR/A and E266I cells showed multiple turns and clear reversals in direction. KR/A cells traveled between 25 and 70 pixel units with an average distance of 50 units for four representative cells, and E266I cells traveled between 30 and 60 pixel units, with an average distance of 40 units for four representative cells. These data indicate that although KR/A and E266I cells are motile, their migratory rate was at least in part impaired by loss of directionality.
Figure 3.

Cell tracking demonstrates that both ion translocation and cytoskeletal anchoring by NHE1 are required for directionality. The paths and distance traveled by representative cells at the wound edge were plotted as a function of time over 16 h. WT cells moved farthest with few turns and no reversals. KR/A and E266I cells turned frequently and reversed direction.
Loss of NHE1-dependent cytoskeletal anchoring and ion translocation impairs polarity
Acquiring spatial asymmetry is an early event necessary for directed cell movement, and in time-lapse recordings of the wounding assay, both KR/A and E226I cells appeared to have impaired polarity. To obtain a more direct measure of cell polarity, we examined two markers of polarity during migration: the orientation of the Golgi apparatus relative to the nucleus and distribution of the serine/threonine kinase Akt at the leading-edge plasma membrane. In migrating cells, the Golgi apparatus polarizes to the leading-edge face of the nucleus (Nobes and Hall, 1999; Kulkarni et al., 2000). Costaining for the Golgi matrix protein Giantin and nuclei indicated that 85 ± 2% of WT cells displayed this orientation (Fig. 4 A). In contrast, only 32 ± 2% of KR/A cells and 28 ± 3% of E266I cells had a front-facing Golgi apparatus, and the majority of both cell types had a mislocalized Golgi apparatus oriented away from the direction of migration (Fig. 4 A, asterisks). Cells displaying a complete reversal of orientation with the Golgi apparatus behind the nucleus were frequently observed in both mutant cell lines, but never in WT cells.
Figure 4.

Both ion translocation and cytoskeletal anchoring by NHE1 regulate cell polarity. (A) Cells migrating in a wounding assay were fixed after 10 h and processed for immunolocalization of the Golgi complex (anti-Giantin antibody) and nuclei (DAPI). Representative fields are shown, with arrows indicating the direction of migration and asterisks indicating cells with misoriented Golgi apparatus. The large cluster of nuclei in the bottom left panel of E266I cells was not included in determining the percentage of cells with correct Golgi apparatus orientation. (B) GFP-PH-Akt was detected in the leading-edge membrane of WT, but not KR/A or E266I cells moving at the front of a migrating monolayer. GFP-PH-Akt was primarily cytosolic in both mutant cell lines. Images are representative of 90% of cells at the wound edge expressing GFP-PH-Akt in three separate cell preparations. Arrows indicate the direction of migration. (C) Immunoblotting with antibodies to GFP indicated that the relative expression level of GFP-PH-Akt was similar in WT and KR/A cells, but higher in E266I cells. There was no GFP signal in WT cells transfected with a vector control. Immunoblotting for β-actin confirmed equivalent loading of protein in all samples. Bars: (A) 5 μm; (B) 2 μm.
In migrating cells, internal signaling asymmetries, which contribute to establishing cell polarity, are amplified by localized activation of PI3-kinases at the leading edge (Parent and Devreotes, 1999; Chung et al., 2001). The lipid products of PI3-kinases contain binding sites for pleckstrin homology (PH) domain-containing proteins, such as the serine/threonine kinase Akt/PKB, which is recruited to the leading-edge membrane of migrating cells. When expressed in cells, a GFP fusion protein containing the PH domain of Akt is localized similarly to native Akt and has been used as a marker for the leading edge of migrating fibroblasts (Haugh et al., 2000), neutrophils (Servant et al., 2000), and Dictyostelium (Meili et al., 1999). Therefore, we monitored cells transiently expressing a GFP-tagged PH domain of Akt during the wounding assay to verify the loss of polarity indicated by our findings on Golgi apparatus orientation. This second assay not only confirmed the previous results, but also indicated that the mechanism of impaired polarity differs between the two mutant cell lines. In WT cells, GFP-PH-Akt was detected at the leading-edge membrane of cells at the migrating front (Fig. 4 B). In KR/A cells GFP-PH-Akt was more diffuse, although a weak signal was detected at the membrane. In 80% of the cells expressing GFP-PH-Akt, this weak, membrane-localized signal, however, was not at the leading edge facing the wound, but misoriented by >90° relative to the direction of migration (Fig. 4 B). Hence, although KR/A cells were able to establish polarized lamellipodia, these membrane protrusions had impaired directionality. This finding supports the observation from time-lapse microscopy that in the absence of cytoskeletal anchoring, KR/A cells spent more time moving parallel to the wound and the observation from cell tracking that these cells were less efficient in their forward movement. In E266I cells lacking NHE1 activity, GFP-PH-Akt retained a diffuse, cytosolic distribution and was not recruited to the plasma membrane. Immunoblotting of cell lysates indicated that the expression level of GFP-PH-Akt was similar in WT and KR/A cells, but higher in E266I cells (Fig. 4 C). These findings suggest that both cytoskeletal anchoring and ion translocation by NHE1 are necessary for establishing or maintaining polarity in migrating cells, although the mechanisms mediating their effects are likely to be distinct.
Loss of cytoskeletal anchoring by NHE1 impairs development of a primary lamellipod
One difference in the impaired polarity associated with both mutant NHE1 proteins was the inability of KR/A cells, but not E266I cells, to establish a primary lamellipod. Previously, we found that compared with the morphology of WT and E266I cells, loss of cytoskeletal anchoring by NHE1 in KR/A cells results in a more fusiform cell shape and smaller lamellipodia (Denker et al., 2000). A KR/A-specific effect on cell morphology was also observed in migrating cells. At the leading edge of the migrating front, KR/A cells had markedly more and smaller protrusions compared with WT and E266I cells (Fig. 5 A). At the wound edge, 55 ± 2% (mean ± SEM; n = 79) of the KR/A cells had three or more distinct protrusions, in contrast to 13 ± 3% (n = 73) and 24 ± 4% (n = 65) of WT and E266I cells, respectively. An increased number of membrane protrusions in KR/A cells was dramatically shown in a modified invasion assay of cells plated on a Transwell filter coated with a Matrigel™ substrate. At 4 h after plating, before invasion had begun, WT and E266I cells developed one major protrusion (Fig. 5 B). In marked contrast, KR/A cells developed multiple protrusions (>6 per cell) that persisted over time (Fig. 5 B). These distinct phenotypes were maintained for up to 12 h after plating.
Figure 5.

ERM binding and cytoskeletal anchoring by NHE1 are required for development of a primary lamellipod. (A) Migrating WT and E266I cells exhibit one broad lamellipod (arrows), whereas KR/A cells have multiple, small protrusions (arrowheads). Bar, 5 μm. (B) When plated on Matrigel™ for 4 h, WT and E266I cells develop one primary protrusion (arrowhead), whereas KR/A cells develop multiple protrusions. (A and B) TRITC-phalloidin staining.
Cytoskeletal anchoring and ion translocation by NHE1 regulate focal adhesion remodeling through distinct mechanisms
Although the independent loss of either NHE1 functions impaired cell polarity and directionality, KR/A and E266I cells had distinct phenotypes and migratory rates because of differences in focal adhesion remodeling. In KR/A cells, the assembly of focal adhesions was impaired, and in E266I cells, de-adhesion at the front and rear was inhibited. Immunostaining for the focal adhesion–associated protein paxillin revealed that compared with WT cells, migrating KR/A cells had fewer and smaller focal adhesions (Fig. 6, A and B), consistent with our previous findings that ERM binding and cytoskeletal anchoring by NHE1 are required for integrin- and Rho-dependent assembly of focal adhesions and actin stress fibers (Tominaga and Barber, 1998; Denker et al., 2000). In contrast, E266I cells had an increased intensity of paxillin labeling at both the front and the rear compared with WT cells, indicating an increase in the number and size of focal adhesions (Fig. 6, A and B). E266I cells have abundant focal adhesions most likely because cytoskeletal anchoring by NHE1 is intact and focal adhesions assemble, but in the absence of NHE1 activity disassembly may be impaired. In migrating fibroblasts, a front-facing protrusion is transiently stabilized by newly formed focal contacts, which then dissociate or are displaced rearward to facilitate pseudopod retraction and translocation (Lauffenburger and Horwitz, 1996). The larger and more abundant focal adhesions at the front of E266I cells likely inhibited lamellipod retraction, resulting in persistent ruffling (Fig. 2 B and Video 3). Increased adhesion at the rear likely impaired de-adhesion, resulting in long tail extensions that failed to retract (Fig. 2 B and Video 3). Therefore, the two functions of NHE1 act cooperatively to remodel focal adhesions in migrating cells, with cytoskeletal anchoring being necessary for the assembly of focal complexes and ion translocation activity promoting de-adhesion.
Figure 6.
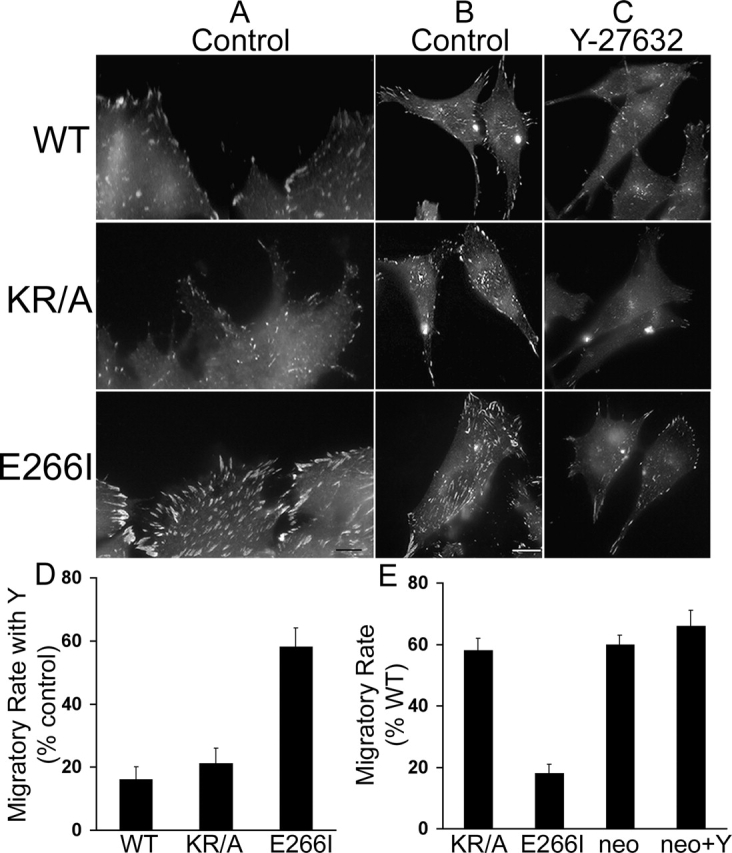
Inhibition of focal adhesion turnover in the absence of NHE1 activity is rate-limiting for migration speed. Cells migrating in the absence (Control) or presence of 10 μM Y-27632 for 15 h were fixed and labeled for paxillin. (A–C). Paxillin labeling of control cells at the wound edge (A) and of representative cells that had migrated into the wound (B) to visualize focal contacts at the front and rear of the cell. Compared with WT cells, KR/A cells have reduced paxillin labeling, whereas E266I cells have increased paxillin labeling. (C) The abundance and intensity of paxillin labeling is decreased in cells treated with 10 μM Y-27632. (D) Migratory rate of cells in the absence or presence of Y-27632 was determined between 0 and 12 h and expressed as a percentage of increase induced by Y-27632 over control rate. (E) Migratory rates of KR/A cells, E266I cells, and NHE1-null PS120neo cells in the absence or presence of Y-27632 were determined between 0 and 12 h and expressed as percentage of the migratory rate of WT cells. Data represent the mean ± SEM of three separate wounding preparations. Bars: (A) 1 μm; (B) 5 μm.
Impaired de-adhesion is rate-limiting for the migration of cells lacking NHE1 activity
In slowly moving fibroblasts, the rate-limiting step for forward migration is de-adhesion and retraction of the trailing edge (Palecek et al., 1998). If the slower migratory rate of E266I cells compared with KR/A cells was due to impaired turnover of focal adhesions, then inhibiting focal adhesion assembly in E266I cells should increase migratory rate. Activation of Rho and its downstream kinase ROCK plays a central role in focal adhesion assembly (Frame and Brunton, 2002), and direct phosphorylation of NHE1 by ROCK is necessary for focal adhesion assembly in response to integrin activation (Tominaga and Barber, 1998; Tominaga et al., 1998). Pharmacological inhibition of ROCK with 10 μM of the pyridine derivative Y-27632 decreased the abundance and intensity of paxillin labeling in all three NHE1 cell lines, indicating a decrease in the number and size of focal adhesions (Fig. 6 C). In WT cells, the intensity of paxillin labeling in peripheral focal contacts decreased, but punctate, intracellular labeling increased, suggesting impaired recruitment of paxillin to the cell edge. Paxillin labeling was only slightly decreased in KR/A cells, which already had small, less organized focal contacts, indicating that cytoskeletal anchoring by NHE1 is a major (but not exclusive) component of ROCK-induced focal adhesion assembly. The effect of inhibiting ROCK activity on paxillin labeling was most dramatic in E266I cells, resulting in the loss of abundant and intense staining. Consistent with impaired de-adhesion being rate-limiting in E226I cells, inhibiting ROCK activity with Y-27632 caused a 58 ± 6% increase in the migratory rate of these cells compared with a 16 ± 4% and 21 ± 5% increase in WT and KR/A cells, respectively (Fig. 6 D). Conflicting findings have been reported for the role of Rho and ROCK in determining migratory rate. When Rho is inhibited with C3 toxin or ROCK activity is blocked with Y-27632, migratory rate is increased in fibroblasts (Nobes and Hall, 1999), but decreased in leukocytes (Alblas et al., 2001; Worthylake et al., 2001). These differences likely reflect cell-specific effects of Rho and ROCK on the assembly of focal adhesions. In fibroblasts, Rho and ROCK play a critical role in assembling focal adhesions, but in leukocytes, focal adhesions are not formed. Therefore, in fibroblasts, inhibiting Rho and ROCK activity decreases focal adhesion assembly, which removes a rate-limiting adhesive component. PS120neo cells lacking NHE1 have impaired focal adhesion assembly (Tominaga and Barber, 1998; Denker et al., 2000) and a migratory rate that is faster than E266I cells and similar to KR/A cells (Fig. 6 E). Like KR/A cells, the decreased migratory rate of PS120neo cells compared with WT cells was likely due to impaired polarity because PS120neo cells also had a misoriented Golgi apparatus and loss of GFP-Akt at the plasma membrane (unpublished data). Moreover, like KR/A and WT cells, inhibiting ROCK activity with Y-27632 increased the migratory rate of PS120neo cells only slightly (Fig. 6 E; 15 ± 5%). Together, these findings suggest that impaired de-adhesion, most likely due to an inhibition of focal adhesion disassembly, is a predominant rate-limiting factor in the migration of E266I cells.
The impaired focal adhesion remodeling seen with both the KR/A and E266I mutations and with inhibiting ROCK activity was associated with decreased activity of the cysteine protease calpain. Calpain activity regulates rear de-adhesion and migratory rate by cleaving integrin–cytoskeletal linkages (Huttenlocher et al., 1997; Glading et al., 2000). Fibroblasts treated with inhibitors of calpain activity have elongated tails and larger peripheral focal adhesions (Huttenlocher et al., 1997), like the morphology of E266I cells. Using a live cell assay (Glading et al., 2000), we found that calpain was activated in WT cells at the wound edge (Fig. 7 A). All WT cells at the migratory front displayed an intense fluorescence signal of calpain activity that was consistently detected three to four cell layers back into the wound. In E266I cells lacking NHE1 activity, a markedly reduced fluorescence signal was observed along the wound edge (Fig. 7 A). Importantly, fluorescence was detected only in cells that had moved into the wound, immediately at the front of the migrating monolayer. Although loss of cytoskeletal anchoring by NHE1 was not associated with impaired rear de-adhesion, calpain activity was also reduced in KR/A cells. WT cells often displayed a punctate fluorescence signal that was not observed in either KR/A or E266I cells, although the physiological significance of this pattern is unknown. Immunoblotting of cell lysates for both μ-calpain and its endogenous inhibitor calpastatin (Fig. 7 B) indicated that the decreased calpain activity in KR/A and E266I cells was not due to changes in the expression of these proteins. Although calpain activity regulates focal adhesion disassembly (Huttenlocher et al., 1997), recent findings suggest that it is also necessary for focal adhesion formation (Bialkowska et al., 2000). Consistent with a role for calpain activity in focal adhesion assembly, ROCK inhibition caused a marked decrease in calpain activity in WT and E266I cells, but had little effect in KR/A cells (Fig. 7 A).
Figure 7.

NHE1 and ROCK regulate calpain activity. (A) Top, calpain activity in control cells, indicated by bright punctate and cytosolic fluorescence, was detected along the front of migrating WT (but not E266I) cells. In KR/A cells, the diffuse, but not the punctate, fluorescence pattern was observed. Bottom, calpain activity in cells treated with Y-27632. (B) Immunoblotting indicated equal abundance of μ-calpain and calpastatin in the three cell lines. Images are representative of cells at the wound edge in four separate wounding assays for each cell type. Bars, 20 μm.
Discussion
Directed cell movement is an integrated multi-step process requiring an initial spatial asymmetry that establishes a functional and morphological polarity and the formation of a distinct front and rear. Once polarity has been established, subsequent steps include protrusion and adhesion of a leading-edge membrane, traction of the cell body, and de-adhesion of focal complexes at the trailing edge followed by retraction (Lauffenburger and Horwitz, 1996). Our current findings indicate that the localized ion transport activity of NHE1 at the leading edge is necessary for a number of stages in the migratory response, including polarization, protrusion, and adhesion at the front and rear of the cell. Due to its localization and interaction with ERM proteins in the lamellipod, together with its ability to respond to changes in both extracellular and intracellular environments, NHE1 is a prime candidate to sense, transmit, and integrate signals at the leading edge of migrating cells. Although pharmacological inhibition of NHE1 was previously shown to inhibit chemotaxis of neutrophils (Simchowitz and Cragoe, 1986) and leukocytes (Ritter et al., 1998) and the migratory rate of endothelial cells (Bussolino et al., 1989) and epithelial cells (Klein et al., 2000; Lagana et al., 2000; Reshkin et al., 2000), distinct effects of NHE1 on stages of the migratory response have not been determined. Additionally, the relative contributions of the two functions of NHE1 in cytoskeletal anchoring and ion translocation have not previously been examined.
Localized ion translocation by NHE1 at the leading edge is necessary for either generating or maintaining polarity. Cell polarity was impaired in fibroblasts expressing an ion translocation–defective NHE1-E266I, which retains cytoskeletal anchoring and localization in lamellipodia, or an anchoring-defective NHE1-KR/A, which retains ion translocation but is uncoupled from actin filaments and is mislocalized along the plasma membrane. Impaired polarity in both cell types was indicated by loss of directional movement perpendicular to the wound and misorientation of the Golgi apparatus away from the direction of migration. Moreover, both functions of NHE1 impaired PI3-kinase signaling, which is a well-established early polarity signal (Meili et al., 1999; Servant et al., 2000). Membrane recruitment of Akt, a downstream target of PI3-kinases, was completely abolished with loss of transport activity, and was decreased and misoriented away from the leading edge with loss of cytoskeletal anchoring. Hence, ion translocation by NHE1 promotes the recruitment of Akt, and localized ion transport at the leading edge directs recruitment to the correct plasma membrane domain.
Loss of cytoskeletal anchoring by NHE1, but not loss of ion translocation, impaired formation of a primary lamellipod and resulted in multiple pseudopodia extending in all directions. The extension of multiple pseudopodia is also seen in cells null for Akt (Meili et al., 1999); however, loss of membrane recruitment of Akt was probably not the exclusive cause of multiple pseudopodia in KR/A cells because E266I cells also had impaired Akt recruitment but retained a primary protrusion. Loss of cytoskeletal anchoring by NHE1 could remove a restrictive signal for lamellipodia formation or, alternatively, with loss of cytoskeletal anchoring the redistribution of NHE1 from a distinct clustered localization at the leading-edge membrane to a more uniform localization along the plasma membrane could create multiple sites for membrane protrusion. Although localized proton fluxes have not been detected in fibroblasts (Grinstein et al., 1993), activation of NHE1 likely increases cytoplasmic pH at the leading edge, which could promote the pH-dependent activation of proteins, such as ADF/cofilin (Bernstein et al., 2000) and gelsolin (Maciver et al., 1998), that regulate actin polymerization at the cell periphery and hence, membrane protrusion. An important future direction is to determine whether localized NHE1 activity regulates F-actin assembly at the leading edge.
Ion translocation and cytoskeletal anchoring by NHE1 act coordinately to regulate focal adhesion remodeling. Previously, we reported that cytoskeletal anchoring is necessary for focal adhesion assembly (Denker et al., 2000), and our current findings indicate that ion translocation promotes the remodeling of focal contacts at both the cell front and rear. These distinct actions were confirmed by inhibiting ROCK activity, which attenuates focal adhesion assembly. In KR/A cells, inhibiting ROCK activity had little effect on the abundance of focal adhesions or migratory rate. In contrast, blocking ROCK activity in E266I cells caused a marked attenuation of focal adhesions and a 50% increase in migratory rate. Hence, in E266I cells, NHE1 anchoring is intact and focal adhesions assemble but are not remodeled in the absence of ion translocation. In KR/A cells, which have impaired actin filament assembly (Denker et al., 2000), focal adhesion formation is reduced, and because ion translocation is intact, remodeling reduces the net abundance of focal contacts.
In migrating fibroblasts, a front-facing protrusion is transiently stabilized by newly formed focal contacts, which then dissociate or are displaced rearward to facilitate pseudopod retraction and translocation (Lauffenburger and Horwitz, 1996). In E266I cells, the formation of large fan-shaped lamellipodia that failed to retract was likely due to impaired remodeling of leading-edge focal contacts. Loss of ion translocation by NHE1 also inhibited focal adhesion disassembly at the trailing edge, resulting in elongated tails and impaired retraction. In slowly migrating cells, like fibroblasts, with intermediate and high levels of adhesiveness, rear retraction is rate-limiting for the speed of movement and at the rear of the cell large fractions of integrin structures are released from the cytoskeleton and remain on the substratum (Palecek et al., 1998). Increased activity of the cysteine protease calpain regulates rear de-adhesion and migratory rate by cleaving integrin–cytoskeletal linkages (Huttenlocher et al., 1997; Glading et al., 2000). Consistent with these findings, calpain activity was decreased with loss of ion translocation by NHE1. It is currently unclear how ion translocation by NHE1 regulates calpain activity. Increases in pH stimulate calpain activity (Zhao et al., 1998), and the cytosolic pH of E266I cells is 0.2–0.3 pH units lower than in WT cells (Denker et al., 2000). However, calpain activity was also attenuated in KR/A cells that had few focal contacts. Because calpain activity is down-regulated in the absence of extensive focal contact remodeling (Bialkowska et al., 2000), the decreased calpain activity associated with loss of both NHE1 activity and anchoring could be mediated by a feedback mechanism involving the focal contact itself. Loss of NHE1 activity markedly decreases migratory rate, hence the need for remodeling is reduced, focal adhesions are more stable, and calpain activity inhibited. Loss of cytoskeletal anchoring by NHE1 results in the assembly of fewer and smaller focal contacts compared with WT cells, which limits the activation of calpain.
Together, our findings indicate that NHE1 acts as a spatially restricted integrator linking migratory cues to the activation of polarity signals at the leading edge of migrating cells (Fig. 8). NHE1 is activated by receptors responding to migratory cues, including receptor tyrosine kinases (Wakabayashi et al., 1992; Ma et al., 1994; Yan et al., 2001), integrins (Schwartz et al., 1991; Tominaga and Barber, 1998), and G protein–coupled receptors (Bertrand et al., 1994; Hooley et al., 1996; Tominaga et al., 1998), and it acts upstream of early polarity signals. Moreover, the ability of NHE1 to regulate de-adhesion at the leading and trailing edge indicates that it integrates spatially distinct events at the front and rear. The current challenge is to determine precisely how the localized activity of NHE1 regulates polarity signals and focal adhesion remodeling. Increases in cytosolic pH or cell volume in response to NHE1 activity are likely important, and because cytoskeletal anchoring restricts the activity of NHE1 to the leading edge, the impaired migratory response of KR/A cells could simply reflect the loss of localized ion translocation. However, we cannot rule out migratory effects of the KR/A mutation due to loss of ERM binding and actin filament assembly. Disrupting the binding of ERM proteins to NHE1 alters their localization from the leading edge of lamellipodia to a more uniform distribution along the cell membrane (Denker et al., 2000), and the activation and localization of ERM proteins are critical for focal adhesion assembly (Mackay et al., 1997) and cell migration (Crepaldi et al., 1997; Ng et al., 2001). Additionally, the KR/A mutation disrupts the tethering of actin filaments to the leading-edge membrane (Denker et al., 2000), which could impair actin remodeling in lamellipodia. Distinguishing the relative contributions of ERM mislocalization, loss of actin filament tethering to the leading-edge membrane, and lack of NHE1 activity in lamellipodia to the migratory response of KR/A cells will require approaches that localize NHE1 to lamellipodia in the absence of ERM binding. The ability to anchor the actin cytoskeleton is shared by several classes of ion transport proteins, including ion channels, ion exchangers, and P-type ATPases (Denker and Barber, 2002). In neurons, cytoskeletal anchoring by voltage-dependent Na+ channels restricts their localization and clustering at axon initial segments, which is necessary to generate local current and to propagate a self-generating action potential (Zhou et al., 1998), and in epithelial cells, cytoskeletal anchoring by the Na+-K+ ATPase maintains its basolateral localization, which is necessary for maintaining cell polarity (Piepenhagen and Nelson, 1998). We now show that in fibroblasts, cytoskeletal anchoring by NHE1 localizes NHE1 to lamellipodia where it spatially integrates migratory cues to regulate cell polarity and adhesion.
Figure 8.

NHE1 integrates migratory cues to spatially amplify early polarity signals and to regulate focal adhesion remodeling. Membrane receptors, including receptor tyrosine kinases (RTK), integrins, and G protein–coupled receptors (GPCR), receive migratory cues. All three classes of receptors activate NHE1, which is spatially restricted to the leading-edge membrane of lamellipodia by cytoskeletal anchoring through the binding of ERM proteins. Localized NHE1 activity integrates signals from diverse membrane receptors to coordinate early polarity signals and promote focal adhesion remodeling. The two functions of NHE1 have distinct effects on focal adhesion remodeling. Cytoskeletal anchoring is necessary for focal adhesion assembly and ion translocation is necessary for de-adhesion.
Materials and methods
Cell culture
NHE1-null fibroblasts stably expressing either WT NHE1 (WT cells) or NHE1 mutants (KR/A and E266I cells) were described previously (Denker et al., 2000). All NHE1 constructs contained an HA-epitope tag at the COOH terminus. Cells were maintained in DME-H21 medium containing 25 mM NaHCO3 and supplemented with 5% FBS and Pen-Strep (growth medium; GIBCO BRL) at 5% CO2. Cell stocks were periodically subjected to G418 selection (WT, KR/A, and E266I) and acid loading in a Hepes buffer (WT and KR/A) to maintain expression of NHE1. Immunoprecipitation of WT and mutant NHE1 was performed to ensure their equal expression in the cell lines, and immunolabeling of NHE1-HA in all cell types was periodically performed to verify expression and localization.
Wounding assays
Before wounding and throughout the wounding assay, cells were maintained at 5% CO2 in growth medium to ensure the function of anion exchangers. Fibroblasts were grown to confluence in 6-well plates and wounded with a P200 pipette tip. Wounded monolayers were washed three times with growth medium and returned to the incubator to recover from wounding before experiments were begun. Plates were placed on a motorized stage equipped with an environmental chamber (Carl Zeiss MicroImaging, Inc.) and maintained at 37°C and 5% CO2. Time-lapse images were collected with a SPOT-RT CCD camera (Molecular Dynamics) every 5 min using the Openlab™ software system (Improvision). Images were compiled every 0.1 s to generate QuickTime movies. Migratory rates in Fig. 2 were determined for cells plated and wounded on gridded coverslips (Eppendorf), and maintained in an incubator at 5% CO2. At the indicated times, plates were removed and monolayers were photographed using the grid as a marker. Wound width was measured on hard copy prints of the images. Migratory rates were determined from time-lapse recordings by using the Openlab™ software region-of-interest measurement tool to determine the wound width on layers selected at 0 and 12 h. Individual cell paths were determined for leading-edge cells at four uniformly spaced points along the wound edge. Cells were tracked using the point counter on every 150th layer, and selected layers were compiled into a separate file to obtain consecutive layers for measurements. To determine the number of membrane protrusions, cells were fixed at 10 h after wounding and stained with phalloidin. Cells at the leading edge of the wound were scored as having three or more protrusions in two fields of three separate wounding assays (six total fields; >50 cells).
For treatment with the ROCK inhibitor Y-27632 (provided by T. Hamashaki, Yoshitomi Pharmaceutical Industries, Inc., Osaka, Japan), wounded monolayers in 6-well plates were returned to the incubator for 1 h before drug treatment. Y-27632 was added to cultures at a final concentration of 10 μM from a 30-mM stock in DMSO. Control plates received 0.01% DMSO. Time-lapse imaging was performed as described above, using a magnification of 10. High magnification images (63×) were collected from separate experiments.
Immunostaining
For detection of HA-tagged NHE1, cells were processed as described previously (Denker et al., 2000). Unless otherwise noted, all other immunocytochemical procedures were performed on cells fixed in 2% PFA in PBS and permeabilized either with 0.1% Triton X-100 for 10 min or with −20°C acetone for 3 min. The following antibodies and dilutions were used: anti-HA mAb (12CA5; 1/1,000; Roche Molecular Biochemicals), TRITC-conjugated phalloidin (1/10,000; Molecular Probes, Inc.), and anti–paxillin mAb (clone Z035, 1/200; Zymed Laboratories). FITC- or Texas red–conjugated secondary antibodies (Molecular Probes, Inc.) were used at 1/200.
Golgi apparatus orientation
Orientation of the Golgi apparatus relative to the nucleus was scored as described previously (Kupfer et al., 1982). At 10 h after wounding, cells were fixed in 2% PFA and stained for the Golgi matrix protein Giantin (anti-Giantin pAb [1/200; Biomol]) and with DAPI (1/10,000; Molecular Probes, Inc.) to visualize nuclei. Cells were scored as having a mislocalized Golgi apparatus if more than 50% of the Giantin immunofluoresence was outside of a 60° sector from a line bisecting the nucleus and perpendicular to the wound edge. All cells (>100) in four random fields of three separate wounding assays were examined.
GFP transfections
Cells plated in 100-mm dishes to obtain 60% confluence after 24 h were transfected with 2.0 μg of a GFP fusion protein containing the PH domain of the serine/threonine kinase Akt (GFP-PH-Akt; provided by T. Balla, National Institutes of Health, Bethesda, MD) by using the Effectene Reagent (QIAGEN) according to the manufacturer's recommendations. Transfections were stopped after 8–10 h, and the cells were washed with growth medium and allowed to recover overnight. Cells were trypsinized and replated on coverslips placed in 6-well plates at a density to achieve confluence in 12 h. Monolayers were wounded and live-cell imaging of GFP at the leading edge was analyzed after 12 h of migration. Images were collected with a SPOT-RT CCD camera. Identical exposure settings were used for each cell line. All post-acquisition processing was performed in Adobe Photoshop®.
Matrigel™ assay
2,000 cells were plated in the inner wells of 8-μm Transwell filters (Costar) coated with 10 μl of Matrigel™ (Becton Dickinson). Growth medium was added to the outer well and plated cells were returned to the incubator for 4 h. Filters were fixed in 2% PFA, permeabilized, and stained with TRITC-phalloidin.
Calpain activity assay
Calpain activity was determined in live cells as described previously (Glading et al., 2000). Cells were plated on coverslips and wounded at confluence. 12 h after wounding, CMAC, t-BOC-Leu-Met dye (Molecular Probes, Inc.) from a 1-mM stock in H2O was added to cell cultures for a final concentration of 1.0 μM. Cultures were returned to the incubator for 20 min. Individual coverslips were then washed quickly in PBS, placed face down on glass slides, and images were immediately collected using identical exposure settings. Images were collected from random fields of cells at the wound edge in four separate wounding assays for each cell type.
Immunoblotting
Cell monolayers at 95% confluence were lysed in buffer on ice for 30 min. Lysates were centrifuged (850 g for 10 min at 4°C) to obtain post-nuclear supernatant and nuclear pellet fractions. 80 μg of post-nuclear supernatant was processed for 10% SDS-PAGE. Separated proteins were transferred to polyvinylidene difluoride membranes and processed for immunoblotting as described previously (Denker et al., 2000). Membranes were probed with anti-HA mAb (12CA5; Roche Molecular Biochemicals) for NHE1-HA, with anti-GFP (Roche) for GFP-PH-AKT, with anti–actin (C4; CHEMICON International), with anti-calpain IgG (BIOMOL Research Laboratories, Inc.), and with anti-calpastatin IgG (BIOMOL Research Laboratories, Inc.). Immune complexes were detected with ECL (Amersham Biosciences).
Online supplemental material
Time-lapse video microscopy (Fig. 2) shows wounded cell monolayers placed in an environmental chamber controlled by Improvision's Openlab™ software. Images were collected into a layered image file format every 5 min over a 30-h period. Layers were compiled into QuickTime movies at regular 0.1-s intervals from t = 2 h (to allow for cells to stabilize in the chamber) to the time the cells just reestablished a monolayer (∼20 h for WT cells and ∼30 h for KR/A cells). For E266I cells, which failed to establish a monolayer, layers from t = 2 h to t = 30 h were used. Movies were compressed using Sorenson codex. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200208050/DC1.
Acknowledgments
We thank Henry Bourne and Eric Brown for comments and suggestions, and Mary McKenney for editing the manuscript.
This work was supported by National Institutes of Health grants GM58642 to D.L. Barber and T32 DE07204 to S.P. Denker.
The online version of this article includes supplemental material.
S.P. Denker's present address is Xenogen Corporation, Alameda, CA 94501.
Footnotes
Abbreviations used in this paper: ERM, ezrin/radixin/moesin; NHE1, Na-H exchanger isoform 1; PH, pleckstrin homology; PI, phosphoinositide; WT, wild type.
References
- Alblas, J., L. Ulfman, P. Hordijk, and L. Koenderman. 2001. Activation of RhoA and ROCK are essential for detachment of migrating leukocytes. Mol. Biol. Cell. 12:2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly, M., J. Wyckoff, B. Bouzahzah, R. Hammerman, V. Sylvestre, M. Cammer, R. Pestell, and J.E. Segall. 2000. Epidermal growth factor receptor distribution during chemotactic responses. Mol. Biol. Cell. 11:3873–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, B.W., W.B. Painter, H. Chen, L.S. Minamide, H. Abe, and J.R. Bamburg. 2000. Intracellular pH modulation of ADF/cofilin proteins. Cell Motil. Cytoskeleton. 47:319–336. [DOI] [PubMed] [Google Scholar]
- Bertrand, B., S. Wakabayashi, T. Ikeda, J. Pouyssegur, and M. Shigekawa. 1994. The Na+/H+ exchanger isoform 1 (NHE1) is a novel member of the calmodulin-binding proteins. Identification and characterization of calmodulin-binding sites. J. Biol. Chem. 269:13703–13709. [PubMed] [Google Scholar]
- Bialkowska, K., S. Kulkarni, X. Du, D.E. Goll, T.C. Saido, and J.E. Fox. 2000. Evidence that beta3 integrin-induced Rac activation involves the calpain-dependent formation of integrin clusters that are distinct from the focal complexes and focal adhesions that form as Rac and RhoA become active. J. Cell Biol. 151:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisy, G.G., and T.M. Svitkina. 2000. Actin machinery: pushing the envelope. Curr. Opin. Cell Biol. 12:104–112. [DOI] [PubMed] [Google Scholar]
- Bussolino, F., J.M. Wang, F. Turrini, D. Alessi, D. Ghigo, C. Costamagna, G. Pescarmona, A. Mantovani, and A. Bosia. 1989. Stimulation of the Na+/H+ exchanger in human endothelial cells activated by granulocyte- and granulocyte-macrophage-colony-stimulating factor. Evidence for a role in proliferation and migration. J. Biol. Chem. 264:18284–18287. [PubMed] [Google Scholar]
- Chung, C.Y., S. Funamoto, and R.A. Firtel. 2001. Signaling pathways controlling cell polarity and chemotaxis. Trends Biochem. Sci. 26:557–566. [DOI] [PubMed] [Google Scholar]
- Crepaldi, T., A. Gautreau, P.M. Comoglio, D. Louvard, and M. Arpin. 1997. Ezrin is an effector of hepatocyte growth factor-mediated migration and morphogenesis in epithelial cells. J. Cell Biol. 138:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker, S.P., and D.L. Barber. 2002. Ion transport proteins anchor and regulate the cytoskeleton. Curr. Opin. Cell Biol. 14:214–220. [DOI] [PubMed] [Google Scholar]
- Denker, S.P., D.C. Huang, J. Orlowski, H. Furthmayr, and D.L. Barber. 2000. Direct binding of the Na–H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H+ translocation. Mol. Cell. 6:1425–1436. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S., and A. Hall. 2001. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 106:489–498. [DOI] [PubMed] [Google Scholar]
- Frame, M.C., and V.G. Brunton. 2002. Advances in Rho-dependent actin regulation and oncogenic transformation. Curr. Opin. Genet. Dev. 12:36–43. [DOI] [PubMed] [Google Scholar]
- Glading, A., P. Chang, D.A. Lauffenburger, and A. Wells. 2000. Epidermal growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J. Biol. Chem. 275:2390–2398. [DOI] [PubMed] [Google Scholar]
- Grinstein, S., M. Woodside, T.K. Waddell, G.P. Downey, J. Orlowski, J. Pouyssegur, D.C. Wong, and J.K. Foskett. 1993. Focal localization of the NHE-1 isoform of the Na+/H+ antiport: assessment of effects on intracellular pH. EMBO J. 12:5209–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugh, J.M., F. Codazzi, M. Teruel, and T. Meyer. 2000. Spatial sensing in fibroblasts mediated by 3′ phosphoinositides. J. Cell Biol. 151:1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooley, R., C.Y. Yu, M. Symons, and D.L. Barber. 1996. Gα13 stimulates Na+-H+ exchange through distinct Cdc42-dependent and RhoA-dependent pathways. J. Biol. Chem. 271:6152–6158. [DOI] [PubMed] [Google Scholar]
- Huttenlocher, A., S.P. Palecek, Q. Lu, W. Zhang, R.L. Mellgren, D.A. Lauffenburger, M.H. Ginsberg, and A.F. Horwitz. 1997. Regulation of cell migration by the calcium-dependent protease calpain. J. Biol. Chem. 272:32719–32722. [DOI] [PubMed] [Google Scholar]
- Klein, M., P. Seeger, B. Schuricht, S.L. Alper, and A. Schwab. 2000. Polarization of Na+/H+ and Cl−/HCO3 − exchangers in migrating renal epithelial cells. J. Gen. Physiol. 115:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfer, A., D. Louvard, and S.J. Singer. 1982. Polarization of the Golgi apparatus and the microtubule-organizing center in cultured fibroblasts at the edge of an experimental wound. Proc. Natl. Acad. Sci. USA. 79:2603–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni, S.V., G. Gish, P. van der Geer, M. Henkemeyer, and T. Pawson. 2000. Role of p120 Ras-GAP in directed cell movement. J. Cell Biol. 149:457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagana, A., J. Vadnais, P.U. Le, T.N. Nguyen, R. Laprade, I.R. Nabi, and J. Noel. 2000. Regulation of the formation of tumor cell pseudopodia by the Na+/H+ exchanger NHE1. J. Cell Sci. 113:3649–3662. [DOI] [PubMed] [Google Scholar]
- Lauffenburger, D.A., and A.F. Horwitz. 1996. Cell migration: a physically integrated molecular process. Cell. 84:359–369. [DOI] [PubMed] [Google Scholar]
- Ma, Y.H., H.P. Reusch, E. Wilson, J.A. Escobedo, W.J. Fantl, L.T. Williams, and H.E. Ives. 1994. Activation of Na+/H+ exchange by platelet-derived growth factor involves phosphatidylinositol 3′-kinase and phospholipase C gamma. J. Biol. Chem. 269:30734–30739. [PubMed] [Google Scholar]
- Maciver, S.K., B.J. Pope, S. Whytock, and A.G. Weeds. 1998. The effect of two actin depolymerizing factors (ADF/cofilins) on actin filament turnover: pH sensitivity of F-actin binding by human ADF, but not of Acanthamoeba actophorin. Eur. J. Biochem. 256:388–397. [DOI] [PubMed] [Google Scholar]
- Mackay, D.J., F. Esch, H. Furthmayr, and A. Hall. 1997. Rho- and rac-dependent assembly of focal adhesion complexes and actin filaments in permeabilized fibroblasts: an essential role for ezrin/radixin/moesin proteins. J. Cell Biol. 138:927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meili, R., C. Ellsworth, S. Lee, T.B. Reddy, H. Ma, and R.A. Firtel. 1999. Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. EMBO J. 18:2092–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, T., M. Parsons, W.E. Hughes, J. Monypenny, D. Zicha, A. Gautreau, M. Arpin, S. Gschmeissner, P.J. Verveer, P.I. Bastiaens, and P.J. Parker. 2001. Ezrin is a downstream effector of trafficking PKC-integrin complexes involved in the control of cell motility. EMBO J. 20:2723–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes, C.D., and A. Hall. 1999. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144:1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek, S.P., A. Huttenlocher, A.F. Horwitz, and D.A. Lauffenburger. 1998. Physical and biochemical regulation of integrin release during rear detachment of migrating cells. J. Cell Sci. 111:929–940. [DOI] [PubMed] [Google Scholar]
- Parent, C.A., and P.N. Devreotes. 1999. A cell's sense of direction. Science. 284:765–770. [DOI] [PubMed] [Google Scholar]
- Piepenhagen, P.A., and W.J. Nelson. 1998. Biogenesis of polarized epithelial cells during kidney development in situ: roles of E-cadherin-mediated cell-cell adhesion and membrane cytoskeleton organization. Mol. Biol. Cell. 9:3161–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshkin, S.J., A. Bellizzi, V. Albarani, L. Guerra, M. Tommasino, A. Paradiso, and V. Casavola. 2000. Phosphoinositide 3-kinase is involved in the tumor-specific activation of human breast cancer cell Na+/H+ exchange, motility, and invasion induced by serum deprivation. J. Biol. Chem. 275:5361–5369. [DOI] [PubMed] [Google Scholar]
- Ritter, M., P. Schratzberger, H. Rossmann, E. Woll, K. Seiler, U. Seidler, N. Reinisch, C.M. Kahler, H. Zwierzina, H.J. Lang, et al. 1998. Effect of inhibitors of Na+/H+-exchange and gastric H+/K+ ATPase on cell volume, intracellular pH and migration of human polymorphonuclear leucocytes. Br. J. Pharmacol. 124:627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, M.A., C. Lechene, and D.E. Ingber. 1991. Insoluble fibronectin activates the Na/H antiporter by clustering and immobilizing integrin alpha 5 beta 1, independent of cell shape. Proc. Natl. Acad. Sci. USA. 88:7849–7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant, G., O.D. Weiner, E.R. Neptune, J.W. Sedat, and H.R. Bourne. 1999. Dynamics of a chemoattractant receptor in living neutrophils during chemotaxis. Mol. Biol. Cell. 10:1163–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant, G., O.D. Weiner, P. Herzmark, T. Balla, J.W. Sedat, and H.R. Bourne. 2000. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 287:1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simchowitz, L., and E.J. Cragoe, Jr. 1986. Regulation of human neutrophil chemotaxis by intracellular pH. J. Biol. Chem. 261:6492–6500. [PubMed] [Google Scholar]
- Stowers, L., D. Yelon, L.J. Berg, and J. Chant. 1995. Regulation of the polarization of T cells toward antigen-presenting cells by Ras-related GTPase CDC42. Proc. Natl. Acad. Sci. USA. 92:5027–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga, T., and D.L. Barber. 1998. Na-H exchange acts downstream of RhoA to regulate integrin-induced cell adhesion and spreading. Mol. Biol. Cell. 9:2287–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga, T., T. Ishizaki, S. Narumiya, and D.L. Barber. 1998. p160ROCK mediates RhoA activation of Na-H exchange. EMBO J. 17:4712–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vexler, Z.S., M. Symons, and D.L. Barber. 1996. Activation of Na+-H+ exchange is necessary for RhoA-induced stress fiber formation. J. Biol. Chem. 271:22281–22284. [DOI] [PubMed] [Google Scholar]
- Wakabayashi, S., P. Fafournoux, C. Sardet, and J. Pouyssegur. 1992. The Na+/H+ antiporter cytoplasmic domain mediates growth factor signals and controls “H+-sensing”. Proc. Natl. Acad. Sci. USA. 89:2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthylake, R.A., S. Lemoine, J.M. Watson, and K. Burridge. 2001. RhoA is required for monocyte tail retraction during transendothelial migration. J. Cell Biol. 154:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, Z., N. Zhang, D.B. Murphy, and P.N. Devreotes. 1997. Dynamic distribution of chemoattractant receptors in living cells during chemotaxis and persistent stimulation. J. Cell Biol. 139:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, W., K. Nehrke, J. Choi, and D.L. Barber. 2001. The Nck-interacting kinase (NIK) phosphorylates the Na+-H+ exchanger NHE1 and regulates NHE1 activation by platelet-derived growth factor. J. Biol. Chem. 276:31349–31356. [DOI] [PubMed] [Google Scholar]
- Zhao, X., J.K. Newcomb, R.M. Posmantur, K.K. Wang, B.R. Pike, and R.L. Hayes. 1998. pH dependency of μ-calpain and m-calpain activity assayed by casein zymography following traumatic brain injury in the rat. Neurosci. Lett. 247:53–57. [DOI] [PubMed] [Google Scholar]
- Zhou, D., S. Lambert, P.L. Malen, S. Carpenter, L.M. Boland, and V. Bennett. 1998. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J. Cell Biol. 143:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]