

Abstract
Ghrelin is an acyl-peptide gastric hormone acting on the pituitary and hypothalamus to stimulate growth hormone (GH) release, adiposity, and appetite. Ghrelin endocrine activities are entirely dependent on its acylation and are mediated by GH secretagogue (GHS) receptor (GHSR)-1a, a G protein–coupled receptor mostly expressed in the pituitary and hypothalamus, previously identified as the receptor for a group of synthetic molecules featuring GH secretagogue (GHS) activity. Des-acyl ghrelin, which is far more abundant than ghrelin, does not bind GHSR-1a, is devoid of any endocrine activity, and its function is currently unknown. Ghrelin, which is expressed in heart, albeit at a much lower level than in the stomach, also exerts a cardio protective effect through an unknown mechanism, independent of GH release. Here we show that both ghrelin and des-acyl ghrelin inhibit apoptosis of primary adult and H9c2 cardiomyocytes and endothelial cells in vitro through activation of extracellular signal–regulated kinase-1/2 and Akt serine kinases. In addition, ghrelin and des-acyl ghrelin recognize common high affinity binding sites on H9c2 cardiomyocytes, which do not express GHSR-1a. Finally, both MK-0677 and hexarelin, a nonpeptidyl and a peptidyl synthetic GHS, respectively, recognize the common ghrelin and des-acyl ghrelin binding sites, inhibit cell death, and activate MAPK and Akt.
These findings provide the first evidence that, independent of its acylation, ghrelin gene product may act as a survival factor directly on the cardiovascular system through binding to a novel, yet to be identified receptor, which is distinct from GHSR-1a.
Keywords: ghrelin; apoptosis; heart; ERKs; Akt
Introduction
Ghrelin is a newly discovered hormone, which induces the release of growth hormone (GH)* and stimulates food intake and adiposity (Kojima et al., 1999; Tschop et al., 2000; Bowers, 2001; Inu, 2001; Nakazato et al., 2001). Although ghrelin is essentially a gastric hormone, it is expressed ubiquitously, although at low level (Gnanapavan et al., 2002). Endocrine activities of ghrelin are mediated by GH secretagogue (GHS) receptor (GHSR)-1a, a G protein–coupled receptor mainly expressed in the pituitary and hypothalamus, identified previously as the receptor for GH secretagogues (GHSs), a group of synthetic molecules endowed with strong GH release activity (Howard et al., 1996). Ghrelin is a 28 amino acid acyl-peptide esterified with octanoic acid on Ser 3. Acylation of ghrelin is required for GHSR-1a activation, whereas des-acyl ghrelin, which is far more abundant than ghrelin (Hosoda et al., 2000), does not bind GHSR-1a and is devoid of any endocrine activity (Kojima et al., 1999; Muccioli et al., 2002; Torsello et al., 2002). The function of des-acyl ghrelin is currently unknown.
Recent evidence indicates that ghrelin and synthetic GHS feature a variety of cardiovascular activities, including increase of myocardial contractility (Bisi et al., 1999; Nagaya et al., 2001a), vasodilatation (Okumura et al., 2002), and protection from myocardial infarction-induced heart failure in vivo (Locatelli et al., 1999; Tivesten et al., 2000; King et al., 2001; Nagaya et al., 2001b). Some indirect evidence suggests that ghrelin and GHS cardioprotective activity is independent from GH secretion (Locatelli et al., 1999; Tivesten et al., 2000). In addition, ghrelin receptor, GHSR-1a, is expressed in the myocardium (Gnanapavan et al., 2002), and both ghrelin and peptidyl GHS recognize high affinity binding sites in the heart (Papotti et al., 2000; Bodart et al., 2002; Katugampola et al., 2001). However, the cellular and molecular mechanisms underlying ghrelin and synthetic GHS cardioprotective activities have not been investigated.
Based on these observations, we raised the hypothesis that ghrelin may directly prevent cell death of cultured cardiomyocytes and/or endothelial cells in vitro through activation of a specific receptor expressed in these cells. The data reported here support and extend such hypothesis, providing the first evidence that both ghrelin and des-acyl ghrelin inhibit apoptosis of cardiomyocytes and endothelial cells through activation of a survival intracellular signaling pathway. In addition, these data indicate that a novel, yet to be identified receptor, distinct from GHSR-1a, mediates ghrelin and des-acyl ghrelin antiapoptotic activity, suggesting a novel role for the ghrelin gene product independent of its acylation.
Results
Ghrelin inhibits doxorubicin-induced cell death in vitro
To investigate whether ghrelin may act as survival factor for cardiovascular cells, we have treated H9c2 cardiomyocytes with doxorubicin, which induces apoptotic cell death of cardiomyocytes both in vitro and in vivo (Arola et al., 2000; Wu et al., 2000). After 24 h of treatment, 1 μM doxorubicin induced cell death of H9c2 cardiomyocytes as observed by phase–contrast microscopy (Fig. 1 A). Dying cells became light refractive and detached off the plate. However, in the presence of a saturating concentration of ghrelin doxorubicin-treated H9c2 cells maintained their morphology and were still attached to the plate. In the absence of doxorubicin, ghrelin did not affect cell morphology. Ghrelin cytoprotective activity was also assayed by measuring cell viability of H9c2 cardiomyocytes. After 24 h of treatment with doxorubicin, 50% of the cells were not viable as measured by the 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. However, doxorubicin-induced cell death was almost completely abolished in the presence of a saturating concentration of ghrelin (Fig. 1 B). The cytoprotective activity of ghrelin was concentration dependent, 0.1 nM being the lowest active concentration (Fig. 1 C). Similar results were obtained when cell viability was measured as loss of membrane impermeability. Upon 48 h of treatment, doxorubicin induced lactate dehydrogenase (LDH) release in the medium, which was completely prevented in the presence of a saturating concentration of ghrelin (Fig. 1 D). Similar results were obtained in PAE endothelial cells (unpublished data). Ghrelin alone did not affect cell survival of H9c2 cardiomyocytes as measured by either MTT or the release of LDH. Thus, these data provide the first indication that ghrelin acts directly on cardiomyocytes to inhibit experimentally induced cell death.
Figure 1.

Ghrelin protects H9c2 cardiomyocytes from doxorubicin-induced cell death. H9c2 cardiomyocytes were treated with 1 μM doxorubicin (grey bar) in the presence or absence of 1 μM ghrelin. (A) Phase–contrast images (200×; bar, 0.1 mm) after 24 h of treatment: untreated (a), ghrelin (b), doxorubicin (c), and doxorubicin + ghrelin (d). (B) Cell death measured by MTT assay after 24 h of treatment is expressed as the percentage of cell death. Values are the mean ± SE of eight samples (*t test, P < 0.01). (C) Dose response of ghrelin in MTT cell death assay after 24 h of treatment. Values are the mean ± SE of eight samples. (D) Cell death measured by LDH release after 48 h of treatment. Values are the mean ± SE of eight samples (*t test, P < 0.01).
Ghrelin prevents apoptosis induced in vitro by both FAS activation and serum starvation
Apoptotic cell death of cardiomyocytes occurs during the development of heart failure, whereas up-regulation of myocardial expression of FAS correlates with cell death of cardiomyocytes in ischemic-dilated and doxorubicin-induced cardiomyopathies (Yamamura et al., 1999; Jeremias et al., 2000; Nakamura et al., 2000). Activation of FAS receptor stimulates apoptosis of terminally differentiated adult cardiomyocytes both in vitro and in vivo (Takemura et al., 2001; Hayakawa et al., 2002). In addition, the inhibition of apoptosis of cardiomyocytes by survival factors has been shown to prevent cardiac heart failure (Parrizas et al., 1997). Here we report that ghrelin treatment of primary adult cardiomyocytes prevented apoptosis stimulated by anti-FAS agonist antibodies (Fig. 2 A) in a concentration-dependent manner. Apoptosis was measured as nucleosomal DNA fragmentation determined by FACS® analysis.
Figure 2.
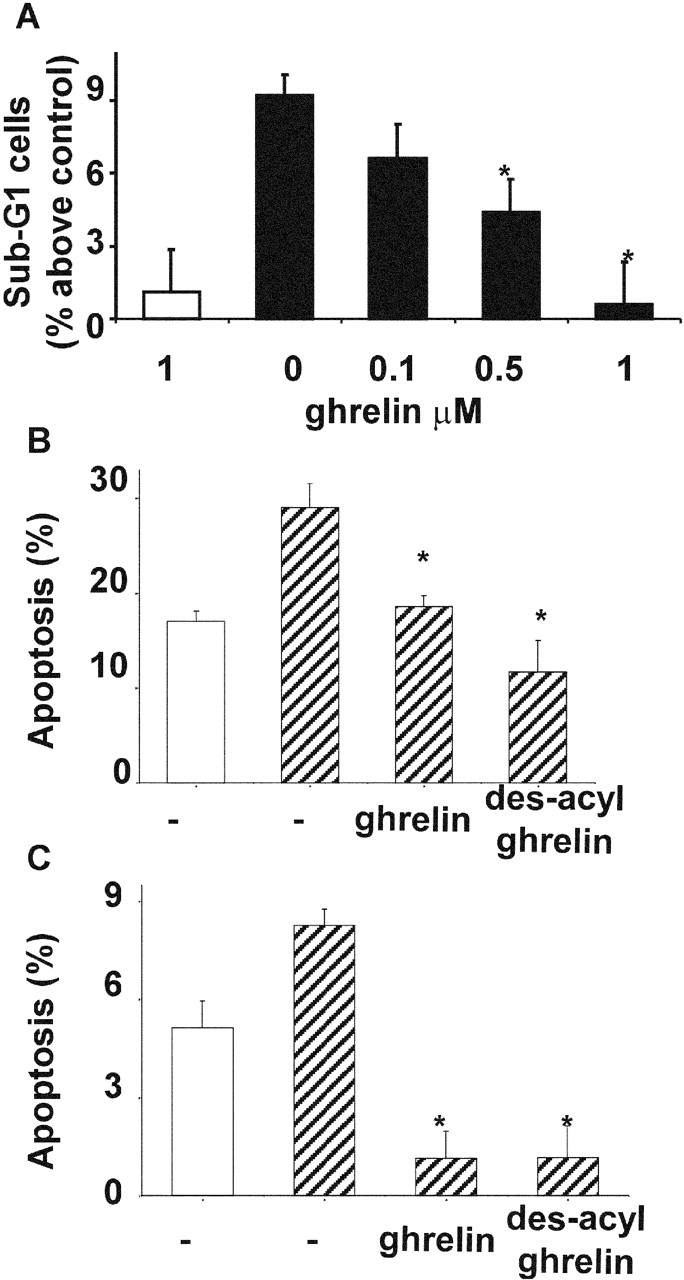
Ghrelin and des-acyl ghrelin inhibit apoptosis. (A) Primary cultures of adult cardiomyocytes were pretreated with either 0.1, 0.5, or 1 μM ghrelin for 16 h. Then FAS was stimulated (black bars), and sub-G1 cells were measured by FACS® analysis after 6 h. Data were calculated as the percentage of apoptotic nuclei induced by FAS stimulation above untreated control. Values are the mean ± SE of three independent experiments (*t test, P < 0.01). H9c2 cardiomyocytes (B) and PAE cells (C) cultured in either 10% FCS (white bar) or 0% FCS (hatched bars) were pretreated with 1 μM ghrelin or des-acyl ghrelin for 48 h. Apoptosis was measured by FACS® analysis with fluorescent annexin V; values are mean ± SE of three samples (*t test, P < 0.02).
In addition, we have assayed ghrelin's ability to prevent apoptosis induced by serum withdrawal in H9c2 and PAE cells. Apoptosis was measured by externalization of phosphatidylserine, an early apoptotic event. Both H9c2 cardiomyocytes and PAE cells underwent apoptosis after 48 h of serum withdrawal. However, a saturating concentration of ghrelin prevented apoptosis induced by the absence of serum (Fig. 2, B and C). Thus, these data strongly indicate that ghrelin acts as a survival factor in cardiomyocytes and endothelial cells by inhibiting apoptosis induced through different stimuli.
Ghrelin inhibits apoptosis by activation of antiapoptotic intracellular signaling pathways
To provide further biochemical evidence supporting the hypothesis that ghrelin may act as a survival factor, we have investigated ghrelin's ability to stimulate intracellular signaling pathways leading to inhibition of apoptosis. Survival factors, such as insulin growth factor-1 (IGF-1), inhibit apoptosis by stimulating several intracellular signaling pathways, including protein-tyrosine phosphorylation and activation of extracellular signal–regulated kinase (ERK)-1/2 and Akt (Parrizas et al., 1997). In H9c2 cardiomyocytes, ghrelin induced tyrosine phosphorylation of two intracellular proteins of ∼100 and 60 kD, suggesting that it may activate a protein-tyrosine kinase (Fig. 3 A). Similar results were obtained in PAE cells (unpublished data). Ghrelin activated both ERK1/2 and Akt in H9c2 cardiomyocytes (Fig. 3, B and C) and in endothelial cells (unpublished data). Activation of ERK1/2 and Akt, measured as serine residue-specific phosphorylation, occurred as early as 2 min after ghrelin stimulation, peaked between 5 and 10 min, and lasted at least 30 min. These data suggest that early activation of ERK1/2 and Akt may be involved in the transduction of the antiapoptotic signaling triggered by ghrelin. To verify such hypothesis, we have investigated whether pharmacological inhibition of either ERK1/2 or Akt impairs ghrelin cytoprotective activity after doxorubicin treatment. Activation of Akt was prevented by inhibition of phosphatidylinositol (PI) 3-kinase, whose enzymatic product PI(3,4,5)P3 mediates growth factor–induced activation of Akt (Alessi et al., 1996; unpublished data). Cell treatment of both H9c2 cardiomyocytes and PAE cells either with 30 μM PD98059, a highly specific ERK1/2 inhibitor, or 100 nM wortmannin, a highly specific inhibitor of PI 3-kinase, abolished ghrelin's cytoprotective activity after 20 h of treatment with 1 μM doxorubicin (Fig. 3, D and E). Under these conditions, neither PD98059 nor wortmannin affected doxorubicin-induced cell death. Thus, these data suggest that ghrelin inhibits cell death by activating at least two prosurvival signaling pathways conveyed by ERK1/2 and PI 3-kinase/Akt.
Figure 3.

Ghrelin stimulates protein-tyrosine phosphorylation and activates ERK1/2 and Akt. Overnight serum-starved H9c2 cells were stimulated with 1 μM ghrelin and lysed at different times. Total lysates were analyzed by Western blot with antiphosphotyrosine antibodies (A), specific anti-phosphoERK1/2 antibodies (B, top) and anti-ERK1/2 antibodies (B, bottom), and specific anti-phosphoAkt antibodies (C, top) and anti-Akt antibodies (C, bottom). H9c2 cardiomyocytes (D) and PAE cells (E) were treated with 1 μM doxorubicin for 20 h (black bars) in the presence or absence of 1 μM ghrelin and in the presence or absence of 100 nM wortmannin or 30 μM PD98059. Cell death was measured by MTT assay. Values are the mean ± SE of eight samples (*t test, P < 0.01).
Ghrelin and des-acyl ghrelin bind to a common receptor, distinct from GHSR-1a, and activate a common biochemical and biological response in H9c2 cardiomyocytes
Although GHSR-1a is expressed in the heart, proof that it mediates ghrelin cardioprotective activity is still lacking. We have assayed the expression of GHSR-1a in H9c2 cardiomyocytes compared with the whole heart and the pituitary. Expression was measured by RT-PCR of GHSR-1a second exon as described previously (Nagaya et al., 2001a). To our surprise, no expression was detected in H9c2 cardiomyocytes, whereas a positive signal was detected from both the whole heart and the pituitary (Fig. 4). Thus, these data suggest that GHSR-1a is not expressed in H9c2 cardiomyocytes and raise the hypothesis that ghrelin antiapoptotic activity may not be mediated by GHSR-1a.
Figure 4.

GHSr-1a is expressed in the whole heart but not in H9c2 cardiomyocytes. RT-PCR was performed on total RNA extracted from H9c2 cardiomyocytes and rat heart with primers specific for GHSR-1a (top) or GAPDH (bottom).
To further investigate the identity of the receptor mediating ghrelin antiapoptotic activity, we have performed binding studies of radio-labeled ghrelin to membranes of H9c2 cardiomyocytes. 125I-Tyr4-ghrelin recognized a single class of high affinity, saturable, and specific binding sites (Fig. 5 A) (K d = 4.0 ± 0.9 nM; Bmax = 20 ± 1.8 fmol/mg protein; data are the mean ± SE of four separate experiments). The binding of radio-labeled ghrelin was displaced by unlabeled ghrelin and, surprisingly, though with lower affinity by des-acyl ghrelin, the deacylated form of ghrelin, which does not bind GHSR-1. The IC50 values for ghrelin and des-acyl ghrelin were 8.1 ± 1.0 nM and 22.5 ± 1.5 nM, respectively (Fig. 5 B and Table I). Consistently, 125I-Tyr4–des-acyl ghrelin recognized high affinity binding sites in H9c2 cardiomyocytes (Fig. 5 C). Binding analysis demonstrated the existence of a single class of binding sites that show Bmax (18.8 ± 1.4 fmol/mg protein) and K d (4.6 ± 0.60 nM) values very close to those detected by radio-labeled ghrelin. In addition, both unlabeled des-acyl ghrelin and ghrelin displaced bound radio-labeled des-acyl ghrelin with IC50 values of 17.5 ± 1.7 nM and 11.5 ± 1.3 nM, respectively (Fig. 5 D and Table I).
Figure 5.

H9c2 cardiomyocytes express a ghrelin receptor distinct from GHSR-1a. (A and C) Specific binding was determined by incubation of crude membranes with increasing concentrations (0.25–20 nM) of either radio-labeled ghrelin (A) or radio-labeled des-acyl ghrelin (C) in the presence or absence of 1 μM unlabeled ghrelin (A) or unlabeled des-acyl ghrelin (C), respectively: total binding (▪), specific binding (▾), and nonspecific binding (○). Data are the average of duplicate assay determinants. Similar results were obtained in at least two other independent experiments. (B and D) Displacement curves of radio-labeled ghrelin (B) or radio-labeled des-acyl ghrelin (D) binding by unlabeled ghrelin (○), des-acyl ghrelin (▪), hexarelin (▴), and MK-0677 (▵). Displacement binding values are the mean ± SE of four separate experiments.
Table I.
IC50 for 125I-Tyr4-ghrelin and 125 I-Tyr4–des-acyl ghrelin
| IC50 (nM) | 125I-Tyr4-ghrelin displacement | 125I-Tyr4–des-acyl ghrelin displacement |
|---|---|---|
| Ghrelin | 8.1 ± 1.0 | 11.5 ± 1.3 |
| Des-acyl ghrelin | 22.5 ± 1.5 | 17.5 ± 1.7 |
| Hexarelin | 13.8 ± 1.2 | 14.9 ± 1.1 |
| MK-0677 | 20.0 ± 1.8 | 20.9 ± 1.7 |
A direct implication of these findings is that des-acyl ghrelin might also be biologically active on H9c2 cardiomyocytes. Furthermore, the findings support our previous hypothesis that a novel ghrelin receptor, distinct from GHSR-1a, is expressed in these cells. Thus, we have assayed des-acyl ghrelin-induced signaling and antiapoptotic activity in H9c2 cardiomyocytes.
Des-acyl ghrelin significantly inhibited apoptosis induced by serum withdrawal in both H9c2 and PAE (Fig. 2, B and C), inhibited doxorubicin-induced cell death in both cell lines (Fig. 6 A), and stimulated ERK-1/2 and Akt in H9c2 cardiomyocytes (Fig. 6, B and C). Together, these data strongly suggest that ghrelin and des-acyl ghrelin stimulate an antiapoptotic pathway through binding and activation of a common receptor.
Figure 6.

Des-acyl ghrelin inhibits cell death and activates ERK1/2 and Akt in H9c2 cardiomyocytes and PAE. (A) H9c2 cardiomyocytes and PAE cells were treated with 1 μM doxorubicin (grey bar) for 24 h in the presence or absence of either 1 μM ghrelin or des-acyl ghrelin. Cell death was assessed by the MTT assay. Values are mean ± SE of eight samples (*t test, P < 0.01). (B and C) H9c2 cardiomyocytes were serum starved overnight, stimulated with 1 μM des-acyl ghrelin, and lysed at different times. Total lysates were analyzed by Western blot with specific anti-phosphoERK1/2 antibodies (B, top) and anti-ERK1/2 antibodies (B, bottom) and specific anti-phosphoAkt antibodies (C, top) and anti-Akt antibodies (C, bottom).
In an attempt to further characterize such receptor pharmacologically, we have investigated the ability of synthetic GHS to recognize it and to activate antiapoptotic signaling pathways. Hexarelin and MK-0677, respectively, a peptidyl and a nonpeptidyl GHS, displace the binding of both 125I-Tyr4-ghrelin and 125I-Tyr4–des-acyl ghrelin with similar affinities (Fig. 5, B and D). IC50 values are summarized in Table I.
Consistent with the binding data, both hexarelin and MK-0677 inhibited doxorubicin-induced cell death of H9c2 cardiomyocytes as observed by phase–contrast microscopy (Fig. 7 A) and measured by MTT assay (Fig. 7 B). Hexarelin and MK-0677 inhibited doxorubicin apoptosis also in PAE endothelial cells as measured by MTT assay (Fig. 7 B).
Figure 7.
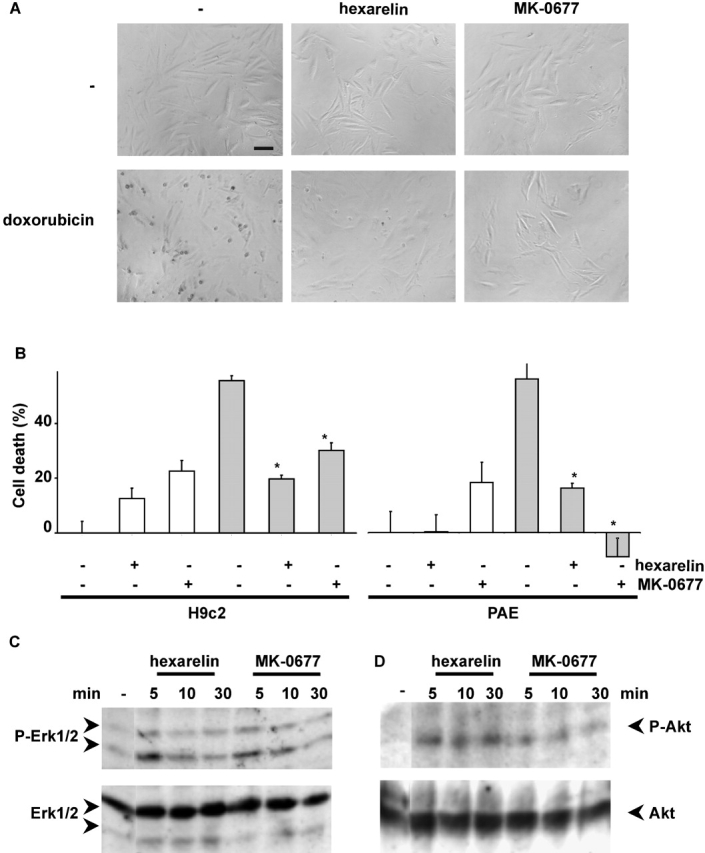
Hexarelin and MK-0677 inhibit cell death and activate ERK1/2 and Akt in H9c2 cardiomyocytes and PAE. (A) H9c2 cardiomyocytes were treated with 1 μM of either doxorubicin, hexarelin, and/or MK-0677 as indicated. Phase–contrast images (200×; bar, 0.1 mm) were captured after 24 h of treatment. Representative fields are shown. (B) H9c2 cardiomyocytes and PAE cells were treated with 1 μM doxorubicin (grey bar) for 24 h in the presence or absence of either 1 μM hexarelin or MK-0677. Cell death was assessed by the MTT assay. Values are mean ± SE of eight samples (*t test, P < 0.01). (C and D) H9c2 cardiomyocytes were serum starved overnight, stimulated with either 1 μM hexarelin or MK-0677, and lysed at different times. Total lysates were analyzed by Western blot with specific anti-phosphoERK1/2 antibodies (C, top) and anti-ERK1/2 antibodies (C, bottom) and specific anti-phosphoAkt antibodies (D, top) and anti-Akt antibodies (D, bottom).
Furthermore, hexarelin and MK-0677 stimulate enzymatic activity of both ERK-1/2 and Akt in H9c2 cardiomyocytes with similar time course and potency of ghrelin and des-acyl ghrelin (Fig. 7, C and D). Together, the data are consistent with previous observations that hexarelin features cardioprotective activity in a GH-independent manner (Locatelli et al., 1999; Tivesten et al., 2000).
Discussion
Ghrelin is a gastric acyl-peptidic hormone acting on the hypothalamus and pituitary to induce GH release, food intake, and adiposity (Kojima et al., 1999; Tschop et al., 2000; Nakazato et al., 2001). In addition to its endocrine activities, ghrelin and synthetic GHS protect heart function from experimentally induced cardiac heart failure in vivo (Locatelli et al., 1999; Tivesten et al., 2000; King et al., 2001; Nagaya et al., 2001b). However, the cellular and molecular mechanisms underlying ghrelin cardioprotective activity are not known. Based on these observations, we have raised the hypothesis that ghrelin may feature a direct cytoprotective activity toward myocardial cells by inhibiting cell death of cardiomyocytes and endothelial cells. In the heart, antiapoptotic factors, such as IGF-1 and cardiotrophin-1 (CT-1), play a crucial role in maintaining cardiomyocytes survival and myocardial function after ischemia- and pressure overload–induced cardiomyopathies (Buerke et al., 1995; Hirota et al., 1999). The data reported here showing the ability of ghrelin to inhibit apoptosis induced by either doxorubicin serum withdrawal or FAS activation suggest that it may act as a survival factor in cardiomyocytes and endothelial cells. The data also supports the hypothesis that ghrelin may play a role in myocardial homeostasis.
Survival factors inhibit cell death by activating specific signaling pathways, including stimulation of protein-tyrosine phosphorylation and activation of ERK-1/2 and PI 3-kinase/Akt, which then lead to the inhibition of the apoptotic signaling cascade (Wu et al., 2000). We have shown that ghrelin stimulates tyrosine phosphorylation and activates ERK-1/2 and Akt and that activation of each of the two pathways is required for ghrelin-induced inhibition of apoptosis. Activation of ERK-1/2 by ghrelin was reported previously in HepG2 cells, which express GHSR-1, although in these cells ghrelin inhibits rather than activates Akt (Murata et al., 2002). The receptor-mediating ghrelin intracellular signaling and antiapoptotic activity is not known. Here we provide three pieces of evidence suggesting that ghrelin antiapoptotic activity is not mediated by GHSR-1a. (i) In contrast to the whole heart, no expression of GHSR-1a was detected in H9c2 cardiomyocytes by RT-PCR. (ii) Both ghrelin and its deacylated form, des-acyl ghrelin, recognize a common high affinity binding site, although only ghrelin and not des-acyl ghrelin bind to GHSR-1a (Kojima et al., 1999). Furthermore, affinity constant of ghrelin binding sites on H9c2 cardiomyocytes are ∼10-fold higher than affinity constant of GHSR-1a as measured on pituitary and hypothalamus membranes (Muccioli et al., 2001). (iii) Des-acyl ghrelin, which is devoid of any GH-releasing activity even in vitro (Kojima et al., 1999), is as effective as ghrelin in inhibiting apoptosis and in activating intracellular signaling pathways, i.e., ERK-1/2, Akt, and protein-tyrosine phosphorylation (unpublished data). Thus, these findings suggest that ghrelin and des-acyl ghrelin bind to a novel receptor, distinct from GHSR-1a. The putative novel receptor is expected to be highly similar to GHSR-1a, since it differs only in its lack of ability to discriminate between the esterified and unesterified ghrelin peptide. Moreover, the demonstration that two synthetic ligands of GHSR-1a, hexarelin and MK-0677, bind and activate the novel receptor provides further support to the hypothesis that the two receptors are highly related.
Whether such receptor is encoded by alternative splicing of GHSR-1 gene or by a distinct gene still remains to be determined. GHSR-1b, a GHSR-1–truncated splicing variant, although expressed in the heart, does not appear to be functional (Howard et al., 1996). We have reported previously that both ghrelin and des-acyl ghrelin bind to a common receptor in breast cancer cells (Cassoni et al., 2001). In these cells, ghrelin and des-acyl ghrelin inhibit cell proliferation, whereas, on the other hand, GHSR-1a appears to mediate a proliferative signal (Murata et al., 2002). Whether the same ghrelin receptor mediates the inhibition of both growth and apoptosis in breast cancer cells and in cardiomyocytes and endothelial cells, respectively, it is not determined. Although most survival factors also display a proliferative activity, members of the transforming growth factor-β family inhibit both cell growth and cell death in myoblasts (Chen et al., 2001; Rios et al., 2001).
In addition, the finding that GHSR-1a is expressed in the whole heart but not in H9c2 cardiomyocytes provides further support to the hypothesis that multiple ghrelin receptors may be expressed in the cardiovascular system (Papotti et al., 2000; Bodart et al., 2002; Gnanapavan et al., 2002; Katugampola et al., 2001). Each receptor may then contribute independently to mediate the wide array of cardiovascular activities induced by both ghrelin and synthetic GHSs. CD36, a fatty acid receptor, has been identified recently as a myocardial receptor for hexarelin, a synthetic peptidyl GHS. However, neither ghrelin nor the synthetic nonpeptidyl GHS, MK-0677, recognize such receptor (Bodart et al., 2002). Interestingly, similar hexarelin-binding sites, which are not displaced by MK-0677, were also reported in H9c2 cardiomyocytes (Filigheddu et al., 2001).
In summary, the data presented here suggest that inhibition of apoptosis of cardiomyocytes and endothelial cells may contribute to the reported cardioprotective activity of ghrelin and of synthetic GHS in vivo. Ghrelin and des-acyl ghrelin are active in vitro even at subnanomolar concentration, whereas their total serum concentration in the rat is 0.22 ± 0.07 nM (Hosoda et al., 2000). In addition, these data suggest that des-acyl ghrelin, which does not induce GH release, may feature a cardioprotective activity in vivo.
Thus, we may speculate that the ghrelin gene product, which is ubiquitously expressed, in addition to its endocrine activity dependent on gastric release of its acylated form may also be a trophic local factor acting in the cardiovascular system in an autocrine/paracrine fashion.
Materials and methods
Cell cultures, chemicals, peptides, and reagents
H9c2 and PAE cells were obtained from American Type Culture Collection and cultured as described previously (Cutrupi et al., 2000; Filigheddu et al., 2001). Terminally differentiated cardiomyocytes were prepared from adult guinea pig as described previously (Gallo et al., 2001). Ghrelin, des-acyl ghrelin, radioiodinated Tyr4-des-Phe4-ghrelin (125I-Tyr4-ghrelin), and Tyr4-des-Phe4–nonoctanoylated ghrelin (125I-Tyr4–des-acyl ghrelin) were provided by Europeptides. Antibodies were from cell signaling except 4G10 antiphosphotyrosine antibodies from UBI. Molecular biology reagents were from GIBCO-BRL except Powerscript reverse transcriptase from CLONTECH Laboratories, Inc. and DyNAzyme EXT taq and PCR reagents from Finzymes. Doxorubicin (Amersham Biosciences and Upjohn) was prepared as a 1 mM solution in PBS and then sonicated and diluted to 1 μM in 5% FCS. Similar data were obtained when Doxorubicin was prepared in unbuffered physiological solution and used without sonication at 0.1 μM.
Cell death and apoptosis
Cells were plated in 96-well slides (∼25,000–30,000 cells/cm2) for MTT and LDH assays and in 10-cm dishes for other studies. Cell death was induced by incubating the cells with 1 μM doxorubicin in 5% FCS (H9c2) or 0% FCS (PAE) for the indicated time. FAS-mediated apoptosis of adult cardiomyocytes was obtained by 30-min cell incubation on ice in the presence of 1 μg/ml anti-FAS agonist antibodies (Bender MedSystem) and 1 μg/ml protein A (Sigma-Aldrich). Cells were then placed for 6 h at RT in tyrode medium as described previously (Gallo et al., 2001). Cell viability was measured by MTT assay (Sigma-Aldrich) as described previously (Filigheddu et al., 2001) and by Cytotoxicity Detection Kit (Roche), which measures LDH released in the medium. Percentage cytotoxicity was calculated according to the manufacturer's instructions. Apoptosis was evaluated in H9c2 and PAE cells by FACS® analysis (Becton Dickinson) of the proportion of cells displaying shrunken/hypergranular morphology or those stained with Annexin-V Fluos (Alexis) above the untreated controls. Briefly, 0.5 × 105 cells were stained with annexin V (1:50) and 1 μg/ml propidium iodide in 10 mM Hepes-NaOH (pH 7.4), 140 mM NaCl, 5 mM CaCl2, and incubated for 10–15 min at RT in the dark. Data were analyzed by flow citometry software (Becton Dickinson). Apoptosis in adult cardiomyocytes was detected as nucleosomal DNA fragmentation measured as the percentage of sub-G1 cells. After treatment, cardiomyocytes were fixed with ice-cold 70% ethanol, stored at −20°C overnight, and then incubated for 10 min in phosphate-citric acid buffer at pH 7.8. Pelleted cells were then stained with 1 ml of 25 μg/ml propidium iodide, 400 mg/ml Rnase A, and 1% Triton X-100 at RT for 15 min, and analyzed by FACS®. The percentage of sub-G1 cells was calculated by CellFit software (Becton Dickinson) and defined as apoptotic. Direct microscopical observation confirmed the presence of nuclear fragmentation and apoptotic bodies.
Western Blot and binding studies
Western blots of solubilized proteins were performed as described previously (Cutrupi et al., 2000). Binding of 125I-Tyr4-ghrelin and 125I-Tyr4–des-acyl ghrelin to crude H9c2 cardiomyocytes membranes (30,000 g pellet) and Scatchard analysis were determined as described previously (Muccioli et al., 2001; Torsello et al., 2002). IC50 values of specific radioligand binding were determined by radiolabeled ghrelin and des-acyl ghrelin displacement curves with increasing concentrations of unlabeled ghrelin and des-acyl ghrelin. The maximal number of binding sites (Bmax) and the dissociation constant (K d) and IC50 values were calculated with the iterative curve-fitting Prism3 program (GradPad Software, Inc.).
RT-PCR
Total RNA was extracted by Rneasy mini kit (QIAGEN) from either cultured cells or rat heart and pituitary tissue mechanically triturated in liquid nitrogen and retrotranscribed (1 μg) in 20 μl of RT-buffer in the presence of 500 ng oligodT and 1 μl Powerscript reverse transcriptase. Reverse transcriptase reaction was performed at 42°C for 1.5 h and at 72°C for 15 min. One fifth of the reverse transcriptase product (4 μl of cDNA) was amplified in 50 μl PCR buffer (1× buffer, 1.5 mM MgCl2, 200 μM dNTP, 1 μM of each primer, 1 U DyNAzyme EXT taq). PCR was performed by a 5-min incubation at 95°C followed by 5 cycles (30 s at 94°C, 30 s at 55°C, and 3 min at 72°C) and 35 cycles (30 s at 94°C, 30 s at 60°C, and 3 min at 72°C). Primers sequences for GHSR-1a were the rat homologues to the human sequences described previously by Nagaya et al. (2001a): 5′-CGGAGAGATGGGATGTGCTG-3′ (forward) and 5′-CTCTGCTGGCTGCCCTTCCA-3′ (reverse).
Acknowledgments
The authors wish to thank Prof. Paola Cassoni and Drs. Tiziana Marrocco, Marianna Notario, Federica Chianale, and Marco Coscia for their participation in the study.
This study was supported by grants from University A. Avogadro of Piemonte Orientale and Regione Piemonte (to A. Graziani), Ministero dell'Università e della Ricerca Scientifica e Tecnologica (1999 and 2000 to A. Graziani and to E. Ghigo), University of Torino (to A. Graziani, A. Fubini, and E. Ghigo), Consiglio Nazionale delle Richerche (98.03040.CP04 to E. Ghigo and to F. Broglio), Fondazione per le Studio delle Malattie Endocrino-Metaboliche, and Europeptides. G. Baldanzi was supported by a fellowship from Fondazione Italiana per la Ricerca sul Cancro, and N. Filigheddu was a recipient of the Ibsen/SIE award.
Footnotes
Abbreviations used in this paper: CT-1 cardiotrophin-1; ERK, extracellular signal–regulated kinase; GH, growth hormone, GHS, GH secretagogue, GHSR, GH secretagogue receptor; IGF-1, insulin-like growth factor-1; LDH, lactate dehydrogenase; MTT, 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide; PI, phosphatidylinositol.
References
- Alessi, D.R., M. Andjelkovic, B. Caudwell, P. Cron, N. Morrice, P. Cohen, and B.A. Hemmings. 1996. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Arola, O.J., A. Saraste, K. Pulkki, M. Kallajoki, M. Parvinen, and L.M. Voipio-Pulkki. 2000. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 60:1789–1792. [PubMed] [Google Scholar]
- Bisi, G., V. Podio, M.R. Valetto, F. Broglio, G. Bertuccio, G. Aimaretti, E. Pelosi, G. Del Rio, G. Muccioli, H. Ong, et al. 1999. Cardiac effects of hexarelin in hypopituitary adults. Eur. J. Pharmacol. 381:31–38. [DOI] [PubMed] [Google Scholar]
- Bodart, V., M. Febbraio, A. Demers, N. McNicoll, P. Pohankova, A. Perreault, T. Sejlitz, E. Escher, R.L. Silverstein, D. Lamontagne, and H. Ong. 2002. CD36 mediates the cardiovascular action of growth hormone-releasing peptides in the heart. Circ. Res. 90:844–849. [DOI] [PubMed] [Google Scholar]
- Bowers, C.Y. 2001. Unnatural growth hormone-releasing peptide begets natural ghrelin. J. Clin. Endocrinol. Metab. 86:1464–1469. [DOI] [PubMed] [Google Scholar]
- Buerke, M., T. Murohara, C. Skurk, C. Nuss, K. Tomaselli, and A.M. Lefer. 1995. Cardioprotective effect of insulin-like growth factor I in myocardial ischemia followed by reperfusion. Proc. Natl. Acad. Sci. USA. 92:8031–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassoni, P., M. Papotti, C. Ghe, F. Catapano, A. Sapino, A. Graziani, R. Deghenghi, T. Reissmann, E. Ghigo, and G. Muccioli. 2001. Identification, characterization, and biological activity of specific receptors for natural (ghrelin) and synthetic growth hormone secretagogues and analogs in human breast carcinomas and cell lines. J. Clin. Endocrinol. Metab. 86:1738–1745. [DOI] [PubMed] [Google Scholar]
- Chen, S., D.C. Guttridge, E. Tang, S. Shi, K. Guan, and C.Y. Wang. 2001. Suppression of tumor necrosis factor-mediated apoptosis by nuclear factor kappaB-independent bone morphogenetic protein/Smad signaling. J. Biol. Chem. 276:39259–39263. [DOI] [PubMed] [Google Scholar]
- Cutrupi, S., G. Baldanzi, D. Gramaglia, A. Maffe, D. Schaap, E. Giraudo, W. van Blitterswijk, F. Bussolino, P.M. Comoglio, and A. Graziani. 2000. Src-mediated activation of alpha-diacylglycerol kinase is required for hepatocyte growth factor-induced cell motility. EMBO J. 19:4614–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filigheddu, N., A. Fubini, G. Baldanzi, S. Cutrupi, C. Ghe, F. Catapano, F. Broglio, A. Bosia, M. Papotti, G. Muccioli, et al. 2001. Hexarelin protects H9c2 cardiomyocytes from doxorubicin-induced cell death. Endocrine. 14(1):113–119. [DOI] [PubMed] [Google Scholar]
- Gallo, M.P., D. Malan, I. Bedendi, C. Biasin, G. Alloatti, and R.C. Levi. 2001. Regulation of cardiac calcium current by NO and cGMP-modulating agents. Pflugers Arch. 441:621–628. [DOI] [PubMed] [Google Scholar]
- Gnanapavan, S., B. Kola, S.A. Bustin, D.G. Morris, P. McGee, P. Fairclough, S. Bhattacharya, R. Carpenter, A.B. Grossman, and M. Korbonits. 2002. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J. Clin. Endocrinol. Metab. 87:2988. [DOI] [PubMed] [Google Scholar]
- Hayakawa, K., G. Takemura, M. Koda, Y. Kawase, R. Maruyama, Y. Li, S. Minatoguchi, T. Fujiwara, and H. Fujiwara. 2002. Sensitivity to apoptosis signal, clearance rate, and ultrastructure of fas ligand-induced apoptosis in in vivo adult cardiac cells. Circulation. 105:3039–3045. [DOI] [PubMed] [Google Scholar]
- Hirota, H., J. Chen, U.A. Betz, K. Rajewsky, Y. Gu, J. Ross, Jr., W. Muller, and K.R. Chien. 1999. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 97:189–198. [DOI] [PubMed] [Google Scholar]
- Hosoda, H., M, Kojima., H. Matsuo, and K. Kangawa. 2000. Ghrelin and des-acyl ghrelin: two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem. Biophys. Res. Comm. 279:909–913. [DOI] [PubMed] [Google Scholar]
- Howard, A.D., S.D. Feighner, D.F. Cully, J.P. Arena, P.A. Liberator, C.I. Rosenblum, M. Hamelin, D.L. Hreniuk, O.C. Palyha, J. Anderson, et al. 1996. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 273:974–977. [DOI] [PubMed] [Google Scholar]
- Inu, A. 2001. Ghrelin: an orexigenic and somatotrophic signal from the stomach. Nat. Rev. Neurosci. 2:551–560. [DOI] [PubMed] [Google Scholar]
- Jeremias, I., C Kupatt, A. Martin-Villalba, H. Habazettl, J. Schenkel, P. Boekstegers, and K.M. Debatin. 2000. Involvement of CD95/Apo1/Fas in cell death after myocardial ischemia. Circulation. 102:915–920. [DOI] [PubMed] [Google Scholar]
- Katugampola, S.D., Z. Pallikaros, and A.P. Davenport. 2001. [125I-His(9)]-ghrelin, a novel radioligand for localizing GHS orphan receptors in human and rat tissue: up-regulation of receptors with athersclerosis. Br. J. Pharmacol. 134:143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, M.K., D.M. Gay, L.C. Pan, J.H. McElmurray, III, J.W. Hendrick, C. Pirie, A. Morrison, C. Ding, R. Mukherjee, and F.G. Spinale. 2001. Treatment with a growth hormone secretagogue in a model of developing heart failure: effects on ventricular and myocyte function. Circulation. 103:308–313. [DOI] [PubMed] [Google Scholar]
- Kojima, M., H. Hosoda, Y. Date, M. Nakazato, H. Matsuo, and K. Kangawa. 1999. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 402:656–660. [DOI] [PubMed] [Google Scholar]
- Locatelli, V., G. Rossoni, F. Schweiger, A. Torsello, V. De Gennaro Colonna, M. Bernareggi, R. Deghenghi, E.E. Muller, and F. Berti. 1999. Growth hormone-independent cardioprotective effects of hexarelin in the rat. Endocrinology. 140:4024–4031. [DOI] [PubMed] [Google Scholar]
- Muccioli, G., M. Papotti, V. Locatelli, E. Ghigo, and R. Deghenghi. 2001. Binding of 125I-labeled ghrelin to membranes from human hypothalamus and pituitary gland. J. Endocrinol. Invest. 24:RC7–RC9. [DOI] [PubMed] [Google Scholar]
- Muccioli, G., M. Tschöp, M. Papotti, R. Deghenghi, M. Heiman, and E. Ghigo. 2002. Neuroendocrine and peripheral activities of ghrelin: implications in metabolism and obesity. Eur. J. Pharmacol. 440:235–254. [DOI] [PubMed] [Google Scholar]
- Murata, M., Y. Okimura, K. Iida, M. Matsumoto, H. Sowa, H. Kaji, M. Kojima, K. Kangawa, and K. Chihara. 2002. Ghrelin modulates the downstream molecules of insulin signaling in hepatoma cells. J. Biol. Chem. 277:5667–5674. [DOI] [PubMed] [Google Scholar]
- Nagaya, N., M. Kojima, M. Uematsu, M. Yamagishi, H. Hosoda, H. Oya, Y. Hayashi, and K. Kangawa. 2001. a. Hemodynamic and hormonal effects of human ghrelin in healthy volunteers. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280:R1483–R1487. [DOI] [PubMed] [Google Scholar]
- Nagaya, N., M. Uematsu, M. Kojima, Y. Ikeda, F. Yoshihara, W. Shimizu, H. Hosoda, Y. Hirota, H. Ishida, H. Mori, and K. Kangawa. 2001. b. Chronic administration of ghrelin improves left ventricular dysfunction and attenuates development of cardiac cachexia in rats with heart failure. Circulation. 104:1430–1435. [DOI] [PubMed] [Google Scholar]
- Nakamura, T., Y. Ueda, Y. Juan, S. Katsuda, H. Takahashi, and E. Koh. 2000. Fas-mediated apoptosis in adriamycin-induced cardiomyopathy in rats: in vivo study. Circulation. 102:572–578. [DOI] [PubMed] [Google Scholar]
- Nakazato, M., N. Murakami, Y. Date, M. Kojima, H. Matsuo, K. Kangawa, and S. Matsukura. 2001. A role for ghrelin in the central regulation of feeding. Nature. 409:194–198. [DOI] [PubMed] [Google Scholar]
- Okumura, H., N. Nagaya, M. Enomoto, E. Nakagawa, H. Oya, and K. Kangawa. 2002. Vasodilatory effect of ghrelin, an endogenous peptide from the stomach. J. Cardiovasc. Pharmacol. 39:779–783. [DOI] [PubMed] [Google Scholar]
- Papotti, M., C. Ghe, P. Cassoni, F. Catapano, R. Deghenghi, E. Ghigo, and G. Muccioli. 2000. Growth hormone secretagogue binding sites in peripheral human tissues. J. Clin. Endocrinol. Metab. 85:3803–3807. [DOI] [PubMed] [Google Scholar]
- Parrizas, M., A.R. Saltiel, and D. LeRoith. 1997. IGF-1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J. Biol. Chem. 272:154–161. [DOI] [PubMed] [Google Scholar]
- Rios, R., I. Carneiro, V.M. Arce, and J. Devesa. 2001. Myostatin regulates cell survival during C2C12 myogenesis. Biochem. Biophys. Res. Commun. 280:561–566. [DOI] [PubMed] [Google Scholar]
- Takemura, G., S. Kato, T. Aoyama, Y. Hayakawa, M. Kanoh, R. Maruyama, M. Arai, K. Nishigaki, S. Minatoguchi, K. Fukuda, et al. 2001. Characterization of ultrastructure and its relation with DNA fragmentation in Fas-induced apoptosis of cultured cardiac myocytes. J. Pathol. 193:546–556. [DOI] [PubMed] [Google Scholar]
- Tivesten, A., E. Bollano, K. Caidahl, V. Kujacic, X.Y. Sun, T. Hedner, A. Hjalmarson, B.A. Bengtsson, and J. Isgaard. 2000. The growth hormone secretagogue hexarelin improves cardiac function in rats after experimental myocardial infarction. Endocrinology. 141:60–66. [DOI] [PubMed] [Google Scholar]
- Torsello, A., C. Ghè, E. Bresciani, F. Catapano, E. Ghigo, R. Deghenghi, V. Locatelli, and G. Muccioli. 2002. Short ghrelin peptides neither displace ghrelin binding in vitro nor stimulate GH release in vivo. Endocrinology. 143:1968–1971. [DOI] [PubMed] [Google Scholar]
- Tschop, M., D.L. Smiley, and M.L. Heiman. 2000. Ghrelin induces adiposity in rodents. Nature. 407:908–913. [DOI] [PubMed] [Google Scholar]
- Wu, W., W.L. Lee, Y.Y. Wu, D. Chen, T.J. Liu, A. Jang, P.M. Sharma, and P.H. Wang. 2000. Expression of constitutively active phosphatidylinositol 3-kinase inhibits activation of caspase 3 and apoptosis of cardiac muscle cells. J. Biol. Chem. 275:40113–40119. [DOI] [PubMed] [Google Scholar]
- Yamamura, T., H. Nakamura, T. Yamamoto, S. Umemoto, T. Fujii, N. Kobayashi, and M. Matsuzaki. 1999. Fas expression and apoptosis correlate with cardiac dysfunction in patients with dilated cardiomyopathy. Jpn. Circ. J. 63:149–154. [DOI] [PubMed] [Google Scholar]