

Abstract
Mechanisms for activating the actin-related protein 2/3 (Arp2/3) complex have been the focus of many recent studies. Here, we identify a novel mode of Arp2/3 complex regulation mediated by the highly conserved actin binding protein coronin. Yeast coronin (Crn1) physically associates with the Arp2/3 complex and inhibits WA- and Abp1-activated actin nucleation in vitro. The inhibition occurs specifically in the absence of preformed actin filaments, suggesting that Crn1 may restrict Arp2/3 complex activity to the sides of filaments. The inhibitory activity of Crn1 resides in its coiled coil domain. Localization of Crn1 to actin patches in vivo and association of Crn1 with the Arp2/3 complex also require its coiled coil domain. Genetic studies provide in vivo evidence for these interactions and activities. Overexpression of CRN1 causes growth arrest and redistribution of Arp2 and Crn1p into aberrant actin loops. These defects are suppressed by deletion of the Crn1 coiled coil domain and by arc35-26, an allele of the p35 subunit of the Arp2/3 complex. Further in vivo evidence that coronin regulates the Arp2/3 complex comes from the observation that crn1 and arp2 mutants display an allele-specific synthetic interaction. This work identifies a new form of regulation of the Arp2/3 complex and an important cellular function for coronin.
Keywords: actin; yeast; coronin; Arp2/3 complex; coiled coil
Introduction
Many cellular processes, including cell locomotion, vesicle and organelle transport, endocytosis, cytokinesis, and polarized cell growth, require dynamic remodeling of the actin cytoskeleton. All of these processes involve spatially controlled assembly and reorganization of actin networks in response to cellular cues. However, the exact mechanisms regulating these spatial and temporal changes are not well understood. In recent years, the actin-related protein 2/3 (Arp2/3)* complex has emerged as a central effector of actin assembly that receives multiple signal inputs (for review see Higgs and Pollard, 2001).
The Arp2/3 complex is composed of seven evolutionarily conserved subunits: two actin-related proteins (Arp2 and Arp3) and five other subunits, which in yeast are called Arc40, Arc35, Arc18, Arc19, and Arc15. In all organisms examined, the Arp2/3 complex localizes to sites of dynamic actin assembly. In yeast, the Arp2/3 complex localizes to cortical actin patches, highly motile filamentous actin structures (for reviews see Pruyne and Bretscher, 2000; Goode and Rodal, 2001). Mutations in different subunits of the yeast Arp2/3 complex disrupt actin organization, actin patch motility, and actin-dependent processes such as endocytosis, cell polarity development, and organelle inheritance (for review see Goode and Rodal, 2001).
The Arp2/3 complex has two established and apparently coupled activities, actin nucleation and actin filament branching (for reviews see Cooper et al., 2001; Borths and Welch, 2002; Kreishman-Deltrick and Rosen, 2002). The Arp2/3 complex can bind to the side of an existing (mother) filament and nucleate the formation of a new (daughter) filament at a 70° angle, leading to the formation of branched filament networks (Mullins et al., 1998). Alone, the Arp2/3 complex has relatively weak actin nucleation activity. Activation is achieved by two complementary mechanisms: (1) association of the complex with the side of an actin filament and (2) interactions with an activator protein, such as SCAR/WASp, myosin I, Abp1, cortactin, and Pan1 (for reviews see Olazabal and Machesky, 2001; Schafer, 2002).
Coronin is a conserved component of the actin cytoskeleton found in all eukaryotes examined from yeast to mammals, where it localizes to sites of dynamic actin assembly (for review see de Hostos, 1999). In budding yeast, coronin-null mutants have no overt phenotype, but overexpression of the coronin gene (CRN1) is lethal and disrupts actin organization. In addition, genetic interactions with act1-159 and cof1-22 suggest that Crn1 regulates some aspect of actin assembly and/or turnover (Goode et al., 1999). In Dictyostelium discoideum, coronin mutants display defects in cell migration, cytokinesis, phagocytosis, and fluid phase endocytosis (de Hostos et al., 1993). In cultured Xenopus cells, overexpression of a coronin fragment causes severe defects in cell migration and spreading (Mishima and Nishida, 1999).
The biochemical properties of coronin support the notion that it regulates actin assembly and organization. In vitro, purified coronin binds specifically to filamentous actin, bundles actin filaments, and weakly promotes actin assembly (Goode et al., 1999; Asano et al., 2001). The amino terminus of coronin contains five to six β-propeller–like WD repeats that form the actin binding domain (Goode et al., 1999). The carboxy terminus is comprised of a “unique” region, which is highly variable among species, and a short conserved coiled coil domain (residues 603–651 in yeast coronin). The coiled coil domain is required for coronin dimerization and actin filament bundling in vitro (Goode et al., 1999; Asano et al., 2001). In Xenopus cells, deletion of the coiled coil domain causes mislocalization of coronin, suggesting that dimerization, or other interactions of the coiled coil domain, is necessary for its proper localization and function (Mishima and Nishida, 1999). However, the exact function of coronin within the actin cytoskeleton has remained unclear.
Here, we identify a molecular function for yeast coronin (Crn1). We provide multiple lines of biochemical and genetic evidence that Crn1 associates with and regulates the Arp2/3 complex through an interaction of its coiled coil domain. These studies reveal an important cellular function for Crn1 and novel aspects of Arp2/3 complex regulation.
Results
Crn1 physically associates with the Arp2/3 complex
To better understand the cellular function of yeast coronin (Crn1), we sought to identify Crn1-interacting partners. Wild-type cell extracts were fractionated on sucrose gradients by velocity sedimentation and the migration patterns of actin, Crn1, and numerous other actin-associated proteins were determined by immunoblotting. Fig. 1 A shows the data for actin, Crn1, and Arp2p (a component of the Arp2/3 complex). The vast majority of actin (43 kD) migrated to a position consistent with actin monomers. Crn1 migration had two distinct peaks, with ∼40% of the Crn1 peaking at a position consistent with Crn1 monomers (72 kD) and ∼60% of the Crn1 peaking at a position suggesting a complex of 250–300 kD. Immunoblotting with antibodies against numerous actin-associated proteins (Aip1, Cof1, Cap2, Pfy1, Sac6, Sla2, Srv2, Tpm1, and Twf1; unpublished data) revealed that only one, Arp2, comigrated with Crn1 in the 250–300-kD range. This raised the possibility that Crn1 and the Arp2/3 complex physically associate.
Figure 1.
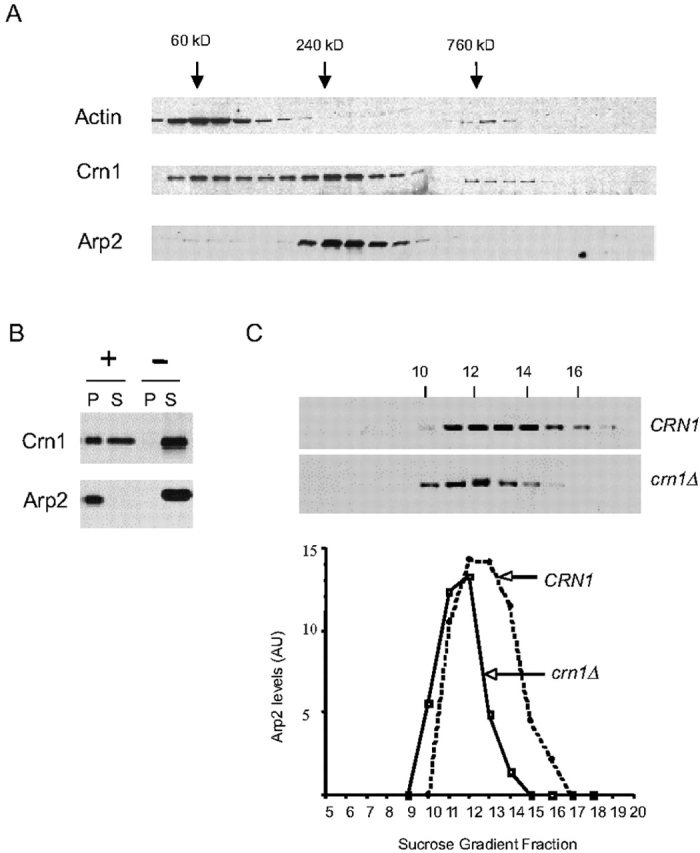
Crn1 physically associates with the Arp2/3 complex. (A) Comigration of Crn1 and Arp2 by sedimentation velocity. Yeast cell lysates were fractionated on sucrose gradients by overnight high speed centrifugation, and then fractions were collected. Samples of each fraction were blotted and probed with antibodies for yeast actin, Crn1, and Arp2. Size standards were fractionated in parallel: BSA (60 kD), Catalase (240 kD), and Thyroglobulin (760 kD). (B) Coimmunoprecipitation of Crn1 with Arp2–HA. Yeast cell lysates expressing a carboxy-terminal–tagged Arp2–HA fusion protein were incubated with anti-HA antibody–coated beads (+) or control beads with no antibody (−). Beads were pelleted, and equivalent loads of pellets and supernatants were blotted and probed with anti-Crn1 or anti-Arp2 antibodies. (C) Comparison of Arp2 migration in wild-type and crn1-null cell extracts fractionated by sedimentation velocity. Arp2 signal was quantified by immunoblotting and densitometry. The distribution was compared for wild-type and crn1-null yeast extracts fractionated on sucrose gradients.
Next, we tested the ability of Crn1 to coimmunoprecipitate with the Arp2/3 complex from cell lysates. We integrated an epitope tag (3xHA) at the carboxy terminus of ARP2. The tagged protein was the only source of Arp2p in cells and fully complemented growth at 16–37°C with no visible defects in actin organization (unpublished data). To control for potential nonspecific interactions from coprecipitation of actin filaments with the Arp2/3 complex, we included 40 μM latrunculin A in the immunoprecipitation reactions. Immunoblotting confirmed the absence of actin in the pellets (unpublished data). As shown in Fig. 1 B, over half of the Crn1 in cells coimmunoprecipitated with the Arp2/3 complex, similar to the fraction of Crn1 that comigrated with the Arp2/3 complex in sucrose gradients (Fig. 1 A). Thus, by two independent approaches (velocity sedimentation and coimmunoprecipitation), Crn1 was found to associate with the Arp2/3 complex.
As an additional test of the interaction, we compared Arp2 migration in extracts from wild-type and crn1-null cells fractionated on sucrose gradients. Arp2 migration exhibited a substantial shift and narrowing of its peak in the crn1-null lysate, consistent with a loss of mass from a large subset of the Arp2/3 complex in cells (Fig. 1 C). We have determined that Crn1 and the Arp2/3 complex have a similar abundance in yeast (Arp2/3 complex is slightly more abundant than Crn1; unpublished data). This, combined with the data in Fig. 1 C, indicates that >25% of the cellular pool of the Arp2/3 complex is stably associated with Crn1.
Next, we tested if the Crn1–Arp2/3 complex interaction is direct. To accomplish this, we purified HA-tagged Arp2/3 complex on HA antibody–coated beads (Fig. 2). The beads were washed in high salt to remove Arp2/3 complex–associated factors, such as coronin, Abp1, and Las17. The purified material has the characteristic gel band pattern of the Arp2/3 complex subunits. Further, mass spectrometry analysis of the complex released from beads verifies that it is the Arp2/3 complex, and the released complex is active in promoting actin nucleation (unpublished data). As shown in Fig. 2, purified Crn1 binds to HA–Arp2/3 complex beads, but not to control beads (HA antibody, but no Arp2/3 complex). This demonstrates that the Crn1–Arp2/3 complex interaction is direct. The binding saturated at a molar stoichiometry of ∼1:1 Crn1 to Arp2/3 complex, and the addition of higher concentrations of Crn1 to the reactions did not increase the amount of Crn1 bound (unpublished data).
Figure 2.

Direct interactions between Crn1, Arp2/3 complex, and actin filaments. Coomassie-stained gel of HA–Arp2/3 complex isolated on HA antibody–coupled beads (lane 1); 1 μM Crn1 cosediments with HA–Arp2/3 complex beads (lane 2), but not with control beads (lane 3). Gel migration positions are labeled for Crn1 (arrow) and Arp2/3 complex subunits (lines).
The coiled coil domain of Crn1 is required for association with the Arp2/3 complex and Crn1 localization in vivo
In a two hybrid screen using the Arc35/p35 subunit of the Arp2/3 complex as bait, we identified a specific interaction with a carboxy-terminal fragment of Crn1. Sequencing of two independently selected plasmids revealed the same fragment of Crn1, encoding residues 466–651 (see Materials and methods). This raised the possibility that the carboxy terminus of Crn1 might be important for mediating physical interactions with the Arp2/3 complex. To test this hypothesis, we examined the ability of Crn1 fragments to coimmunoprecipitate with the Arp2/3 complex. Low copy plasmids expressing fragments of Crn1 were transformed into a crn1-null strain carrying a 3xHA epitope tag integrated at the carboxy terminus of ARP2. Cell lysates from these strains were used for immunoprecipitation assays. Full-length Crn1, Crn1 (1–600), and Crn1 (400–651) were expressed to similar levels (Fig. 3 A; whole cell extract blot). As shown in Fig. 3 A, Crn1 and Crn1 (400–651) coimmunoprecipitated with the Arp2/3 complex, but Crn1 (1–600) did not. These data show that the coiled coil domain–containing carboxy terminus of Crn1 is both required and sufficient for association with the Arp2/3 complex in vivo.
Figure 3.
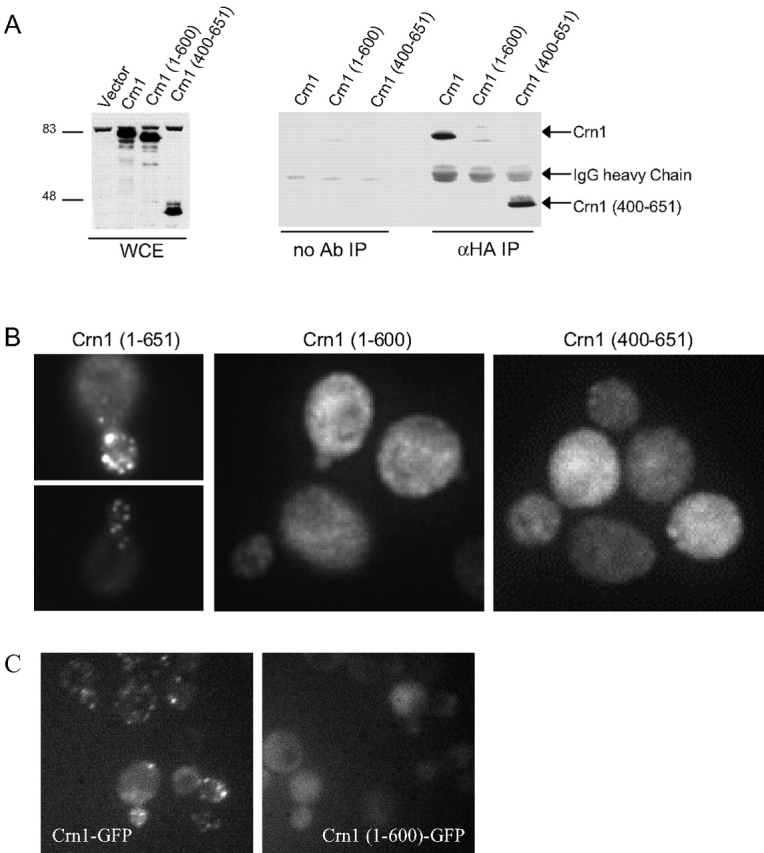
Domain requirements of Crn1 for association with the Arp2/3 complex and localization to actin patches in vivo. (A) Coimmunoprecipitation of Crn1 with the Arp2/3 complex is dependent on the Crn1 coiled coil domain. Arp2–HA was immunoprecipitated using anti-HA antibodies from lysates of crn1-null cells transformed with low copy plasmids expressing full-length Crn1, Crn1 (1–600), and Crn1 (400–651). Whole cell extracts and pellets from the immunoprecipitations were blotted and probed with anti-Crn1 antibodies. (B) Localization of full-length Crn1, Crn1 (1–600), or Crn1 (400–651) in the same cells by immunofluorescence microscopy using anti-Crn1 antibodies. (C) Localization of Crn1–GFP and Crn1 (1–600)–GFP fusion proteins in live cells.
Next, we examined the localization of Crn1 and Crn1 fragments in cells by immunofluorescence with anti-Crn1 antibodies. Crn1 localized to actin patches, as expected, but Crn1 (1–600) and Crn1 (400–651) localized primarily to the cytoplasm, with only faint residual actin patch staining (Fig. 3 B). The localization of Crn1 (1–600) to the cytoplasm was unexpected, given that this construct binds to actin filaments in vitro (Goode et al., 1999), and prompted us to examine the localization patterns of Crn1 and Crn1 (1–600) by an independent approach. We integrated GFP tags at the CRN1 locus carboxy terminus after the codons for residues 600 and 650, generating strains that express Crn1–GFP and Crn1 (1–600)–GFP fusion proteins, respectively. Immunoblotting with Crn1 and GFP antibodies confirmed that these constructs were expressed at normal levels and were the only source of Crn1 in cells (unpublished data). As shown in Fig. 3 C, Crn1–GFP localizes to cortical actin patches, and Crn1 (1–600)–GFP localizes primarily to the cytoplasm, confirming the immunofluorescence data. These results demonstrate that neither the actin binding domain nor the Arp2/3 complex–interacting carboxy terminus of Crn1 is sufficient for localization in vivo.
The coiled coil domain is required for defects in actin organization and cell growth caused by Crn1 overproduction
Deletion of the CRN1 gene in yeast causes no overt growth phenotype or defects in actin organization (Heil-Chapdelaine et al., 1998; Goode et al., 1999). However, as shown in Fig. 4 A, galactose promoter–driven overexpression of untagged Crn1 causes severe defects in actin organization and arrest of cell growth. Cells overproducing Crn1 are swollen, have depolarized actin patches, and form spiraled or looped actin structures (Fig. 4 B). The actin loops do not appear to be cable like, because they do not label with tropomyosin antibodies (a cable-specific marker) and they form in the absence of any functional formin proteins, Bnr1 and Bni1 (unpublished data). The actin loops also are distinct from the actin bars formed in cells overproducing a GST–Crn1 fusion protein (Goode et al., 1999), because unlike the bars, the loops label with rhodamine phalloidin. These aberrant actin loops were detected in 36% of cells overproducing Crn1, but never in control cells (>100 cells scored in three separate experiments).
Figure 4.

Overexpression of Crn1 is lethal and causes the formation of aberrant actin loop structures. (A) Growth on glucose- and galactose-containing media of cells carrying empty vector or plasmids expressing CRN1 and CRN1 (1–600) under control of the GAL promoter. Cells were serially diluted, spotted onto plates, and grown for 3 d. (B) Rhodamine phalloidin staining of abnormal actin structures in cells overproducing Crn1. Cells carrying pGAL-CRN1 were grown to log phase in glucose and the expression of CRN1 was induced by growth in galactose-containing medium for 4 h. Then, cells were fixed and actin organization was examined by rhodamine phalloidin staining. (C) Immunoblot of total cellular extracts from strains expressing Crn1 from a low copy plasmid (see Fig. 3) and induced to overexpress different Crn1 constructs by growth in galactose-containing medium. The blot was probed with rabbit anti-Crn1 antibodies.
To define the part of Crn1 that mediates these defects, we examined cells overproducing different Crn1 fragments from the galactose-inducible promoter. Whereas cells overexpressing full-length Crn1 showed growth arrest on galactose media, cells carrying vector alone or pGAL-Crn1 (1–600) were viable (Fig. 4 A). Further, these cells did not contain the aberrant actin loop structures found in cells overexpressing full-length Crn1 (unpublished data). Immunoblotting confirmed that Crn1 and Crn1 (1–600) were overexpressed to similar levels in these strains, well above endogenous Crn1 expression levels (Fig. 4 C). Thus, the coiled coil domain is required for the growth arrest and formation of actin loop structures caused by Crn1 overexpression. This finding raises the possibility that interactions between the coiled coil domain of Crn1 and the Arp2/3 complex lead to these defects in cell growth and actin loop formation. We were unable to determine if overproduction of Crn1 (400–651) was sufficient to cause the defects, because this construct was not successfully overproduced; the overexpression levels of this construct were similar to endogenous Crn1 in wild-type cells (Fig. 4 C).
To test whether Crn1 becomes mislocalized upon overproduction, we examined Crn1 localization by immunofluorescence in the Crn1-overexpressing cells (Fig. 5). In cells carrying an empty vector, endogenous Crn1 colocalized with actin patches as expected. However, in cells overproducing Crn1, Crn1 was found to associate with actin patches and loop structures (Fig. 5 A). Treatment of these cells with latrunculin A, an actin monomer sequestering agent, caused Crn1 staining to shift to the cytoplasm, demonstrating that the localization of Crn1 to both structures depends on filamentous actin (Fig. 5 B). Costaining with actin and Crn1 antibodies confirmed that Crn1 localizes to the same aberrant actin loops that form as a result of Crn1 overexpression (Fig. 5 C).
Figure 5.
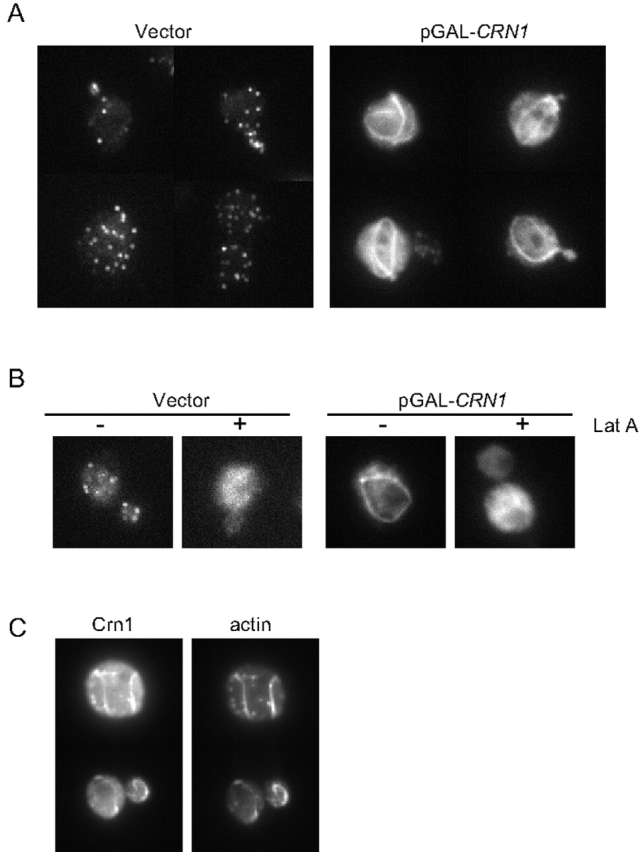
Localization of overproduced Crn1 to aberrant actin loop structures. (A) Cells carrying pGAL-CRN1 or empty vector were grown to log phase in glucose medium, and then cultures were shifted to galactose-containing medium for 4 h to induce overexpression of CRN1. The cells were fixed and Crn1 localization was determined by immunofluorescence using rabbit anti-Crn1 antibodies. (B) Localization of Crn1 in latrunculin A–treated cells. Cells carrying pGAL-CRN1 or empty vector grown as above and shifted to galactose-containing medium for 4 h were treated for 15 min with 100 μM Lat A dissolved in DMSO or an equal volume of DMSO. Crn1 localization was determined by immunofluorescence as above. (C) Coimmunofluorescence of Crn1 and actin in cells overexpressing CRN1 under control of the GAL promoter.
We also examined the localization of Arp2–YFP in cells overexpressing Crn1 (Fig. 6). In control cells, Arp2–YFP localized to actin patches, similar to Arp2 immunostaining (Moreau et al., 1996). However, in strains overproducing Crn1, Arp2–YFP also localized to looped structures. Importantly, two other actin patch components, Abp1 and capping protein, remained localized to actin patches in cells overexpressing Crn1 (Fig. 6). Similarly, Las17–GFP remained localized to actin patches and was not recruited to the actin loops in cells overexpressing Crn1 (unpublished data). These data demonstrate that recruitment of Arp2 to Crn1-induced looped structures is specific. They also provide further in vivo support for a physical interaction between Crn1 and the Arp2/3 complex.
Figure 6.

Localization of Arp2–YFP in cells overexpressing CRN1. (A) Cells expressing an integrated Arp2–YFP fusion protein and carrying empty vector or pGAL-CRN1 were grown to log phase, and expression of CRN1 was induced for 4 h in galactose-containing medium. Arp2–YFP localization was examined in live cells by fluorescence microscopy. (B) Cells expressing integrated Abp1–GFP or Cap2–GFP fusion proteins and carrying empty vector or pGAL-CRN1 were grown to log phase. Then, expression of CRN1 was induced for 4 h in galactose-containing medium. Abp1–GFP and Cap2–GFP localization was examined in live cells by fluorescence microscopy.
Genetic interactions between CRN1, ARC35, and ARP2
Given that the lethality caused by CRN1 overexpression requires its coiled coil domain and that this region of Crn1 interacts with Arc35 in the two hybrid assay, we reasoned that mutations in arc35 might be able to suppress the lethal effects of CRN1 overexpression. To test this hypothesis, we used a collection of temperature-sensitive arc35 alleles generated by random mutagenesis (unpublished data). We transformed the integrated arc35 mutant strains with a GAL–CRN1 overexpression plasmid or vector alone. The transformed cells were diluted serially, spotted on glucose and galactose media, and grown for 3 d at a range of temperatures. Most of the arc35 mutant strains, independent of CRN1 overexpression, exhibited normal growth at 28°C on glucose medium but grew poorly, if at all, on galactose medium relative to an isogenic wild-type ARC35 strain (Fig. 7 A, top). One allele carrying pGAL-CRN1, arc35-26 grew well on galactose. Fig. 7 A shows the data for wild-type ARC35, arc35-26, and one of the many nonsuppressing arc35 alleles, arc35-12. This result shows that arc35-26 strongly suppresses the CRN1 overexpression defects, and, reciprocally, CRN1 overexpression suppresses arc35-26 growth defects on galactose (compare with arc35-26 carrying empty vector on galactose; Fig. 7 A, top right).
Figure 7.

Genetic interactions between Crn1 and the Arp2/3 complex. (A) Suppression of CRN1 overexpression growth defects by arc35-26. A collection of 17 mutant arc35 alleles and a congenic wild-type strain were transformed with pGAL-CRN1 or empty vector alone. Transformants were serially diluted, spotted onto glucose- and galactose-containing media, and grown for 3 d at 28°C. Data are shown for wild-type ARC35, arc35-26, and arc35-12 strains. (B) Synthetic growth defects between a crn1-null mutation and the arp2-21 temperature-sensitive mutant. Cells were grown to log phase, serially diluted, spotted onto glucose-rich plates, and grown for 3 d at 30°C or 34°C.
We next explored the possibility of genetic interactions between CRN1 and the genes encoding other subunits of the Arp2/3 complex. We performed directed crosses between a crn1-null and a number of published arp2 alleles, the arc40-40 allele (Tong et al., 2001), and several unpublished arp2 alleles (a gift from H. Xu and C. Boone, University of Toronto). These crosses revealed an allele-specific genetic interaction between the crn1-null mutant and arp2-21, a temperature-sensitive mutant (Fig. 7 B). arp2-21 mutant cells exhibit normal growth at 30°C, are partially compromised for growth at 34°C, and are dead at 37°C, whereas crn1Δ-null cells exhibit normal growth at all temperatures. However, crn1Δ arp2-21 double mutant cells are severely compromised for growth at 34°C. Rhodamine phalloidin staining showed that crn1Δ arp2-21 cells have similar defects in actin organization (highly depolarized actin patches) to arp2-21 cells (unpublished data). These data, combined with the suppression analysis above, strongly support an in vivo functional interaction between Crn1 and the Arp2/3 complex in a similar physiological process.
The carboxy terminus of Crn1 inhibits actin nucleation of the Arp2/3 complex
To investigate the biochemical basis of our in vivo observations, we compared the nucleation activities of purified Arp2/3 complex in the presence and absence of Crn1. The polymerization of actin alone is slow (Fig. 8 A, curve F), reflecting an inherently poor nucleation activity of purified actin monomers. However, the addition of 20 nM Arp2/3 complex plus 200 nM activating (WA) fragment of Las17/Bee1 (the yeast homologue of WASp) stimulated rapid actin nucleation (Fig. 8 A, curve B). The addition of 500 nM Crn1 greatly extended the lag phase and reduced the rate of WA-activated Arp2/3 complex–mediated actin assembly (Fig. 8 A, curve D). This effect is not the result of interactions of Crn1 with actin, because the addition of 500 nM Crn1 to actin alone caused a modest increase in the rate of actin assembly, consistent with previous reports (Goode et al., 1999). These data show that Crn1 directly inhibits the actin nucleation activity of WA-activated Arp2/3 complex.
Figure 8.


The coiled coil domain of Crn1 inhibits Arp2/3 complex–mediated actin nucleation. (A) Inhibition of Arp2/3 complex by the carboxy terminus of Crn1. Assembly kinetics for 2 μM monomeric actin in the presence or absence of 20 nM Arp2/3 complex, 200 nM WA fragment of Las17/Bee1, and 500 nM Crn1 or Crn1 fragments. Curve A, Arp2/3 complex, WA, and Crn1 (1–600); curve B, Arp2/3 complex and WA; curve C, Arp2/3 complex, WA, and 500 nM Crn1 (400–651); curve D, Arp2/3 complex, WA, and 500 nM Crn1 (1–651); curve E, Arp2/3 complex; curve F, actin alone. (B) Dose-responsive inhibition of the Arp2/3 complex by Crn1 (400–651). The graph shows assembly of 2 μM monomeric actin (10% pyrene labeled) in the presence of 20 nM Arp2/3 complex, 200 nM WA, and 0–1,000 nM Crn1 (400–651). Curve A, no Crn1 added; curve B, 67.5 nM Crn1 (400–651); curve C, 250 nM Crn1 (400–651); curve D, 500 nM Crn1 (400–651); curve E, 1,000 nM Crn1 (400–651); curve F, actin alone. (C) Graph showing the concentration-dependent effects of Crn1 (400–651) on the rate of Arp2/3 complex–nucleated actin assembly; the data is from B (see Materials and methods). (D) Crn1 inhibits Abp1-stimulated Arp2/3 complex activity. Monomeric actin (2 μM) assembled with 50 nM Arp2/3 complex, 500 nM Crn1 (400–651), 50 nM Abp1, or 200 nM WA. Curve A, Arp2/3 and Abp1; curve B, Arp2/3 and WA; curve C, Arp2/3, WA, and Crn1 (400–651); curve D, Arp2/3, Abp1, and Crn1 (400–651); curve E, Arp2/3; curve F, actin alone.
To define the part of Crn1 that inhibits the Arp2/3 complex activity, we tested the effects of Crn1 fragments in these assays. Truncation of the coiled coil domain (residues 601–651) abolished the effects (Fig. 8 A, curve A), whereas a carboxy-terminal fragment of Crn1 (residues 400–651) showed a similar activity to full-length Crn1 (Fig. 8 A, curve C). As shown in Fig. 8 B, the effects of Crn1 (400–651) are dose responsive, with a half-maximal concentration of ∼100 nM (Fig. 8 C). Crn1 (400–651) also inhibited Abp1-activated Arp2/3 complex (Fig. 8 D). Importantly, this Crn1 fragment has no detectable affinity for actin (Goode et al., 1999), indicating that inhibition is direct.
Crn1 recruits the Arp2/3 complex to the sides of actin filaments
What is the mechanism of Arp2/3 complex inhibition by Crn1? We observed a two hybrid interaction between Crn1 (466–651) and Arc35/p35, and there is strong evidence that this subunit mediates binding of the Arp2/3 complex to the sides of actin filaments (Mullins et al., 1997; Bailly et al., 2001; Gournier et al., 2001). Therefore, we considered the possibility that Crn1 (400–651) interactions with Arc35 might block Arp2/3 complex association with the sides of actin filaments to delay nucleation. However, we detected no difference in Arp2/3 complex affinity for actin filaments in a cosedimentation assay in the presence and absence of Crn1 (400–651) (unpublished data). This suggests that inhibition does not result from blocking Arp2/3 complex interactions with the sides of actin filaments. Further, inhibition does not appear to result from Crn1 interference with activator binding to the Arp2/3 complex, because Crn1 inhibits the basal nucleation activity of the Arp2/3 complex, in the absence of any activators (Fig. 9 A).
Figure 9.



Crn1 recruits the Arp2/3 complex to the sides of actin filaments. (A) Assembly kinetics of 2 μM monomeric actin (10% pyrene labeled) in the presence or absence of 50 nM Arp2/3 complex and 500 nM Crn1 (400–651). Curve A, Arp2/3 complex; curve B, actin alone; curve C, Crn1 (400–651); curve D, Arp2/3 complex and Crn1 (400–651). (B) Coomassie-stained gel of pellets and supernatants from cosedimentation assays containing 2 μM F-actin, 0.5 μM Arp2/3 complex, and/or 2 μM full-length Crn1. (C) The addition of preformed actin filaments overrides Crn1 inhibition. Assembly kinetics for 2 μM monomeric actin plus 0.5 μM preformed actin filaments, with 20 nM Arp2/3 complex, 200 nM WA fragment of Las17/Bee1, and/or 500 nM Crn1 (400–651). Curve A, F-actin, Arp2/3, and WA; curve B, Arp2/3, WA, and Crn1 (400–651); curve C, 500 nM Crn1 (400–651); curve D, F-actin.
In contrast to Crn1 (400–651), full-length Crn1 actually increased the association of the Arp2/3 complex with the sides of actin filaments (Fig. 9 B). Purified Crn1 binds strongly to actin filaments (Kd ∼10 nM) through its amino terminus (Goode et al., 1999), whereas yeast Arp2/3 complex binds weakly to the sides of actin filaments (Kd ∼2–3 μM), similar to Arp2/3 complex isolated from other species (discussed in Goode et al., 2001). As shown in Fig. 9 B, Arp2/3 complex cosedimentation with 2 μM actin filaments increases significantly in the presence of 2 μM Crn1. Thus, Crn1 recruits the Arp2/3 complex to the sides of actin filaments. Further, these effects do not result from actin filament bundling by Crn1, because a different actin bundling protein (Sac6/fimbrin) had no effect in this assay (unpublished data).
We also tested the ability of Crn1 to inhibit Arp2/3-mediated actin assembly in the presence of preformed actin filaments. When 500 nM preassembled actin filaments was added to 2 μM actin monomers and WA-activated Arp2/3 complex, the lag phase was nearly eliminated (Fig. 9 C, curve A), consistent with previous reports (Machesky et al., 1997). When we further added 500 nM Crn1 (400–651), a concentration that dramatically inhibits Arp2/3 complex in the absence of filaments (Fig. 8 A, compare curves B and C), there was little, if any, inhibition detected (Fig. 9 C, curve B). Thus, Crn1 suppresses the Arp2/3 complex specifically in the absence of actin filaments.
Discussion
Coronin is a ubiquitous component of the eukaryotic actin cytoskeleton and localizes to sites of dynamic actin assembly in cells (for review see de Hostos, 1999). However, since its identification almost 10 yr ago, the precise function of coronin in the actin cytoskeleton has remained poorly understood. Here, we have made an important step toward defining the cellular function of yeast coronin (Crn1). We show that (a) Crn1 and the Arp2/3 complex physically and functionally interact in vivo, (b) the interaction is mediated by the coiled coil domain of Crn1, which is evolutionarily conserved, and (c) Crn1 strongly modulates Arp2/3 complex activity, inhibiting actin nucleation in the absence of preformed actin filaments. This work defines a new mode of regulation for the Arp2/3 complex and identifies Crn1 as the first direct inhibitor of the Arp2/3 complex.
Physical and genetic interactions between Crn1 and the Arp2/3 complex
We have found a strong physical association between Crn1 and the Arp2/3 complex, as demonstrated by a variety of assays, including comigration on sucrose gradients, coimmunoprecipitation, two hybrid analysis, and direct binding of purified proteins. These interactions and the effects of Crn1 on Arp2/3 complex activity are mediated by the coiled coil domain of Crn1. Our attempts by blot overlay assays to map the specific subunit(s) of the Arp2/3 complex that binds Crn1 have been unsuccessful thus far (unpublished data). However, our two hybrid data suggest that Arc35 may be an important target of Crn1 binding, and this is supported by the observation that an arc35 allele suppresses the growth defects caused by CRN1 overexpression.
Over 50% of the cellular Crn1 is bound to the Arp2/3 complex, suggesting that the cellular functions of Crn1 and the Arp2/3 complex are closely linked. This is supported by in vivo evidence, including synthetic defects between crn1-null mutants and arp2-21 and suppression by arc35-26 of defects caused by CRN1 overexpression. Further, localization of Crn1 to cortical actin patches in vivo depends on both its actin binding domain and its Arp2/3 complex–interacting coiled coil domain. A similar observation was made in cultured Xenopus cells, where deletion of the coiled coil domain caused mislocalization of coronin (Mishima and Nishida, 1999). Because coronin forms coiled coil–dependent homodimers in vitro (Goode et al., 1999; Asano et al., 2001), it was postulated that coronin dimerization may be necessary for localization. However, our findings raise the possibility that interactions of the coiled coil domain with the Arp2/3 complex may contribute to localization. Importantly, these models are not mutually exclusive; coronin localization may require both homodimerization and interactions with the Arp2/3 complex. Another important point raised here and in the above-mentioned study is that actin binding alone is not sufficient to localize coronin to actin filament structures in vivo. Therefore, associations between coronin and actin may be regulated in vivo.
A mechanism for Crn1 inhibition of Arp2/3 complex activity
We found that Crn1 inhibits WA-activated Arp2/3-mediated actin nucleation and that these effects are dose responsive and mediated by the Crn1 coiled coil domain. The addition of 0.5 μM Crn1 (400–651) virtually abolishes WA- and Abp1-activated Arp2/3 complex activity. Importantly, this activity is independent of Crn1 interactions with actin, because Crn1 (400–651) has no actin binding affinity (Goode et al., 1999). Therefore, the inhibition of the Arp2/3 complex by Crn1 is direct.
To explore the mechanism of inhibition, it is helpful to review current models for Arp2/3 complex activation (for reviews see Cooper et al., 2001; Borths and Welch, 2002; Kreishman-Deltrick and Rosen, 2002). Recently, the crystal structure of the inactive Arp2/3 complex and cryo-electron micrograph structures of the activated complex alone and at filament branch points were reported (Robinson et al., 2001; Volkmann et al., 2001). Together, these studies suggest that association of the Arp2/3 complex with the sides of actin filaments and interactions with an activator converge, inducing allosteric changes in the complex that reposition Arp2 and Arp3 into a nucleation-competent actin-like dimer. The p35/Arc35 subunit is strongly implicated in physically linking the Arp2/3 complex to the sides of actin filaments (Mullins et al., 1997; Bailly et al., 2001; Gournier et al., 2001). Thus, interactions between p35 and the side of a filament may transduce one set of conformational changes, while interactions between an activator and other subunits of the complex may transduce a complementary set of conformational changes.
Initially, we considered a simple model for inhibition by Crn1, in which Crn1 competes with and displaces activators from the Arp2/3 complex. However, our results are inconsistent with this model. First, Crn1 suppresses the inherent actin nucleation of the Arp2/3 complex alone, in the absence of any activators. Second, Crn1 does not have an acidic “A” motif, found in and required for association with the Arp2/3 complex in all known activators (for review see Cooper et al., 2001). Third, Crn1 interacts genetically and by two hybrid assay with p35/Arc35, in contrast to activators, which are implicated in binding to four different subunits: Arp2, Arp3, p40/Arc40, and p21/Arc18 (Mullins et al., 1997; Machesky and Insall, 1998; Zalevsky et al., 2001). Fourth, increasing the concentration of WA in the reactions fails to override Crn1 inhibition (unpublished data). Thus, all of our data point to a functional interaction between the coiled coil domain of Crn1 and the p35 subunit of the Arp2/3 complex, via a distinct interface from the activators.
A second model we considered for inhibition was that Crn1 (400–651) might interfere with Arp2/3 complex binding to the sides of actin filaments. However, Crn1 (400–651) did not affect Arp2/3 complex association with filament sides, and, in fact, full-length Crn1 increased association of the Arp2/3 complex with the sides of actin filaments (Fig. 2 B).
What does the arc35-26 suppression data tell us about the mechanism of inhibition? The allele-specific inhibition of CRN1 overexpression defects strengthens our hypothesis that Crn1 interactions with the Arp2/3 complex occur through the Arc35/p35 subunit. Intriguingly, arc35-26 suppresses the growth defects associated with Crn1 overexpression, and, reciprocally, CRN1 overexpression suppresses the growth defects of arc35-26. This cosuppression suggests a highly specific functional interaction between Crn1 and Arc35. In future work, isolating the Arp2/3 complex from arc35-26 mutant cells and studying its activities may provide valuable insights into Crn1 action. In addition, defining the residues in p35/Arc35 that mediate Crn1 interactions may lend important clues to the mode of inhibition.
The cellular role of Crn1 in regulating the Arp2/3 complex
We have shown that Crn1 inhibits the actin nucleation activity of the Arp2/3 complex specifically in the absence of actin filaments via its coiled coil domain and recruits the Arp2/3 complex to the sides of actin filaments via its actin binding domain. Both of these activities may be used in vivo to direct the Arp2/3 complex activity to the sides of preexisting actin filaments, promoting the formation of filament networks. Such a function might be important during cellular processes that rely on the rapid formation of actin networks, including cell locomotion and intracellular transport of vesicles and organelles. Consistent with this possibility, loss of coronin function in Dictyostelium and Xenopus cells has been shown to cause defects in cell migration and/or endocytosis (de Hostos et al., 1993; Mishima and Nishida, 1999). Next, it will be important to assess whether coronin–Arp2/3 complex interactions are conserved in other organisms and determine the cellular consequences of disrupting such interactions. Already, there are indications that the interaction may be conserved, because substoichiometric amounts of coronin have been shown to copurify with the Arp2/3 complex from human neutrophils (Machesky et al., 1997). Perhaps the most significant challenge for the future will be to determine how the Arp2/3 complex integrates so many different signals, from (a) multiple activators, (b) coronin, and (c) binding to the side of an actin filament, to spatially and temporally control actin nucleation in the cell.
Materials and methods
Strains and media
The Saccharomyces cerevisiae strains used in this study are listed in Table I. Standard methods were used to generate strains with integrated tags (GFP and HA epitope) at the carboxy termini of CRN1 and ARP2 and a strain with a GFP tag integrated after residue 600 in CRN1 (Longtine et al., 1998). A strain with a CRN1 gene deletion and an HA epitope tag integrated at the carboxy terminus of ARP2 (BGY704) was generated by crossing strains BGY26 and BAY1412. The resulting diploids were sporulated, tetrads were dissected, and haploids with the BGY704 genotype were selected. The presence of the ARP2–HA epitope tag and loss of the CRN1 gene were confirmed by immunoblotting with anti-Crn1 and anti-HA antibodies. Standard methods were used for growth and transformation of yeast (Guthrie and Fink, 1991). For yeast growth assays, cultures were grown to log phase, serially diluted, spotted on plates, and grown for 3 d.
Table I. Strains used in this study.
| Name | Genotype | Source |
|---|---|---|
| BGY006 | MATα, his3-11,15, ura3-52, leu2-3,112, ade2-1, trp1-1, GAL+, CRN1-GFP::HIS3MX6 | This study |
| BGY008 | MATα, his3-11,15, ura3-52, leu2-3,112, ade2-1, trp1-1, GAL+, CRN1(1-600)-GFP::HIS3MX6 | This study |
| BGY026 | MATα, his3-11,15, ura3-52, leu2-3,112, ade2-1, trp1-1, GAL+, ARP2-3xHA::HIS3MX6 | This study |
| BGY029 | MATα, his3-11,15, leu2-3,112, ade2-1, trp1-1, GAL+, ura3Δ::ACT1pr-ABP1-GFP::URA3 | This study |
| BGY046 | MATa, leu2-3,112, ura3-52, ade2-101, ade3-130, crn1Δ::LEU2 | This study |
| BGY654 | MATα, ARP2-TEV-3xHA::HIS3MX6, leu2D::ARC35::LEU2, arc35Δ::KANMX6, ura3-52, his3Δ200 | D. Robinsa |
| BGY661 | MATα, ARP2-TEV-3xHA::HIS3MX6, leu2Δ::arc35-12::LEU2, arc35Δ::KANMX6, ura3-52, his3Δ200 | D. Robinsa |
| BGY670 | MATα, ARP2-TEV-3xHA::HIS3MX6, leu2Δ::arc35-26::LEU2, arc35Δ::KANMX6, ura3-52, his3Δ200 | D. Robinsa |
| BGY704 | MATα, his3-11,15, ura3-52, leu2-3,112, ade2-1, trp1-1, psi+, ssd−, GAL+, ARP2-3xHA::HIS3MX6, crn1Δ:: KANMX6 | This study |
| BGY714 | MATa, ARP2-YFP::HIS3MX6, ura3-52, his3Δ200, ade2-1, leu2-3,112 | This study |
| BGY721 | MATα, arp2-21::URA3, crn1Δ::KANMX6, leu2Δ0 | This study |
| BAY263 | MATa, trp1Δ63, GAL2+, ura3-52, lys2-801, ade2-107, his3Δ200, leu2-Δ1 | Measday et al., 1994 |
| BAY1355 | MATa, crn1Δ::URA3, trpΔ63, GAL2+, ura3-52, lys2-801, ade2-107, his3Δ200, leu2-Δ1 | This study |
| BAY1412 | MATa, crn1Δ::KANMX6, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 | Research Genetics |
| BAY1674 | MATα, arp2-21::URA3, mfa1Δ::MFA1pr-HIS3, can1Δ, his3Δ1, leu2Δ0, MET15+, lys2Δ0 | C. Booneb |
| BAY3029 | MATα, arc40-40::URA3, mfa1Δ::MFAprHIS3, can1Δ, his3Δ1, leu2Δ0, MET15+, lys2Δ0 | Tong et al., 2001 |
| YJC1265 | Ura3-52/ura3-53, leu2-3,112/leu2-3,112, trp1Δ::CAP2-GFP::TRP1, trp1Δ::CAP2-GFP::TRP1 | Waddle et al., 1996 |
Brandeis University.
University of Toronto.
Plasmid construction
All plasmids used in this study are listed in Table II. To express full-length Crn1 and Crn1 (1–600) on CEN plasmids under the control of the CRN1 promoter, we constructed pBG290 and pBG291 and pBG296, respectively. The designated regions of the CRN1 open reading frame with 300 bp of 5′ untranslated sequence upstream were PCR amplified using high fidelity polymerase and subcloned into the BamH1–Not1 sites of pRS316. To express different parts of Crn1 in yeast, we constructed pBG226, pBG289, pBG290, and pBG291 by subcloning BamH1–Nsi1 CRN1 fragments from plasmids pBG203 and pBG206, respectively, into p425GAL1 and p415MET25. For overexpression of full-length Crn1, Crn1 (1–600), or Crn1 (400–651) under control of the GAL promoter, we constructed pBG222, pBG223, and pBG224. The designated regions of the CRN1 open reading frame were amplified by PCR using high fidelity polymerase and subcloned into BamH1–Not1 sites of p425GAL1 or p426GAL1. For two hybrid analysis, an Arc35 insert was excised as a BamH1–Xho1 fragment from pEG202-END9 (a gift from C. Schaerer-Brodbeck, University of Basel, Basel, Switzerland) and cloned into BamH1–Sal1 sites of pAS2 to yield pAS2-ARC35. The plasmid was shown to express a functional fusion protein by complementation of the 37°C growth defect of arc35-1 (Schaerer-Brodbeck and Riezman, 2000). All plasmids were sequenced to confirm that the CRN1 coding sequences contained no mutations.
Table II. Plasmids used in this study.
| Name | Insert | Vector | Reference |
|---|---|---|---|
| pBG203 | GST–CRN1 | pGAT2 | Goode et al., 1999 |
| pBG205 | GST–CRN1 (400–651) | pGAT2 | Goode et al., 1999 |
| pBG206 | GST–CRN1 (1–600) | pGAT2 | Goode et al., 1999 |
| pBG222 | CRN1 | pRS426GAL1 | This study |
| pBG223 | CRN1 (1–600) | pRS426GAL1 | This study |
| pBG224 | CRN1 (400–651) | pRS426GAL1 | This study |
| pBG290 | CRN1 | pRS415MET25 | This study |
| pBG291 | CRN1 (1–600) | pRS415MET25 | This study |
| pBG294 | CRN1 | pRS316 | This study |
| pBG295 | CRN1 (1–600) | pRS316 | This study |
| pBG298 | CRN1 (400–651) | pRS415MET25 | This study |
| pAS2ΔΔ::ARC35 | ARC35 | pAS2ΔΔ | This study |
Antibody preparation and immunoblotting
Two different Crn1 antibodies were used, a mouse polyclonal anti-Crn1 antibody previously described (Goode et al., 1999) and a rabbit polyclonal anti-Crn1 antibody generated here (Faculty of Medicine, University of Toronto). The rabbits were immunized with a GST–Crn1 fusion protein expressed and purified from Escherichia coli (Goode et al., 1999), and the antibodies were affinity purified from serum (Measday et al., 1994). Like the mouse anti-Crn1 antibody, the rabbit anti-Crn1 antibody recognizes an 85-kD band on blots of wild-type total cellular protein that is absent from crn1-null lanes (not depicted). Proteins were detected on blots using 1:1,000 mouse anti-Crn1, 1:5,000 rabbit anti-Crn1, and 1:1,000 rabbit anti-Arp2 (Moreau et al., 1996). HA–Arp2 was detected using 1:10,000 mouse anti-HA antibody conjugated to HRP (Covance; Denver, CO). For all other blots, 1:10,000 HRP-conjugated secondary antibody was used. Signals were detected by ECL from Amersham Biosciences.
Protein purification
The Arp2/3 complex was purified from yeast (Goode et al., 2001). Crn1, Crn1 (1–600), and Crn1 (400–651) were purified from E. coli (Goode et al., 1999). The carboxy-terminal WA fragment of Las17/Bee1 was purified from E. coli (Winter et al., 1999). Unlabeled and pyrene-labeled rabbit skeletal muscle actin were purchased from Cytoskeleton, Inc. Monomeric actin was reconstituted from a lyophilized state as per the manufacturer's instructions, diluted to 25–50 μM in G-buffer (5 mM Tris-HCl , pH 7.5, 0.2 mM DTT, 0.2 mM ATP, 0.2 mM CaCl2), incubated overnight on ice, and cleared by centrifugation for 1 h at 4°C, 90,000 rpm in a TLA100 rotor (Beckman Coulter).
Sucrose gradient fractionation of yeast lysates
11-ml sucrose gradients (3–30%) were poured in 12-ml ultra clear tubes for an SW41 rotor (Beckman Coulter). Crude cell lysates were prepared from wild-type and crn1-null yeast as previously described (Goode et al., 1999). Lysates were precleared by centrifugation for 15 min, 70,000 rpm, 4°C in a TLA100.3 rotor (Beckman Coulter). 400 μl supernatant or high molecular weight gel filtration size standards (Amersham Biosciences) were layered over each gradient. Samples were centrifuged for 15 h at 34,000 rpm, 4°C in an SW41 rotor, and 0.4-ml fractions were collected. Samples of each fraction were run on SDS-PAGE gels, blotted, and probed with antibodies to determine the positions of proteins in the gradients.
Coimmunoprecipitation assays
For coimmunoprecipitation assays, we used yeast strains with an integrated carboxy-terminal 3xHA epitope tag on ARP2. Cells were grown to log phase, washed, frozen, and lysed as previously described (Goode et al., 1999). 1 g of cell lysate was added to 1 ml HEK buffer (20 mM Hepes, pH 7.5, 1 mM EDTA, 50 mM KCl), supplemented with 1mM DTT, 0.1% NP-40 detergent, and a standard cocktail of protease inhibitors (Goode et al., 1999). The lysate was thawed to 4°C and precleared by centrifugation for 15 min at 4°C, 80,000 rpm in a TLA100.3 rotor. The supernatant was harvested and preabsorbed with CL4B protein A–Sepharose (Amersham Biosciences) for 1 h at 4°C. The beads were pelleted and 500 μl of supernatant was added to 2.5 μl of HA.11 monoclonal antibody (5–7 mg/ml ascites fluid; Covance) and incubated for 1 h at 4°C. Next, we added 20 μl of CL4B protein A–Sepharose (preswollen in HEK buffer) and incubated for 1 h at 4°C with mixing. The beads were washed twice in HEK buffer and then once in HEK buffer supplemented with 0.5% NP-40, and samples were prepared in SDS sample buffer for immunoblotting.
Binding interactions between Crn1 and the Arp2/3 complex
To test direct binding between Crn1 and the Arp2/3 complex, we assayed the cosedimentation of purified Crn1 with purified HA-tagged yeast Arp2/3 complex immobilized on beads. The Arp2/3 complex–loaded beads were prepared as previously described (Goode et al., 2001), yielding a bead suspension of 1 μM Arp2/3 complex. 10 μl of Arp2/3-loaded beads or control beads (no Arp2/3) was included in a 100-μl reaction in HEK buffer containing 1 μM Crn1. The final concentration of the Arp2/3 complex in the reactions was 0.1 μM. Reactions were incubated for 20 min at 4°C, the beads were washed, and the bound proteins were removed with SDS sample buffer (without reducing agent). Samples were run on 12% SDS-PAGE gels and stained with Coomassie blue.
Two hybrid analysis
A yeast cDNA library in pGAD-GH was transformed into the Y190 yeast strain containing pAS2-ARC35, and a nonsaturating Gal two hybrid screen was performed as previously described (Madania et al., 1999). Transformants were selected on −TRP, −LEU, −HIS, +30 mM 3-aminotriazole medium, and 24 clones were isolated. After a series of secondary tests, four clones remained, two of which were found to contain a fragment of Crn1 encoding its carboxy-terminal 186 residues (466–651).
Fluorescence light microscopy
Actin and Crn1 organization was examined in cells overproducing full-length Crn1, Crn1 (1–600), and Crn1 (400–651) under control of the GAL1/10 promoter, from plasmids pBG222, pBG223, and pBG224. Cells were grown at 30°C in selective glucose medium to early log phase, washed, transferred to selective galactose medium, grown for 4 h at 30°C, fixed, and prepared for immunofluorescence (Ayscough and Drubin, 1997; Lee et al., 1998). To disrupt the actin cytoskeleton in cells, log-phase yeast cultures were treated with 100 μM latrunculin A for 15 min before chemical fixation. We also determined the localization of full-length Crn1, Crn1 (1–600), and Crn1 (400–651) expressed in cells from low copy plasmids (pBG290, pBG291, and pBG298) under the control of the MET25 promoter (Fig. 3 B). Similar results were obtained for low copy plasmids expressing full-length Crn1 and Crn1 (1–600) under the control of its own promoter: pBG294 and pBG295 (not depicted). For immunofluorescence detection of Crn1 and actin, we used a 1:500 dilution of rabbit anti-Crn1 and a 1:2,000 dilution of guinea pig anti-Act1 (Mulholland et al., 1994). Cells were imaged on a Leica DM-LB microscope. Images were captured with a Micromax 1300y high-speed digital camera (Princeton Instruments) and analyzed with Metaview software (Universal Imaging Corp.). The localization of GFP and YFP was examined in yeast cells grown to log phase.
Actin assembly kinetics
Actin assembly was monitored by the pyrene–actin fluorescence assay as previously described (Goode et al., 2001), using a final concentration of 2 μM actin in 70-μl reactions unless otherwise indicated. In brief, 56.5 μl of monomeric actin (10% pyrene-labeled, 90% unlabeled) in G buffer was mixed with 10 μl HEKG5 buffer (HEK buffer + 5% glycerol) or different combinations of proteins in HEKG5 buffer. The reaction was mixed immediately with 3.5 μl 20× initiation buffer (1 M KCl, 40 mM MgCl2, 10 mM ATP) in a quartz fluorometry cuvette (3-mm light path; Hellma). Pyrene–actin fluorescence was monitored by excitation at 365 nM and emission at 407 nM in a fluorescence spectrophotometer (Photon Technology International) held at the constant temperature of 25°C. For seeded reactions, actin was preassembled to steady state for 1 h. Then, 2 μM monomeric actin (10% pyrene labeled) was mixed with 500 nM preassembled actin filaments in the presence or absence of different proteins.
Actin filament recruitment assays
Binding of the Arp2/3 complex to the sides of actin filaments was measured as previously described (Goode et al., 2001). In Fig. 9 B, 2 μM full-length Crn1 and/or 0.5 μM Arp2/3 complex were added to 2 μM preassembled actin filaments. After a 20-min incubation, the reactions were centrifuged for 20 min in a TLA100 rotor (Beckman Coulter). Equal loads of pellets and supernatants were fractionated on 12% SDS-PAGE gels and stained with Coomassie blue.
Acknowledgments
We are very grateful to A. Rodal for advice throughout the study, C. Boone, S. Hippenmeyer and H. Riezman (University of Basel), D. Robins, and H. Xu for sharing unpublished reagents, J. Wong, P. Dumoulin, and S. Thuault for technical assistance, and C. Boone, F. Chang, A. Goodman, D. Pellman, A. Rodal, and I. Sagot for helpful comments on the manuscript.
This work was supported by an award from the Canadian Institutes of Health Research to C. Humphries, an operating grant from the National Cancer Institute of Canada with funds from the Canadian Cancer Society to B. Andrews, support from the Centre National de Recherche Scientifique and the Association pour la Recherche sur le Cancer to B. Winsor, and a grant from the National Institutes of Health (NIIH) to G. Barnes (GM47842). B. Goode was supported by a Pew Scholars award, a Basil O'Conner award, and the NIH (GM63691).
C.L. Humphries and H.I. Balcer contributed equally to this work.
Footnotes
Abbreviation used in this paper: Arp2/3, actin-related protein 2/3.
References
- Asano, S., M. Mishima, and E. Nishida. 2001. Coronin forms a stable dimer through its C-terminal coiled coil region: an implicated role in its localization to cell periphery. Genes Cells. 6:225–235. [DOI] [PubMed] [Google Scholar]
- Ayscough, K.R., and D.G. Drubin. 1997. Immunofluorescence microscopy of yeast cells. Cell Biology, A Laboratory Handbook. Vol. 2. J. Celis, editor. Academic Press. San Diego, CA. 477–485.
- Bailly, M., I. Ichetovkin, W. Grant, N. Zebda, L.M. Machesky, J.E. Segall, and J. Condeelis. 2001. The F-actin side binding activity of the Arp2/3 complex is essential for actin nucleation and lamellipod extension. Curr. Biol. 11:620–625. [DOI] [PubMed] [Google Scholar]
- Borths, E.L., and M.D. Welch. 2002. Turning on the Arp2/3 complex at atomic resolution. Structure (Camb). 10:131–135. [DOI] [PubMed] [Google Scholar]
- Cooper, J.A., M.A. Wear, and A.M. Weaver. 2001. Arp2/3 complex: advances on the inner workings of a molecular machine. Cell. 107:703–705. [DOI] [PubMed] [Google Scholar]
- de Hostos, E.L. 1999. The coronin family of actin-associated proteins. Trends Cell Biol. 9:345–350. [DOI] [PubMed] [Google Scholar]
- de Hostos, E.L., C. Rehfuess, B. Bradtke, D.R. Waddell, R. Albrecht, J. Murphy, and G. Gerisch. 1993. Dictyostelium mutants lacking the cytoskeletal protein coronin are defective in cytokinesis and cell motility. J. Cell Biol. 120:163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode, B.L., and A.A. Rodal. 2001. Modular complexes that regulate actin assembly in budding yeast. Curr. Opin. Microbiol. 4:703–712. [DOI] [PubMed] [Google Scholar]
- Goode, B.L., J.J. Wong, A.C. Butty, M. Peter, A.L. McCormack, J.R. Yates, D.G. Drubin, and G. Barnes. 1999. Coronin promotes the rapid assembly and cross-linking of actin filaments and may link the actin and microtubule cytoskeletons in yeast. J. Cell Biol. 144:83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode, B.L., A.A. Rodal, G. Barnes, and D.G. Drubin. 2001. Activation of the Arp2/3 complex by the actin filament binding protein Abp1p. J. Cell Biol. 153:627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gournier, H., E.D. Goley, H. Niederstrasser, T. Trinh, and M.D. Welch. 2001. Reconstitution of human Arp2/3 complex reveals critical roles of individual subunits in complex structure and activity. Mol. Cell. 8:1041–1052. [DOI] [PubMed] [Google Scholar]
- Guthrie, C., and R. Fink. 1991. Guide to yeast genetics and molecular biology. Methods Enzymol. 194:1–933. [PubMed] [Google Scholar]
- Heil-Chapdelaine, R.A., N.K. Tran, and J.A. Cooper. 1998. The role of Saccharomyces cerevisiae coronin in the actin and microtubule cytoskeletons. Curr. Biol. 8:1281–1284. [DOI] [PubMed] [Google Scholar]
- Higgs, H.N., and T.D. Pollard. 2001. Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu. Rev. Biochem. 70:649–676. [DOI] [PubMed] [Google Scholar]
- Kreishman-Deltrick, M., and M.K. Rosen. 2002. Ignition of a cellular machine. Nat. Cell Biol. 4:E31–E33. [DOI] [PubMed] [Google Scholar]
- Lee, J., K. Colwill, V. Aneliunas, C. Tennyson, L. Moore, Y. Ho, and B. Andrews. 1998. Interaction of yeast Rvs167 and Pho85 cyclin-dependent kinase complexes may link the cell cycle to the actin cytoskeleton. Curr. Biol. 8:1310–1321. [DOI] [PubMed] [Google Scholar]
- Longtine, M.S., A. McKenzie III, D.J. Demarini, N.G. Shah, A. Wach, A. Brachat, P. Philippsen, and J.R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 14:953–961. [DOI] [PubMed] [Google Scholar]
- Machesky, L.M., and R.H. Insall. 1998. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 8:1347–1356. [DOI] [PubMed] [Google Scholar]
- Machesky, L.M., E. Reeves, F. Wientjes, F.J. Mattheyse, A. Grogan, N.F. Totty, A.L. Burlingame, J.J. Hsuan, and A.W. Segal. 1997. Mammalian actin-related protein 2/3 complex localizes to regions of lamellipodial protrusion and is composed of evolutionarily conserved proteins. Biochem. J. 328:105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madania, A., P. Dumoulin, S. Grava, H. Kitamoto, C. Scharer-Brodbeck, A. Soulard, V. Moreau, and B. Winsor. 1999. The Saccharomyces cerevisiae homologue of human Wiskott-Aldrich syndrome protein Las17p interacts with the Arp2/3 complex. Mol. Biol. Cell. 10:3521–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Measday, V., L. Moore, J. Ogas, M. Tyers, and B. Andrews. 1994. The PCL2 (ORFD)-PHO85 cyclin-dependent kinase complex: a cell cycle regulator in yeast. Science. 266:1391–1395. [DOI] [PubMed] [Google Scholar]
- Mishima, M., and E. Nishida. 1999. Coronin localizes to leading edges and is involved in cell spreading and lamellipodium extension in vertebrate cells. J. Cell Sci. 112:2833–2842. [DOI] [PubMed] [Google Scholar]
- Moreau, V., A. Madania, R.P. Martin, and B. Winson. 1996. The Saccharomyces cerevisiae actin-related protein Arp2 is involved in the actin cytoskeleton. J. Cell Biol. 134:117–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland, J., D. Preuss, A. Moon, A. Wong, D. Drubin, and D. Botstein. 1994. Ultrastructure of the yeast actin cytoskeleton and its association with the plasma membrane. J. Cell Biol. 125:381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins, R.D., W.F. Stafford, and T.D. Pollard. 1997. Structure, subunit topology, and actin-binding activity of the Arp2/3 complex from Acanthamoeba. J. Cell Biol. 136:331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins, R.D., J.A. Heuser, and T.D. Pollard. 1998. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Adad. Sci. USA. 95:6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olazabal, I.M., and L.M. Machesky. 2001. Abp1p and cortactin, new “hand-holds” for actin. J. Cell Biol. 154:679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruyne, D., and A. Bretscher. 2000. Polarization of cell growth in yeast. J. Cell Sci. 113:571–585. [DOI] [PubMed] [Google Scholar]
- Robinson, R.C., K. Turbedsky, D.A. Kaiser, J.B. Marchand, H.N. Higgs, S. Choe, and T.D. Pollard. 2001. Crystal structure of Arp2/3 complex. Science. 294:1679–1684. [DOI] [PubMed] [Google Scholar]
- Schafer, D.A. 2002. Coupling actin dynamics and membrane dynamics during endocytosis. Curr. Opin. Cell Biol. 14:76–81. [DOI] [PubMed] [Google Scholar]
- Schaerer-Brodbeck, C., and H. Riezman. 2000. Saccharomyces cerevisiae Arc35p works through two genetically separable calmodulin functions to regulate the actin and tubulin cytoskeletons. J. Cell Sci. 113:521–532. [DOI] [PubMed] [Google Scholar]
- Tong, A.H., M. Evangelista, A.B. Parsons, H. Xu, G.D. Bader, N. Page, M. Robinson, S. Raghibizadeh, C.W. Hogue, H. Bussey, et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 294:2364–2368. [DOI] [PubMed] [Google Scholar]
- Volkmann, N., K.J. Amann, S. Stoilova-McPhie, C. Egile, D.C. Winter, L. Hazelwood, J.E. Heuser, R. Li, T.D. Pollard, and D. Hanein. 2001. Structure of Arp2/3 complex in its activated state and in actin filament branch junctions. Science. 293:2456–2459. [DOI] [PubMed] [Google Scholar]
- Waddle, J.A., T.S. Karpova, R.H. Waterston, and J.A. Cooper. 1996. Movement of cortical actin patches in yeast. J. Cell Biol. 132:861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter, D.C., E.Y. Choe, and R. Li. 1999. Genetic dissection of the budding yeast Arp2/3 complex: a comparison of the in vivo and structural roles of individual subunits. Proc. Natl. Acad. Sci. USA. 96:7288–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalevsky, J., L. Lempert, H. Kranitz, and R.D. Mullins. 2001. Different WASP family proteins stimulate different Arp2/3 complex-dependent actin-nucleating activities. Curr. Biol. 11:1903–1913. [DOI] [PubMed] [Google Scholar]