

Abstract
The yeast silent information regulator (Sir)2 protein links cellular metabolism and transcriptional silencing through its nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase activity. We report that mitochondria from mammalian cells contain intrinsic NAD-dependent deacetylase activity. This activity is inhibited by the NAD hydrolysis product nicotinamide, but not by trichostatin A, consistent with a class III deacetylase. We identify this deacetylase as the nuclear-encoded human Sir2 homologue hSIRT3, and show that hSIRT3 is located within the mitochondrial matrix. Mitochondrial import of hSIRT3 is dependent on an NH2-terminal amphipathic α-helix rich in basic residues. hSIRT3 is proteolytically processed in the mitochondrial matrix to a 28-kD product. This processing can be reconstituted in vitro with recombinant mitochondrial matrix processing peptidase (MPP) and is inhibited by mutation of arginines 99 and 100. The unprocessed form of hSIRT3 is enzymatically inactive and becomes fully activated in vitro after cleavage by MPP. These observations demonstrate the existence of a latent class III deacetylase that becomes catalytically activated upon import into the human mitochondria.
Keywords: HDAC; chromatin; 11p15.5; apoptosis; acetylation
Introduction
Reversible protein acetylation is emerging as a critical posttranslational modification involved in the regulation of many biological processes. Although most of the pioneering experiments focused on the role of histone acetylation in transcriptional control, recent findings have generalized the concept of reversible protein acetylation to many nonhistone proteins (Sterner and Berger, 2000). Histone proteins are acetylated at lysines in their NH2-terminal tails under the control of competing histone acetyltransferases and histone deacetylases (HDACs).* Eighteen distinct human histone deacetylases have been identified and are grouped in three classes based on their homology to Saccharomyces cerevisiae histone deacetylases: RPD3 (class I), HDA1 (class II), and silent information regulator (Sir)2 (class III).
Sir2 protein participates in transcriptional silencing at telomeres, mating-type loci, and the ribosomal RNA locus. Sir2 has also been implicated in the repair of chromosomal double-strand breaks, in cell cycle progression, and in the control of chromosome stability in yeast (Gottschling et al., 1990; Martin et al., 1999). Increased dosage of the Sir2 gene increases life span in yeast and in Caenorhabditis elegans (Kaeberlein et al., 1999; Lin et al., 2000; Tissenbaum and Guarente, 2001). Yeast Sir2 has nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylase activity that links Sir2 functions to cellular metabolism (Guarente, 2000). This activity is conserved from bacteria to humans and is also exhibited by mammalian Sir2 homologues (Imai et al., 2000; Smith et al., 2000). The NAD dependency of Sir2-like enzymes distinguishes them from the class I and class II HDACs, which use a zinc-catalyzed mechanism (Finnin et al., 1999). Seven human Sir2 homologues have been identified in humans and are designated hSIRT1–7 (Frye, 1999, 2000). hSIRT1, 2, 3, and 5 have NAD-dependent deacetylase activity (unpublished data; Luo et al., 2001; Vaziri et al., 2001). Although a silencing function of SIRT proteins can be anticipated by analogy to their S. cerevisiae homologues, little is known about their biological activities. It is likely that the deacetylase activity of this family of enzymes is not restricted to histone proteins. Indeed, a distant homologue of Sir2 called CobB is found in Salmonella typhimurium, which do not have histones, where it can compensate for the loss of the phosphoribosyltransferase CobT, suggesting a ribosyltransferase activity (Tsang and Escalante-Semerena, 1998). Recent findings also support the concept that nonhistone proteins can serve as substrates for Sir2-like proteins in mammalian cells. hSIRT1 deacetylates the transcription factor p53 and inhibits its activation in response to DNA damage and oxidative stress (Luo et al., 2001; Vaziri et al., 2001). Mouse Sir2α deacetylates the TAFI68 subunit of the TATA-box binding protein–containing factor, leading to the repression of RNA polymerase I transcription (Muth et al., 2001). Here, we provide further evidence for the role of Sir2-like proteins on nonhistone substrates by demonstrating that hSIRT3 is an exclusive mitochondrial NAD-dependent deacetylase.
Results
Mitochondria contain Sir2-like deacetylase activity
A systematic survey of subcellular fractions for the presence of histone deacetylase activities led to the detection of a deacetylase activity in human mitochondrial fractions prepared from HEK293T cells (Fig. 1 A). This activity was strictly dependent on NAD and was suppressed by nicotinamide, a product of NAD hydrolysis that inhibits Sir2-like proteins (Landry et al., 2000a, 2000b; Tanner et al., 2000; Tanny and Moazed, 2001; Fig. 1 A). In contrast, trichostatin A (TSA), a specific inhibitor of class I and class II deacetylases, had no effect (Fig. 1 A). Under the same conditions, TSA inhibited the activity of a prototypic class II HDAC, HDAC6 (unpublished data). These findings indicated the presence of Sir2-like class III protein deacetylases in mitochondria.
Figure 1.
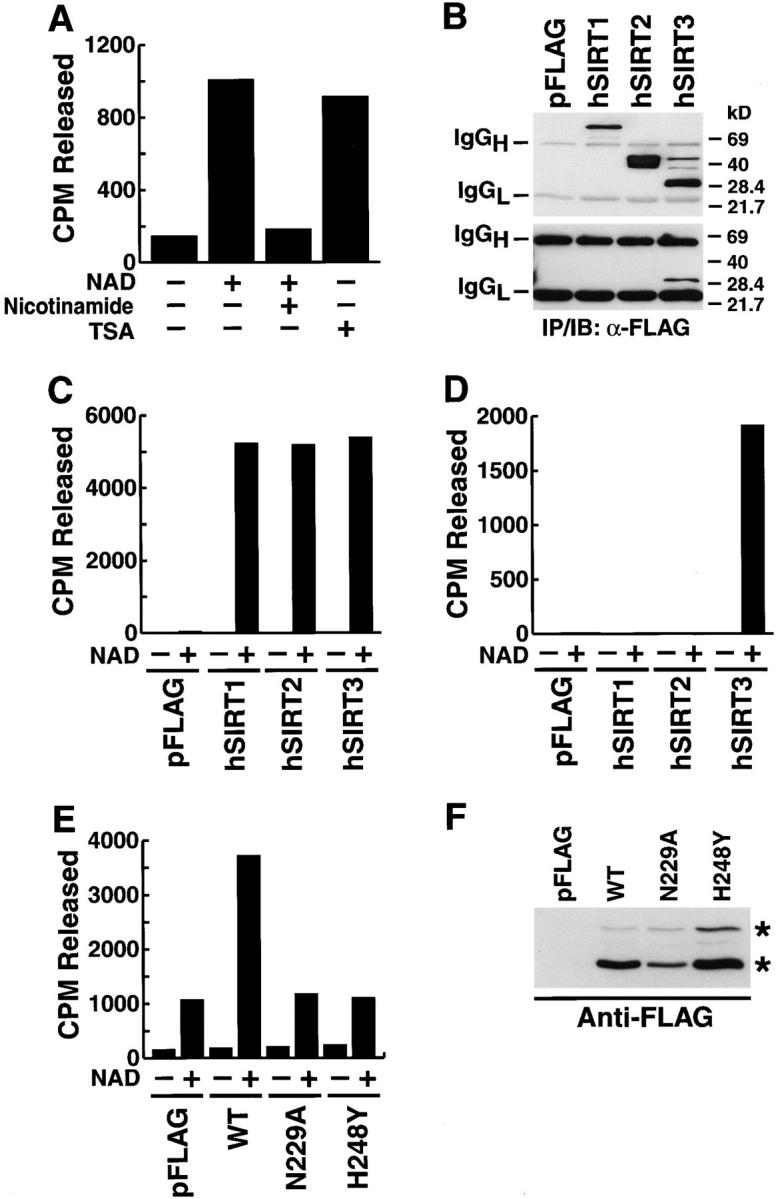
Mitochondria contain Sir2-like deacetylase activity. (A) Mitochondrial lysates were prepared from HEK293T cells and assayed for deacetylase activity on a histone H4 peptide in the presence or absence of NAD (1 mM) or in combination with nicotinamide (5 mM) or TSA (400 nM) for 2 h at 25°C. Released acetyl was quantitated as described in Materials and methods. A representative experiment is shown. (B) Western blot analysis of anti-FLAG immunoprecipitates obtained from whole-cell lysates (top) or purified mitochondria (bottom) after transfection of hSIRT3. Because different amounts of cellular and mitochondrial lysates were used in the immunoprecipitation, the top and bottom panels cannot be compared quantitatively. (C) hSIRT proteins were immunoprecipitated from whole-cell lysates of transfected HEK293T cells with anti-FLAG antibodies and assayed in the presence or absence of NAD (1 mM). (D) Purified mitochondria from HEK293T cells transfected with hSIRT proteins were lysed and FLAG-tagged proteins were immunoprecipitated and analyzed for in vitro deacetylase activity. (E) Mitochondria were isolated from HEK293T cells transfected with hSIRT3–FLAG (WT), hSIRT3N229A–FLAG, hSIRT3H248Y–FLAG, or control vector (pFLAG). Lysates were prepared and tested for deacetylase activity. (F) Mitochondria were analyzed for wild-type and mutant hSIRT3 by Western blotting. Two hSIRT3–FLAG specific forms (asterisks) were detected.
hSIRT3 mediates NAD-dependent deacetylase activity in the mitochondria
In transfected mammalian cells, hSIRT1, 2, 3, and 5 exhibit bona fide NAD-dependent deacetylase activity, while hSIRT4, 6, and 7 do not (unpublished data). To determine which hSIRT protein contributed to the mitochondrial activity, we transfected expression vectors for hSIRT1, 2, and 3 (epitope tagged with FLAG at the COOH terminus) or a control vector into HEK293T cells. Cells were harvested, whole-cell and mitochondrial lysates were prepared, and hSIRT proteins were immunoprecipitated with anti-FLAG antibodies. In whole-cell lysates, all three proteins were detected by Western blotting, including two forms of hSIRT3: a 44-kD product of the expected size given the cDNA sequence (predicted molecular mass, 43.6 kD) and a smaller, 28-kD product (Fig. 1 B, top). However, in the mitochondrial lysates, only hSIRT3 (28-kD product) was detected (Fig. 1 B, bottom). All three hSIRTs showed robust NAD-dependent enzymatic activity after immunoprecipitation from whole-cell lysates (Fig. 1 C), but only anti-FLAG immunoprecipitates from cells transfected with hSIRT3 showed mitochondrial deacetylase activity (Fig. 1 D). These results suggest that hSIRT3 mediates NAD-dependent deacetylase activity specifically within mitochondria.
Transfection of an expression vector for hSIRT3 increased the NAD-dependent HDAC activity of mitochondrial lysates in comparison to cells transfected with the empty vector (Fig. 1 E). Mitochondrial lysates from hSIRT3 and control cells had similar sensitivities to nicotinamide and TSA (unpublished data). In contrast, transfection of two catalytically inactive mutants, hSIRT3-N229A and hSIRT3-H248Y, did not increase basal HDAC activity of mitochondrial lysates (Fig. 1 E). Both mutants were shown to have lost all HDAC activity after immunoprecipitation in separate experiments (unpublished data). Importantly, both mutants were efficiently targeted to mitochondria, were equally well expressed after transfection, and were processed to the smaller 28-kD product produced as wild-type hSIRT3 (Fig. 1 F). These observations are consistent with the selective targeting of hSIRT3 to mitochondria.
Visualization of hSIRT3 in mitochondria
To further determine the subcellular localization of hSIRT3 in cells, we generated a GFP fusion protein (hSIRT3–GFP). Confocal laser scanning microscopy of HeLa cells transfected with hSIRT3–GFP revealed that it localized exclusively to cytoplasmic substructures. Costaining with a mitochondria-specific dye (MitoTracker red) showed total overlapping of the two signals (Fig. 2 A), indicating that hSIRT3 exclusively localizes to mitochondria. Similar results were obtained with an epitope-tagged (FLAG) hSIRT3 recombinant protein using indirect immunofluorescence (unpublished data).
Figure 2.

Subcellular localization of hSIRT3 in mitochondria. (A) hSIRT3–GFP was transfected into HeLa cells grown on coverslips. Cells were stained with the mitochondrial marker MitoTracker, embedded, and analyzed by confocal laser scanning microscopy. (Left) Fluorescence from the hSIRT3–GFP fusion protein. (Middle) Fluorescence from the MitoTracker–stained mitochondria in the same focal plane. (Right) Merged image showing complete overlap of the two staining patterns. (B) HEK293T cells transfected with hSIRT3–FLAG were homogenized and fractionated by differential centrifugation. Equal amounts (30 μg) of heavy membranes (HM), light membranes (LM) and cytosolic proteins (S-100) fraction were analyzed by immunoblotting. hSIRT3–FLAG was revealed by detection with monoclonal M2 anti-FLAG antibodies. Two hSIRT3–FLAG specific forms (asterisks) were detected. Nitrocellulose membranes were stripped and reprobed with antibodies against cytochrome c (cyt c) and Hsp90α. (C) Mitochondria were prepared from HEK293T cells and lysates were analyzed by Western blotting with a polyclonal rabbit hSIRT3 antiserum or preimmune serum obtained from the same rabbit. (D) hSIRT3 was immunoprecipitated from HEK293T cells with hSIRT3 antiserum (0.35 mg/ml), preimmune serum (0.35 mg/ml), or protein G Sepharose. Equal amounts of immunoprecipitate were analyzed for in vitro deacetylase activity in the absence (−) or presence (+) of NAD.
This observation was further verified in cell fractionation experiments with hSIRT3-FLAG–transfected cells. Equal amounts of protein from each subcellular fraction were subjected to SDS-PAGE and immunoblotting with an anti-FLAG antibody. hSIRT3-FLAG and cytochrome c were detected only in the heavy membrane fraction representing mitochondria; two FLAG-reactive bands of ∼44 and 28 kD were detected in the mitochondrial fraction (Fig. 2 B). Immunoblotting of subfractions prepared from untransfected cells confirmed that both bands were specific for hSIRT3-FLAG (unpublished data).
hSIRT3 is a mitochondrial protein with NAD-dependent deacetylase activity
The subcellular localization of endogenous hSIRT3 was examined with a specific antiserum against a peptide corresponding to the last 15 amino acids of hSIRT3 (N-DLVQRETGKLDGPDK-C). This antiserum recognized two peptides (∼44 and ∼28 kD) in the mitochondrial fractions, whereas the preimmune antiserum obtained from the same rabbit was unreactive to these proteins (Fig. 2 C). These two bands corresponded in size to the fragments detected after transfection of the FLAG-tagged hSIRT3. Immunoprecipitation of the mitochondrial fraction with this antiserum showed the presence of a specific NAD-dependent deacetylase activity that was not identified with the preimmune serum or with protein G Sepharose alone (Fig. 2 D). These experiments demonstrate that endogenous hSIRT3 is located in the mitochondria and is associated with NAD-dependent deacetylase activity in that compartment.
The NH2 terminus of hSIRT3 is required for mitochondrial import
Mitochondrial targeting signals frequently contain an amphipathic α-helix and tend to contain positively charged hydrophobic and hydroxylated amino acids (Roise et al., 1986, 1988; von Heijne et al., 1989; Abe et al., 2000). Secondary structure predictions of hSIRT3 revealed that an NH2-terminal peptide corresponding to residues 1–25 has a high probability of containing an amphipathic α-helix (Fig. 3 A, middle). When plotted as a helical wheel (Fig. 3 A, right), residues 4–21 showed a cluster of positively charged arginine residues on one side of the helix opposed by hydrophobic residues on the other side, a typical feature of mitochondrial presequences (for review see Pfanner and Geissler, 2001). To test the importance of this putative α-helix in hSIRT3 mitochondrial import, we deleted amino acids 1–25 from hSIRT3 and fused it to GFP (hSIRT3Δ1–25-GFP). Expression of this construct in HeLa cells showed pancellular distribution (Fig. 3 B). No significant colocalization between the fusion protein and MitoTracker–stained mitochondria could be observed. This localization was in sharp contrast to the subcellular localization observed after expression of full-length hSIRT3 fused to GFP (Fig. 2 A) and indicated that the NH2-terminal 25 amino acids of hSIRT3 are necessary for mitochondrial targeting.
Figure 3.

The NH 2 -terminal region of hSIRT3 is required for mitochondrial import. (A) Schematic diagram of hSIRT3. The orange box illustrates the region involved in mitochondrial targeting (left). Parts of the NH2-terminal region show a high probability of forming an amphiphatic α-helix (middle). A helical wheel plot of residues 4–21 reveals a cluster of basic amino acids (black) on one side of the putative helix (right). (B) HeLa cells grown on coverslips were transfected with hSIRT3Δ1–25–GFP for 36 h, stained with MitoTracker, and analyzed by confocal laser scanning microscopy. (left) GFP fluorescence emitted by the fusion protein (green). (Middle) MitoTracker signal (red). (Right) Merged image.
To further define the requirements for mitochondrial import of hSIRT3, we used cell-free mitochondrial in vitro import assays. Similar assays have been used to elucidate the import requirements of a variety of mitochondrial proteins. [35S]-labeled hSIRT3 or hSIRT3Δ1–25 was synthesized in rabbit reticulocyte lysates and incubated with isolated mammalian mitochondria at 30°C for 2, 5, or 15 min in the presence of succinate and ATP. Mitochondria were reisolated from the mixture by centrifugation, and cosedimenting proteins were analyzed by SDS-PAGE and autoradiography. We observed a time-dependent accumulation of hSIRT3, but not of hSIRT3Δ1–25, into mitochondria (Fig 4, A and B) . The import of hSIRT3 across the mitochondrial membrane was dependent on the mitochondrial transmembrane potential (ΔΨm), as import was inhibited by antimycin (8 μM), oligomycin, (20 μM), and valinomycin (1 μM). (Fig. 4 A, lane 4). Because ΔΨm is involved in the translocation of proteins across the inner mitochondrial membrane, this finding suggests that hSIRT3 is imported across the inner mitochondrial membrane (Martin et al., 1991). When the proteinase K digestion performed at the end of the import reaction was omitted, both hSIRT3 and hSIRT3Δ1–25 could bind to the outer surface of mitochondria in vitro, indicating that adhesion to mitochondria was not dependent on the NH2-terminal 25 amino acids of hSIRT3 (unpublished data). To exclude the possibility that proteins had aggregated and cosedimented nonspecifically, similar experiments were carried out in the absence of mitochondria, but no nonspecific sedimentation occurred (unpublished data).
Figure 4.

Mitochondrial import of hSIRT3 in vitro.(A) [35S]-labeled hSIRT3–FLAG or hSIRT3Δ1–25–FLAG synthesized in rabbit reticulocyte lysate was imported into isolated mammalian mitochondria at 30°C. Import in the absence of Δψm (lane 4), was arranged by adding valinomycin (1 μM), antimycin (8 μM), and oligomycin (20 μM) to mitochondria 5 min before the addition of proteins. Import was stopped after 2, 5, or 15 min by dissipating Δψm (addition of 1 μM valinomycin) and incubation at 0°C. Samples were treated with proteinase K to remove nonimported proteins. Imported proteins were visualized by autoradiography after reisolation of mitochondria and SDS-PAGE. (B) Quantitation of data from panel A by phosphorimaging. (C, left) Mutants were generated to assess the role of the amphipathic helix of hSIRT3 in mitochondrial import. [35S]-labeled hSIRT3 wild-type or mutants were imported into isolated mitochondria for 20 min at 30°C. Import was stopped as described above and nonimported proteins were removed by proteinase K treatment. Reisolated and washed mitochondria were lysed in SDS sample buffer and analyzed by SDS-PAGE. Standards representing 50% of the input used in the individual import reactions were loaded adjacent to each import sample. (Right) Import efficiency of individual hSIRT3 mutants was quantitated in relation to their standards by phosphorimaging. The import efficiency of hSIRT3 (WT) was set to 100%.
Next, we generated a series of point mutations in the first 25 amino acids. We used two strategies. First, we disrupted the α-helix by introducing prolines at positions 12 and 13. Second, we modified the charge of the amphipathic helix by replacing arginines with glycines or glutamines. The polar but uncharged glutamine residues were predicted to preserve the α-helical conformation while changing the amphipathic character of the α-helix. To study the import efficiency, wild-type hSIRT3 and mutants were synthesized in rabbit reticulocyte lysates in the presence of [35S]-methionine and assayed using the in vitro import assay described above. Mutation of R7 and R13 to glycines or glutamines resulted in a loss of mitochondrial import. In contrast, mutation of R17 and R21 reduced import by ∼50% (Fig. 4 C). Mutating all four arginines to glutamines or glycines reduced import efficiency even further. Disrupting the putative helical structure by introducing two prolines reduced mitochondrial import to about the same extent as the R7/13G mutation. These results demonstrate that the positively charged residues and the α-helical structure of region 1–25 are important for the import of hSIRT3 into mitochondria.
hSIRT3 is a mitochondrial matrix protein
To determine the exact localization of hSIRT3 in the mitochondria, we isolated mitochondria from HEK293T cells expressing hSIRT3-FLAG and incubated them in hypotonic MOPS-buffer. This treatment leads to the rupture of the outer mitochondrial membrane (mitoplast formation) and to the release of soluble proteins in the intermembrane space. Mitoplasts and mitochondria were reisolated by centrifugation and analyzed by Western blotting (Fig. 5 A). The ∼28-kD form of hSIRT3 was not affected by rupture of the outer mitochondrial membrane and subsequent proteinase K digestion (Fig. 5 A). To exclude the possibility that hSIRT3-FLAG had formed a protease-stable aggregate, mitochondria from cells transfected with hSIRT3-FLAG were lysed in 0.5% Triton X-100 and digested with proteinase K. Under these conditions, hSIRT3 was completely degraded (unpublished data). In this respect, hSIRT3 behaved similarly to the matrix protein Hsp60 (Cheng et al., 1990; Fig. 5 A). Confirmation of the rupture of the outer membrane by the hypotonic treatment was obtained by blotting against the intermembrane space protein cytochrome c. In contrast to hSIRT3, cytochrome c was lost after protease treatment of mitoplasts (Fig. 5 A). These results were consistent with three possible locations for hSIRT3: (a) the mitochondrial matrix; (b) peripherally attached to the inner side of the inner mitochondrial membrane; and (c) in the inner mitochondrial membrane.
Figure 5.

hSIRT3 is localized in the mitochondrial matrix. (A) Mitochondria were isolated from HEK293T cells transfected with hSIRT3–FLAG and treated with proteinase K to remove proteins bound to the outer mitochondrial surface. Mitochondrial preparations were divided, and one half was diluted with hypotonic buffer to create mitoplasts (MP), while the other half was maintained under isotonic conditions (M). After incubation (20 min at 0°C), mitochondria and mitoplasts were treated again with proteinase K and reisolated by centrifugation followed by Western blotting. Rupture of the outer mitochondrial membrane was confirmed by detection of endogenous intermembrane space protein cytochrome c (cyt c). Integrity of the inner mitochondrial membrane was determined with the matrix protein Hsp60 as a marker. hSIRT3-FLAG was detected using anti-FLAG M2 antibodies. (B) Mitochondria were isolated from HEK293T cells transfected with hSIRT3–FLAG and treated with proteinase K. The preparation was divided, and one half was resuspended in SDS sample buffer (Total, left lane). The other half of the preparation was resuspended in sodium carbonate (Na2CO3) buffer. The extract was centrifuged at 100,000 g at 4°C, and the mitochondrial membranes (Pellet, middle lane) were resuspended in SDS sample buffer. The supernatant containing the soluble and peripheral membrane proteins (Soluble, right lane ) was precipitated with TCA. Samples were analyzed by Western blotting. hSIRT3 was detected with anti–FLAG antibodies. Alkaline extraction was controlled by detection of the marker proteins COXIV and mtHsp70.
To differentiate between these possibilities, we extracted mitochondria with sodium carbonate, pH 11.5. This treatment releases soluble and peripheral membrane proteins into the supernatant, while integral membrane proteins sediment with the membranes in the pellet (Fujiki et al., 1982). The ∼28 kD form of hSIRT3 was found in the supernatant, indicating that it is either a soluble matrix protein or is peripherally attached to the inner face of the inner membrane (Fig. 5 A). The ∼44-kD form of hSIRT3 was detected mostly in the pellet, suggesting that it is associated with the inner mitochondrial membrane. As expected, the soluble matrix chaperonin mtHsp70 was detected in the supernatant, whereas the inner-membrane protein COXIV was associated with the membrane fraction (Fig. 5 B). These experiments indicate that the 28-kD form of hSIRT3 is a soluble matrix protein.
Proteolytic processing of hSIRT3
As discussed above, the majority of hSIRT3 is present in mitochondria as a truncated 28-kD protein. Because this form is reactive to the anti-FLAG antibody after transfection of a COOH-terminal FLAG fusion protein, we concluded that hSIRT3 is proteolytically cleaved at its NH2 terminus. Most mitochondrial proteins carrying NH2-terminal targeting signals are processed by matrix processing peptidase (MPP) after import into the mitochondrial matrix (Jensen and Johnson, 2001). Incubation of radiolabeled hSIRT3 with recombinant yeast MPP yielded a 28-kD cleavage product, undistinguishable in size from the product detected in vivo in mitochondria (Fig. 6 A). Cleavage of a fusion protein between subunit 9 of F0/F1-ATPase and DHFR (Su9-DHFR) by MPP in vitro resulted in the appearance of digestion products similar to what has been previously reported, confirming the specificity of the MPP enzyme preparation used (Geli, 1993). Based on the size of the processed hSIRT3 protein, we scanned the primary sequence of hSIRT3 for putative MPP recognition motifs. MPP specifically processes many mitochondrial precursor proteins but no consensus processing site has emerged. However, an arginine at –2 relative to the cleavage site and additional aromatic or hydrophobic residues in position 1 relative to the cleavage site appear to be necessary for cleavage (Hartl et al., 1989; Hendrick et al., 1989; Gavel and von Heijne, 1990). Several hSIRT3 mutants targeting arginines at positions 99, 100, 133, 135, 139, and 158 were constructed by site-directed mutagenesis and synthesized in rabbit reticulocyte lysates in the presence of [35S]-methionine. A mutant carrying two glycines substituted for arginines 99 and 100 was not cleaved by MPP in vitro (Fig. 6 B), whereas other mutants were unaffected (unpublished data). These results indicate that residues R99/100 are critical for the processing of hSIRT3 by MPP. Transfection of this construct into mammalian cells led to a partial inhibition of the processing of hSIRT3 into the 28-kD fragment and a new fragment of higher molecular weight was detected (unpublished data).
Figure 6.
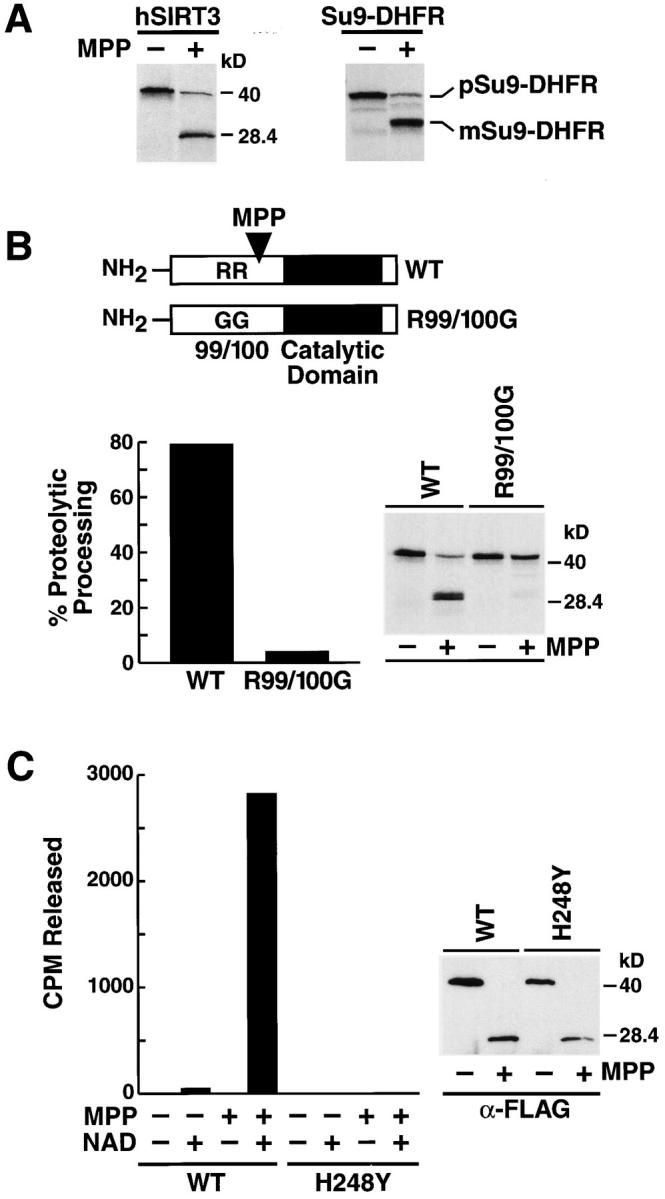
Proteolytic processing of hSIRT3 by MPP leads to enzymatic activation. (A) [35S]-labeled hSIRT3–FLAG (left) or pSu9–DHFR (right) was incubated with purified recombinant yeast MPP for 45 min at 27°C. Samples were analyzed by SDS-PAGE and autoradiography. m, mature form of pSu9–DHFR; p, precursor form. (B) [35S]-labeled hSIRT3–FLAG (WT) and hSIRT3R99/100G–FLAG (R99/1006) were incubated with MPP and analyzed as in A. (Left) Efficiency of proteolytic processing by recombinant yeast MPP was quantitated by phosphorimaging. (Right) Autoradiography of the same experiment. (C) Unlabeled hSIRT3–FLAG (WT) or hSIRT3H248Y–FLAG (H2484) synthesized in vitro in rabbit reticulocyte lysates was incubated with recombinant yeast MPP for 45 min at 27°C. FLAG-tagged proteins were immunoprecipitated with anti-FLAG M2-agarose beads and analyzed for deacetylase activity in vitro with the H4 histone peptide assay in the presence or absence of NAD (1 mM; left). Western blot analysis of immunoprecipitates used in the deacetylase assay (right).
Catalytic activation of a latent hSIRT3 by MPP-mediated proteolytic processing
We had noted that the in vitro translated hSIRT3 protein was catalytically inactive in our in vitro deacetylase assay. Similarly, hSIRT3 expressed in Escherichia coli was not processed and was enzymatically inactive (unpublished data). This led us to test the hypothesis that proteolytic processing of hSIRT3 might lead to its catalytic activation. Unlabeled hSIRT3 was synthesized in vitro in rabbit reticulocyte lysates. Samples were split in half and one half was subjected to cleavage by recombinant MPP in vitro while the other half was incubated in the same buffer in the absence of MPP. hSIRT3 was immunoprecipitated and assayed for deacetylase activity in the presence or absence of NAD. Remarkably, the hSIRT3 processed by MPP showed NAD-dependent deacetylase activity, whereas the full-length uncleaved hSIRT3 remained inactive (Fig. 6 C). These results linked processing of hSIRT3 to the activation of its NAD-dependent deacetylase activity. To exclude the possibility that unspecific factors or MPP itself had caused the NAD-dependent deacetylase activity, we tested the catalytically inactive hSIRT3-H248Y mutant for deacetylase activity after incubation and cleavage with MPP. No NAD-dependent deacetylase activity was detected (Fig. 6 C, left and right). These results demonstrate that proteolytic processing of hSIRT3 by MPP leads to the activation of its latent enzymatic activity.
Discussion
The identification of a Sir2-related enzyme in the mammalian mitochondrion raises a number of interesting questions related to the NAD-dependent enzymatic activity associated with this family of enzymes and the pivotal role played by NAD in mitochondrial metabolism. In almost every respect, hSIRT3 behaves as a classical mitochondrial matrix protein. Its dependency on an NH2-terminal cleavable presequence has been reported for other mitochondrial matrix proteins (Roise et al., 1986, 1988; von Heijne et al., 1989; Abe et al., 2000). Mitochondrial targeting sequences are characterized by the presence of positively charged and hydrophobic residues (negative charged residues are very rare) (Roise et al., 1988) and tend to adopt a helical, frequently amphipathic, conformation. Mutational analysis of an amphipathic helix within the NH2 terminus of hSIRT3 showed that eliminating the positive charges by substituting arginines with glycines or glutamines led to a loss of mitochondrial import. Disrupting the putative helical conformation of the targeting signal by introducing proline residues within the helix also suppressed import despite the conservation of three charged residues. Thus, both the net charge and the helical nature of the targeting region are necessary for import of hSIRT3 into mitochondria. Whereas the NH2 terminus of hSIRT3 is critical for mitochondrial import, preliminary experiments indicate that a fusion protein consisting of the NH2-terminal 25 amino acids of hSIRT3 and GFP is not targeted to mitochondria. This result indicates that other domains of hSIRT3, most likely the first NH2-terminal 100 amino acids are involved in its mitochondrial import.
The hSIRT3 cDNA predicts a protein product of ∼43.6 kD; however, an antiserum specific for the COOH terminus of hSIRT3 detected a smaller form of 28 kD as the major form in cultured cells. A product of similar size was observed as the major protein after transfection of hSIRT3 with COOH-terminal FLAG tag. Both experiments are consistent with the deletion of a leader sequence at the NH2 terminus of hSIRT3. The observation that a deletion mutant of hSIRT3 lacking the first 25 amino acids was not targeted to the mitochondria and was also not processed to a smaller product suggests either that mitochondrial targeting is necessary for proteolytic processing or that targeting of the protease to hSIRT3 is dependent on its first 25 amino acids.
Most proteins with NH2-terminal mitochondrial targeting sequences are processed after import into the mitochondrial matrix by MPP (Arretz et al., 1991). MPP processed hSIRT3 in vitro to a new product similar in size to the 28-kD processed endogenous hSIRT3 protein, suggesting that MPP is responsible for hSIRT3 cleavage within the mitochondrial matrix. Mutational analysis of hSIRT3 revealed that arginines 99 and 100 are necessary for cleavage by MPP in vitro. The presence of critical arginine residues within the cleavage site recognized by MPP has been described previously (Ogishima et al., 1995; Song et al., 1996; Shimokata et al., 1997). Although these mutations completely suppressed cleavage by MPP in vitro, we observed residual cleavage of the same mutant (hSIRT3-R99/100G) in vivo after transfection. These results are likely to reflect the limiting digestion conditions imposed by our in vitro assay or, less likely, the presence of an alternative mitochondrial processing enzyme. In addition, transfection of hSIRT3/R99–100G into mammalian cell led to the appearance of novel cleavage products, suggesting additional processing events. Interestingly, 30% of mitochondrial precursor proteins processed by MPP are further processed by the mitochondrial intermediate peptidase (MIP) (Kalousek et al., 1988; Isaya et al., 1991). MIP cleavage removes an additional octapeptide from the NH2 terminus of the MPP-processed precursor. It is possible that cleavage by MIP or another mitochondrial protease is responsible for the processing of hSIRT3 when MPP processing has been inhibited. Finally, we noted in our alkaline fractionation experiments that the incompletely processed form of hSIRT3 was associated with the membrane fraction, indicating that entry of hSIRT3 into the matrix compartment is likely to represent a critical step in its proteolytic processing, consistent with the matrix localization of MPP.
We had initially observed that hSIRT3 expressed in E. coli or after in vitro translation systems was catalytically inactive. Remarkably, MPP cleavage of hSIRT3 synthesized in vitro resulted in its catalytic activation. This was not observed with a catalytically inactive hSIRT3-H248Y, although this mutant protein was cleaved to the same extent as wild-type hSIRT3. This observation indicates that enzymatic activation of hSIRT3 by proteolytic processing by MPP is not an artifact linked to the MPP preparation but is strictly dependent on the intrinsic enzymatic activity of hSIRT3. This control also indicates indirectly that enzymatic activity of hSIRT3 is not necessary for its proteolytic processing.
These observations suggest that hSIRT3 is synthesized as an inactive precursor within the cytoplasm, transported to the mitochondrial matrix, where it is proteolytically processed to activate its enzymatic potential. This model would allow the safe transfer of a latent enzyme and its selective activation when the proper destination in the mitochondrial matrix has been reached. While our in vitro evidence strongly supports this model, we have had difficulties evaluating the role of hSIRT3 cleavage on its enzymatic activity in vivo. When the mutant hSIRT3-R99/100G harboring the mutated MPP recognition site was transfected into cells, cleavage efficiency was reduced, but a certain amount of processed hSIRT3 protein could still be immunoprecipitated and exhibited NAD-dependent deacetylase activity. Therefore, whether uncleaved full-length hSIRT3 can exert NAD-dependent deacetylation in vivo remains an unanswered question. However, our experimental evidence indicates that the processed form of hSIRT3, either endogenous or after transfection, is indeed enzymatically active.
Although we have demonstrated that hSIRT3 can deacetylate a histone H4 peptide, mitochondria lack histone proteins. Therefore, other nonhistone mitochondrial proteins are likely to be substrate(s) of hSIRT3. Two recent reports have described the specific deacetylation of the transcription factor p53 by another human Sir2 homologue protein, hSIRT1 (Luo et al., 2001; Vaziri et al., 2001). We have also obtained evidence that a third human Sir2 protein, hSIRT2, is predominantly cytoplasmic (unpublished data). These observations are in agreement with previous reports that several mammalian Sir2 homologues are found in nonnuclear subcellular localizations (Zemzoumi et al., 1998; Afshar and Murnane, 1999; Yang et al., 2000; Perrod et al., 2001). As an alternative, hSIRT3 could also target a nonprotein substrate for deacetylation.
The selective targeting of a mammalian Sir2-like protein to mitochondria is intriguing because mitochondria represent the bioenergetic and metabolic centers of eukaryotic cells. It had been speculated that the phylogenetically conserved family of Sir2 proteins is involved in sensing cellular energy and redox states (Smith et al., 2000). Given its mitochondrial matrix localization, hSIRT3 might differ from other Sir2 proteins in its sensitivity to metabolic activity. In contrast to the cytoplasm where NAD levels can change in response to ATP abundance, the mitochondrial matrix content of NAD is believed to be stable and not subject to changes caused by varying ATP levels (Vinogradov et al., 1972; Devin et al., 1997; Tischler et al., 1977; Di Lisa et al., 2001). The presence of stable NAD levels might ensure the constitutive activity of hSIRT3 in the mitochondrial matrix leading to the constitutive deacetylation of one or several mitochondrial proteins.
In contrast to class I and II HDACs, the deacetylation reaction catalyzed by Sir2-like proteins does not lead to the production of acetate. Rather, Sir2-like enzymes catalyze a unique reaction in which the cleavage of NAD and the deacetylation of substrate are coupled with the formation of O-acetyl-ADP-ribose, a novel metabolite (Tanner et al., 2000; Borra et al., 2002; Jackson and Denu, 2002). This metabolite has intrinsic biological activity and causes a delay/block in oocyte maturation (Borra et al., 2002). These observations imply the existence of cellular enzymes that can efficiently utilize O-acetyl-ADP-ribose. We will explore the possibility that hSIRT3 functions as an NAD-dependent sensor and controls a variety of metabolic activities through the formation of O-acetyl-ADP-ribose.
In addition, certain conditions leading to an abrupt decrease in mitochondrial NAD might shut off or severely decrease the activity of hSIRT3, leading to a relative hyperacetylation of its substrates and to a decrease in O-acetyl-ADP-ribose production. These conditions include the opening of the mitochondrial permeability transition pore (mtPTP), a high-conductance channel within the inner mitochondrial membrane. Adding calcium to isolated mitochondria leads to a rapid depletion of mitochondrial NAD, most likely due to the opening of mtPTP (Vinogradov et al., 1972). NAD released from the matrix is hydrolyzed by NAD glycohydrolases (NADases) in the intermembrane space. Hydrolysis of NAD by NADases leads to the formation of ADP-ribose and nicotinamide, itself an inhibitor of hSIRT3 enzymatic activity. Reactive oxygen species (ROS) can also trigger mtPTP opening and NADase stimulation (Bernardi, 1999; Bernardi et al., 1999; Ziegler et al., 1997). Interestingly, as we show in this article, disruption of Δψm inhibits mitochondrial import of hSIRT3, thereby reducing its mitochondrial content. Therefore, mtPTP opening caused by apoptotic stimuli, ROS or calcium elevation is likely to inhibit hSIRT3 function by several independent mechanisms including matrix NAD depletion, increased NAD hydrolysis, formation of nicotinamide and inhibition of hSIRT3 import into the matrix. According to this model, constitutive hSIRT3 activity could play an important role in protection against apoptosis, and its inhibition might lead to increased acetylation of factors directly involved in apoptotic pathways.
HSIRT3 could also play a pathogenic role in cancer. The gene encoding hSIRT3 maps to chromosome 11p15.5, a region close to the telomere subjected to genomic imprinting. This region contains a major yet unidentified tumor suppressor gene (Henry et al., 1991; Weksberg et al., 1993; for review see Feinberg, 2000) and a locus associated with the Beckwith-Wiedemann syndrome (BWS), which causes prenatal overgrowth and predisposition to cancer (Lee et al., 1999).
The exact function of hSIRT3 remains to be elucidated. However, the localization of this enzyme to the mitochondrial matrix already gives important clues to its potential substrates and biological functions. Future experiments will address these important questions.
Materials and methods
Plasmid construction
Plasmids expressing hSIRT3 were constructed by PCR amplification of the hSIRT3 coding sequence using primers containing EcoRI sites and pCR2.1-SIRT3 as a template. The SIRT3 sequence was PCR amplified from human spleen Marathon cDNA library (CLONTECH Laboratories, Inc.) and cloned into pCR2.1 (Invitrogen). Amplified sequences were digested with EcoRI and cloned into a modified pcDNA3.1 + vector (Invitrogen) to yield hSIRT3 with a COOH-terminal FLAG tag. hSIRT3Δ1–25–FLAG was constructed by using modified NH2-terminal PCR primers to introduce EcoRI sites and a methionine start codon before amino acid 26 of the wild-type protein. Site-directed mutagenesis (QuikChange Mutagenesis Kit; Stratagene) was used to construct hSIRT3N229A-FLAG, hSIRT3H248Y-FLAG, hSIRT3R7/13G-FLAG, hSIRT3R17/21G-FLAG, hSIRT3R7/13/17/21G-FLAG, hSIRT3R7/13Q-FLAG, hSIRT3R17/21Q-FLAG, hSIRT3R7/13/17/21Q-FLAG, hSIRT3L12P/R13P-FLAG, and hSIRT3R99/100G-FLAG. All constructs were verified by DNA sequencing. pSu9-DHFR was provided by J. Brix and N. Pfanner (Institut fuer Biochemie und Molekularbiologie, Freiburg, Germany).
GFP fusion constructs
To generate fusion proteins of GFP with wild-type hSIRT3 or with amino acids 26–399 of hSIRT3, corresponding coding sequences were amplified by PCR and cloned into pEGFP-N1 (CLONTECH Laboratories, Inc.).
Cell culture and transfection
HEK293T and HeLa cells were cultured in DME supplemented with 10% FCS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin and grown in 5% CO2 at 37°C. Calcium phosphate transfection was used to transfect HEK293T cells (Chen and Okayama, 1987). HeLa cells were transfected with Lipofectamine (Life Technologies).
Immunoblot analysis
Antibodies used for immunoblotting included anti-mtHsp70 (Clone JG1; Affinity Bioreagents), anti-Hsp60 (Clone 4B9/89; Affinity Bioreagents), anti-Hsp90α (StressGen), anti–cytochrome c oxidase subunit IV (Clone 20E8–C12; Molecular Probes), anti-FLAG M2 (Sigma-Aldrich), and anti–cytochrome c (clone 7H8.2C12; Pharmingen). hSIRT3 antisera were raised in rabbits against a COOH-terminal peptide (H2N-DLVQRETGKLDGPDK-COOH). Western blots were revealed with enhanced chemiluminescence (Amersham Biosciences). Membranes were either nitrocellulose (Hybond ECL; Amersham Biosciences) or polyvinylidene fluoride (Immun-Blot; Bio-Rad Laboratories).
Confocal microscopy
HeLa cells grown on coverslips were incubated for 45 min with 30 nM MitoTracker red (CMXRos; Molecular Probes) in DME at 37°C, transferred to fresh DME, and were incubated for 60 min. Cells on coverslips were rinsed in PBS, fixed in 3.7% formaldehyde/PBS for 30 min, washed again in PBS, and mounted. Images were acquired on a BioRad Radiance 2000 laser scanning microscope equipped with an Olympus B×60 microscope and an Olympus PlanApo 60×/1.40 oil objective. Excitation laser line was 488 nm for EGFP and 578 nm for MitoTracker.
Preparation of subcellular fractions
Subcellular fractionation was performed as described with minor modifications (Yang et al., 1997; Condorelli et al., 2001). All steps were performed at 4°C. In brief, cells were homogenized in ice-cold buffer A (250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiotreithol, 0.1 mM phenylmethylsulfonyl fluoride, 20 mM Hepes-KOH, pH 7.5) and homogenized in a Dounce homogenizer (Wheaton). Homogenization was checked by phase-contrast microscopy. The homogenate was centrifuged twice at 800 g to remove nuclei and unbroken cells. Mitochondria were sedimented by centrifugation at 7,000 g for 15 min, washed twice with buffer A, and resuspended in TXIP-1 buffer (1% Triton X-100 [vol/vol], 150 mM NaCl, 0.5 mM EDTA, 50 mM Tris-HCl, pH 7.4) supplemented with protease inhibitors. Postmitochondrial supernatants were fractionated by ultracentrifugation at 100,000 g for 30 min. The supernatant constituting the cytosolic S-100 fraction was removed, and the pellet was resuspended in TXIP-1 buffer. Protein concentrations of the fractions were determined (DC Protein Assay; Bio-Rad Laboratories) and equal amounts of each fraction were separated by sodium dodecylsufate–polyacrylamide gel electrophoresis (SDS-PAGE) and blotted to nitrocellulose membranes.
Isolation of mitochondria from mammalian cells
Mitochondria were isolated by differential centrifugation as described (Yang et al., 1997). After several washes in SEM buffer (250 mM sucrose, 1 mM EDTA, 10 mM MOPS–KOH, pH 7.2), mitochondria were resuspended in SEM buffer. Mitochondria were further purified by layering a crude mitochondrial fraction on a discontinuous sucrose gradient (1–1.5 M) in T10E1 buffer (1 mM EDTA, 10 mM Tris-HCl, pH 7.5). After centrifugation for 20 min at 60,000 g at 4°C, mitochondria were recovered from the 1.0 M/1.5 M interface, carefully adjusted to 250 mM sucrose, and washed twice in SEM buffer.
Immunoprecipitation
Cells or isolated mitochondria were lysed in ice-cold TXIP-1 buffer containing either PMSF or protease inhibitor cocktail (Roche). Lysates were centrifuged at 16,000 g for 5 min at 4°C, and anti-FLAG monoclonal M2 antibody covalently coupled to agarose was added. Samples were incubated at 4°C for 12 h, centrifuged, and washed four times in TXIP-1 buffer. For the deacetylation assays, the fourth wash was carried out in SIRT deacetylase buffer (4 mM MgCl2, 0.2 mM dithiothreitol, 50 mM Tris-HCl, pH 9.0).
Import of radiolabeled proteins into isolated mitochondria
Proteins were imported into isolated mitochondria as previously reported (Wiedemann et al., 2001). Proteins were synthesized in the presence of [35S]-methionine by coupled transcription–translation in rabbit reticulocyte lysate (Promega; Pelham and Jackson, 1976). In vitro translation reactions were centrifuged at 108,000 g, for 15 min at 2°C and adjusted to 250 mM sucrose. Import reactions contained 5% (vol/vol) reticulocyte lysate in import buffer (3% [wt/vol] fatty acid–free BSA, 250 mM sucrose, 80 mM KCl, 5 mM MgCl2, 2 mM KH2PO4, 5 mM l-methionine, 10 mM 3–[N–morpholino]propanesulfonic acid–KOH, pH 7.2). In each import reaction, 50 μg of freshly isolated mammalian mitochondria was mixed with radiolabeled proteins and incubated at 30°C. ATP (2 mM) and sodium succinate (10 mM) were added to maintain coupling of isolated mitochondria. Import was stopped by adding valinomycin (1 μM) and placing the mixture on ice. Where indicated, samples were treated with proteinase K (50 μg/ml) for 10 min on ice. Protease treatment was stopped by adding 2 mM PMSF. Mitochondria were reisolated by centrifugation at 10,000 g for 5 min at 4°C, washed in SEM buffer, and recentrifuged as above. Mitochondrial pellets were resuspended in SDS sample buffer containing DTT and heated to 95°C for 5 min. Samples were subjected to SDS-PAGE. Dried gels were exposed to Biomax MR film (Kodak) at –70°C and analyzed on a Fuji FUJIX BAS 1000 phosphorimager. Where indicated, mitochondrial transmembrane potential was disrupted by blocking complex III of the respiratory chain (antimycin, 8 μM), F0/F1-ATPase (oligomycin, 20 μM), and potassium flux (valinomycin, 1 μM).
Swelling experiments were performed according to published protocols (Ryan et al., 2001). Mitochondria were isolated from hSIRT3-FLAG transfected HEK293T cells, washed, and treated with proteinase K (150 μg/ml) to remove nonimported protein. Mitochondria were reisolated by centrifugation at 10,000 g for 5 min, washed with SEM buffer, and recentrifuged. Mitochondrial pellets were resuspended in SM buffer (250 mM sucrose, 10 mM MOPS-KOH, pH 7.2) diluted tenfold into M buffer (10 mM MOPS-KOH, pH 7.2) to induce swelling, and incubated on ice for 15 min. Mitoplasts and nonswollen mitochondria were treated with proteinase K (150 μg/ml) for 10 min at 0°C. Protease digestion was stopped by adding 2 mM PMSF, and mitoplasts and mitochondria were reisolated by centrifugation, washed, and lysed in sample buffer. Samples were separated by SDS-PAGE and blotted onto polyvinylidene fluoride membrane.
Fractionation of mitochondrial proteins by alkaline treatment
Mitochondrial proteins were fractionated as described (Fujiki et al., 1982; Honlinger et al., 1996). In brief, washed mitochondrial pellets were resuspended in freshly prepared 0.1 M sodium carbonate, pH 11.5, and incubated at 0°C for 30 min. Mitochondrial membranes were sedimented by ultracentrifugation at 100,000 g for 30 min at 4°C. The pellet was resuspended in SDS sample buffer, and proteins in the supernatant were concentrated by trichloracetate precipitation and resuspended in sample buffer.
In vitro deacetylase assay
Deacetylase assays were performed in 100 μl of SIRT deacetylase buffer (4 mM MgCl2, 0.2 mM dithiothreitol, 50 mM Tris-HCl, pH 9.0) containing immunoprecipitated proteins or mitochondrial lysates and a peptide corresponding to the first 23 amino acids of histone 4 chemically acetylated in vitro. (Emiliani et al., 1998). The histone peptide was acetylated in vitro by overnight incubation with [3H]-acetate (5 mCi, 5.3 Ci/mmol; NEN) in 500 μl of EtOH in the presence of 0.24 M benzotriazole-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate (Sigma-Aldrich) and 0.2 M triethylamine followed by reverse-phase HPLC purification. Where indicated, 1 mM NAD, 5 mM nicotinamide, or 400 nM TSA (WAKO) was added. Deacetylation reactions were stopped after 2 h of incubation at room temperature by adding 25 μl of stop solution (0.1 M HCl, 0.16 M acetic acid). Released acetate was extracted into 500 μl ethyl acetate, and samples were vigorously shaken for 15 min. After centrifugation for 5 min, 400 μl of the ethyl acetate fraction was mixed with 5 ml of scintillation fluid (Packard), and the released radioactivity was measured with a liquid scintillation counter.
MPP cleavage assay
Purified recombinant yeast MPP (Geli, 1993) was obtained from G. Isaya (Mayo Clinic Foundation, Rochester, MN). Cleavage of radiolabeled in vitro–translated proteins was carried out in reaction buffer (1 mM dithiothreitol, 1 mM MnCl2, 10 mM Hepes-KOH, pH 7.4). Purified MPP or reaction buffer was added to each sample followed by incubation at 27°C for 45 min. Reactions were stopped by adding SDS sample buffer and boiling at 95°C for 5 min. Samples were separated by SDS-PAGE and analyzed by phosphorimaging.
Acknowledgments
We thank G. Isaya for providing MPP and helpful comments, and J. Brix and N. Pfanner for providing the Su9–DHFR plasmid and technical advice. We thank John C.W. Carroll and Stephen Gonzales for graphics, Heather Gravois for manuscript preparation, and Stephen Ordway and Gary Howard for editorial assistance.
This work was supported by unrestricted funds from the Gladstone Institutes (to E. Verdin) and by a grant from the Sandler Family Foundation (to E. Verdin). B. Schwer was supported by scholarships from the German Academic Exchange Service and the Biomedical Science Exchange Program.
Footnotes
Abbreviations used in this paper: BWS, Beckwith-Wiedemann syndrome; HDAC, histone deacetylase; MIP, mitochondrial intermediate peptidase; MPP, matrix processing peptidase; mtPTP, mitochondrial permeability transition pore; NAD, nicotinamide adenine dinucleotide; NADase, NAD glycohydrolase; Sir, silent information regulator; TSA, trichostatin A.
References
- Abe, Y., T. Shodai, T. Muto, K. Mihara, H. Torii, S. Nishikawa, T. Endo, and D. Kohda. 2000. Structural basis of presequence recognition by the mitochondrial protein import receptor Tom20. Cell. 100:551–560. [DOI] [PubMed] [Google Scholar]
- Afshar, G., and J.P. Murnane. 1999. Characterization of a human gene with sequence homology to Saccharomyces cerevisiae SIR2. Gene. 234:161–168. [DOI] [PubMed] [Google Scholar]
- Arretz, M., H. Schneider, U. Wienhues, and W. Neupert. 1991. Processing of mitochondrial precursor proteins. Biomed. Biochim. Acta. 50:403–412. [PubMed] [Google Scholar]
- Bernardi, P. 1999. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 79:1127–1155. [DOI] [PubMed] [Google Scholar]
- Bernardi, P., L. Scorrano, R. Colonna, V. Petronilli, and F. Di Lisa. 1999. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur. J. Biochem. 264:687–701. [DOI] [PubMed] [Google Scholar]
- Borra, M.T., F.J. O'Neill, M.D. Jackson, B. Marshall, E. Verdin, K.R. Foltz, and J.M. Denu. 2002. Conserved enzymatic production and biological effect of O-acetyl ADP ribose by Sir2-like NAD+-dependent deacetylases. J. Biol. Chem. 277:12632–12641. [DOI] [PubMed] [Google Scholar]
- Chen, C., and H. Okayama. 1987. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7:2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, M.Y., F.U. Hartl, and A.L. Horwich. 1990. The mitochondrial chaperonin hsp60 is required for its own assembly. Nature. 348:455–458. [DOI] [PubMed] [Google Scholar]
- Condorelli, F., P. Salomoni, S. Cotteret, V. Cesi, S.M. Srinivasula, E.S. Alnemri, and B. Calabretta. 2001. Caspase cleavage enhances the apoptosis-inducing effects of BAD. Mol. Cell. Biol. 21:3025–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devin, A., B. Guerin, and M. Rigoulet. 1997. Cytosolic NAD+ content strictly depends on ATP concentration in isolated liver cells. FEBS Lett. 410:329–332. [DOI] [PubMed] [Google Scholar]
- Di Lisa, F., R. Menabo, M. Canton, M. Barile, and P. Bernardi. 2001. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 276:2571–2575. [DOI] [PubMed] [Google Scholar]
- Emiliani, S., W. Fischle, C. Van Lint, Y. Al-Abed, and E. Verdin. 1998. Characterization of a human RPD3 ortholog, HDAC3. Proc. Natl. Acad. Sci. USA. 95:2795–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg, A.P. 2000. DNA methylation, genomic imprinting and cancer. Curr. Top. Microbiol. Immunol. 249:87–99. [DOI] [PubMed] [Google Scholar]
- Finnin, M.S., J.R. Donigian, A. Cohen, V.M. Richon, R.A. Rifkind, P.A. Marks, R. Breslow, and N.P. Pavletich. 1999. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 401:188–193. [DOI] [PubMed] [Google Scholar]
- Frye, R.A. 1999. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 260:273–279. [DOI] [PubMed] [Google Scholar]
- Frye, R.A. 2000. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 273:793–798. [DOI] [PubMed] [Google Scholar]
- Fujiki, Y., A.L. Hubbard, S. Fowler, and P.B. Lazarow. 1982. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavel, Y., and G. von Heijne. 1990. Cleavage-site motifs in mitochondrial targeting peptides. Protein Eng. 4:33–37. [DOI] [PubMed] [Google Scholar]
- Geli, V. 1993. Functional reconstitution in Escherichia coli of the yeast mitochondrial matrix peptidase from its two inactive subunits. Proc. Natl. Acad. Sci. USA. 90:6247–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschling, D.E., O.M. Aparicio, B.L. Billington, and V.A. Zakian. 1990. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell. 63:751–762. [DOI] [PubMed] [Google Scholar]
- Guarente, L. 2000. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 14:1021–1026. [PubMed] [Google Scholar]
- Hartl, F.U., N. Pfanner, D.W. Nicholson, and W. Neupert. 1989. Mitochondrial protein import. Biochim. Biophys. Acta. 988:1–45. [DOI] [PubMed] [Google Scholar]
- Hendrick, J.P., P.E. Hodges, and L.E. Rosenberg. 1989. Survey of amino-terminal proteolytic cleavage sites in mitochondrial precursor proteins: leader peptides cleaved by two matrix proteases share a three-amino acid motif. Proc. Natl. Acad. Sci. USA. 86:4056–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, I., C. Bonaiti-Pellie, V. Chehensse, C. Beldjord, C. Schwartz, G. Utermann, and C. Junien. 1991. Uniparental paternal disomy in a genetic cancer-predisposing syndrome. Nature. 351:665–667. [DOI] [PubMed] [Google Scholar]
- Honlinger, A., U. Bomer, A. Alconada, C. Eckerskorn, F. Lottspeich, K. Dietmeier, and N. Pfanner. 1996. Tom7 modulates the dynamics of the mitochondrial outer membrane translocase and plays a pathway-related role in protein import. EMBO J. 15:2125–2137. [PMC free article] [PubMed] [Google Scholar]
- Imai, S., C.M. Armstrong, M. Kaeberlein, and L. Guarente. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD- dependent histone deacetylase. Nature. 403:795–800. [DOI] [PubMed] [Google Scholar]
- Isaya, G., F. Kalousek, W.A. Fenton, and L.E. Rosenberg. 1991. Cleavage of precursors by the mitochondrial processing peptidase requires a compatible mature protein or an intermediate octapeptide. J. Cell Biol. 113:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, M.D., and J.M. Denu. 2002. Structural identification of 2'- and 3′-O-acetyl-ADP ribose as novel metabolites derived from the Sir2 family of beta-NAD+-dependant histone/protein deacetylases. J. Biol. Chem. 277:18535–18547. [DOI] [PubMed] [Google Scholar]
- Jensen, R.E., and A.E. Johnson. 2001. Opening the door to mitochondrial protein import. Nat. Struct. Biol. 8:1008–1010. [DOI] [PubMed] [Google Scholar]
- Kaeberlein, M., M. McVey, and L. Guarente. 1999. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13:2570–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalousek, F., J.P. Hendrick, and L.E. Rosenberg. 1988. Two mitochondrial matrix proteases act sequentially in the processing of mammalian matrix enzymes. Proc. Natl. Acad. Sci. USA. 85:7536–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry, J., J.T. Slama, and R. Sternglanz. 2000. a. Role of NAD(+) in the deacetylase activity of the SIR2-like proteins. Biochem. Biophys. Res. Commun. 278:685–690. [DOI] [PubMed] [Google Scholar]
- Landry, J., A. Sutton, S.T. Tafrov, R.C. Heller, J. Stebbins, L. Pillus, and R. Sternglanz. 2000. b. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. USA. 97:5807–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, M.P., M.R. DeBaun, K. Mitsuya, H.L. Galonek, S. Brandenburg, M. Oshimura, and A.P. Feinberg. 1999. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. USA. 96:5203–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.J., P.A. Defossez, and L. Guarente. 2000. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 289:2126–2128. [DOI] [PubMed] [Google Scholar]
- Luo, J., A.Y. Nikolaev, S. Imai, D. Chen, F. Su, A. Shiloh, L. Guarente, and W. Gu. 2001. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 107:137–148. [DOI] [PubMed] [Google Scholar]
- Martin, J., K. Mahlke, and N. Pfanner. 1991. Role of an energized inner membrane in mitochondrial protein import. Delta psi drives the movement of presequences. J. Biol. Chem. 266:18051–18057. [PubMed] [Google Scholar]
- Martin, S.G., T. Laroche, N. Suka, M. Grunstein, and S.M. Gasser. 1999. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 97:621–633. [DOI] [PubMed] [Google Scholar]
- Muth, V., S. Nadaud, I. Grummt, and R. Voit. 2001. Acetylation of TAF(I)68, a subunit of TIF-IB/SL1, activates RNA polymerase I transcription. EMBO J. 20:1353–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogishima, T., T. Niidome, K. Shimokata, S. Kitada, and A. Ito. 1995. Analysis of elements in the substrate required for processing by mitochondrial processing peptidase. J. Biol. Chem. 270:30322–30326. [DOI] [PubMed] [Google Scholar]
- Pelham, H.R., and R.J. Jackson. 1976. An efficient mRNA-dependent translation system from reticulocyte lysates. Eur. J. Biochem. 67:247–256. [DOI] [PubMed] [Google Scholar]
- Perrod, S., M.M. Cockell, T. Laroche, H. Renauld, A.L. Ducrest, C. Bonnard, and S.M. Gasser. 2001. A cytosolic NAD-dependent deacetylase, Hst2p, can modulate nucleolar and telomeric silencing in yeast. EMBO J. 20:197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanner, N., and A. Geissler. 2001. Versatility of the mitochondrial protein import machinery. Nat. Rev. Mol. Cell Biol. 2:339–349. [DOI] [PubMed] [Google Scholar]
- Roise, D., S.J. Horvath, J.M. Tomich, J.H. Richards, and G. Schatz. 1986. A chemically synthesized pre-sequence of an imported mitochondrial protein can form an amphiphilic helix and perturb natural and artificial phospholipid bilayers. EMBO J. 5:1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roise, D., F. Theiler, S.J. Horvath, J.M. Tomich, J.H. Richards, D.S. Allison, and G. Schatz. 1988. Amphiphilicity is essential for mitochondrial presequence function. EMBO J. 7:649–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, M.T., W. Voos, and N. Pfanner. 2001. Assaying protein import into mitochondria. Mitochondria, Methods In Cell Biology. Vol. 65. L.A. Pon and E.A. Schon, editors. Academic Press, San Diego, CA. 189–216. [DOI] [PubMed]
- Shimokata, K., T. Nishio, M.C. Song, S. Kitada, T. Ogishima, and A. Ito. 1997. Substrate recognition by mitochondrial processing peptidase toward the malate dehydrogenase precursor. J. Biochem. 122:1019–1023. [DOI] [PubMed] [Google Scholar]
- Smith, J.S., C.B. Brachmann, I. Celic, M.A. Kenna, S. Muhammad, V.J. Starai, J.L. Avalos, J.C. Escalante-Semerena, C. Grubmeyer, C. Wolberger, and J.D. Boeke. 2000. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc. Natl. Acad. Sci. USA. 97:6658–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, M.C., K. Shimokata, S. Kitada, T. Ogishima, and A. Ito. 1996. Role of basic amino acids in the cleavage of synthetic peptide substrates by mitochondrial processing peptidase. J. Biochem. 120:1163–1166. [DOI] [PubMed] [Google Scholar]
- Sterner, D.E., and S.L. Berger. 2000. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64:435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner, K.G., J. Landry, R. Sternglanz, and J.M. Denu. 2000. Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. USA. 97:14178–14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny, J.C., and D. Moazed. 2001. Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: Evidence for acetyl transfer from substrate to an NAD breakdown product. Proc. Natl. Acad. Sci. USA. 98:415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler, M.E., D. Friedrichs, K. Coll, and J.R. Williamson. 1977. Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch. Biochem. Biophys. 184:222–236. [DOI] [PubMed] [Google Scholar]
- Tissenbaum, H.A., and L. Guarente. 2001. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 410:227–230. [DOI] [PubMed] [Google Scholar]
- Tsang, A.W., and J.C. Escalante-Semerena. 1998. CobB, a new member of the SIR2 family of eucaryotic regulatory proteins, is required to compensate for the lack of nicotinate mononucleotide:5,6-dimethylbenzimidazole phosphoribosyltransferase activity in cobT mutants during cobalamin biosynthesis in Salmonella typhimurium LT2. J. Biol. Chem. 273:31788–31794. [DOI] [PubMed] [Google Scholar]
- Vaziri, H., S.K. Dessain, E. Ng Eaton, S.I. Imai, R.A. Frye, T.K. Pandita, L. Guarente, and R.A. Weinberg. 2001. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 107:149–159. [DOI] [PubMed] [Google Scholar]
- Vinogradov, A., A. Scarpa, and B. Chance. 1972. Calcium and pyridine nucleotide interaction in mitochondrial membranes. Arch. Biochem. Biophys. 152:646–654. [DOI] [PubMed] [Google Scholar]
- von Heijne, G., J. Steppuhn, and R.G. Herrmann. 1989. Domain structure of mitochondrial and chloroplast targeting peptides. Eur. J. Biochem. 180:535–545. [DOI] [PubMed] [Google Scholar]
- Weksberg, R., I. Teshima, B.R. Williams, C.R. Greenberg, S.M. Pueschel, J.E. Chernos, S.B. Fowlow, E. Hoyme, I.J. Anderson, D.A. Whiteman, et al. 1993. Molecular characterization of cytogenetic alterations associated with the Beckwith-Wiedemann syndrome (BWS) phenotype refines the localization and suggests the gene for BWS is imprinted. Hum. Mol. Genet. 2:549–556. [DOI] [PubMed] [Google Scholar]
- Wiedemann, N., N. Pfanner, and M.T. Ryan. 2001. The three modules of ADP/ATP carrier cooperate in receptor recruitment and translocation into mitochondria. EMBO J. 20:951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J., X. Liu, K. Bhalla, C.N. Kim, A.M. Ibrado, J. Cai, T.I. Peng, D.P. Jones, and X. Wang. 1997. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 275:1129–1132. [DOI] [PubMed] [Google Scholar]
- Yang, Y.H., Y.H. Chen, C.Y. Zhang, M.A. Nimmakayalu, D.C. Ward, and S. Weissman. 2000. Cloning and characterization of two mouse genes with homology to the yeast Sir2 gene. Genomics. 69:355–369. [DOI] [PubMed] [Google Scholar]
- Zemzoumi, K., D. Sereno, C. Francois, E. Guilvard, J.L. Lemesre, and A. Ouaissi. 1998. Leishmania major: cell type dependent distribution of a 43 kDa antigen related to silent information regulatory-2 protein family. Biol. Cell. 90:239–245. [DOI] [PubMed] [Google Scholar]
- Ziegler, M., D. Jorcke, and M. Schweiger. 1997. Identification of bovine liver mitochondrial NAD+ glycohydrolase as ADP-ribosyl cyclase. Biochem. J. 326:401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]