

Abstract
Unregulated FGF signaling affects endochondral ossification and long bone growth, causing several genetic forms of human dwarfism. One major mechanism by which FGFs regulate endochondral bone growth is through their inhibitory effect on chondrocyte proliferation. Because mice with targeted mutations of the retinoblastoma (Rb)-related proteins p107 and p130 present severe endochondral bone defects with excessive chondrocyte proliferation, we have investigated the role of the Rb family of cell cycle regulators in the FGF response. Using a chondrocyte cell line, we found that FGF induced a rapid dephosphorylation of all three proteins of the Rb family (pRb, p107, and p130) and a blockade of the cells in the G1 phase of the cell cycle. This cell cycle block was reversed by inactivation of Rb proteins with viral oncoproteins such as polyoma large T (PyLT) antigen and Adenovirus E1A. Expression of a PyLT mutant that efficiently binds pRb, but not p107 and p130, allowed the cells to be growth inhibited by FGF, suggesting that pRb itself is not involved in the FGF response. To investigate more precisely the role of the individual Rb family proteins in FGF-mediated growth inhibition, we used chondrocyte micromass culture of limb bud cells isolated from mice lacking Rb proteins individually or in combination. Although wild-type as well as Rb −/− chondrocytes were similarly growth inhibited by FGF, chondrocytes null for p107 and p130 did not respond to FGF. Furthermore, FGF treatment of metatarsal bone rudiments obtained from p107 −/ −;p130 − / − embryos failed to inhibit proliferation of growth plate chondrocytes, whereas rudiments from p107-null or p130-null embryos showed only a slight inhibition of growth. Our findings indicate that p107 and p130, but not pRb, are critical effectors of FGF-mediated growth inhibition in chondrocytes.
Keywords: cell cycle; growth factors; pocket proteins; knockout mice; bone development
Introduction
Signaling through FGF receptors (FGFR)* plays a major role in development by eliciting a variety of responses in FGF-targeted cells, ranging from induction of proliferation and differentiation to growth inhibition. FGF signaling plays a role in bone development and affects the process of endochondral ossification, which accounts for the formation of most of the skeletal bones (Goldfarb, 1996; De Luca and Baron, 1999; Karsenty, 1999; Powers et al., 2000). In endochondral ossification, the initial formation of a cartilage anlage by the condensation of mesenchymal stem cells is followed by proliferation and differentiation of chondrocytes that provide the template for bone formation. Considerable evidence from human and mouse genetics indicates that longitudinal bone growth is regulated by FGF signaling. Gain of function mutations in FGFR3, which is expressed in growth plate chondrocytes, are responsible for several forms of human dwarfism including achondroplasia, whereas in mice, knockout of the FGFR3 gene causes overgrown and deformed long bones and unregulated FGF signaling produces a variety of chondrodysplasias (Deng et al., 1996; Naski et al., 1996; Webster and Donoghue, 1997; Karsenty, 1999).
In line with the genetic evidence that excessive FGF signaling impairs long bone growth (Naski and Ornitz, 1998), we previously showed that FGF signaling inhibited chondrocyte proliferation in vitro and in vivo (Sahni et al., 1999), suggesting that one major mechanism by which FGFs regulate bone development is through their inhibitory effect on chondrocyte proliferation. We also showed, through the use of Stat1 knockout mice, that this phenomenon required the intervention of STATl, a signal transducing molecule originally identified in the interferon pathway (Sahni et al., 1999). Furthermore, we showed that FGF signaling induced chondrocyte apoptosis both in vitro and in vivo, and this also required STAT1 function (Sahni et al., 2001). Although FGF induces a proliferative arrest through a STAT1-dependent process in chondrocytes, in most other cell types FGFs elicit increased proliferation (Basilico and Moscatelli, 1992). Presently, the basis for the FGF growth inhibition of chondrocytes, versus the FGF-proliferative effect in most other cell types, is not defined.
To gain information on the mechanisms through which FGF signaling induces growth arrest in chondrocytes, we sought to identify other downstream effectors of FGF-mediated growth inhibition. It had been previously shown that mice with targeted inactivation of two members of the retinoblastoma (Rb) gene family, p107 and p130, died at birth with developmental defects including defective endochondral bone development and abnormally high levels of chondrocyte proliferation (Cobrinik et al., 1996). The Rb family of pocket proteins includes three members (pRB, p107, and p130) that are known to be major regulators of cell cycle progression in essentially all cells and tissues (Ewen, 1998; Mulligan and Jacks, 1998). In the active underphosphorylated form, the Rb family proteins bind the E2F family of transcription factors, resulting in transcriptional inhibition of many cell cycle progression and DNA synthesis genes. Inactivation of Rb family proteins by phosphorylation allows freeing of E2Fs factors which then positively influence transcription of cell cycle progression genes (Dyson, 1998).
We have investigated the hypothesis that the high levels of chondrocyte proliferation observed in p107 −/− ;p130 −/− murine embryos could have been due, at least in part, to an impaired response to the growth-inhibitory effects of FGF signaling. To this end, we have expressed in cultured chondrocytes the adenovirus E1A (E1A) and polyoma large T (PyLT)-Ag proteins, that are known to bind and inactivate Rb proteins (Dyson et al., 1990; Classon and Dyson, 2001), and studied the response to FGF treatment of chondrocyte micromass cultures and cartilaginous bone rudiments from pRb, p130, and p107 knockout mouse embryos. Here, we show that expression of E1A or PyLT in rat chondrosarcoma (RCS) chondrocytes produces cells that no longer respond to FGF with growth inhibition. Experiments with chondrocyte micromass cultures and bone rudiments from mutant embryos further revealed that p107 and p130, but not pRb, are required for the growth inhibitory effects of FGF. Our data demonstrate that p107 and p130 are essential downstream mediators of the FGF-induced growth arrest of chondrocytes.
Results
FGF-induced growth inhibition of RCS cells is associated with dephosphorylation of Rb family proteins
Treatment of RCS chondrocytes with FGF-1 induces a potent growth inhibitory response (Sahni et al., 1999; see Fig. 2 A). FACScan™ analysis revealed that FGF-1 treatment of RCS cells promotes an accumulation of cells in the G0/G1 phase of the cell cycle (Fig. 1 A). Because Rb proteins constitute major regulators of the G1 to S phase transition (Ewen, 1998), we examined the phosphorylation status of these proteins as a read-out of their activities. This was done by Western blotting and immunodetection of the different phosphorylated forms of the Rb proteins, based on their characteristic electrophoretic mobilities. Treatment of RCS cells with FGF-1 for different times over a 24-h period showed that all three Rb family members (pRb, p107, and p130) became dephosphorylated after FGF treatment (Fig. 1 B). The kinetics of dephosphorylation was rapid, particularly in the case of p107, which is totally dephosphorylated as early as 9 h after FGF treatment. After 18 h of FGF treatment, all three Rb proteins were mainly present in their underphosphorylated forms. The G1 arrest induced by FGF-1 in RCS chondrocytes is reversible. Resumption of cell proliferation is accompanied by an increase in Rb proteins phosphorylation (unpublished data).
Figure 2.

Functional inactivation of Rb proteins interferes with FGF-induced growth arrest. (A) BrdU incorporation assay of FGF-treated (10 ng/ml) or untreated RCS cells expressing either PyLT, E1A, or mutant of these proteins defective in Rb family proteins interaction (PYLTΔ141–158, E1A-12S.928). (B) BrdU incorporation assay of dexamethasone-treated parental and PyLT-expressing RCS cells. Note that proliferation assays presented in Figs. 2 and 3 were conducted at least three times and gave similar results. Quantification was evaluated by counting the number of BrdU-positive cells among the total number of cells (200–400 cells) from several representative fields (±SD).
Figure 1.


FGF induces G1 arrest of RCS cells and dephosphorylation of Rb proteins. (A) FACScan™ analysis of propidium iodide–labeled cells, treated with 10 ng/ml FGF-1 (or left untreated) for 24 h. Percentage of cells in each phase of the cell cycle is indicated. (B) Time course analysis (Western blot) of the phosphorylation status of Rb protein family members (pRb, p107, and p130) after FGF treatment (10 ng/ml) of RCS cells. Black or white arrows indicate the position of the hyper- or hypophosphorylated forms of Rb proteins, respectively. (C) Western blot analysis showing the expression levels of Cdk inhibitors of the Cip/Kip (p21, p27, and p57) and Ink4 (p16 and p18) families, after 0, 12, and 24 h of FGF treatment. Equal amount of protein loading was confirmed by Grb2 immunodetection.
Because Rb proteins phosphorylation is mediated by Cdk, whose activity in turn is regulated by several Cdk inhibitors, we measured the expression of several members of the Cip/Kip and Ink4 families of Cdk inhibitors in RCS cells after FGF treatment. As previously shown by us and others (Sahni et al., 1999; Aikawa et al., 2001), the expression of p21 was up-regulated rather rapidly after FGF addition. On the other hand, expression of p27 and p57 was increased slightly and at later times. No changes in the expression of p16 and p18 were detected (Fig. 1 C).
Inactivation of Rb family proteins abolishes FGF-mediated growth inhibition of RCS cells
The rapid dephosphorylation of Rb proteins after FGF treatment suggested that their dephosphorylation could be a cause and not an effect of the growth arrest. To investigate whether the function of Rb proteins was required for FGF-induced growth arrest of RCS cells, we expressed by retroviral-mediated gene transfer two viral oncoproteins, PyLT antigen and E1A, which are known to interact directly with Rb proteins, and as a consequence, inactivate their function (Classon and Dyson, 2001). We measured the frequency of DNA-synthesizing cells by counting cells that had incorporated BrdU from 18 to 24 h after FGF treatment, and compared parental RCS cells with RCS that stably expressed PyLT or E1A. As shown in Fig. 2 A, expression of PyLT or E1A into RCS cells led to a marked decrease in FGF-induced growth inhibition in comparison with wild-type or vector-transfected RCS cells. In the case of PyLT-expressing RCS cells, we further confirmed that the early effects of FGF signaling were unaffected by the introduction of PyLT protein as judged by FGF-induced tyrosine phosphorylation of FGFR3 (the major FGF receptor expressed in these cells) as well as tyrosine phosphorylation of several endogenous proteins (unpublished data).
To confirm that the Rb family proteins binding ability of PyLT or E1A was implicated in the loss of FGF response, we used well defined mutants of these proteins carrying either a specific deletion or a point mutation of the Rb binding site (PyLT-Δ141–158 and E1A-12S.928, respectively; Wang et al., 1993; Pilon et al., 1996), and having impaired binding to each of the pRb family proteins. As shown in Fig. 2 A, expression of these mutants did not affect the growth inhibitory response observed in FGF-treated RCS cells, indicating that inactivation of Rb proteins by PyLT or E1A was responsible for the lack of FGF response. To exclude the possibility that the effect produced by the expression of PyLT and E1A could be triggered by the introduction of any oncogene into RCS cells, we also expressed another unrelated oncogene, an activated form of H-ras (Dotto et al., 1985), in these cells. Proliferation of v-H-ras–expressing RCS cells was inhibited by FGF to the same extent as RCS-treated cells (unpublished data).
Next, we wanted to exclude the possibility that PyLT- or E1A-expressing RCS cells were resistant to all growth inhibitory signals. Dexamethasone, a glucocorticoid that has been shown to inhibit chondrocyte growth both in vitro and in vivo (Jux et al., 1998; Klaus et al., 2000), induced a dose-dependent inhibition of proliferation of RCS cells (Fig. 2 B). Importantly, PyLT-expressing RCS cells treated with dexamethasone were comparably growth inhibited. Thus, the expression of PyLT does not make RCS cells refractory to all growth inhibitory signals.
Inactivation of pRb is not sufficient to block FGF-mediated growth inhibition of RCS cells
As discussed in the Introduction, studies of knockout mice have indicated an important role of the pRb-related proteins (p107 and p130) during chondrogenesis and particularly in the growth control of chondrocytes. To determine whether pRb, p107, and p130 equally contributed to FGF-induced growth arrest, we took advantage of a PyLT deletion mutant (PyLT-Δ256–272) that has been described to possess a reduced ability to bind p107, while maintaining both the potential to interact with pRb and to immortalize fibroblasts in vitro (Pilon et al., 1996). Because the binding characteristics of this mutant were determined in vitro using recombinant proteins, we first asked whether this PyLT mutant behaved similarly when expressed in cells. We expressed PyLT-Δ256–272 in RCS cells and examined the interaction of this mutant with Rb proteins in comparison to wild-type PyLT in coimmunoprecipitation experiments. For these experiments as well as for the proliferation experiments, we selected clones expressing similar amounts of PyLT and PyLT-Δ256–272 proteins, as judged by Western blot analysis (Fig. 3 B, see inset). As shown in Fig. 3 A, immunoprecipitation of both p107 and p130 revealed a strong reduction in the amount of PyLT-Δ256–272 in the immune complexes (Fig. 3 A, lanes 3), in comparison with wild-type PyLT (Fig. 3 A, lanes 2). On the other hand, immunoprecipitation of pRb produced bands of similar intensity for both wild-type and mutant PyLT. Thus, these results were in agreement with the published data regarding the pRb/p107 binding abilities of the mutant PyLT-Δ256–272 protein (Pilon et al., 1996). They also revealed that the ability of PyLT-Δ256–272 protein to interact with p130 is affected in parallel with its ability to bind p107, a finding perhaps predictable given the extensive protein sequence similarities between p130 and p107.
Figure 3.

RCS cells expressing a mutant PyLT protein that does not bind efficiently to p107 and p130 are growth inhibited by FGF. (A) Analysis of the interactions between Rb protein family members and PyLT versus PyLTΔ256–272 deletion mutant in RCS cells by coimmunoprecipitation. Rb protein family members were individually immunoprecipitated from lysates of RCS cells (lane 1), PyLT- expressing cells (lane 2), and PyLTΔ256–272–expressing cells (lane 3). After SDS-PAGE and Western blotting, membranes were probed with an anti-PyLT mAb (top panels). The same nitrocellulose membranes were stripped and reprobed with specific antibodies against pRb proteins (bottom panels). Black arrows indicate the position of wild-type PyLT (slower migrating) and mutant PyLT_256–272 (faster migrating). The asterisk indicates a nonspecific contaminant immunoprecipitated by the anti-pRb antibody and recognized by PyLT antibody. (B) BrdU incorporation assay of control RCS cells and RCS expressing either wild-type or mutant (PyLT_256–272) PyLT treated for 24 h with increasing concentration of FGF. (inset) Western blot analysis of the expression level of wild-type and mutant PyLT proteins in the cells used for these experiments.
BrdU incorporation assays (Fig. 3 B) showed that PyLT-Δ256–272–expressing RCS cells were growth-inhibited by FGF treatment in a dose-dependent manner, similarly to control RCS cells, whereas wild-type PyLT-expressing RCS cells were consistently resistant to FGF growth inhibition. The inability of a PyLT mutant that can bind pRb, but not p107 and p130, to block the FGF response indicates that inactivation of pRb alone is not sufficient to interfere with FGF-mediated growth inhibition of RCS cells, and that the FGF response is likely mediated through p107 and p130 function.
p107 and p130 mediate the FGF-induced growth arrest in micromass chondrocytes
Although the above analyses suggested the importance of p107 and p130 to FGF-induced arrest in RCS cells, they did not exclude the possibility that any one of the Rb family proteins was sufficient to mediate the FGF response. Moreover, because RCS are tumor cells, it was of interest to define the role of each of the Rb family proteins in the FGF response in nontransformed cells. Thus, to define the importance of p107, p130, or pRb to FGF-induced growth arrest, we prepared primary chondrocytes that specifically lacked those proteins. The chondrocytes were obtained using high density micromass cultures derived from prechondrogenic limb bud mesenchyme from embryos of different p107, p130, and Rb genotypes. These mesenchyme cultures form multiple condensations in which both chondrogenic differentiation and chondrocyte proliferation take place (Ahrens et al., 1979).
The initial behavior of micromass cultures of the different genotypes was consistent with that previously described (Ahrens et al., 1979). The cells initially formed a densely packed layer, then formed foci, and the size of these foci increased with time (Fig. 4 D). By day three, chondrogenesis had taken place within the foci, as detected by Alcian blue staining of the cartilage matrix and by collagen II and FGFR3 protein expression (Fig. 4 and data not shown). This is in keeping with the finding that early steps of chondrogenic differentiation were not blocked in p107/p130-deficient embryos (Rossi et al., 2002). Furthermore, no differences in FGFR3 expression were found among the different genotypes (unpublished data).
Figure 4.
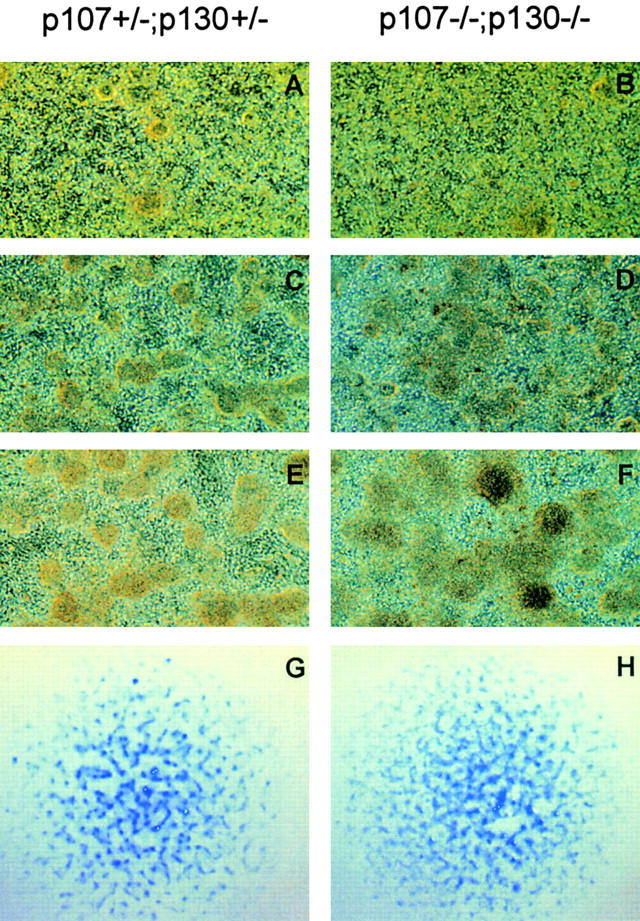
Production of p107 +/− ;p130 +/− and p107 −/− ;p130 −/− chondrocytes in high density micromass cultures. (A–F) Phase-contrast image of micromass cultures from p107 +/− ;p130 +/− (A, C, and E) and p107 −/− ;p130 −/− (B, D, and F) cultures at day 1 (A and B), day 3 (C and D), and day 4 (E and F) after plating. (G and H) Alcian blue staining of chondrocyte foci in p107 +/− ;p130 +/− (G) and p107 −/− ;p130 −/− (H) cultures 3 d after plating, and shown at lower magnification than in A–F. Each blue spot represents a chondrocyte focus.
To determine the effect of FGF on chondrocytes proliferation, day 3 cultures were treated with FGF for 24 h, and BrdU was added over the last 2 h. The cultures were stained simultaneously with an antibody against collagen II (to identify chondrocytes) and with an antibody against BrdU (to identify S phase cells), and the proportion of BrdU-positive chondrocytes was determined by confocal microscopy. This technique allows individual chondrocytes within the cartilage nodules to be examined by focusing on a single plane.
In wild-type cultures, FGF induced a 50% decrease in chondrocyte BrdU incorporation, with chondrocytes in the middle of the foci responding more strongly to FGF than those at the edge (Fig. 5) . Interestingly, Rb −/− chondrocytes responded to FGF to the same extent as wild-type chondrocytes (Fig. 5), indicating that pRb was not needed for FGF to inhibit proliferation. Similarly, p107 +/−;p130 +/− chondrocytes underwent an FGF-induced growth arrest (Fig. 5), implying that a single wild-type p107 and p130 allele is sufficient for the full FGF response. In contrast, p107 +/− ;p130 −/− chondrocytes displayed a partial inhibition (Fig. 5), suggesting that p130 can mediate, at least in part, the FGF growth arrest response. Strikingly, p107/p130 double mutants chondrocytes did not arrest in response to FGF. Indeed, neither did p107 −/− ;p130 +/− nor p107 −/− chondrocytes (Fig. 5), indicating that p107 was crucial to the FGF-induced growth arrest in this system.
Figure 5.


Loss of p107 and p130 impairs FGF-induced growth arrest in micromass chondrocytes. (A) Day 3 micromass cultures of the indicated genotypes were treated with FGF or vehicle for 24 h, and allowed to incorporate BrdU over the final 2 h. Chondrocytes were stained with anti–collagen II and an FITC-conjugated secondary antibody (green), and with anti-BrdU and a Cy3-conjugated secondary antibody (red), and analyzed by confocal microscopy. (B) Quantification of the percentage of BrdU(+) chondrocytes in a series of confocal sections from at least five foci for each genotype and each condition. Error bars represent SD of the percentages of BrdU(+) cells in each focus. Note that in FGF-treated wild-type (WT), Rb −/− and p107 +/− ;p130 +/− cultures, the majority of the BrdU(+) nuclei were at the periphery of the foci, and that growth inhibition was more substantial for cells within the cores of the foci.
p107 and p130 mediate FGF-induced growth inhibition in mouse bone rudiments
Next, we sought to determine whether p107 and p130 were needed for the FGF response not only in cultured chondrocytes, but also in chondrocytes that are within developing bones that have a normal cartilage architecture. For this purpose, we used organ culture of metatarsal bone rudiments of mice with targeted p107 and/or p130 inactivation. In brief, bone rudiments from E15 mouse embryos were isolated and cultivated in vitro for 24 h and then treated with FGF-1 or left untreated for an additional 24 h. BrdU was added to the culture medium for the last 6 h. The rudiments were processed for immunohistochemistry, and BrdU incorporation was evaluated by immunostaining of the rudiment sections.
The frequency of chondrocytes incorporating BrdU in the proliferative zone of untreated rudiments was similar (Fig. 6) in all the different genotypes that we examined. Metatarsals from double heterozygotes mice (p107 +/− ;p130 +/−) showed strong inhibition of DNA synthesis upon FGF treatment (Fig. 6), similar to that observed in wild-type bone rudiments (not depicted). FGF treatment produced essentially no inhibition of DNA synthesis in p107 −/− ;p130 +/− rudiments, and a slight but reproducible decrease in p107 +/− ;p130 −/− rudiments, similar to that observed in chondrocyte micromass cultures. FGF treatment of bone rudiments from p107/p130-null rudiments resulted in no inhibition in DNA synthesis, and actually caused a small but reproducible increase in the rate of proliferation (Fig. 6 B), suggesting that blocking the growth inhibitory effects of FGF signaling in these cells could perhaps unmask its growth stimulatory potential. These results show that in developing bone rudiments, removal of p107 activity essentially abolishes chondrocyte growth inhibition by FGF, and that chondrocytes become completely resistant when p130 is also absent. pRb, which is not mutated in these embryos, fails to compensate for the loss of p107 and p130. Thus, p107 and p130 are the major mediators of FGF growth inhibition in growth plate chondrocytes.
Figure 6.


Absence of FGF-induced growth inhibition in bone rudiment chondrocytes lacking p107 and/or p130. Metatarsal bone rudiments from E15 embryos were isolated and cultivated in vitro for 48 h with or without FGF treatment (100 ng/ml for 24 h). BrdU was added during the last 6 h of the culture. After fixation, rudiments were processed for immunodetection of BrdU. Counterstaining with Alcian blue was done in order to stain the cartilage-specific sulfate proteoglycans present in the extracellular matrix. (A) Representative pictures of BrdU incorporation in the proliferative zone of the growth plate for the different genotypes analyzed. R, reserve zone; P, proliferating zone; H, hypertrophic zone. (B) Quantification. Note that error bars in Fig. 6 B represent the SD calculated from at least six counts (percentage of cells positive for BrdU) of 80–160 cells each. Organ culture experiments were performed twice and gave similar results.
Discussion
The results described in this paper indicate that two members of the Rb family of pocket proteins (p107 and p130), but surprisingly not the prototype of the family (pRb), are essential effectors of the FGF signal cascade in chondrocytes that leads to inhibition of cell proliferation. Expression of viral proteins such as E1A and PyLT-Ag, which bind and inactivate Rb proteins, essentially suppress the growth inhibitory response of RCS chondrocytes to FGF. Not only was this effect not manifested in cells expressing mutant forms of PyLT and E1A incapable of binding to all Rb proteins, but more specifically a PyLT mutant that is selectively impaired in binding to p107 and p130, but not to pRb, was also incapable of blocking the FGF response. Notably, the loss of Rb family proteins' function did not affect the response to other growth inhibitory agents.
The most conclusive demonstration of the p107 and p130 role in mediating the FGF effect in cell proliferation was provided by experiments with chondrocyte micromass cultures or cartilaginous metatarsal bone rudiments from murine embryos with targeted mutations of pRb, p107, and p130. Although the absence of pRb had no apparent effect on the FGF response, the loss of either p107 or p130 resulted in almost no inhibition of chondrocyte proliferation. Chondrocytes lacking both p107 and p130 showed no growth inhibition in response to FGF, and actually a small but reproducible increase in the rate of proliferation. This effect could be due to an “unmasking” of the growth stimulatory effects of FGF when growth inhibition is removed, and is reminiscent of what we previously observed in bone rudiments from STAT1 knockout mice (Sahni et al., 1999).
Although our analysis indicates that pRb is dispensable for FGF-induced growth arrest, it is difficult at this point to determine whether p130 or p107 is the major mediator of the FGF response. Although the experiments performed with micromass chondrocytes seem to suggest that p107 plays the major role, it is clear from the deregulated proliferation of p107 +/− ;p130 −/− but not p107 +/− ;p130 +/− chondrocytes in micromass and bone rudiments that p130 also mediates the FGF growth inhibitory effect. These findings are consistent with roles of p107 and p130 that were deduced from the phenotype of p107- and p130-deficient mice. The original investigation of p107 knockout mice showed only a subtle bone phenotype (Cobrinik et al., 1996), and that chondrocyte proliferation was severely deregulated only in the p107/p130-deficient growth plates. However, more recent studies indicated that p107 was essential to chondrocyte proliferation control at early times during chondrogenesis, when p130 expression is minimal (Rossi et al., 2002). This also militates in favor of an overlapping, redundant role of p107 and p130 in chondrocyte proliferation. It is possible that the micromass and bone rudiments organ culture conditions make the chondrocytes less susceptible to FGF-mediated growth inhibition, so that removal of either p130 or p107 is sufficient to almost abolish the effect of FGF treatment, whereas loss of both is needed to substantially deregulate proliferation in vivo. Furthermore, other factors may counteract the action of FGF in vivo, which would have to be completely blocked to produce a distinct phenotype.
Although further work will be needed to resolve this issue, it is perhaps more interesting to ask why pRb does not seem to participate in the FGF response of chondrocytes. pRb is equally dephosphorylated in RCS cells upon FGF treatment, but alone it does not seem to be capable of affecting FGF-mediated growth inhibition. pRb may not be an important regulator of cell cycle progression in chondrocytes. It has been long suspected that the function of the Rb family proteins is overlapping, but also shows tissue specificity (Lipinski and Jacks, 1999). The loss of all three Rb proteins is necessary to fully immortalize fibroblasts (Dannenberg et al., 2000; Sage et al., 2000), but loss of pRb alone is sufficient to deregulate proliferation of liver, lens, neural, muscle, and pituitary cells (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992). Similarly, the loss of pRb, but not of p107 or p130, inhibits late osteoblast differentiation in vitro (Thomas et al., 2001). On the other hand, in the Ba/F3 hematopoietic cell line, cell cycle progression appears to be mainly controlled by p130 (Hoshikawa et al., 1998). An alternative hypothesis is that our results could reflect a more specific role of p107 and p130 in the FGF response, such as if FGF signaling targeted functions of p107 and/or p130 that are not possessed by pRb. p107 and p130 are known to form complexes with E2F4 and E2F5, whereas pRb preferentially regulates E2F1, E2F2, and E2F3 (Sardet et al., 1995; Dyson, 1998; Classon and Dyson, 2001). E2F/p107 and p130 complexes bind specific DNA sequences and inhibit the transcription of targeted genes (Hurford et al., 1997). It is possible that FGF-mediated growth inhibition depends strictly on the repression of specific genes, which would be regulated by the E2F/p107 or p130 complexes. As another possibility, the p107- and p130-dependent inhibition of chondrocyte proliferation may depend upon the ability of these proteins to directly bind and inhibit Cdk2 kinase, a property that is not shared with pRb (Hannon et al., 1993; Classon and Dyson, 2001).
A further issue that is raised by the current work is whether the role of p107 and p130 in the FGF response is linked to their roles in chondrocyte differentiation. Analysis of p107 −/− ;p130 −/− embryos clearly showed a block to chondrocyte hypertrophic differentiation (Rossi et al., 2002), and this defect was also observed in organ cultures of the metatarsal rudiments, regardless of FGF treatment. Similar to the in vivo situation, the appearance of hypertrophic chondrocytes could be easily detected in p107 +/− ;p130 +/− rudiments as well as in p130 −/− ;p107 +/− rudiments, but was rarely detected in p107 −/− ;p130 +/− rudiments and not detected at all in rudiments from p107 −/− ;p130 −/− embryos (data not shown). As shown recently (Rossi et al., 2002), the inhibition of chondrocyte differentiation observed in p130/p107 knockout embryos could be due to the impaired expression of Cbfa 1, a transcription factor that is important for chondrocyte hypertrophic differentiation (Inada et al., 1999; Kim et al., 1999; Takeda et al., 2001). Whether this is in turn related to the resistance of these cells to FGF treatment is currently unclear. The question of whether FGF treatment accelerates or inhibits chondrocyte terminal differentiation and whether this effect may require cell cycle arrest and the intervention of the Rb proteins is presently undergoing investigation.
Finally, our results are consistent with a relatively simple mechanism by which FGF would mediate growth inhibition of chondrocytes. FGF treatment could, through the activation of STAT1, induce the expression of Cdk inhibitors, leading to inhibition of Rb family proteins phosphorylation. Indeed, it was previously shown that p21 is induced by FGF treatment in chondrocytes (Sahni et al., 1999; Aikawa et al., 2001). As shown here, the expression of p27 and p57 is also induced by FGF in chondrocytes, but weakly and with delayed kinetics. On the other hand, we found that p21 is also strongly induced by FGF in many cell types where its major biological effect is growth stimulation, and not growth inhibition. Furthermore, p21 −/− mice have not been reported as having a clear bone phenotype (Deng et al., 1995), and p21 deletion only partially reduced the ability of FGF to inhibit chondrocyte proliferation in organ culture (Aikawa et al., 2001). In addition to inducing expression of Cdk inhibitors, FGF signaling could also induce the expression or the activity of phosphatases specifically targeting the Rb proteins. This activity might be particularly relevant to p107, because p107 appeared to undergo dephosphorylation with kinetics that were more rapid than was the accumulation of G1 cells (our unpublished results). In this regard, p107 dephosphorylation has been reported to be rapidly induced by DNA damaging agents (Voorhoeve et al., 1999; Kondo et al., 2001), suggesting that rapid p107 dephosphorylation might similarly be induced in FGF-treated chondrocytes. At this time, we believe that induction of phosphatases and Cdk inhibitors could participate, probably together with other mechanisms, in producing the profound growth-inhibiting effect of FGF in chondrocytes.
Materials and methods
Cell culture and proliferation assay
RCS cells were cultured on 100-mm cell culture dishes (Corning Incorporated) in DME containing 10% FCS. For proliferation assays, cells either cultivated on glass coverslips or Lab-Tek® chamber slides (Nunc) were incubated for 3 h with 10 μg/ml of BrdU and were fixed with 3.7% PFA. Incorporation of BrdU into cell DNA was detected by immunofluorescence. In brief, fixed cells were permeabilized with 0.5% Triton X-100 and subsequently treated with 1.5 M HCl. After several washes in PBS, cells were incubated with a mouse anti-BrdU mAb (RPN-202; Amersham Biosciences). Cy3-conjugated anti–mouse antibody (Jackson ImmunoResearch Laboratories) was used as a secondary antibody. Cell nuclei were counterstained by incubation with 0.5 μg/ml Hoechst (Sigma-Aldrich) before mounting on slides. Pictures were taken with a microscope (Axioplan 2; Carl Zeiss MicroImaging, Inc.) equipped with a digital camera.
For FACScan™ analysis, trypsinized cells were fixed in 70% ice-cold ethanol. After centrifugation, the cell pellets were washed with PBS and treated overnight with 50 μg/ml propidium iodide in the presence of 100 μg/ml RNase in order to stain the nuclei. Flow cytometry was performed using a FACScan™ (Becton Dickinson) equipped with a doublet discrimination module. Doublets were gated out on a plot of signal area versus signal width and analyzed using ModFit LT™ (Verity Software House).
For proliferation assays, FACScan™ analysis, and biochemical assays, RCS cells cultivated in DME containing 10% FCS were treated with recombinant human FGF-1 at the indicated concentrations together with 10 μg/ml heparin. Dexamethasone was purchased from Sigma-Aldrich.
Cloning and retroviral infections
PyLT antigen cDNA cloned in the pBabe-Puro retroviral expression vector was described previously (Mansukhani et al., 2000). This vector contains a resistance gene for puromycin. Mutant PyLT cDNAs (PyLT-Δ141–158 and PyLT-Δ256–272, a gift from Dr. A. Mes-Masson, McGill University, Montreal, Canada), as well as wild-type E1A and mutant E1A (E1A-12S.928) cDNAs were subcloned in pBabe-Puro. For retroviral infection, 293T cells were first transfected by calcium phosphate precipitation method by mixing equal amounts of retroviral construct together with helper virus vector pCL-eco (Naviaux et al., 1996). 48–72 h after transfection, supernatant was collected and used to infect RCS cells for 16 h in the presence of 10 μg/ml polybrene. Cells were cultivated for two additional days before selection with 2 μg/ml puromycin. PyLT- and PyLT-Δ256–272–expressing RCS cells were further subcloned and at least two clones of each line were analyzed.
Immunoprecipitation and Western blotting
Cell lysis was done either in RIPA buffer or immunoprecipitation lysis buffer (50 mM Tris, pH 7.5, 250 mM NaCl, 0.1% Triton X-100, 1 mM EDTA) in the presence of a cocktail of protease and phosphatase inhibitors. For immunoprecipitation assays, 1 mg of total protein extracts was incubated overnight at 4°C with 10 μg antibodies. Protein G Plus–Agarose (Oncogene Research Products) was added the next day for 2 h at 4°C, and the immune complexes were washed three to four times in immunoprecipitation lysis buffer. Immunoprecipitates and total protein extracts (20 μg) were separated by SDS-PAGE, transferred onto nitrocellulose membranes (Protran®; Schleicher & Schuell) and analyzed by ECL detection system (Amersham Biosciences). The following antibodies were used: anti-pRb mAb (G3–245; BD Biosciences), anti-p107 pAb (C-18; Santa Cruz Biotechnology, Inc.), p130 mAb (R27020; Transduction Laboratories), anti-p16, anti-p18, anti-p21, and anti-p57 pAbs (M-156, N-20, C-19, and H-91, respectively; Santa Cruz Biotechnology, Inc.), anti-p27 mAb (K25020; Transduction Laboratories), anti-Grb2 mAb (G16720; Transduction Laboratories), anti-PyLT antigen mAb (PN116; a gift from Dr. B. Schaffhausen, Tufts University Medical School, Boston, MA), anti-FGFR3 pAb (C-15; Santa Cruz Biotechnology, Inc.), and anti-phosphotyrosine mAb (4G10; Upstate Biotechnology). HRP-conjugated anti–mouse and anti–rabbit secondary antibodies were purchased from Promega.
Preparation of chondrocyte micromass cultures
To generate embryos of the desired genotypes, matings were conducted between p107 −/−;p130+/− and p107 +/−;p130−/− mice or by intercrossing p107 + / − or Rb + / − mice, and were assumed for timing purposes to occur at midnight. Micromass cultures were performed as in Edwall-Arvidsson and Wroblewski (1996) with the following modifications: forelimb and hindlimb buds were removed from E11.5 embryos and pooled in an Eppendorf tube. After two washes in PBS, the limbs were centrifuged for 1 min at 3,000 rpm in a microcentrifuge, resuspended in 1 mg/ml collagenase A (Roche) in Ham's F12 medium and incubated at 37°C for 1 h. The tissues were gently triturated to obtain a single cell suspension, washed two times in Ham's F12 medium, and resuspended at 0.9 × 106 cells/ml, in Ham's F12 medium supplemented with 1% FCS (HyClone). High density (supra-confluent) micromass cultures were initiated by spotting 20-μl drops onto the surface of tissue culture dishes (6-well dish; Nunc), or onto a coverslip for experiments requiring confocal microscopy, and incubated at 37°C for 2 h to allow the cells to attach. Ham's F12 medium containing 1% FCS and antibiotics was then added to the culture. The medium was changed after 2 d of culture or just before FGF treatment.
At various times after plating, micromass cultures were washed in PBS and fixed for 30 min in 4% PFA in PBS. To determine the accumulation of extracellular cartilage matrix, fixed cultures were stained overnight with 1% Alcian blue (Sigma-Aldrich) in 3% acetic acid (Leonard et al., 1991), and were washed for 6 h in 3% acetic acid.
Confocal microscopy analysis
Micromass cultures were washed in PBS and fixed in 100% methanol. After three washes in PBS, the cultures were blocked in 4% horse serum in PBS for 1 h. Antibodies anti-BrdU (Beckton Dickinson) at 1/100 dilution, anti–collagen II (Santa-Cruz Biotechnology, Inc.) at 1/100 dilution, or anti-FGFR3 (sc-123; Santa-Cruz Biotechnology, Inc.) at 1/100 dilution were added in 4% horse serum in PBS, at 4°C overnight. After three washes with PBS, the secondary antibodies (anti–mouse Cy3, anti–goat FITC, and anti–rabbit Cy3; Jackson ImmunoResearch) were added at 1/200 dilution, for 45 min at RT. After three washes with PBS, the nuclei were stained with DAPI, and the coverslip was mounted on a glass slide with Vectashield® (Vector Laboratories). Foci of ∼20 μm diameter were selected and analyzed over a series of 1 μm confocal sections using a confocal laser scanning microscope (model LSM410; Carl Zeiss MicroImaging, Inc.). The percentage of BrdU-positive cells was determined by analysis of each confocal section. The results are a mean of at least five foci for each genotype and each condition.
Organ cultures
Metatarsal long bone rudiments from E15 mouse embryo (generated as described in the chondrocyte micromass section) heterozygotes or nulls for the p107 and/or p130 genes were dissected under sterile conditions. The cartilaginous long bones were left intact. Organ culture medium consisted of MEM without nucleosides (GIBCO BRL) supplemented with 50 μg/ml ascorbic acid, 300 μg/ml l-glutamine, 50 μg/ml gentamicine, 250 μg/ml Fungizone, 1 mM β-glycerophosphate, and 0.2% BSA Cohn fraction V (complete medium; Sigma-Aldrich). Each long bone was cultured individually in Transwells® (Costar) containing 400 μl of complete medium in the presence or absence of 100 ng/ml recombinant human FGF-1 and 10 μg/ml heparin. The metatarsal cultures were maintained at 37°C for 48 h. For the proliferation assays, the long bones were treated with FGF-1 for 24 h and BrdU (1 μg/ml final concentration) was added during the last 6 h of treatment. The experiments were set up for left/right paired observations, with one metatarsal serving as a control to the other.
Metatarsals were fixed in 4% PFA overnight at 4°C and embedded in paraffin, and 4-μm tissue sections were performed. Sections were deparaffinized by treatment with xylene and rehydrated with successive washes of 100, 95, and 70% ethanol, followed by washes in TS buffer (100 mM Tris-HCl, pH 7.4, 150 mM NaCl) and permeabilization with 0.25% Triton X-100. After 3 h of blocking with 3% goat serum, the endogenous peroxidase activity was inactivated by incubating in 5% H2O2 in methanol for 10 min. Anti-BrdU mAb was used with a Vectastain Elite ABC Kit (Vector Laboratories). Preparations were subsequently counterstained with 0.1% Alcian blue 8GX in 3% acetic acid. After dehydration, samples were mounted with Permount® (Fisher Scientific International).
Acknowledgments
We wish to thank Lisa Dailey, Alka Mansukhani, and H. Skopicki for advice and helpful discussion, Milton Gomez for help with the histology preparations, and Anne Marie Mes-Masson and Paolo Dotto for the gift of plasmids.
This investigation was supported by National Institutes of Health grants DE13745 (to C. Basilico) and HD35156 (to D. Cobrinik). The Columbia University Optical Microscopy Facility is supported by grant P30CA13696.
Emmanuel Laplantine and Ferdinand Rossi contributed equally to this work.
Footnotes
Abbreviations used in this paper: E1A, adenovirus E1A; FGFR, FGF receptors; PyLT, polyoma large T; Rb, retinoblastoma; RCS, rat chondrosarcoma.
References
- Ahrens, P.B., M. Solursh, R.S. Reiter, and C.T. Singley. 1979. Position-related capacity for differentiation of limb mesenchyme in cell culture. Dev. Biol. 69:436–450. [DOI] [PubMed] [Google Scholar]
- Aikawa, T., G.V. Segre, and K. Lee. 2001. Fibroblast growth factor inhibits chondrocytic growth through induction of p21 and subsequent inactivation of cyclin E-Cdk2. J. Biol. Chem. 276:29347–29352. [DOI] [PubMed] [Google Scholar]
- Basilico, C., and D. Moscatelli. 1992. The FGF family of growth factors and oncogenes. Adv. Cancer Res. 59:115–165. [DOI] [PubMed] [Google Scholar]
- Clarke, A.R., E.R. Maandag, M. van Roon, N.M. van der Lugt, M. van der Valk, M.L. Hooper, A. Berns, and H. te Riele. 1992. Requirement for a functional Rb-1 gene in murine development. Nature. 359:328–330. [DOI] [PubMed] [Google Scholar]
- Classon, M., and N. Dyson. 2001. p107 and p130: versatile proteins with interesting pockets. Exp. Cell Res. 264:135–147. [DOI] [PubMed] [Google Scholar]
- Cobrinik, D., M.H. Lee, G. Hannon, G. Mulligan, R.T. Bronson, N. Dyson, E. Harlow, D. Beach, R.A. Weinberg, and T. Jacks. 1996. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 10:1633–1644. [DOI] [PubMed] [Google Scholar]
- Dannenberg, J.H., A. van Rossum, L. Schuijff, and H. te Riele. 2000. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 14:3051–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca, F., and J. Baron. 1999. Control of bone growth by fibroblast growth factors. Trends Endocrinol. Metab. 10:61–65. [DOI] [PubMed] [Google Scholar]
- Deng, C., P. Zhang, J.W. Harper, S.J. Elledge, and P. Leder. 1995. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 82:675–684. [DOI] [PubMed] [Google Scholar]
- Deng, C., A. Wynshaw-Boris, F. Zhou, A. Kuo, and P. Leder. 1996. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 84:911–921. [DOI] [PubMed] [Google Scholar]
- Dotto, G.P., L.F. Parada, and R.A. Weinberg. 1985. Specific growth response of ras-transformed embryo fibroblasts to tumour promoters. Nature. 318:472–475. [DOI] [PubMed] [Google Scholar]
- Dyson, N. 1998. The regulation of E2F by pRB-family proteins. Genes Dev. 12:2245–2262. [DOI] [PubMed] [Google Scholar]
- Dyson, N., R. Bernards, S.H. Friend, L.R. Gooding, J.A. Hassell, E.O. Major, J.M. Pipas, T. Vandyke, and E. Harlow. 1990. Large T antigens of many polyomaviruses are able to form complexes with the retinoblastoma protein. J. Virol. 64:1353–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwall-Arvidsson, C., and J. Wroblewski. 1996. Characterization of chondrogenesis in cells isolated from limb buds in mouse. Anat. Embryol. (Berl). 193:453–461. [DOI] [PubMed] [Google Scholar]
- Ewen, M.E. 1998. Regulation of the cell cycle by the Rb tumor supressor family. In Cell Cycle Control. M. Pagano, editor. Springer-Verlag, New York. 149–179. [DOI] [PubMed]
- Goldfarb, M. 1996. Functions of fibroblast growth factors in vertebrate development. Cytokine Growth Factor Rev. 7:311–325. [DOI] [PubMed] [Google Scholar]
- Hannon, G.J., D. Demetrick, and D. Beach. 1993. Isolation of the Rb-related p130 through its interaction with CDK2 and cyclins. Genes Dev. 7:2378–2391. [DOI] [PubMed] [Google Scholar]
- Hoshikawa, Y., A. Mori, K. Amimoto, K. Iwabe, and M. Hatakeyama. 1998. Control of retinoblastoma protein-independent hematopoietic cell cycle by the pRB-related p130. Proc. Natl. Acad. Sci. USA. 95:8574–8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurford, R.K., Jr., D. Cobrinik, M.H. Lee, and N. Dyson. 1997. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 11:1447–1463. [DOI] [PubMed] [Google Scholar]
- Inada, M., T. Yasui, S. Nomura, S. Miyake, K. Deguchi, M. Himeno, M. Sato, H. Yamagiwa, T. Kimura, N. Yasui, et al. 1999. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn. 214:279–290. [DOI] [PubMed] [Google Scholar]
- Jacks, T., A. Fazeli, E.M. Schmitt, R.T. Bronson, M.A. Goodell, and R.A. Weinberg. 1992. Effects of an Rb mutation in the mouse. Nature. 359:295–300. [DOI] [PubMed] [Google Scholar]
- Jux, C., K. Leiber, U. Hugel, W. Blum, C. Ohlsson, G. Klaus, and O. Mehls. 1998. Dexamethasone impairs growth hormone (GH)-stimulated growth by suppression of local insulin-like growth factor (IGF)-I production and expression of GH- and IGF-I-receptor in cultured rat chondrocytes. Endocrinology. 139:3296–3305. [DOI] [PubMed] [Google Scholar]
- Karsenty, G. 1999. The genetic transformation of bone biology. Genes Dev. 13:3037–3051. [DOI] [PubMed] [Google Scholar]
- Kim, I.S., F. Otto, B. Zabel, and S. Mundlos. 1999. Regulation of chondrocyte differentiation by Cbfa1. Mech Dev. 80:159–170. [DOI] [PubMed] [Google Scholar]
- Klaus, G., C. Jux, P. Fernandez, J. Rodriguez, R. Himmele, and O. Mehls. 2000. Suppression of growth plate chondrocyte proliferation by corticosteroids. Pediatr. Nephrol. 14:612–615. [DOI] [PubMed] [Google Scholar]
- Kondo, T., H. Higashi, H. Nishizawa, S. Ishikawa, S. Ashizawa, M. Yamada, Z. Makita, T. Koike, and M. Hatakeyama. 2001. Involvement of pRB-related p107 protein in the inhibition of S phase progression in response to genotoxic stress. J. Biol. Chem. 276:17559–17567. [DOI] [PubMed] [Google Scholar]
- Lee, E.Y., C.Y. Chang, N. Hu, Y.C. Wang, C.C. Lai, K. Herrup, W.H. Lee, and A. Bradley. 1992. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 359:288–294. [DOI] [PubMed] [Google Scholar]
- Leonard, C.M., H.M. Fuld, D.A. Frenz, S.A. Downie, J. Massague, and S.A. Newman. 1991. Role of transforming growth factor-beta in chondrogenic pattern formation in the embryonic limb: stimulation of mesenchymal condensation and fibronectin gene expression by exogenous TGF-beta and evidence for endogenous TGF-beta-like activity. Dev. Biol. 145:99–109. [DOI] [PubMed] [Google Scholar]
- Lipinski, M.M., and T. Jacks. 1999. The retinoblastoma gene family in differentiation and development. Oncogene. 18:7873–7882. [DOI] [PubMed] [Google Scholar]
- Mansukhani, A., P. Bellosta, M. Sahni, and C. Basilico. 2000. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J. Cell Biol. 149:1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan, G., and T. Jacks. 1998. The retinoblastoma gene family: cousins with overlapping interests. Trends Genet. 14:223–229. [DOI] [PubMed] [Google Scholar]
- Naski, M.C., and D.M. Ornitz. 1998. FGF signaling in skeletal development. Front. Biosci. 3:D781–D794. [DOI] [PubMed] [Google Scholar]
- Naski, M.C., Q. Wang, J. Xu, and D.M. Ornitz. 1996. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat. Genet. 13:233–237. [DOI] [PubMed] [Google Scholar]
- Naviaux, R.K., E. Costanzi, M. Haas, and I.M. Verma. 1996. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70:5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon, A.A., P. Desjardins, J.A. Hassell, and A.M. Mes-Masson. 1996. Functional implications of mutations within polyomavirus large T antigen Rb-binding domain: effects on pRb and p107 binding in vitro and immortalization activity in vivo. J. Virol. 70:4457–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers, C.J., S.W. McLeskey, and A. Wellstein. 2000. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer. 7:165–197. [DOI] [PubMed] [Google Scholar]
- Rossi, F., H.E. MacLean, W. Yuan, R.O. Francis, E. Semenova, C.S. Lin, H.M. Kronenberg, and D. Cobrinik. 2002. p107 and p130 coordinately regulate proliferation, Cbfa1 expression, and hypertrophic differentiation during endochondral bone development. Dev. Biol. 247:271–285. [DOI] [PubMed] [Google Scholar]
- Sage, J., G.J. Mulligan, L.D. Attardi, A. Miller, S. Chen, B. Williams, E. Theodorou, and T. Jacks. 2000. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 14:3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni, M., D.C. Ambrosetti, A. Mansukhani, R. Gertner, D. Levy, and C. Basilico. 1999. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 13:1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni, M., R. Raz, J.D. Coffin, D. Levy, and C. Basilico. 2001. STAT1 mediates the increased apoptosis and reduced chondrocyte proliferation in mice overexpressing FGF2. Development. 128:2119–2129. [DOI] [PubMed] [Google Scholar]
- Sardet, C., M. Vidal, D. Cobrinik, Y. Geng, C. Onufryk, A. Chen, and R.A. Weinberg. 1995. E2F-4 and E2F-5, two members of the E2F family, are expressed in the early phases of the cell cycle. Proc. Natl. Acad. Sci. USA. 92:2403–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda, S., J.P. Bonnamy, M.J. Owen, P. Ducy, and G. Karsenty. 2001. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 15:467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, D.M., S.A. Carty, D.M. Piscopo, J.S. Lee, W.F. Wang, W.C. Forrester, and P.W. Hinds. 2001. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol. Cell. 8:303–316. [DOI] [PubMed] [Google Scholar]
- Voorhoeve, P.M., R.J. Watson, P.G. Farlie, R. Bernards, and E.W. Lam. 1999. Rapid dephosphorylation of p107 following UV irradiation. Oncogene. 18:679–688. [DOI] [PubMed] [Google Scholar]
- Wang, H.G., Y. Rikitake, M.C. Carter, P. Yaciuk, S.E. Abraham, B. Zerler, and E. Moran. 1993. Identification of specific adenovirus E1A N-terminal residues critical to the binding of cellular proteins and to the control of cell growth. J. Virol. 67:476–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, M.K., and D.J. Donoghue. 1997. FGFR activation in skeletal disorders: too much of a good thing. Trends Genet. 13:178–182. [DOI] [PubMed] [Google Scholar]